

Background: The ATPase NSF works with the adaptor protein αSNAP to disassemble SNARE protein complexes involved in membrane fusion.
Results: Kinetic assays demonstrate that three αSNAP and 10 ATP molecules are required to disassemble a SNARE complex.
Conclusion: The energy requirements and functional stoichiometry of αSNAP in SNARE complex disassembly are established.
Significance: The results suggest models of NSF action.
Keywords: ATPase, ATPases Associated with Diverse Cellular Activities (AAA), Intracellular Trafficking, Membrane Trafficking, Soluble NSF Attachment Protein Receptor (SNARE), NSF, alpha-SNAP
Abstract
The fusion of intracellular membranes is driven by the formation of a highly stable four-helix bundle of SNARE proteins embedded in the vesicle and target membranes. N-Ethylmaleimide sensitive factor recycles SNAREs after fusion by binding to the SNARE complex through an adaptor protein, αSNAP, and using the energy of ATP hydrolysis to disassemble the complex. Although only a single molecule of αSNAP binds to a soluble form of the SNARE complex, we find that three molecules of αSNAP are used for SNARE complex disassembly. We describe an engineered αSNAP trimer that supports more efficient SNARE complex disassembly than monomeric αSNAP. Using the trimerized αSNAP, we find that N-ethylmaleimide-sensitive factor hydrolyzes 10 ATP molecules on average to disassemble a single SNARE complex.
Introduction
The eukaryotic cell moves cargo between membrane-bound compartments using vesicles that bud from one membrane and move to, dock, and finally fuse with a target membrane to effect delivery of luminal cargo and integral membrane components. Soluble NSF5 attachment protein receptors (SNAREs) are membrane-associated proteins present on both vesicle (v-SNARE) and target (t-SNARE) membranes that are central to the process of membrane fusion (1, 2). After a vesicle docks with its target membrane, the cytoplasmic regions of SNAREs associate into a coiled-coil of four α-helices, designated the SNARE complex (SC), that is thought to provide the free energy required for bilayer fusion (3). Force measurements of SNARE complex disassembly, using the neuronal t-SNAREs Syx1a (syntaxin 1a) and SNAP-25 (synaptosome-associated protein of 25 kDa), and the v-SNARE VAMP2 (vesicle-associated membrane protein 2)/synaptobrevin lacking their transmembrane anchors, indicate that the complex is stabilized by ∼39 kcal mol−1 relative to the free SNARE proteins (4).
After bilayer fusion, N-ethylmaleimide-sensitive factor (NSF; EC: 3.6.4.6) disassembles the SNARE complex in order to recycle the SNARE proteins for further rounds of fusion (5–8) (Fig. 1A). NSF utilizes the energy from ATP hydrolysis to dissociate the SNARE complex in the postfusion membrane, and it also disassembles potentially non-productive t-SNARE complexes to “prime” them for bilayer fusion (9). NSF is a hexameric ATPase that is a member of the protein family of ATPases associated with various cellular activities (AAA+) (10). These enzymes are generally involved in unfolding, disassembly, or remodeling of proteins and their complexes, and are typically pentamers or hexamers in their active state. The NSF monomer comprises a substrate-binding N-terminal domain (N-domain), followed by two AAA+ ATPase domains, designated D1 and D2. Experiments in which either D1 or D2 is deleted, and others in which key catalytic residues are mutated in the active sites, suggest that D1 is the more active ATPase and indicate that its activity is essential for SNARE complex disassembly. Although D2 has little or no ATPase activity as an isolated domain, ATP binding to D2 is essential for hexamer formation and SNARE disassembly (11).
FIGURE 1.

NSF uses the energy of ATP hydrolysis and the αSNAP adaptor to disassemble the SNARE complex. A, NSF binds to the membrane-bound SC via αSNAP, which is probably membrane-associated. Using the energy from ATP hydrolysis, the SC is disassembled into its component proteins, and NSF and αSNAP are available for subsequent rounds of disassembly. B, simplified model for the dissociation of the SC by NSF bound to 3 αSNAP, in which either αSNAP binds SC before NSF (red pathway) or SC binds to αSNAP·NSF to form the 20 S complex (blue pathway in B and C). C, kinetic modeling scheme for the association of n αSNAP molecules with SC and disassembly of the SC by NSF. The red branch represents binding of n αSNAP molecules to the SC to form the complete NSF substrate before interaction with NSF; the blue branch corresponds to n αSNAP molecules binding to NSF before association with the SC. Potential intermediate, non-productive assemblies lacking sufficient αSNAP molecules to support SC disassembly are also shown. For a given model that uses n αSNAP molecules, the 20 S complex will be (αSNAP)n·SC·NSF, which disassembles into n αSNAPs, the individual SNARE proteins, and free NSF (curved green arrows). D, kinetic modeling scheme for the association of n αSNAP3 molecules with SC and disassembly of the SC by NSF. The red branch represents binding of n αSNAP3 molecules to the SC to form the complete NSF substrate before interaction with NSF; the blue branch corresponds to n αSNAP3 molecules binding to NSF before association with the SC. Potential intermediate, non-productive assemblies lacking sufficient αSNAP3 molecules to support SC disassembly are also shown. For a given model that uses n αSNAP3 molecules, the 20 S complex will be (αSNAP3)n·SC·NSF, which disassembles into n αSNAP3s, the individual SNARE proteins, and free NSF (curved green arrows).
