

Background: ArgBP2 is a cytoskeletal adaptor protein down-regulated in tumors.
Results: ArgBP2 binds to α-actinin through a conserved protein domain. ArgBP2 expression is associated with cross-linked F-actin stress fibers.
Conclusion: ArgBP2 inhibits cell migration via its interaction with α-actinin, which links it to the actomyosin network; this is negatively regulated by PKA.
Significance: We propose a reason for reduced ArgBP2 expression in cancer metastasis.
Keywords: 14-3-3 Protein, Cell Migration, Cytoskeleton, Protein kinase A (PKA), Tumor Metastasis, Actin Stress Fibers, Actinin, ArgBP2
Abstract
Cell migration requires dynamic remodeling of the actomyosin network. We report here that an adapter protein, ArgBP2, is a component of α-actinin containing stress fibers and inhibits migration. ArgBP2 is undetectable in many commonly studied cancer-derived cell lines. COS-7 and HeLa cells express ArgBP2 (by Western analysis), but expression was detectable only in approximately half the cells by immunofluorescence. Short term clonal analysis demonstrated 0.2–0.3% of cells switch ArgBP2 expression (on or off) per cell division. ArgBP2 can have a fundamental impact on the actomyosin network: ArgBP2 positive COS-7 cells, for example, are clearly distinguishable by their denser actomyosin (stress fiber) network. ArgBP2γ binding to α-actinin appears to underlie its ability to localize to stress fibers and decrease cell migration. We map a small α-actinin binding region in ArgBP2 (residues 192–228) that is essential for these effects. Protein kinase A phosphorylation of ArgBP2γ at neighboring Ser-259 and consequent 14-3-3 binding blocks its interaction with α-actinin. ArgBP2 is known to be down-regulated in some aggressively metastatic cancers. Our work provides a biochemical explanation for the anti-migratory effect of ArgBP2.
Introduction
The actomyosin network and associated focal adhesions play important roles in cell adhesion, polarization, and migration (1). The misregulation of proteins involved in these processes are associated with cancer metastasis (2). Arg kinase-binding pprotein 2 (ArgBP2)2 is an adapter protein that can localize to both focal adhesions and actomyosin filaments (3) and has recently emerged as a potential tumor suppressor capable of blocking cancer metastasis (4).
ArgBP2 (also known as Sorbs2) was originally identified as an interactor of Arg kinase (3). Multiple splice isoforms are reported including ArgBP2α, ArgBP2β (3), nArgBP2 (5), ArgBP2γ (6), and ArgBP2δ (7). ArgBP2 mRNA is widely detected and particularly enriched in the pancreas, kidney, heart, and brain although absent in leukocytes (3). Vertebrates express two other ArgBP2 related adapters, known commonly as ponsin and vinexin (8). This family of proteins contain an N-terminal SoHo (Sorbin homology) domain, and three conserved SH3 domains in the C-terminal region (8). Although ArgBP2, vinexin, and ponsin are all focal adhesion proteins (9, 10), ArgBP2 is unique among this family in also localizing to the actomyosin filaments.
The three ArgBP2 SH3 domains are reported to have multiple partners. The Arg/c-Abl kinases bind via the first and third SH3 domains leading to tyrosine phosphorylation of ArgBP2 (3). The nArgBP2 isoform was shown to interact with SAPAP (synapse-associated Protein 90/postsynaptic density-95-associated protein), the focal adhesion protein, vinculin, and the cell junctional protein, Afadin (5). Other interaction partners include c-Cbl (11), Pyk2 (11), synaptojanin (12), dynamin (12), WAVE (12), PKB (6), PAK1 (6), and Palladin (13). ArgBP2 is also reported to interact robustly with the important actin cross-linking protein α-actinin (13), but the binding site is uncertain. The SoHo domain of ArgBP2, vinexin, and ponsin can bind to flotillin, an important membrane-bound lipid raft protein (14).
ArgBP2 localizes to Z-discs in cardiomyocytes (3) and to puncta along stress fibers (analogous to Z-discs (15)) in non-muscle cells (3, 12, 13). Ectopic expression of ArgBP2 in these cells resulted in an obvious reorganization of the actin filaments (3, 12) but the mechanism for this reorganization remains unclear. ArgBP2 interacts with c-Cbl (11), which can lead to Rac1 activation via Crk and DOCK180 (16, 17). Silencing of nArgBP2 expression reportedly affected focal adhesions and increased plasma membrane ruffles in mouse astrocytes (12). Loss of ArgBP2 is generally accompanied by increased cell migration, whereas ectopic expression of ArgBP2 in metastatic cell lines blocked migration (4, 18), suggesting that ArgBP2 is a potential tumor suppressor (19). However, the mechanism for this tumor suppression is unclear.
Here we describe a mosaic pattern of ArgBP2 expression in COS-7 and HeLa cells with only ∼50% of cells expressing detectable levels of ArgBP2. The mosaic expression is likely to be due to HDAC control of ArgBP2 expression recently reported (20). Cells with detectable ArgBP2 expression are readily discriminated by their denser actomyosin network (with ∼2.5-fold more α-actinin localized to it) compared with cells with undetectable levels of ArgBP2. A novel highly conserved region of ArgBP2 found in the N-terminal half of the protein is necessary and sufficient for its localization to the enlarged puncta along actin stress fibers. This domain was also required for ArgBP2 interaction with α-actinin. Moreover, this domain is not found in ponsin and vinexin and can account for the difference in localization observed. We show that ArgBP2γ can bind to 14-3-3 and PKA phosphorylation can enhance this interaction. The Ser-259 of ArgBP2γ is crucial for 14-3-3 binding. The increased 14-3-3 interaction blocks α-actinin association and moves ArgBP2γ off stress fibers. We show that the ArgBP2/α-actinin interaction is required to inhibit cell migration. Finally, we propose a model for the ArgBP2-mediated inhibition of cell migration by increased cross-linking of the actomyosin network.
EXPERIMENTAL PROCEDURES
Chemicals, Reagents, and Antibodies
The following antibodies were used: rabbit anti-ArgBP2 was raised against residues 1–303 (Genemed Synthesis Inc.). Rabbit anti-14-3-3 and anti-MLC2 were from Santa Cruz Biotechnology. Mouse anti-vinculin, M2-anti-FLAG, rabbit anti-FLAG, and anti-GFP were from Sigma. Rabbit anti-actinin, and anti-actin and mouse anti-actinin, and anti-actin were from Abcam. Rabbit anti-ArgBP2 was also from Proteintech. Secondary antibodies Alexa Fluor 488 anti-mouse, 546 anti-mouse, 488 anti-rabbit, and 546 anti-rabbit were from Invitrogen. Chemicals and reagents were as follows: forskolin, cytochalasin, and fibronectin were from Sigma. Lipofectamine 2000, neomycin, rhodamine phalloidin, and Alexa Fluor 633 phalloidin were from Invitrogen. The α-actinin siRNA HSS100130 (Invitrogen) was transfected with Lipofectamine (Invitrogen). Cells were analyzed after 24–36 h after siRNA addition.
Plasmid Constructions
ArgBP2γ was subcloned cloned by PCR from I.M.A.G.E. clone 5093566. Similarly ponsin was subcloned from IMAGE 3602997 and vinexin was cloned from IMAGE 6201273. Full-length GFP-α-actinin was a kind gift from Dr. Dong Jing Ming. The constructs were cloned into either pXJ-FLAG or pXJ-GFP (21).
Cell Culture and Transfection
COS-7, HeLa, and U2-OS cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum and 4.5 g/liter of glucose at 5% CO2. Transient transfections were conducted with Lipofectamine (Invitrogen) or TransIT (Mirrus) according to the manufacturer's instructions. Typically, COS-7 cells were grown on 96-mm culture dishes and transfected with 6 μg of plasmid DNA. Typically, HeLa cells or U2-OS lines were grown on 18 × 18-mm glass coverslips (Marienfeld) and transfected with 1 μg of plasmid DNA. U2-OS stable lines were obtained by cotransfecting U2-OS cells with GFP-tagged or GFP-BirA-tagged constructs and pXJ-41 (with neomycin marker) and selected with neomycin (1 mm) for 1 week to obtain clonal populations of cells. These were isolated and cultured further by passaging. To isolate clonal populations of COS-7 and HeLa cells, they were diluted 1:1000 and fixed at 1, 3, 6, and 9 days after plating on fibronectin-coated coverslips.