NSF does not directly disassemble SNARE complexes, but instead requires adaptor proteins known as soluble NSF attachment proteins (SNAPs) that bind to both the SNARE complex and NSF (Fig. 1B). αSNAP binds to NSF in an ATP-dependent manner and accelerates the ATPase activity of NSF (12–14). αSNAP is ubiquitously expressed and binds to all ternary SNARE complexes as well as binary complexes containing only t-SNAREs (15). The crystal structure of the yeast αSNAP homolog shows that the protein is an α-helical solenoid that is capped by a C-terminal globular helical domain (16). Mutations of positively charged residues along the solenoid disrupt binding to the SNARE complex (17), and electron microscopy shows that binding of αSNAP to the rodlike SNARE complex increases the width but not the length of the rod, suggesting that the αSNAP solenoid binds along the length of the SNARE complex (18) (Fig. 1A). In the absence of NSF, αSNAP binds to the neuronal SNARE complex lacking transmembrane anchors in a 1:1 stoichiometry (15).
The NSF·αSNAP·SC assembly, also known as the “20 S complex” (Fig. 1A), has been studied biochemically and structurally. The molecular mass of the 20 S complex obtained by electron microscopy and quantitative amino acid analysis of NSF and αSNAP bands isolated by SDS-PAGE of purified 20 S complex suggested the presence of three αSNAP molecules (19). Cryoelectron microscopy (cryo-EM) reconstruction of the 20 S complex is also consistent with the presence of three αSNAP molecules bound to the SNARE complex (20, 21). In qualitative pull-down assays, a trimeric αSNAP produced by adding an N-terminal coiled-coil domain was shown to bind NSF in the absence of SNARE complex, whereas monomeric αSNAP bound NSF only in the presence of the SNARE complex (19), but the effect of trimerization on NSF ATPase or SNARE complex disassembly activities was not tested. The NSF N-domain interacts with the C-terminal domain of αSNAP (19, 22), and αSNAP contains a membrane-associated loop near its N terminus that probably orients the C terminus of αSNAP toward the cytosol (23) (Fig. 1A).
Here we have addressed two key mechanistic aspects of NSF-mediated SNARE disassembly. First, understanding the role of αSNAP as the adaptor between NSF and the SNARE complex has been complicated by the lack of a clear understanding of the functional stoichiometry of the 20 S complex. Second, the number of ATPs needed to disassemble a SNARE complex, which sets boundaries on potential models and efficiencies of action, is not known. We addressed both of these unknowns using equilibrium binding and kinetic analysis of SNARE disassembly and ATPase activity. Although only one αSNAP appears to bind to the SNARE complex in solution (15), we determined that three molecules of αSNAP are required for full activity of NSF on its substrate. We describe an engineered αSNAP trimer that enhances the dissociation of the SNARE complex by NSF, beyond that expected by simply providing multiple copies of αSNAP in a single molecule. Using pre-steady-state kinetics with the αSNAP trimer, we found that 10 ATP molecules are required to disassemble a SNARE complex.
EXPERIMENTAL PROCEDURES
Molecular Biology
DNA sequences encoding the proteins of interest were cloned into plasmids for expression in Escherichia coli. NSF from Cricetulus longicaudatus with an N-terminal His6 tag was cloned into a pQE9 plasmid and transformed into E. coli M15 cells containing the pREP4 repressor plasmid (Qiagen Inc., Valencia, CA). The DNA sequence for Bos taurus αSNAP was cloned into a pGEX vector, yielding a construct with a cleavable N-terminal glutathione S-transferase (GST) tag.
The T4 fibritin foldon domain (encoding amino acid residues 457–486) (24, 25) was inserted at the 5′-end of the B. taurus αSNAP to induce the formation of a stable αSNAP trimer (αSNAP3). PCR was used to assemble an oligonucleotide containing the DNA sequence of the T4 fibritin foldon domain with 5′- and 3′-ends that are complementary to the N terminus of GST-αSNAP (26, 27), using the following sequences: assembly PCR target, CGGGAATTTCCGGTGGTGGTGGTGGAGGCAGCGGCTATATTCCGGAAGCGCCGCGCGATGGC CAGGCGTATGTGCGCAAAGATGGCGAATGGGTGCTGCTGAGCACCTTTCTGATTCCTATGGACAACTCCGGGAAGGAGGCG; assembly primer 1, CGGGAATTTCCGGTGGTGGTGGTGGAGGC AGCGGCTATATTCCG; assembly primer 2, TGCGCACATACGCCTGGCCATCGCGCGGCGCTTCCGGAATATAGCCGCTGC; assembly primer 3, CAGGCGTATGTGCGCAAAGATGGCGAATGGGTGCTGCTGAGCACCTTTCTGATT; assembly primer 4, CGCCTCCTTCCCGGAGTTGTCCATAGGAATCAGAAAGGTGCTCAGCAG; flanking primer 1, CGGGAATTTCCGGTGGTG; flanking primer 2, CGCCTCCTTCCCGGA.
Briefly, assembly primers 1–4 were used in a PCR with Platinum Taq (Invitrogen) and a Tanneal = 50 °C. The crude PCR product was further amplified using flanking primers 1 and 2 in a PCR with Tanneal = 51 °C. After cleanup, the assembly PCR target was utilized in a modified site-directed mutagenesis reaction with GST-αSNAP in a pGEX vector to insert the trimerization domain in frame at the 5′-end of the αSNAP sequence.
Protein Expression and Purification
All fast protein liquid chromatography steps were performed on an ÄKTApurifier system with the following columns: HiLoad Superdex S200 26/60, HiLoad Superdex S200 16/60, MonoQ GL 10/100, and MonoS GL 10/100 (GE Healthcare). All protein concentrations were determined by A280 using the calculated extinction coefficient, or Bradford assay (Bio-Rad).