Immunofluorescence and Microscopy
Cells were plated on coverslips overnight (except for cell spreading assay) and fixed with 3% (w/v) paraformaldehyde in PBS for 20 min at room temperature. The cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min and then blocked with 10% goat serum, 0.1% Triton X-100 in PBS (T-PBS) for 10 min. The primary antibody (1:200) in T-PBS was applied to the coverslip and incubated for 3 h at room temperature. The cells were washed (3 × 5 min) in T-PBS. The secondary antibody (1:100 in T-PBS) and phalloidin (where appropriate) were applied to the coverslip, and incubated for 1 h at room temperature. The coverslips were then mounted onto glass slides with fluorsave (Merck) and visualized on an Olympus laser scanning microscope (FV-1000). All images were visualized with a ×60 objective lens unless otherwise stated. The images were processed using Adobe Photoshop CS4.
Immunoprecipitation and Western Blotting
Cell lysates (92-mm dish) were harvested in 400 μl of lysis buffer: 25 mm Hepes, 150 mm NaCl, 5 mm MgCl2, 0.5% (v/v) Triton X-100, 4% (v/v) glycerol and protease inhibitors mixture (Roche Applied Science). The lysate was centrifuged (13,000 × g for 10 min) and the supernatant incubated with 30 μl of anti-FLAG-Sepharose (Sigma) for 3 h at 4 °C. The beads were washed (three times 300 μl) and resuspended in SDS sample buffer: this was incubated at 80 °C for 10 min to release bound proteins. For in vitro assessment of ArgBP2 binding, Latrunculin-A (1 μm), Ca2+ (10 μm), or phalloidin (0.2 μg/ml) were added to clarified lysates and incubated for 30 min before immunoprecipitation. Proteins were separated by SDS-PAGE using 9% acrylamide gels, and transferred to PVDF membranes (Bio-Rad). Standard Western blots using HRP-conjugated second antibodies were visualized with SuperSignal West Pico (Pierce).
Cell Culture and Migration Assays
Coverslips (18 × 18 or 22 × 22 mm) were incubated with 10 μg/ml of fibronectin for 1 h. Forskolin (20 μm) was added for 30 min. COS-7 or U2-OS stable lines were plated on the coated coverslips and allowed to spread for 30–45 min before fixation. For monolayer migration assays cell lines were grown to 100% confluence in a 4-well Chamlide magnetic chamber (CM-S22-4). The 4-well rubber divider was removed to create a “wound.” The dish was imaged for 30 h by a spinning disc confocal system (Nikon Eclipse Ti with a Yokogawa CSU-22). The control cells and ArgBP2 expressing cells were imaged simultaneously. The area covered by the cells between t = 0 and 25 h was determined by ImageJ after manual outlining of the cell edge. The difference in the area was calculated for multiple wounds (n = 6), and subjected to t test in Prism.
Live Cell Imaging of Tagged Proteins
Cells were plated on glass. GFP fusion protein expressing cells were imaged with the Olympus Laser Scanning Confocal Microscope. Typically the images were acquired at 0.6–0.9% laser power (5.75 milliwatts, 488 nm) or 5–10% (0.86 milliwatts, 546 nm) with acquisition intervals of 15 s. For cell tracking experiments, U2-OS cell lines were freshly plated on glass with 10 μg/ml of fibronectin coating and allowed to attach for 2 h. Cells (∼20 per field) were imaged (Deltavision DIC ×40 objective) for 8 h after being verified as GFP positive. The cell migration data were analyzed and processed using Metamorph. Cells that underwent division were excluded. The nuclear position was mapped over a 5-h window and the resultant tracks were used to calculate distances and speed of migration (μm/min).
TIRF Imaging and Quantification
The TIRF assay was performed on a Deltavision OMX system equipped with a ×100 TIRF objective. U2-OS cells stably expressing GFP-ArgBP2 or GFP-ArgBP2-(S259A) and transfected with mCherry-lifeAct were plated on fibronectin (10 μg/ml) overnight with 2% FBS. Cells positive for lifeAct were selected (n = 13) and imaged for 15 min prior to forskolin addition (20 μm). The cells were then immediately imaged for a further 30 min. ArgBP2 signals were analyzed by ImageJ; a region of interest (ArgBP2 puncta) was chosen at random but excluding focal adhesions (example region shown in figures) and the intensity was measured over different time points. Values were input into Prism and a Student's t test was performed to test statistical significance.
Puncta Quantification
Fields of view with an ArgBP2 “positive” and ArgBP2 “negative” cells were obtained. The α-actinin channel was imported into ImageJ and background subtracted. Regions of the α-actinin staining along the stress fibers (as assayed by phalloidin on another channel) but without focal adhesion structures were cropped for ArgBP2 positive and negative cells. The signal intensity was measured for 9 cells and plotted. The cropped images were “thresholded” to obtain binary images for the puncta. A watershed algorithm was applied to separate objects that were joined by the threshold function. The object sizes were calculated and plotted.
Structured Illumination Microscopy (SIM) Imaging
The SIM assay was performed on a Deltavision OMX system. The U2-OS stably expressing GFP-ArgBP2 were fixed and stained with phalloidin. SIM imaging was done in collaboration with the Institute of Medical Biology Microscopy Unit as per the manufacturer's guidelines.
Peptide Binding Assay
Analysis of 14-3-3 binding to synthetic phosphopeptides has been previously described (22). Peptides were synthesized using standard chemistry on cellulose (PepSpots, Jerini Biotools, Germany). Recombinant biotinylated 14-3-3ζ (10 μg/ml) was incubated for 30 min at room temperature in binding/wash buffer (20 mm Hepes, pH 7.3, 137 mm NaCl, 5 mm KCl, 0.05% Tween 20).
F-actin Co-sedimentation Assay
Rabbit muscle actin (Cytoskeleon Inc.) was polymerized at 1 mg/ml in 5 mm Tris-HCl, pH 8.0, 0.2 mm CaCl2, 50 mm KCl, 2 mm MgCl2, 0.3 mm ATP, and 0.05 mm DTT. Polymerized actin was pelleted at 100,000 × g for 1 h, and then washed with the same buffer. The F-actin was resuspended and incubated (1 mg/ml) with or without 10 μg of purified GST-FLAG-α-actinin(CH1/2) for 30 min at room temperature. The pellet fraction (100,000 × g for 1 h) was washed, resuspended, and incubated in 100 μl with either 5 μg of HA-GST-ArgBP2(194–339) protein or 5 μg of HA-GST-ArgBP2(260–339) and incubated at room temperature for 30 min. The mixture was centrifuged at 100,000 × g for 1 h and 10% of the final supernatant or re-suspected pellet (in the same volume) was loaded into 9% SDS-PAGE gel for analysis.
RESULTS
Identification and Characterization of ArgBP2γ
In a prior proteomic analysis of affinity purified 14-3-3-binding proteins (23), IRSp53 and ArgBP2 were identified as 14-3-3ζ-binding proteins in a 60–70-kDa fraction. ArgBP2 localizes to actin stress fibers and adhesion complexes (3). Multiple kinases can either negatively or positively regulate the actomyosin network such as PAK1 (21), RhoA-ROCK (24), and Cdc42-MRCK (25). Because ArgBP2 is localized to these structures and may be regulated by phosphorylation mediated 14-3-3 binding, we wanted to test if ArgBP2 might play a role in regulating the actomyosin network.
Based on the exon map of the human ArgBP2 from the Ensembl database, ArgBP2 can be expressed as multiple isoforms ranging from 70 to 140 kDa. ArgBP2γ has a translational start site within exon 21, whereas ArgBP2α and ArgBP2β have start sites in exons 14 and 16, respectively (Fig. 1A). The nArgBP2 isoform contains a 526-amino acid insert encoded by exon 38 not present in other ArgBP2 isoforms (Fig. 1A). We assessed the abundance of various isoforms based on the presence or absence of exons 33, 34, 35, and 38 and found that ArgBP2γ and nArgBP2 represent the most abundant isoforms in the human EST database. We isolated a cDNA clone (IMAGE clone 5093566) encoding the ArgBP2γ isoform (573 amino acids with mobility of ∼70 kDa).
FIGURE 1.

ArgBP2 localizes to actin stress fibers. A, schematic exon map of ArgBP2α, ArgBP2β, nArgBP2, and ArgBP2γ. Exon numbers are as indicated. The sorbin homology and SH3 domains, and conserved regions denoted A, B, and C as described in this study are marked accordingly. B, Western analysis of ArgBP2 expression in various cell lines. FLAG-ArgBP2γ was immunoprecipitated and run on 9% SDS-PAGE, then stained with Coomassie Blue to determine its mobility (top of panel). ArgBP2γ (red arrow) and ArgBP2α/β (blue arrow) were detected in HeLa and COS-7, whereas MDCK cell express only ArgBP2α/β. Typical staining patterns of F-actin and ArgBP2 in COS-7 cells (C) and HeLa cells (D) with detectable (yellow plus) or undetectable (blue dot) ArgBP2 expression are shown. Cells that are ArgBP2 positive cells present with thicker actin stress fibers. ArgBP2 is localized in distinct puncta along the dorsal and ventral actin stress fibers in HeLa cells. The fluorescent signal was inverted (white to black) for greater clarity. E and F, control U2OS cell (E) and GFP-ArgBP2γ localization (F) in U2-OS imaged by SIM. Distribution of the GFP-tagged protein closely resembles that of endogenous ArgBP2 in HeLa cells. GFP-ArgBP2γ expressing cells are characterized by denser and more cross-linked actin stress fibers. The red box marks the region enlarged to highlight the increased bundling at the puncta. The fluorescent signal is inverted (white to black) for greater clarity.
Evolutionary conservation of protein sequences generally indicates functionally important domains. Aligning ArgBP2 sequences across vertebrate species (human, rat, pufferfish, and zebrafish), we observed a high conservation in three regions between the N-terminal SoHo domain and the three SH3 domains. These are designated as regions A, B, and C (Fig. 1A, sequence conservation for B and C shown in Fig. 6A). Region A is proline-rich sequence, whereas region C is basic and was described as a potential nuclear localization signal (3). It also overlaps with a 14-3-3 binding site (see later). Region “B” (previously undescribed) exhibits high evolutionary sequence conservation but exhibits no sequence homology to other proteins. Regions A–C are present in all ArgBP2 isoforms (Fig. 1A).
FIGURE 6.

ArgBP2γ region B interacts with α-actinin. A, the schematic illustrates the constructs used and amino acid sequence alignment of regions that are conserved (B in red and C in orange) comparing human, rat, and zebrafish ArgBP2. B, proteins encoding FLAG-tagged ArgBP2γ, ponsin, Vinexin, ArgBP2γ-ΔB, ArgBP2γ-ΔC, or ArgBP2γ-(S259A) as indicated were transfected into COS-7 cells. The 2% SDS-soluble cell lysate and 2% Triton X-100-soluble cell lysate were probed for either FLAG or α-actinin. The solubility of α-actinin was reduced only in the presence of ArgBP2γ and ArgBP2γ-ΔC. C, FLAG-tagged ArgBP2γ, ArgBP2γ-ΔB, ArgBP2γ-C, ArgBP2γ-(S259A), or ponsin were mixed with an equal volume of cell lysate containing GFP-α-actinin and subjected to anti-FLAG immunoprecipitation. GFP-α-actinin bound FLAG-ArgBP2γ only in the presence of domain B. Ponsin displayed poor binding to GFP-α-Actinin despite its ∼4-fold higher expression compared with ArgBP2γ. D, the schematic shows the domain structure of α-actinin. Various domains of α-actinin were tested for their ability to interact with endogenous ArgBP2; only CH1 appeared necessary to bind ArgBP2. E, FLAG-ArgBP2γ binding to α-actinin is enhanced by F-actin. The Triton X-100-soluble lysates were treated latrunculin A, phalloidin, or Ca2+ (10 μm) for 30 min prior to anti-FLAG immunoprecipitation (IP). F, the lysates from COS-7 cells expressing FLAG-ArgBP2, U2-OS cells transfected with either control siRNA or actinin siRNA were probed for actin and actinin levels. The actinin siRNA reduced the level of actinin by 70%. FLAG-ArgBP2γ containing lysates were mixed with those from control or α-actinin siRNA knockdown cells. The level of actin co-immunoprecipitated with ArgBP2γ was reduced when lysates contained less α-actinin. G, ArgBP2 was tested for F-actin co-sedimentation (see “Experimental Procedures”) with or without recombinant α-actinin (CH1 + 2). The top panel shows the blot for the GST-HA-ArgBP2 protein (as indicated) that includes the B domain, and the bottom panel shows recombinant GST-HA-ArgBP2 lacking domain B. The recombinant protein was clarified before incubation with buffer alone, with F-actin alone, or with F-actin + α-actinin(CH1 + 2) as indicated by the horizontal bar. Equal volumes of supernatant (S) or re-suspended pellet (P) fractions (100,000 × g, 1 h) were analyzed by SDS-PAGE and Western blotting.
We generated a polyclonal antibody against recombinant N-terminal portion of ArgBP2γ (present in all isoforms) and assessed the protein expression in a panel of commonly studied cell lines (Fig. 1B). Although COS-7 and HeLa cells expressed ArgBP2, it was undetectable in U2-OS, NIH3T3, and 293T cells. Based on their predicted sizes, the two bands detected in HeLa and COS-7 represent the ArgBP2α/β and ArgBP2γ isoforms. We analyzed the ArgBP2 cDNAs amplified from COS-7 and HeLa cells (data not shown) and found that ArgBP2γ was the dominant isoform in these cells, consistent with our Western analysis.
COS-7 cells do not express myosin IIa (26) and thus have a rudimentary actomyosin system (compared with other fibroblastic cells) based exclusively on myosin IIb. In COS-7 cells, endogenous ArgBP2 localized to both actin stress fibers and associated focal adhesions (Fig. 1C). This pattern is similar to that reported for ArgBP2α in PtK2 cells (3). However, not all cells expressed detectable ArgBP2 (details in next section). COS-7 cells with detectable ArgBP2 expression (yellow plus sign) consistently exhibited denser stress fibers than cells lacking (detectable) ArgBP2. Indeed, one can differentiate cells with ArgBP2 expression based on enhanced F-actin staining (Fig. 1C, right panel). HeLa cells (which express both myosin IIa and IIb) have a well developed actomyosin system and, in these cells, ArgBP2 was localized to distinct F-actin-rich puncta along actin stress fibers (Fig. 1D). The localization of GFP-ArgBP2γ in U2-OS was similar to that of endogenous ArgBP2 in HeLa (Fig. 1e). Furthermore, ectopic ArgBP2 expression correlated with an increased size of these actin puncta as imaged by super resolution/SIM. The increased puncta size correlated with an actin network that appeared more cross-linked.
Plasticity of ArgBP2 Expression
Remarkably, ArgBP2 displayed a mosaic pattern of expression in both COS-7 and HeLa cells (Figs. 1, C and d, and 2A). Many COS-7 cells lacked detectable ArgBP2 expression, whereas ArgBP2 positive cells (denoted by yellow or red dots in Fig. 2A) were easily visualized. The lack of expression was neither related to the cell cycle stage nor the expression level of large T antigen (data not shown). To investigate this further, we plated COS-7 and HeLa cells at a very low density that resulted in 10–20 well separated colonies per coverslip. After 5–6 cell doublings, the colonies were characterized for ArgBP2 expression. The ArgBP2-positive colonies (those with detectable ArgBP2 expression) exhibited more compact morphology and less dispersed cells compared with ArgBP2-negative colonies (illustrated in Fig. 2B). However, beyond 6 doublings, we started to observe that many colonies lacking ArgBP2 expression contained a few cells with detectable ArgBP2 and vice versa. This makes it impossible to derive homogeneous COS-7 and HeLa ArgbP2 positive or negative lines. Based on counting 100 colonies (>2500 cells), we calculated that ∼0.3% of COS-7 cells and ∼0.15% of HeLa cells change their ArgBP2 expression status per cell division. Thus with further passaging, both cell types re-established a mosaic expression pattern. Recently, the epigenetic regulation of ArgBP2 by HDAC7 was demonstrated (20) that may underlie our observation here.
FIGURE 2.
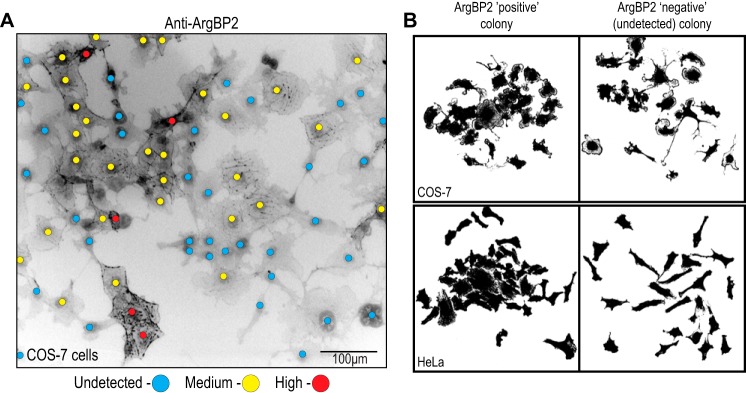
Clonal analysis of ArgBP2 expression in COS-7 and HeLa cells. A, COS-7 cells demonstrate a mosaic expression for ArgBP2 as assessed by indirect immunofluorescence. Red and yellow dots denote ArgBP2 high and medium/low expression, respectively, and blue dots denote cells with undetectable ArgBP2 levels. The fluorescent signal is inverted (white to black) for greater clarity. B, HeLa and COS-7 colonies derived from single cells in culture were fixed and analyzed at day 6. The cell outlines (using binary mask of anti-α-actinin staining) illustrates the typical cell morphology of these clonal populations, with either detectable (ArgBP2 positive) or undetectable (ArgBP2 negative) expression. ArgBP2-positive colonies are more compacted and less dispersed than ArgBP2-negative colonies.