E. coli M15 cells (Qiagen) transformed with the pQE9 plasmid encoding NSF were grown at 37 °C in modified super broth (3.2% tryptone, 2% yeast extract, 0.5% NaCl, 0.02% NaOH (w/v)) until OD = 0.8 and induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at 25–30 °C for 4–5 h. Cells were harvested by centrifugation and stored at −80 °C. Cell pellets were lysed in an EmulsiFlex C-3 (Avestin Inc., Ottawa, Ontario, Canada) in lysis buffer (100 mm NaHEPES, pH 7.0, 500 mm KCl, 10% (w/v) glycerol, 5 mm β-mercaptoethanol, 1 mm PMSF, 1 μm pepstatin, 1 mm ATP). The lysate was clarified by ultracentrifugation, and NSF was purified by nickel-nitrilotriacetic acid-agarose (Qiagen) chromatography and successive runs of size exclusion chromatography (S200 26/60) in a buffer containing 25 mm Tris-HCl, pH 8.5, 300 mm KCl, 5 mm β-mercaptoethanol, 1 mm NaEDTA, 10% (w/v) glycerol, and 1 mm ATP. Fractions containing pure NSF were pooled and concentrated using Ultracel 30K centrifugal filters (Amicon). Concentrated NSF was exchanged into storage buffer containing 1 μm ATP as the penultimate step to flash freezing in liquid nitrogen and storage at −80 °C.
GST-αSNAP was expressed from a pGEX vector in E. coli BL21 RIL CodonPlus cells and purified via GST-Sepharose (Sigma-Aldrich) affinity chromatography. The GST tag was removed on column using thrombin (Sigma-Aldrich), and the cleaved protein was purified by size exclusion chromatography (S200 26/60) and anion exchange (MonoQ) (23). Fractions containing pure αSNAP were pooled and concentrated using Ultracel 10K centrifugal filters. Concentrated αSNAP was flash-frozen in liquid nitrogen and stored at −80 °C. αSNAP containing the “foldon” domain was purified as a cleavable GST fusion protein in the same method as for unmodified αSNAP.
The syntaxin 1a SNARE domain (residues 188–267), a SNAP-25 mutant in which the four cysteines normally palmitoylated were mutated to alanine, and the S79C mutant of VAMP2 lacking the C-terminal transmembrane anchor were expressed in E. coli BL21 RIL CodonPlus cells and purified as described previously (28). The SC was formed by incubating these individual SNARE proteins overnight in 20 mm Tris-HCl, pH 7.4, 100 mm NaCl, and 1 mm dithiothreitol, followed by purification by anion exchange chromatography (MonoQ) (17, 29).
Purification of Fluorescently Labeled VAMP2 and SNARE Complex
Purified VAMP2(S79C) was labeled with Alexa Fluor 488-maleimide (Invitrogen) according to the manufacturer's directions. This labeled VAMP2 (VAMP2-A488) was separated from unlabeled VAMP2 by cation exchange chromatography (MonoS). Fractions containing pure VAMP2-A488 were pooled, concentrated, and flash-frozen in liquid nitrogen prior to storage at −80 °C. The Alexa Fluor 488-labeled SNARE complex was assembled from purified syntaxin 1a SNARE domain, SNAP-25, and VAMP2-A488, followed by purification from free SNARE proteins as described previously (29). In some of the experiments measuring binding to αSNAP or αSNAP3, the purified SNARE complex was further purified using size exclusion chromatography. However, this extra purification step produced no significant difference in these binding assays and was not included in the purification of SNARE complexes used in the disassembly or ATP hydrolysis assays.
Nucleotide Binding Assay
Radioactively labeled nucleotides, [α-32P]ATP (specific activity, 3000 Ci/mmol), [γ-32P]ATP (SA, 10 Ci/mmol) (PerkinElmer Life Sciences) were incubated with NSF at 4 °C or 30 °C in binding buffer (50 mm HEPES, pH 7.4, 300 mm potassium chloride, 10% (w/v) glycerol, 12 mm MgCl2, 2 mm dithiothreitol). Protein·nucleotide complexes were separated from free nucleotide using Zeba spin columns (Thermo Fisher Scientific) equilibrated with binding buffer and spun at 1500 × g for 90 s. NSF concentration was varied at each nucleotide concentration, and the slope of the nucleotide bound versus NSF concentration was used to determine the amount of nucleotide bound specifically to NSF at each concentration.
ATP Hydrolysis Assay
All ATP hydrolysis assays were performed at 30 °C. The reaction mixture contained 50 mm NaHEPES, pH 7.4, 120 mm potassium glutamate, 20 mm potassium acetate, and varying concentrations of [γ-32P]ATP (10 Ci/mmol) diluted in unlabeled ATP to final concentrations of 300–500 μm. The reaction mixture was prepared on ice and incubated at 30 °C for 5 min prior to the addition of Mg2+ (MgCl2 or Mg(CH3COO)2) or NSF to start the reaction. Reaction aliquots (25–50 μl) were removed and quenched using trichloroacetic acid, and released 32Pi was separated by molybdate-Pi extraction and measured by liquid scintillation counting (30). ATP hydrolysis measurements used in steady-state analysis were confined to the first 4–8 min of the reaction to obtain linear initial rates (of the 40 independent experiments, 35 had R2 ≥ 0.95, and 5 had 0.85 < R2 < 0.95) free from substrate depletion and product inhibition. Linear rates were measured in duplicate, and the uncertainty reported the error in ATP hydrolysis as propagated from each of the regression lines establishing linear rates.
Fluorescence Anisotropy Measurements
Fluorescence anisotropy was used to measure the formation and dissolution of protein complexes containing VAMP2 labeled with the fluorophore Alexa Fluor 488 (VAMP2-A488). All measurements were performed in black, flat-bottomed 96-well microplates (Costar 3915, Corning Inc.), and readings were taken in a Synergy 4 Hybrid microplate reader (BioTek, Winooski, VT). Microplates were prewarmed to 30 °C for 10 min prior to the start of the experiment. Time courses of 60–120 min were recorded at 30 °C, with the addition of Mg2+ to start the reaction. Fluorescence anisotropy r was calculated as follows,
 |
where the subscripts indicate intensity measurements parallel and perpendicular to the incident polarization. Once thawed, A488-labeled SNARE complex gives reproducible anisotropy readings for 4–5 days when stored at 4 °C. All experiments where anisotropy values were directly compared were performed within 24 h.
αSNAP·SNARE Complex Binding Assay
SC containing VAMP2-A488 at 25–1000 nm was incubated with varying concentrations of αSNAP or αSNAP3 (25–5000 nm) in 50 mm NaHEPES, pH 7.4, 120 mm potassium glutamate, 20 mm potassium acetate at room temperature prior to incubation at 30 °C. Complex formation at equilibrium was measured by an increase in anisotropy over a 60-min time course. Bovine serum albumin (Sigma-Aldrich) was added in place of αSNAP in parallel experiments as a control for nonspecific binding. ATP and Mg2+ were found to have no effect on αSNAP-SC binding. Equilibrium binding experiments were performed in triplicate, and the error bars represent S.E.
SNARE Complex Disassembly Assay
SC (25–1000 nm) containing VAMP2-A488 was incubated in 50 mm NaHEPES, pH 7.4, 120 mm potassium glutamate, 20 mm potassium acetate with αSNAP, NSF, and 400–500 μm ATP. The plate was prewarmed to 30 °C for 10 min prior to the addition of Mg2+ at a final concentration of 5 mm to start the disassembly reaction. Disassembly of the SC by NSF (1–10 nm) results in a decrease in fluorescence anisotropy that reaches a minimum value identical to free VAMP-A488. The assembled neuronal SNARE complex is resistant to SDS and runs as a single band on SDS-PAGE (31), so disassembly of SNARE complex was confirmed by SDS-PAGE. SC disassembly was monitored over a 60–120-min time course. Initial rates of disassembly were determined from the initial linear phase (R2 > 0.90) of the reaction, when higher SC concentrations were used. Controls lacking NSF, ATP, or Mg2+ showed no disassembly. The SC concentration was maintained below 1 μm to minimize the contribution of reassembly and to keep within the detection range of the fluorescence plate reader. Pre-steady-state reactions included an excess of NSF (2 μm) and low concentrations of αSNAP (50–100 nm). Steady-state measurements to determine Km and kcat for SC disassembly used NSF at 2 nm and SC at 50–250 nm, with αSNAP varied from 25 to 2000 nm. SC concentrations above these values in steady-state conditions resulted in significant SC reassembly and therefore were avoided. Disassembly rate was plotted versus αSNAP concentration, and the data were fit to a hyperbola using the equation, v = Vmax/(1 + (Km/[αSNAP]). Steady-state SC disassembly experiments were performed in triplicate, and error bars depict S.E.; the titration disassembly experiments were performed in duplicate. There was no difference in kcat or KmαSNAP over the range of SC concentrations examined.
Kinetic Modeling
KinTek Explorer was used to simulate the effect of different αSNAP/SC stoichiometries on the kinetics of SC disassembly by NSF with monomeric αSNAP, using the schemes shown in Fig. 1, B and C. Table 1 summarizes the Kd values used in the model shown in Fig. 1C. The Kd values for dissociation of the αSNAP·SC, measured in this work (K1–4), were taken to be independent of the number of αSNAP molecules bound to the SC. Likewise, the Kd values for dissociation of the (αSNAP)n·SC from NSF were set to the value of Km for αSNAP stimulation of SC disassembly and were assumed to be the same regardless of the value of n. The Kd for the binding of αSNAP to NSF was taken as the Km for αSNAP stimulation of the NSF ATPase activity and was assumed to be independent of the number of αSNAP molecules bound to NSF. The remaining Kd values, which describe the formation of intermediate αSNAPi·SC·NSF complexes, were calculated from the above constants via thermodynamic cycles.
TABLE 1.
Summary of Kd values used in kinetic modeling with monomeric αSNAP
| Reaction | Name | Kd | Source |
|---|---|---|---|
| αSNAP + SC ⇌ αSNAP·SC | K1 | 450 nm | Direct measurement (Fig. 2A) |
| αSNAP + αSNAP·SC ⇌ (αSNAP)2·SC | K2 | 450 nm | Direct measurement (Fig. 2A) |
| αSNAP + (αSNAP)2·SC ⇌ (αSNAP)3·SC | K3 | 450 nm | Direct measurement (Fig. 2A) |
| αSNAP + (αSNAP)3·SC ⇌ (αSNAP)4·SC | K4 | 450 nm | Direct measurement (Fig. 2A) |
| αSNAP·SC + NSF ⇌ αSNAP·SC·NSF | K5 | 1200 nm | Km of αSNAP for SC disassembly by NSF (Fig. 2C) |
| (αSNAP)2·SC + NSF ⇌ (αSNAP)2·SC·NSF | K6 | 1200 nm | Km of αSNAP for SC disassembly by NSF (Fig. 2C) |
| (αSNAP)3·SC + NSF ⇌ (αSNAP)3·SC·NSF | K7 | 1200 nm | Km of αSNAP for SC disassembly by NSF (Fig. 2C) |