Given this natural heterogeneity in ArgBP2 expression, we focused on COS-7 and HeLa cells to assay differences in cell behavior based on the presence or absence of detectable ArgBP2. On the other hand, because tagged ArgBP2 might dimerize with endogenous protein (27), we used U2-OS cells (ArgBP2-null) to generate and test various GFP-ArgBP2 mutant expressing cell lines.
Stress Fibers Contain ArgBP2 at F-actin-rich Regions Marked by α-Actinin
The C-terminal half of ArgBP2 contains three SH3 domains responsible for focal adhesion localization. ArgBP2, ponsin, and vinexin contain the conserved SH3 domains and are focal adhesion proteins (8). However, ArgBP2 is asymmetrically enriched at the proximal end of focal adhesions adjacent to actin stress fibers (Fig. 3A, line scan). Unlike ponsin and vinexin, ArgBP2 can to bind α-actinin (13) and is colocalized with α-actinin along stress fibers (Fig. 3B, line scan). ArgBP2 is, however, excluded from regions containing myosin II as illustrated in the super-resolution SIM image (Fig. 3C, line scan). Interestingly, ArgBP2 did not colocalize with α-actinin at the cell periphery (Fig. 3B, blue arrow), suggesting that interaction between ArgBP2 and α-actinin is not constitutive and can be regulated.
FIGURE 3.

ArgBP2 colocalizes with α-actinin-rich puncta along actin stress fibers. As indicated, HeLa cells were stained with (A) anti-ArgBP2 and anti-vinculin to stain focal adhesions or (B) anti-ArgBP2 and anti-α-actinin to stain actin stress fibers. Line scan analysis of signal from each channel was performed along the white arrow in the indicated direction. The white box shows a smaller region enlarged with the signal inverted to highlight the localization more clearly. The blue arrowhead in B highlights a ruffle at the cell edge where α-actinin is enriched but ArgBP2 is not detected. C, GFP-ArgBP2 expressing U2-OS cells were stained for endogenous Myosin IIa and imaged by SIM for better resolution. A line scan was performed along the white arrow in the indicated direction. The individual channels were inverted for greater clarity.
Differences in the level of ArgBP2 protein affected the α-actinin distribution in COS-7 (Fig. 4A) and HeLa cells (Fig. 4B). Those lacking ArgBP2 (as determined by immunostaining) exhibited smaller α-actinin puncta on stress fibers. This effect on α-actinin distribution was also apparent when ArgBP2 was ectopically expressed in U2-OS cells (Fig. 4C). To quantify the changes in α-actinin distribution on stress fibers by endogenous ArgBP2, we measured the signal intensity of α-actinin in identically sized areas of ArgBP2 positive versus ArgBP2 negative HeLa cells (the cell pairs selected from the same image, e.g. Fig. 4B). On average, we found a ∼2.5-fold increase in the α-actinin signal on stress fibers in ArgBP2 positive cells (Fig. 4D). The average α-actinin puncta size was also calculated: expression of ArgBP2 was associated with a ∼25% increase in their average size (Fig. 4E).
FIGURE 4.

ArgBP2 expression increases the level of actinin on stress fibers and increases the cross-linking and organization of the actomyosin network. COS-7 (A), HeLa (B), and U2-OS (C) cells were stained for endogenous α-actinin and for endogenous ArgBP2 in A and B or GFP-ArgbP2 in C. The detection of endogenous ArgBP2 in COS-7 (A) and HeLa (B) correlates with more α-actinin present along the actin stress fibers. C, U2-OS cells stably expressing GFP-ArgBP2 also increased the α-actinin recruitment to the actin stress fibers. Yellow plus signs denote ArgBP2-positive cells and blue dots denote ArgBP2-negative cells. The fluorescent signal was inverted (white to black) for greater clarity. The increase in intensity of α-actinin at these puncta was detected by selecting an identically sized region from ArgBP2 positive and negative cells. The background was subtracted for these regions in ImageJ and the signal intensity was quantified and plotted (D). These regions were then thresholded and objects were separated by a watershed algorithm in ImageJ. The area of the objects were calculated and plotted in E. Both graphs display one S.D.
GFP-ArgBP2γ dynamics was monitored by time lapse microscopy in HeLa and U2-OS cells; the ArgBP2γ containing puncta originated at the cell periphery and underwent retrograde flow to the center of the cell (supplemental Movie S1). This movement is likely to coincide with the movement of myosin IIA and myosin 18a complexes in the lamella region (25), and the retrograde movement of α-actinin containing fibers (28). These ArgBP2γ puncta are present in both the circumferential and dorsal actin stress fibers (28, 29). Smaller puncta often coalesced into larger puncta as the associated stress fibers became more bundled.
ArgBP2γ Localizes to Actin Stress Fibers via Its N-terminal Region
Given the ability of ArgBP2 to associate with actin stress fibers, we wanted to map the domains required for this localization. Various deletion constructs were tested by transient transfection as illustrated in Fig. 5A (a summary of their localization is also given in the table). Domain B comprising just 36 residues (residue 194–228) was necessary and sufficient for stress fiber localization (Fig. 5B). Removal of B yielded an ArgBP2γ mutant that localized predominantly to focal adhesions with only residual localization to actin stress fibers. Neither the SoHo domain nor regions A or C deletions noticeably affected the normal distribution of ArgBP2γ. Therefore, we conclude that domain B is the major domain responsible for localizing ArgBP2 to the actomyosin network with other regions providing an alternate, albeit much weaker, targeting to stress fibers.
FIGURE 5.

ArgBP2γ binds to actin stress fibers through an N terminally located sequence. A, the table summarizes the ArgBP2 construct localization in HeLa cells indicated as “−” (absent), “+” (weak), “++” (moderate), and “+++” (strong). B, the micrographs show representative images of FLAG-tagged proteins in HeLa cells. The full-length ArgBP2γ(1–573), the N-terminal half (1–340), and ArgBP2γ lacking regions A and C localized prominently to puncta along stress fibers. However, ArgBP2γ lacking domain B was concentrated in focal adhesions (blue arrows) with only residual localization to the actin stress fibers with no observable increase in puncta size. Domain B (last panel) was clearly localized to α-actinin-rich puncta on actin stress fibers (red arrows). The fluorescent signal was converted to black on white for clarity.
A Region of 36 Amino Acids Involved in Binding α-Actinin
The F-actin cross-linking protein α-actinin binds to ArgBP2 (13). There was excellent co-localization of α-actinin and ArgBP2γ on actin stress fibers in both epithelial HeLa cells and fibroblastic COS-7 (Fig. 3B and data not shown). Region B is needed for this localization and is well conserved across species (Fig. 6a). ArgBP2γ and various deletions were tested for their ability to immunoprecipitate endogenous α-actinin. However, we discovered that expression of tagged ArgBP2γ greatly reduced the quantity of Triton X-100-soluble α-actinin, whereas total α-actinin (extracted by SDS) remained unchanged (upper panels Fig. 6B). However ArgBP2γ-ΔB (ArgBP2γ lacking the B domain) did not affect Triton X-100-soluble α-actinin levels. Neither ponsin nor vinexin, which do not contain region B, affected α-actinin solubility. To circumvent this problem we expressed ArgBP2γ constructs or GFP-α-actinin separately, and allowed binding of the Triton X-100-solubilized proteins only after detergent extraction. Under these conditions, we show that ArgBP2γ but not ArgBP2γ-ΔB, bound to and co-immunopreciptated with GFP α-actinin (Fig. 6C).