| (αSNAP)4·SC + NSF ⇌ (αSNAP)4·SC·NSF | K8 | 1200 nm | Km of αSNAP for SC disassembly by NSF (Fig. 2C) |
| αSNAP + NSF ⇌ αSNAP·NSF | K9 | 4700 nm | Km of αSNAP for the ATPase activity of NSF (Fig. 2D) |
| αSNAP + αSNAP·NSF ⇌ (αSNAP)2·NSF | K10 | 4700 nm | Km of αSNAP for the ATPase activity of NSF (Fig. 2D) |
| αSNAP + (αSNAP)2·NSF ⇌ (αSNAP)3·NSF | K11 | 4700 nm | Km of αSNAP for the ATPase activity of NSF (Fig. 2D) |
| αSNAP + (αSNAP)3·NSF ⇌ (αSNAP)4·NSF | K12 | 4700 nm | Km of αSNAP for the ATPase activity of NSF (Fig. 2D) |
| αSNAP·NSF + SC ⇌ αSNAP·SC·NSF | K13 | 120 nm | Thermodynamic cycle ((K1 × K5)/K9) |
| (αSNAP)2·NSF + SC ⇌ (αSNAP)2·SC·NSF | K14 | 12 nm | Thermodynamic cycle ((K13 × K17)/K10) |
| (αSNAP)3·NSF + SC ⇌ (αSNAP)3·SC·NSF | K15 | 1.1 nm | Thermodynamic cycle ((K14 × K18)/K11) |
| (αSNAP)4·NSF + SC ⇌ (αSNAP)4·SC·NSF | K16 | 0.11 nm | Thermodynamic cycle ((K15 × K19)/K12) |
| αSNAP·SC·NSF + αSNAP ⇌ (αSNAP)2·SC·NSF | K17 | 450 nm | Thermodynamic cycle ((K2 × K5)/K6) |
| (αSNAP)2·SC·NSF + αSNAP ⇌ (αSNAP)3·SC·NSF | K18 | 450 nm | Thermodynamic cycle ((K3 × K6)/K7) |
| (αSNAP)3·SC·NSF + αSNAP ⇌ (αSNAP)4·SC·NSF | K19 | 450 nm | Thermodynamic cycle ((K4 × K7)/K8) |
The forward rate kon was set at 100 nm−1·min−1, and the reverse rate constant was calculated as kon × Kd. Modeling with these rate constants scaled up or down 1000-fold produced the same general shape of the curve (data not shown). To compensate for the fact that the relative but not absolute values of the rate constants were known, the reaction rates were normalized to the maximum rate of reaction for that model. Because KinTek Explorer only permits the modeling of elementary reaction steps, we were unable to model the dissociation of the (αSNAP)n·SC·NSF complex into free NSF, αSNAP, Syx1a, SNAP-25, and VAMP2 as a single step. Thus, the catalytic step was defined as the conversion of the (αSNAP)n·SC·NSF complex into an (αSNAP)n·product·NSF, with a rate of formation equal to kcat. The (αSNAP)n·product·NSF complex then dissociated into (αSNAP)n·product + NSF and then into n αSNAP + product (in n elementary reaction steps). The rate constants for these subsequent dissociations were set at 1010 min−1 to make them virtually instantaneous.
Disassembly reaction rates were calculated over the αSNAP concentration range 0–7100 nm in 100 nm increments, with SC and NSF concentrations fixed at their experimental concentrations of 1000 nm. In all cases, the time for the modeling was fixed at 0.1 min (6 s) to ensure that the product versus time curves used to obtain the rates remained in the linear range.
The same procedures were followed for modeling trimerized αSNAP (αSNAP3) as for monomeric αSNAP, except that the reaction rates were calculated in the range 0–1710 nm αSNAP3 in 30 nm increments. The Kd values used for modeling SC dissociation by trimeric αSNAP3 (Fig. 1D) are summarized in Table 2. αSNAP3 was treated as a single molecular species, so the experimental concentrations and Kd values, which were calculated on a per monomer basis, were divided by 3 for the modeling. The SC and NSF concentrations were fixed at their experimental concentrations of 280 nm.
TABLE 2.
Summary of Kd values used in kinetic modeling with trimeric αSNAP3
| Reaction | Name | Kd | Source |
|---|---|---|---|
| αSNAP3 + SC ⇌ αSNAP3·SC | K20 | 100 nm | Direct measurement (Fig. 3C) |
| αSNAP3 + αSNAP3·SC ⇌ (αSNAP3)2·SC | K21 | 100 nm | Direct measurement (Fig. 3C) |
| αSNAP3 + (αSNAP3)2·SC ⇌ (αSNAP3)3·SC | K22 | 100 nm | Direct measurement (Fig. 3C) |
| αSNAP3 + (αSNAP3)3·SC ⇌ (αSNAP3)4·SC | K23 | 100 nm | Direct measurement (Fig. 3C) |
| αSNAP3·SC + NSF ⇌ αSNAP3·SC·NSF | K24 | 84 nm | Km of αSNAP3 for SC disassembly by NSF (Fig. 3D) |
| (αSNAP3)2·SC + NSF ⇌ (αSNAP3)2·SC·NSF | K25 | 84 nm | Km of αSNAP3 for SC disassembly by NSF (Fig. 3D) |
| (αSNAP3)3·SC + NSF ⇌ (αSNAP3)3·SC·NSF | K26 | 84 nm | Km of αSNAP3 for SC disassembly by NSF (Fig. 3D) |
| (αSNAP3)4·SC + NSF ⇌ (αSNAP3)4·SC·NSF | K27 | 84 nm | Km of αSNAP3 for SC disassembly by NSF (Fig. 3D) |
| αSNAP3 + NSF ⇌ αSNAP3·NSF | K28 | 390 nm | Km of αSNAP3 for the ATPase activity of NSF (Fig. 3E) |
| αSNAP3 + αSNAP3·NSF ⇌ (αSNAP3)2·NSF | K29 | 390 nm | Km of αSNAP3 for the ATPase activity of NSF (Fig. 3E) |
| αSNAP3 + (αSNAP3)2·NSF ⇌ (αSNAP3)3·NSF | K30 | 390 nm | Km of αSNAP3 for the ATPase activity of NSF (Fig. 3E) |
| αSNAP3 + (αSNAP3)3·NSF ⇌ (αSNAP3)4·NSF | K31 | 390 nm | Km of αSNAP3 for the ATPase activity of NSF (Fig. 3E) |
| αSNAP3·NSF + SC ⇌ αSNAP3·SC·NSF | K32 | 22 nm | Thermodynamic cycle ((K20 × K24)/K28) |
| (αSNAP3)2·NSF + SC ⇌ (αSNAP3)2·SC·NSF | K33 | 5.6 nm | Thermodynamic cycle ((K32 × K36)/K29) |
| (αSNAP3)3·NSF + SC ⇌ (αSNAP3)3·SC·NSF | K34 | 1.4 nm | Thermodynamic cycle ((K33 × K37)/K30) |
| (αSNAP3)4·NSF + SC ⇌ (αSNAP3)4·SC·NSF | K35 | 0.36 nm | Thermodynamic cycle ((K34 × K38)/K31) |
| αSNAP3·SC·NSF + αSNAP3 ⇌ (αSNAP3)2·SC·NSF | K36 | 100 nm | Thermodynamic cycle ((K21 × K24)/K25) |
| (αSNAP3)2·SC·NSF + αSNAP3 ⇌ (αSNAP3)3·SC·NSF | K37 | 100 nm | Thermodynamic cycle ((K22 × K25)/K26) |
| (αSNAP33·SC·NSF + αSNAP3 ⇌ (αSNAP3)4·SC·NSF | K38 | 100 nm | Thermodynamic cycle ((K23 × K26)/K27) |
Calculation of 7 S Complex Concentration
The predicted concentration of αSNAP3·SC complex (7 S complex) in the pre-steady-state experiment was calculated from the input concentrations of SC and αSNAP3 using the measured dissociation constant KdαSNAP·SC = 100 nm and the general form of the binding equation,
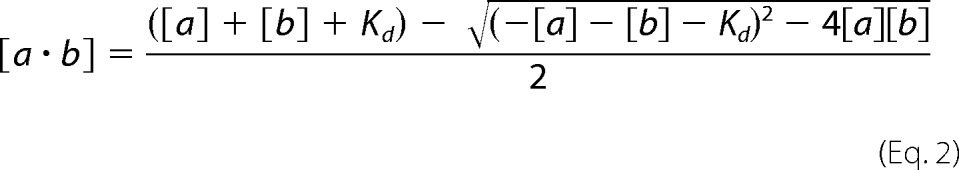 |
where [a] and [b] are the input protein concentrations of SC and αSNAP3.
RESULTS
Multiple αSNAP Molecules Are Required for SNARE Complex Disassembly
Recent biophysical data show that αSNAP forms a 1:1 complex with a variety of SNARE complexes lacking membrane anchors (15) (Fig. 2A), a surprising result given prior biochemical and structural data indicating that the 20 S complex contains three copies of αSNAP (17–21). We incubated a minimal neuronal SC, consisting of the SNARE domain of syntaxin 1a, SNAP-25, and the S79C mutant of VAMP2 labeled with Alexa Fluor 488, with αSNAP. Binding was measured by the increase in fluorescence anisotropy due to the decreased tumbling time of the αSNAP·SC relative to that of the SC (Fig. 2A). The αSNAP·SC equilibrium binding data can be fit to a single-site, 1:1 binding model with KdαSNAP = 450 ± 50 nm (Fig. 2A). This value agrees closely with the reported value of Kd = 470 nm by biolayer interferometry (15). A model in which three αSNAP molecules bind to a single SC with three distinct binding sites and binding affinities did not provide a significantly better fit to the binding data despite the additional fitting parameters. These data are consistent with the observation that αSNAP binds to the SNARE complex lacking transmembrane anchors with a 1:1 stoichiometry (15) but do not in themselves rule out an αSNAP/SC stoichiometry of >1:1.
FIGURE 2.

The role of αSNAP in binding and disassembling the SNARE complex. A, SC containing labeled VAMP2-A488 (50 nm) was combined with increasing concentrations of αSNAP (0–3 μm), and the change in fluorescence anisotropy was measured. The data are shown fit to a single-site binding curve giving KdαSNAP·SNAREd = 450 ± 52 nm. The inset schematic shows a single αSNAP interaction with the SC. B, representative disassembly assay. Alexa Fluor 488-labeled SC (SC*) (50 nm) was combined with excess αSNAP (4 μm) and disassembled by NSF (2 nm) in the presence of ATP (1 mm) and Mg2+ (5 mm) (red). A control experiment was performed in the absence of Mg2+ (blue). C, initial rates of SC disassembly in steady-state conditions were measured with increasing concentrations of αSNAP (0–2500 nm). The data are fit to the equation, v = Vmax/(1 + Km/αSNAP) to give the maximal rate kcat and KmαSNAP for SC (solid black line) (Table 2). D, the ATPase activity of NSF as a function of αSNAP concentration. The data are fit to the equation, ν = Vmax/(1 + (Km/[αSNAP])) (solid black line) to give the KmαSNAP for NSF ATPase activity. E, SC disassembly rates (y axis) were measured with SC and NSF at high, fixed concentrations (1 μm each) with a range of αSNAP concentrations (0.4–7.1 μm). The lines correspond to models with different αSNAP stoichiometry, as indicated in the key. Descriptions of the models can be found under “Experimental Procedures.” F, association of αSNAP (2 μm) with SC (50 nm) as measured by fluorescence anisotropy. A 6-s instrument dead time was accounted for by shifting the data points 6 s in the direction of the positive x axis. Error bars, S.E.