We then mapped the region of α-actinin required to interact with ArgBP2 (Fig. 6d). Both full-length α-actinin and the calponin homology 1 (CH1) domain co-precipitated endogenous ArgBP2, whereas other regions did not. The CH1 domain is also responsible for binding F-actin (30). This raised the question, did ArgBP2γ interact with α-actinin directly or indirectly by binding F-actin. Increasing the level of F-actin in the lysates (by phalloidin treatment prior to immunoprecipitation) increased the yield of α-actinin bound to ArgBP2γ (Fig. 6E), whereas latrunculin (to depolymerize F-actin) decreased immunoprecipitated α-actinin. Adding 10 μm Ca2+ (to disrupt the α-actinin·F-actin complex) also decreased the ArgBP2/α-actinin interaction. Therefore we hypothesized that the α-actinin·actin complex may bind to ArgBP2γ as a trimeric complex. To investigate whether ArgBP2γ interacts with F-actin (without α-actinin), we knocked down α-actinin (Fig. 6F). The loss of α-actinin had a pronounced effect on the amount of actin bound to ArgBP2γ without affecting the total actin levels (Fig. 6F). This suggests that ArgBP2 predominantly interacts with F-actin via α-actinin. To test this hypothesis we conducted an in vitro F-actin co-sedimentation assay (Fig. 6G). Purified polymerized actin was incubated with recombinant GST-ArgBP2 B domain fragments, with or without the two N-terminal CH domains of α-actinin. The GST-ArgBP2(194–339) protein co-sedimented with F-actin 3-fold more prominently in the presence of purified α-actinin (CH1 + 2) (Fig. 6G, top panel). Furthermore, the GST-ArgBP2(260–339) protein that lacks domain B showed no interaction with F-actin in either case (Fig. 6G, bottom panel). We conclude that domain B is responsible for localizing ArgBP2 to stress fibers and also binds α-actinin when it is F-actin associated.
ArgBP2γ Is Regulated by PKA Phosphorylation and 14-3-3 Binding
FLAG-14-3-3ζ copurified with ArgBP2γ in COS-7 cells (23); conversely endogenous 14-3-3 co-immunoprecipitated with FLAG-ArgBP2γ (Fig. 7A, lane 2) at levels that could be detected by Coomassie Blue staining (Fig. 7C, red arrow at 30 kDa). Removal of ArgBP2 region “C” prevented 14-3-3 interaction (Fig. 7A, fifth lane) as did mutation of Ser-259 to Ala (sixth lane). To confirm direct binding of 14-3-3 to this site, we used a synthetic phosphopeptide array containing sequences corresponding to potential 14-3-3 binding sites found in the N-terminal region of ArgBP2. The sequence corresponding to Cdc25c Ser-216 was used as a control (Fig. 7B). The ArgBP2 Ser(P)-207 and Ser(P)-259 sites bound strongly to recombinant 14-3-3ζ. Because Ser-259 is the most commonly phosphorylated site on ArgBP2 (based on PhosphoSite database annotation) and removal of Ser-207 (located within domain B) has no effect on 14-3-3 binding, we conclude that Ser-259 is a key 14-3-3 “gatekeeper” site in ArgBP2 (31). The sequence around Ser-259 resembles a typical protein kinase A site (RRX(S/T)), and indeed forskolin treatment, which activated the kinase (20 μm, 30 min), increased endogenous 14-3-3 binding to FLAG-ArgBP2γ (Fig. 7C). The forskolin effect was blocked when treated together with a PKA inhibitor, H89 (Fig. 7C).
FIGURE 7.

ArgBP2 binding to 14-3-3 is PKA dependent and interferes with α-actinin interaction. A, immunoprecipitated FLAG-tagged ArgBP2γ, ArgBP2γ-ΔA, ArgBP2γ-ΔB, ArgBP2γ-ΔC, or ArgBP2γ-(S259A) were probed for bound 14-3-3. The region C and, in particular, Ser-259 were required for 14-3-3 binding. B, a solid-phase phosphopeptide array contained 13-mer peptides with a central phosphoserine derived from human ArgBP2 with Cdc25c Ser-216 as control was probed for 14-3-3 binding. The sequences surrounding Ser(P)-207 and Ser(P)-259 were more efficient than the control Cdc25c peptide at binding 14-3-3. C, the effects of forskolin (25 μm) or H89 (20 μm) on 14-3-3 binding to FLAG-ArgBP2γ. An increase in 14-3-3 binding induced by forskolin was blocked by prior H89 treatment, confirming the involvement of PKA. Forskolin treatment typically results in a 4-fold increase in 14-3-3 binding (n = 4, error bars are 1 S.D.). D, cells expressing either ArgBP2γ or ArgBP2γ-(S259A) were treated with DMSO or forskolin (25 μm) for 30 min; Triton X-100-soluble lysate recovered from these cells was mixed with those from cells expressing (untreated) GFP-α-actinin, prior to immunoprecipitation (IP). ArgBP2γ-(S259A) did not bind 14-3-3 and, importantly, α-actinin binding was unaffected by forskolin treatment. E, cells transiently expressing either FLAG-ArgBP2γ(1–340) or FLAG-ArgBP2γ(1–340)-(S259A) were treated with DMSO or forskolin (25 μm) for 30 min. The 14-3-3 binding to ArgBP2γ(1–340) was increased by forskolin and the α-actinin binding was correspondingly reduced. The α-actinin binding to ArgBP2γ(1–340)-(S259A) was unaffected by forskolin as this mutant was unable to bind 14-3-3 even with forskolin treatment. F, cells expressing FLAG-ArgBPγ either untreated or treated with DMSO, forskolin (25 μm), or calyculin A (50 μm) for 30 min. Both forskolin and calyculin treatment increased 14-3-3 binding to FLAG-ArgBPγ and reduced α-actinin binding.
As the 14-3-3 binding site is adjacent to the α-actinin binding domain, we reasoned that the binding of 14-3-3 could interfere with the α-actinin interaction. Indeed forskolin treatment increased the ArgBP2γ/14-3-3 interaction and resulted in a loss of α-actinin binding. Significantly the ArgBP2γ-(S259A) mutant was unaffected by this treatment (Fig. 7D). The N-terminal half of ArgBP2γ behaved similarly to the full-length constructs, indicating that 14-3-3 is not regulated by the SH3 domains (Fig. 7E). Calyculin (a potent S/T protein phosphatase inhibitor) treatment for 10 min also increased 14-3-3 binding to ArgBP2, and concomitantly reduced α-actinin binding (Fig. 7f). Thus these effects are phosphorylation-dependent.
Forskolin Treatment Affects ArgBP2γ Localization
Given the PKA effect on α-actinin interaction, we hypothesized that ArgBP2γ phosphorylation by PKA might also affect its localization to stress fibers. We looked at the distribution of endogenous ArgBP2 in HeLa cells before (Fig. 8A) and after (Fig. 8B) forskolin treatment; a clear loss of ArgBP2 from the central region containing stress fibers was observed. To quantify the loss of ArgBP2 from these stress fibers, we imaged either GFP-ArgBP2-wt (Fig. 9A) or the non-phosphorylatable mutant GFP-ArgBP2-(S259A) (Fig. 9B). The analysis focused on the ventral stress fibers using TIRFM, and analyzed the same fibers before and after forskolin treatment. We co-expressed mCherry-lifeAct to mark F-actin. The signal intensity of small ArgBP2 puncta present on actin stress fibers were tracked before and after forskolin treatment. The change in intensity of GFP signals (as a percentage of initial intensity) was quantified along multiple stress fibers (Fig. 9C). In this analysis, ∼25% loss of the GFP-ArgBP2 signal followed forskolin treatment, whereas GFP-ArgBP2-(S259A) was unaffected. Taken together, we concluded that PKA phosphorylates ArgBP2γ and increases 14-3-3 binding, which in turn reduces interaction with α-actinin. A similar role for 14-3-3 is reported for myosin phosphatase targeting subunit (MYPT1) whose phosphorylation blocks its interaction with myosin II (32).
FIGURE 8.

Forskolin treatment leads to loss of ArgBP2 from stress fibers. A and B, a micrograph showing control cells treated with DMSO (A) and the effect of forskolin (B; 25 μm) on HeLa cells. Anti-ArgBP2, anti-α-actinin and phalloidin staining are shown; a loss of ArgBP2 from stress fibers is observed but the protein remains at focal adhesions. The fluorescent signals were inverted for greater clarity. The box marks the enlarged panel where stress fibers are clearly visible.
FIGURE 9.