We next assessed the functional stoichiometry of αSNAP in SC disassembly by NSF by examining the dependence of the rate of SC disassembly on αSNAP concentration. We found, as has been noted by others (32), that the presence of Cl− diminishes NSF activity. Although NSF activity can be measured under very low ionic strength conditions to circumvent this problem (32), we found that a buffer containing carboxylates as the predominant anions, which more closely mimics physiological conditions, supports NSF activity (23). Disassembly of the SC in this buffer was measured by the decrease in anisotropy of labeled VAMP2 (VAMP2-A488) under steady-state conditions (Fig. 2, B and C). Analysis of initial rates of SC disassembly, measured by the dissociation of VAMP2-A488 from the SC as a function of αSNAP concentration, gave KmαSNAP = 1.2 ± 0.2 μm and kcatdisassembly = 1.9 ± 0.1 min−1 at 30 °C (Fig. 2C and Table 3). We also assessed the association of αSNAP with NSF under these conditions by determining its Km for NSF-mediated ATP hydrolysis, which was 4.7 μm (Fig. 2D and Table 1).
TABLE 3.
αSNAP and αSNAP3 in steady-state NSF-mediated SC disassembly
SC disassembly was monitored in steady-state conditions with varying concentrations of αSNAP or αSNAP3 to determine their contribution to disassembly ([NSF] = 2 nm, [ATP] = 400 μm, [SC] = 50 nM, [αSNAP] = 0–2500 nm, [αSNAP3] = 0–590 nm).
| αSNAP + SC | αSNAP3 + SC | |
|---|---|---|
| kcatdisassembly (min−1) | 1.9 ± 0.14 | 1.6 ± 0.04 |
| KmαSNAP3 (μm) | 1.2 ± 0.19 | 0.084 ± 0.010 |
| kcatdisassembly/KmαSNAP3 (μm−1 min−1) | 1.7 ± 0.30 | 19 ± 2.3 |
To determine the number of αSNAPs involved in SNARE complex disassembly, we performed a titration experiment by fixing NSF and SNARE complex at high concentrations and measuring initial SNARE disassembly rates over a range of αSNAP concentrations from substoichiometric to excess (Fig. 2E). At a given NSF concentration, the disassembly rate will be proportional to the amount of the αSNAP·SC, which is the NSF substrate. Considering the red pathway in Fig. 1B, if binding of αSNAP to the SNARE complex is faster than SNARE complex disassembly (i.e. the equilibrium for αSNAP·SNARE complex formation is established faster than the disassembly reaction), then with a fixed concentration of SNARE complex, the rate of disassembly will be a function of the concentration of input αSNAP. In the alternative case that αSNAP binds first to NSF (blue pathway in Fig. 1B), the rate of disassembly will also be a function of the concentration of input αSNAP if the equilibrium for SNARE complex binding to the SNAP·NSF complex is faster than disassembly. Thus, the number of αSNAP molecules required for disassembly can be obtained by comparing the observed disassembly rate with that calculated from models using the equilibrium binding constants for formation of αSNAP·SNARE and αSNAP·NSF complexes, KdαSNAP, and the input concentrations of αSNAP and SNARE complex for different stoichiometries of the αSNAP·SNARE complex interaction.
To assess whether the time for equilibration of the αSNAP-SC binding reaction is faster than SC disassembly (i.e. the condition needed for the titration experiment), we followed the association of αSNAP with the SC by fluorescence anisotropy (Fig. 2F). The t½ values estimated from progress curves of disassembly and αSNAP-SC association measured at the same SC concentration indicate that the latter is ∼40 times faster (Fig. 2, B and F). Therefore, the disassembly rate should be a function of the αSNAP·SC concentration based on the amount of input αSNAP and SC.
The stoichiometry of αSNAP in the (αSNAP)n·SC·NSF complex was determined by modeling the reaction with n = 1, 2, 3, and 4 (red pathway in Fig. 1C) and comparing the predictions from each of these models to the experimental data (Fig. 2E). The experimental data do not follow a model with 1:1 stoichiometry and are best described by a 3:1 αSNAP/SC stoichiometry.
These models assumed that αSNAP binds SC before NSF (red pathway in Fig. 1, B and C). However, αSNAP stimulates NSF ATPase activity in the absence of SNARE complex (Fig. 2D), indicating that it can bind to NSF by itself. Therefore, it was also necessary to consider a second pathway in which SC binds to αSNAP·NSF to form the 20 S complex (blue pathway in Fig. 1, B and C); as we did for binding to free αSNAP, we assumed that the binding of SC to NSF-bound αSNAP reaches equilibrium faster than the rate of 20 S disassembly. Formally, there can be intermediates of the form αSNAPi·SC·NSF (i < n) (Fig. 1C). We generated a model with such intermediates in the 3:1 αSNAP/SC case, which shows a decline in rate at high αSNAP concentrations due to sequestration of αSNAP in (αSNAP)n·SC and (αSNAP)n·NSF complexes rather than (αSNAP)n·SC·NSF complexes by mass action. The absence of a decline in the observed disassembly rate at high αSNAP concentrations suggests that there is no significant accumulation of these intermediate species, although the predicted decline could be due to misestimation of the Kd values describing their formation. Importantly, the data cannot distinguish whether αSNAP binds NSF or SC first (i.e. blue versus red pathways in Fig. 1B).
Engineering a Trimeric αSNAP
Prior studies have shown that fusion of a trimeric coiled-coil sequence to the N terminus of αSNAP qualitatively enhances binding to NSF (19). Given the results of the titration experiment suggesting the involvement of three αSNAP molecules in SNARE complex disassembly, we hypothesized that an αSNAP homotrimer would support more efficient disassembly as well. We fused the bacteriophage T4 fibritin “foldon” domain, which assembles into a very stable β-propeller with an estimated Kd in the low picomolar range (24, 25), to the N terminus of αSNAP. Structural data indicate that αSNAP binds to the outside of the SNARE coiled-coil complex (21), so a (Gly-Ser)6 linker was inserted between the foldon and the N terminus of αSNAP to allow each αSNAP in the trimer to simultaneously contact the SNARE complex. In this orientation, the foldon mimics tethering of individual αSNAP molecules to the membrane through the N-terminal loop (Fig. 3A) (23). Analysis of the foldon-αSNAP fusion protein by size exclusion chromatography (Fig. 3B) and characterization by native gel electrophoresis (data not shown) confirmed that it assembles into a stable trimer with no apparent dissociation or aggregation into higher oligomers. We refer to this fusion protein as αSNAP3.