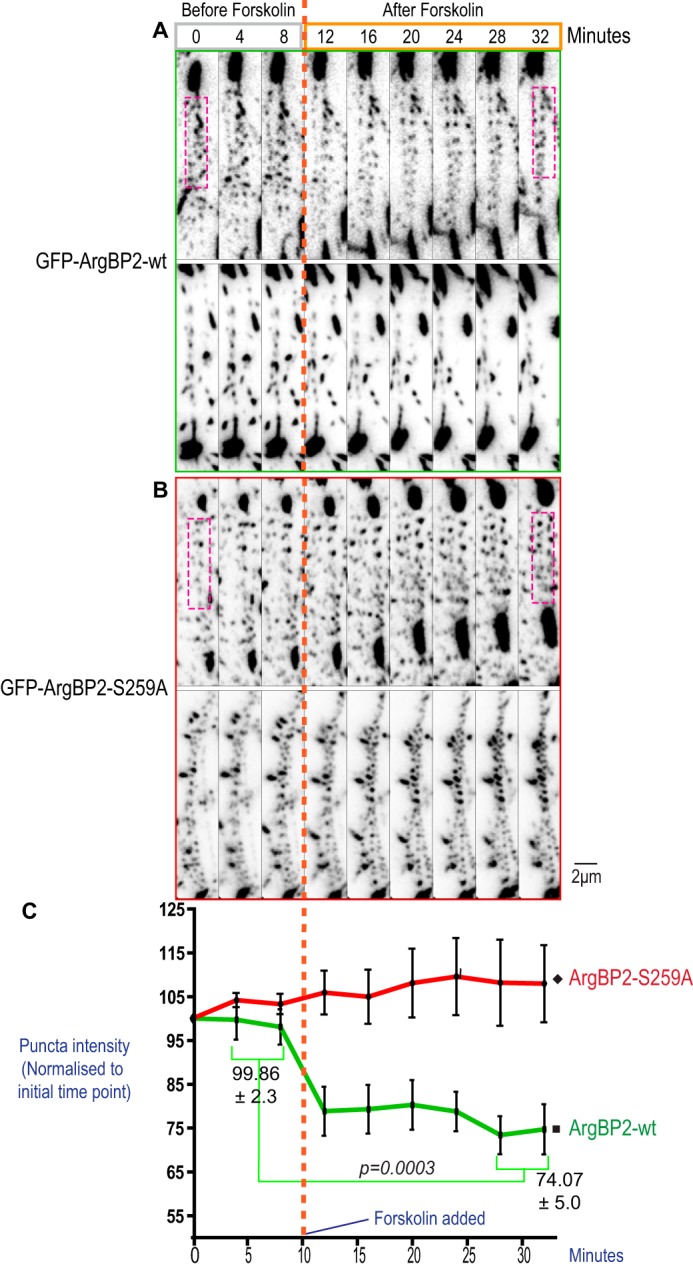
Effect of forskolin on ArgBP2 localization. U2-OS cell lines stably expressing GFP-ArgBP2γ (A) or GFP-ArgBP2γ-(S259A) (B) were imaged with TIRFM to illuminate only the ventral stress fibers. The cells were imaged for 15 min before (gray box) forskolin (25 μm/ml) was added (orange dotted line), and imaged for a further 30 min after treatment (orange box). Two representative cells are shown for each condition. The large structures correspond to focal adhesions, whereas smaller puncta were distributed along stress fibers. Signals corresponding to GFP-ArgBP2-wt puncta in these arrays were often reduced after forskolin treatment, but not with GFP-ArgBP2γ-(S259A) cells. C, the intensity of signal along stress fibers (an example region of interest is highlighted by the magenta dotted box) was expressed as the percentage change from its original intensity and plotted in the graph (calculated across multiple cells, n = 8, error bars are S.E.). To determine the statistical significance of these observations, the intensity was averaged over the first 2 time points (pre-treatment) and the last 2 time points (30 min after treatment) as displayed in the graph. After forskolin treatment, there was a significant difference in the change in ArgBP2-wt (p = 0.0003) but not with the ArgBP2-(S259A) mutant.
ArgBP2γ Inhibits Cell Migration
Cell migration requires a precise orchestration of actomyosin II filament polymerization and depolymerization (33–35). RhoA drives the formation of actin stress fibers, which can inhibit migration (35), however, these effects are driven through a myosin II-associated complex, whereas ArgBP2 acts through actinin/actin. ArgBP2 can interact with the WAVE complex protein CIP4 (18), but the authors noted CIP4 is dispensable for the ArgBP2-mediated inhibition of cell migration. When a confluent monolayer of COS-7 cells were subjected to a typical scratch wound assay ArgBP2 positive cells (∼50% of total) were under-represented in the cells at the leading edge (∼10%) suggesting that ArgBP2 expression correlated with reduced cell migration (data not shown). This is consistent with a link between ArgBP2 expression and reduced cancer cell migration (4).
We analyzed the rate of wound closure for U2-OS monolayers stably expressing either GFP (control) or GFP-ArgBP2γ (Fig. 10A). The control cells migrated into the wound twice as fast as cells expressing ArgBP2 over 25 h. Time-lapse movies indicated control cells often migrated away from others at the wound edge (supplemental Movie S2), whereas ArgBP2 expressing cells stayed tightly packed. To test individual cell migration rates we analyzed U2-OS cells plated a lower dilution on fibronectin (supplemental Movie S3). Over a period of 5 h we found that GFP-ArgBP2γ and GFP-ArgBP2γ-(S259A) expressing cells were impaired with respect to their migration speeds by ∼50% compared with controls (Fig. 10B). By contrast, the migration speed of GFP-ArgBP2γ-ΔB expressing cells was not significantly different than controls. Therefore we suggest that ArgBP2γ expression affects cell migration primarily through interactions of domain B with α-actinin and F-actin. This effect of ArgBP2 expression on cell migration fits with an emerging consensus that ArgBP2 is a tumor suppressor (4, 19, 37). These findings are summarized in Fig. 10C.
FIGURE 10.

ArgBP2γ expression affects stress fiber turnover and cell migration inhibition by ArgBP2γ requires α-actinin binding. A, in our wound healing assay, control U2-OS cells and cells stably expressing GFP-ArgBP2 were plated at 100% confluence into a Chamlide 4-well device. After the cells were attached, the removal of the 4-well rubber separator created an even wound. The wound edges were tracked as depicted in the micrograph and the change in area was plotted for multiple wound areas (n = 6, error bars are 1 S.D.). The control cells invaded this wound at a rate 50% faster than cells expressing ArgBP2 (p < 0.0001). B, random cell migration tracks of U2-OS cell lines stably expressing vector, GFP-ArgBP2γ, GFP-ArgBP2γ-(S259A), GFP-ArgBP2γ-ΔB (a lacking α-actinin binding domain) or control cells. Cells were plated on a fibronectin-coated matrix for 2 h, then migration was monitored over 5 h (n = 40, error bars are 1 S.D.). Both GFP-ArgBP2γ and GFP-ArgBP2γ-(S259A) expressing cells exhibited on average a ∼50% reduction in migration speed relative to control cells (p < 0.0001 for both). The GFP-ArgBP2γ-ΔB cells exhibited migration behavior similar to control cells (p = 0.1948). C, model of ArgBP2 function. ArgBP2 is targeted to stress fibers through an interaction with α-actinin where it is able to increase cross-linking of the actomyosin network. Following PKA phosphorylation, ArgBP2 binds to 14-3-3, which displaces α-actinin interaction and its localization to the actomyosin network. The focal adhesions interaction is unaffected by PKA phosphorylation, which is mediated by the C-terminal SH3 domains. In metastatic cells where ArgBP2 is frequently lost, this regulation of stress fibers is absent causing less cross-linking by α-actinin and allowing for faster cell migration.
DISCUSSION
We detected ArgBP2 as a 14-3-3 interacting partner in a screen in COS-7 cells (23), and cloned the dominant ∼70-kDa ArgBP2γ isoform present in these cells. Many transformed cell lines do not have detectable ArgBP2 expression although HeLa, COS-7, and MDCK appear to contain α- and/or γ-ArgBP2. In HeLa and COS-7 cells, we found a heterogeneous mosaic ArgBP2 expression pattern, but ArgBP2 expression appeared unrelated to cell cycle state. Those cells that are ArgBP2 positive have a denser actomyosin network with increased bundling of actin/α-actinin puncta. This mosaic expression could be under epigenetic regulation by HDAC7 (20). Thus, one of the functions of HDAC7 appears to be as a negative regulator of ArgBP2 expression. Interesting HDAC7 localizes to both the nucleus and cytoplasm of C2C12 myoblasts, but moves out of the nucleus when myotubes differentiate (38). As ArgBP2 is an important component of muscle Z-discs (3, 39), this would be consistent with ArgBP2 levels increasing in response to loss of nuclear HDAC7. Clearly, the regulation of ArgBP2 expression in normal and disease states warrants further investigation.
Here we demonstrate that the 36-residue region B (not found in ponsin and vinexin and no homology to any known actin-binding domain) is an α-actinin binding domain (Fig. 6C) predominantly responsible for its stress fiber localization. This actin cross-linking protein function of α-actinin is essential for the stability of stress fibers (40, 41); α-actinin is also found at focal adhesions. ArgBP2α is localized to muscle Z-discs along with α-actinin, and other signaling proteins (42, 43). α-Actinin has a mechanosensing function (44) and in dilated cardiomyopathy, a condition associated with faulty mechanosensing, ArgBP2 levels are elevated (45). ArgBP2 interactions with palladin (13) and spectrin (12) at this site are likely to further stabilize the various protein interactions.
14-3-3 is one of the most common transducers of serine/threonine phosphorylation events (46, 47). Given that the basic region around the ArgBP2γ 14-3-3ζ binding site is reported as a nuclear localization signal (3), 14-3-3 proteins could play a role in preventing nuclear import of ArgBP2γ, as found in other contexts (48). However, in our analysis, 14-3-3 functions primarily to prevent ArgBP2γ interaction with α-actinin downstream of the cAMP-regulated kinase, PKA. Increasing PKA activity by forskolin treatment leads to increased 14-3-3 binding, which is not seen with ArgBP2γ-(S259A). Binding of 14-3-3 often acts as a reversible steric inhibitor to compete for other local protein interactions (31, 49): for example, PKA phosphorylation and 14-3-3 binding blocks the myopodin/α-actinin interaction (50). We suggest 14-3-3 binding similarly allows ArgBP2γ to be displaced from actin stress fibers by preventing α-actinin interaction, as evidenced by the loss of ArgBP2 from these structures (Figs. 8 and 9). Moreover, ArgBP2 is reported to function as a dimer (27), suggesting the phospho-Ser-259 modification is sufficient for it to bind to 14-3-3 dimers.
It is notable that metastatic pancreatic cancers (4, 51) and cervical carcinoma (19) are associated with loss of ArgBP2 expression. Furthermore, ArgBP2 is lost in malignant metastatic cancers (as annotated in the NCBI GEO database), for example, in GDS1949, ovarian serous carcinomas; GDS1732, papillary thyroid cancer; GDS1713, Ewing family tumors; and GDS2698, synovial sarcoma. Interestingly, a truncated ArgBP2 has been detected in B-cell lymphoma (36), which might have a dominant inhibitory function due to its dimerization. Given the epigenetic switching mechanism apparently at work in cell culture and the recent discovery of HDAC regulation of ArgBP2 expression at a genomic level, strategies to induce re-expression of ArgBP2 via HDAC inhibition (and perhaps other proteins co-regulated in this manner) in metastatic cells could be an alternative approach to limit their migration in vivo.
In summary, we propose that ArgBP2γ expression can affect cell migration through its interaction with α-actinin (Fig. 10C). Cell lines expressing an ArgBP2 or a mutant that constitutively binds α-actinin inhibit migration. In contrast, another mutant lacking the α-actinin-binding domain does not show retarded migration (Fig. 10B). ArgBP2 binding to α-actinin increases in size the F-actin puncta along stress fibers and hence cross-linking of the entire network. Normally the ArgBP2/α-actinin interaction is regulated by phosphorylation and 14-3-3 binding. During metastasis, ArgBP2 expression is lost and the cells no longer have a highly cross-linked actomyosin network and hence migrate faster. Further studies of how ArgBP2 mediates greater cross-linking and which other kinases and phosphatases can regulate it certainly warrants closer investigation.
Supplementary Material
Acknowledgment
We thank Prof. Louis Lim for support.
This work was supported in part by the GSK-Institute of Molecular and Cell Biology research fund.

This article contains supplemental Movies S1–S3.
- ArgBP2
- Arg kinase-binding protein 2
- SIM
- structured illumination microscopy
- CH
- calponin homology
- HDAC
- histone deacetylase
- SH3
- Src homology domain 3.
REFERENCES
- 1. Vicente-Manzanares M., Ma X., Adelstein R. S., Horwitz A. R. (2009) Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 10, 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamaguchi H., Wyckoff J., Condeelis J. (2005) Cell migration in tumors. Curr. Opin. Cell Biol. 17, 559–564 [DOI] [PubMed] [Google Scholar]
- 3. Wang B., Golemis E. A., Kruh G. D. (1997) ArgBP2, a multiple Src homology 3 domain-containing, Arg/Abl-interacting protein, is phosphorylated in v-Abl-transformed cells and localized in stress fibers and cardiocyte Z-disks. J. Biol. Chem. 272, 17542–17550 [DOI] [PubMed] [Google Scholar]
- 4. Taieb D., Roignot J., André F., Garcia S., Masson B., Pierres A., Iovanna J. L., Soubeyran P. (2008) ArgBP2-dependent signaling regulates pancreatic cell migration, adhesion, and tumorigenicity. Cancer Res. 68, 4588–4596 [DOI] [PubMed] [Google Scholar]
- 5. Kawabe H., Hata Y., Takeuchi M., Ide N., Mizoguchi A., Takai Y. (1999) nArgBP2, a novel neural member of ponsin/ArgBP2/vinexin family that interacts with synapse-associated protein 90/postsynaptic density-95-associated protein (SAPAP). J. Biol. Chem. 274, 30914–30918 [DOI] [PubMed] [Google Scholar]
- 6. Yuan Z. Q., Kim D., Kaneko S., Sussman M., Bokoch G. M., Kruh G. D., Nicosia S. V., Testa J. R., Cheng J. Q. (2005) ArgBP2γ interacts with Akt and p21-activated kinase-1 and promotes cell survival. J. Biol. Chem. 280, 21483–21490 [DOI] [PubMed] [Google Scholar]
- 7. Murase K., Ito H., Kanoh H., Sudo K., Iwamoto I., Morishita R., Soubeyran P., Seishima M., Nagata K. (2012) Cell biological characterization of a multidomain adaptor protein, ArgBP2, in epithelial NMuMG cells, and identification of a novel short isoform. Med. Mol. Morphol. 45, 22–28 [DOI] [PubMed] [Google Scholar]
- 8. Kioka N., Ueda K., Amachi T. (2002) Vinexin, CAP/ponsin, ArgBP2: a novel adaptor protein family regulating cytoskeletal organization and signal transduction. Cell Struct. Funct. 27, 1–7 [DOI] [PubMed] [Google Scholar]
- 9. Kioka N., Sakata S., Kawauchi T., Amachi T., Akiyama S. K., Okazaki K., Yaen C., Yamada K. M., Aota S. (1999) Vinexin: a novel vinculin-binding protein with multiple SH3 domains enhances actin cytoskeletal organization. J. Cell Biol. 144, 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mandai K., Nakanishi H., Satoh A., Takahashi K., Satoh K., Nishioka H., Mizoguchi A., Takai Y. (1999) Ponsin/SH3P12: an l-afadin- and vinculin-binding protein localized at cell-cell and cell-matrix adherens junctions. J. Cell Biol. 144, 1001–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haglund K., Ivankovic-Dikic I., Shimokawa N., Kruh G. D., Dikic I. (2004) Recruitment of Pyk2 and Cbl to lipid rafts mediates signals important for actin reorganization in growing neurites. J. Cell Sci. 117, 2557–2568 [DOI] [PubMed] [Google Scholar]
- 12. Cestra G., Toomre D., Chang S., De Camilli P. (2005) The Abl/Arg substrate ArgBP2/nArgBP2 coordinates the function of multiple regulatory mechanisms converging on the actin cytoskeleton. Proc. Natl. Acad. Sci. U.S.A. 102, 1731–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rönty M., Taivainen A., Moza M., Kruh G. D., Ehler E., Carpen O. (2005) Involvement of palladin and α-actinin in targeting of the Abl/Arg kinase adaptor ArgBP2 to the actin cytoskeleton. Exp. Cell Res. 310, 88–98 [DOI] [PubMed] [Google Scholar]
- 14. Baumann C. A., Ribon V., Kanzaki M., Thurmond D. C., Mora S., Shigematsu S., Bickel P. E., Pessin J. E., Saltiel A. R. (2000) CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature 407, 202–207 [DOI] [PubMed] [Google Scholar]
- 15. Otey C. A., Carpen O. (2004) α-Actinin revisited: a fresh look at an old player. Cell Motil. Cytoskeleton 58, 104–111 [DOI] [PubMed] [Google Scholar]
- 16. Scaife R. M., Langdon W. Y. (2000) c-Cbl localizes to actin lamellae and regulates lamellipodia formation and cell morphology. J. Cell Sci. 113, 215–226 [DOI] [PubMed] [Google Scholar]
- 17. Scaife R. M., Courtneidge S. A., Langdon W. Y. (2003) The multi-adaptor proto-oncoprotein Cbl is a key regulator of Rac and actin assembly. J. Cell Sci. 116, 463–473 [DOI] [PubMed] [Google Scholar]
- 18. Roignot J., Taïeb D., Suliman M., Dusetti N. J., Iovanna J. L., Soubeyran P. (2010) CIP4 is a new ArgBP2 interacting protein that modulates the ArgBP2 mediated control of WAVE1 phosphorylation and cancer cell migration. Cancer Lett. 288, 116–123 [DOI] [PubMed] [Google Scholar]
- 19. Backsch C., Rudolph B., Steinbach D., Scheungraber C., Liesenfeld M., Häfner N., Hildner M., Habenicht A., Runnebaum I. B., Dürst M. (2011) An integrative functional genomic and gene expression approach revealed SORBS2 as a putative tumour suppressor gene involved in cervical carcinogenesis. Carcinogenesis 32, 1100–1106 [DOI] [PubMed] [Google Scholar]
- 20. Martin M., Geudens I., Bruyr J., Potente M., Bleuart A., Lebrun M., Simonis N., Deroanne C., Twizere J. C., Soubeyran P., Peixoto P., Mottet D., Janssens V., Hofmann W. K., Claes F., Carmeliet P., Kettmann R., Gerhardt H., Dequiedt F. (2013) PP2A regulatory subunit Bα controls endothelial contractility and vessel lumen integrity via regulation of HDAC7. EMBO J. 32, 2491–2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manser E., Huang H. Y., Loo T. H., Chen X. Q., Dong J. M., Leung T., Lim L. (1997) Expression of constitutively active α-PAK reveals effects of the kinase on actin and focal complexes. Mol. Cell. Biol. 17, 1129–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan P. M., Ng Y. W., Manser E. (2011) A robust protocol to map binding sites of the 14–3-3 interactome: Cdc25C requires phosphorylation of both S216 and S263 to bind 14–3-3. Mol. Cell Proteomics 10, M110.005157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robens J. M., Yeow-Fong L., Ng E., Hall C., Manser E. (2010) Regulation of IRSp53-dependent filopodial dynamics by antagonism between 14–3-3 binding and SH3-mediated localization. Mol. Cell. Biol. 30, 829–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leung T., Chen X. Q., Manser E., Lim L. (1996) The p160 RhoA-binding kinase ROK α is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell. Biol. 16, 5313–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tan I., Yong J., Dong J. M., Lim L., Leung T. (2008) A tripartite complex containing MRCK modulates lamellar actomyosin retrograde flow. Cell 135, 123–136 [DOI] [PubMed] [Google Scholar]
- 26. Tullio A. N., Accili D., Ferrans V. J., Yu Z. X., Takeda K., Grinberg A., Westphal H., Preston Y. A., Adelstein R. S. (1997) Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc. Natl. Acad. Sci. U.S.A. 94, 12407–12412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roignot J., Bonacci T., Ghigo E., Iovanna J. L., Soubeyran P. (2014) Oligomerization and phosphorylation dependent regulation of ArgBP2 adaptive capabilities and associated functions. PLoS One 9, e87130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hotulainen P., Lappalainen P. (2006) Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol. 173, 383–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burnette D. T., Shao L., Ott C., Pasapera A. M., Fischer R. S., Baird M. A., Der Loughian C., Delanoe-Ayari H., Paszek M. J., Davidson M. W., Betzig E., Lippincott-Schwartz J. (2014) A contractile and counterbalancing adhesion system controls the 3D shape of crawling cells. J. Cell Biol. 205, 83–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Way M., Pope B., Weeds A. G. (1992) Evidence for functional homology in the F-actin binding domains of gelsolin and alpha-actinin: implications for the requirements of severing and capping. J. Cell Biol. 119, 835–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yaffe M. B. (2002) How do 14–3-3 proteins work?: gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 513, 53–57 [DOI] [PubMed] [Google Scholar]
- 32. Koga Y., Ikebe M. (2008) A novel regulatory mechanism of myosin light chain phosphorylation via binding of 14–3-3 to myosin phosphatase. Mol. Biol. Cell 19, 1062–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mitchison T. J., Cramer L. P. (1996) Actin-based cell motility and cell locomotion. Cell 84, 371–379 [DOI] [PubMed] [Google Scholar]
- 34. Pollard T. D., Borisy G. G. (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 [DOI] [PubMed] [Google Scholar]
- 35. Nobes C. D., Hall A. (1999) Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144, 1235–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mestre-Escorihuela C., Rubio-Moscardo F., Richter J. A., Siebert R., Climent J., Fresquet V., Beltran E., Agirre X., Marugan I., Marín M., Rosenwald A., Sugimoto K. J., Wheat L. M., Karran E. L., García J. F., Sanchez L., Prosper F., Staudt L. M., Pinkel D., Dyer M. J., Martinez-Climent J. A. (2007) Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood 109, 271–280 [DOI] [PubMed] [Google Scholar]
- 37. Roignot J., Soubeyran P. (2009) ArgBP2 and the SoHo family of adapter proteins in oncogenic diseases. Cell Adh. Migr. 3, 167–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao C., Liu Y., Lam M., Kao H. Y. (2010) Histone deacetylase 7 (HDAC7) regulates myocyte migration and differentiation. Biochim. Biophys. Acta 1803, 1186–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Geiger B., Dutton A. H., Tokuyasu K. T., Singer S. J. (1981) Immunoelectron microscope studies of membrane-microfilament interactions: distributions of α-actinin, tropomyosin, and vinculin in intestinal epithelial brush border and chicken gizzard smooth muscle cells. J. Cell Biol. 91, 614–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lazarides E., Burridge K. (1975) α-Actinin: immunofluorescent localization of a muscle structural protein in nonmuscle cells. Cell 6, 289–298 [DOI] [PubMed] [Google Scholar]
- 41. Sanger J. W., Sanger J. M., Jockusch B. M. (1983) Differences in the stress fibers between fibroblasts and epithelial cells. J. Cell Biol. 96, 961–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pyle W. G., Solaro R. J. (2004) At the crossroads of myocardial signaling: the role of Z-discs in intracellular signaling and cardiac function. Circ. Res. 94, 296–305 [DOI] [PubMed] [Google Scholar]
- 43. Frank D., Kuhn C., Katus H. A., Frey N. (2006) The sarcomeric Z-disc: a nodal point in signalling and disease. J. Mol. Med. 84, 446–468 [DOI] [PubMed] [Google Scholar]
- 44. Hampton C. M., Taylor D. W., Taylor K. A. (2007) Novel structures for α-actinin:F-actin interactions and their implications for actin-membrane attachment and tension sensing in the cytoskeleton. J. Mol. Biol. 368, 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barth A. S., Kuner R., Buness A., Ruschhaupt M., Merk S., Zwermann L., Kääb S., Kreuzer E., Steinbeck G., Mansmann U., Poustka A., Nabauer M., Sültmann H. (2006) Identification of a common gene expression signature in dilated cardiomyopathy across independent microarray studies. J. Am. Coll. Cardiol. 48, 1610–1617 [DOI] [PubMed] [Google Scholar]
- 46. Gohla A., Bokoch G. M. (2002) 14–3-3 regulates actin dynamics by stabilizing phosphorylated cofilin. Curr. Biol. 12, 1704–1710 [DOI] [PubMed] [Google Scholar]
- 47. Mackintosh C. (2004) Dynamic interactions between 14–3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem. J. 381, 329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lopez-Girona A., Furnari B., Mondesert O., Russell P. (1999) Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature 397, 172–175 [DOI] [PubMed] [Google Scholar]
- 49. Muslin A. J., Xing H. (2000) 14–3-3 proteins: regulation of subcellular localization by molecular interference. Cell Signal. 12, 703–709 [DOI] [PubMed] [Google Scholar]
- 50. Faul C., Dhume A., Schecter A. D., Mundel P. (2007) Protein kinase A, Ca2+/calmodulin-dependent kinase II, and calcineurin regulate the intracellular trafficking of myopodin between the Z-disc and the nucleus of cardiac myocytes. Mol. Cell. Biol. 27, 8215–8227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nakamura T., Furukawa Y., Nakagawa H., Tsunoda T., Ohigashi H., Murata K., Ishikawa O., Ohgaki K., Kashimura N., Miyamoto M., Hirano S., Kondo S., Katoh H., Nakamura Y., Katagiri T. (2004) Genome-wide cDNA microarray analysis of gene expression profiles in pancreatic cancers using populations of tumor cells and normal ductal epithelial cells selected for purity by laser microdissection. Oncogene 23, 2385–2400 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.