FIGURE 3.

An engineered trimeric αSNAP binds to the SNARE complex, activates NSF ATPase, and mediates more efficient disassembly of the SNARE complex. A, schematic model of αSNAP interaction with the SC. The membrane is indicated by a gray bar, and the membrane-interacting loop of αSNAP is indicated by the solid red circle. The T4-foldon trimer is indicated by F. The N-terminal fusion of the trimer to αSNAP may mimic the orientation of αSNAP on the membrane. B, comparison of the A280 signal in milliabsorbance units from size exclusion chromatography (S200) elution profiles of αSNAP3 (solid, left y axis) and αSNAP (dashed, right y axis). C, SC containing labeled VAMP2 (A488; 0.05 μm) was combined with increasing concentrations of αSNAP3 (0–0.5 μm), and the change in fluorescence anisotropy was measured. The data are shown fit to a single-site binding curve giving KdαSNAP·SC = 100 ± 4 nm. D, initial rates of SC disassembly in steady-state conditions were measured in increasing concentrations of αSNAP3 (0–590 nm). The data are fit to the equation, ν = Vmax/(1 + (Km/[αSNAP])) to give the maximal rate kcat and KmαSNAP3 for SC disassembly (solid black line) (Table 2). E, the ATPase activity of NSF was measured as a function of αSNAP3 concentration. The data are fit to the equation, ν = Vmax/(1 + (Km/[αSNAP])) (solid black line) to give KmαSNAP3 for NSF ATPase activity. F, SC disassembly rates (y axis) were measured with SC and NSF at fixed concentrations (280 nm) over a range of αSNAP3 concentrations (320–1710 nm). The lines correspond to models with different αSNAP3 stoichiometry, as indicated in the key. The steeper rise for the branched curve as compared with the unbranched curve results from the normalization of the calculated data to the maximum rate. Because the branched curve has a lower absolute maximum rate than the unbranched curve, it reaches its maximum more quickly. Descriptions of the models used can be found under “Experimental Procedures.” Error bars, S.E.
αSNAP3 binds to the SNARE complex with Kd = 100 ± 4 nm (Fig. 3C), 4.5-fold more strongly than monomeric αSNAP, which is consistent with the presence of three copies of αSNAP in the oligomer but provides no indication of any more than a single site on the SNARE complex that is stably occupied with a bound αSNAP molecule. Steady-state measurements of SC disassembly in the presence αSNAP3 gave kcatdisassembly = 1.6 ± 0.1 min−1 and KmαSNAP = 84 ± 10 nm (Fig. 3D and Table 3). As with the αSNAP monomer, we measured the association of αSNAP3 with NSF in the absence of SC by determining its Km for NSF-mediated ATP hydrolysis, which was 390 nm (Fig. 3E and Table 2).
A titration experiment similar to that described above is consistent with αSNAP3 acting unimolecularly in SC disassembly (Fig. 3F). The similarity in kcatdisassembly between αSNAP and αSNAP3 (Table 3) suggests that the reaction is proceeding through the same rate-limiting step. However, the 14-fold decrease in KmαSNAP (Table 3) is significantly larger than the 3-fold decrease that would be expected from the presence of three copies of αSNAP in the trimer. The finding that monomeric αSNAP is required at a greater than 1:1 stoichiometry for SC disassembly, whereas trimeric αSNAP appears to act in a unimolecular manner, provides quantitative evidence that more than a single αSNAP participates in SC disassembly.
Nucleotide Binding and Hydrolysis Activities
The binding of nucleotides to NSF was measured by incubating purified NSF with [α-32P]ATP at 4 °C or 30 °C and measuring the radioactivity of the resulting protein·nucleotide complex (Fig. 4). The data indicate that 11.5 ± 0.8 molecules of ATP bound to NSF with a Kdapp of 20 ± 4.8 μm. When the experiment was repeated with [γ-32P]ATP, no radioactivity was detected at 30 °C. These data indicate that NSF binds to 12 molecules of ATP, which are hydrolyzed to ADP with the release of inorganic phosphate to produce an ADP-bound form at steady state, and suggest that all 12 nucleotide-binding sites are active.
FIGURE 4.

The NSF hexamer binds 12 ADP molecules at saturation. Purified NSF (20 nm) was incubated at 30 °C with varying concentrations of [α-32P]ATP (0.5–2000 μm) in the presence of Mg2+, and nucleotide·protein complexes were quantified for protein and nucleotide content. NSF binds 11.5 ± 0.8 molecules of [α-32P]ATP per hexamer with a Kd of 20 ± 4.8 μm. With [γ-32P]ATP used in the same binding experiment, no 32P signal is detected (data not shown), indicating that NSF binds 12 ADP molecules at equilibrium.
We examined the effects of αSNAP and αSNAP3 on the ATP hydrolysis activity of NSF by steady-state kinetics. NSF alone hydrolyzes ATP with a kcatATP = 5.7 ± 0.2 min−1 and a KmATP = 20 ± 3.6 μm (Table 4). There was no evidence of cooperative or distinct ATPase activities in the hydrolysis kinetics, despite the presence of 12 ATPase sites in the NSF hexamer (i.e. the data are well described by a single KmATP value). In the presence of αSNAP, kcatATP increases 2–3-fold, consistent with previous reports (9, 11–13) (Table 4). αSNAP3 increases the NSF ATPase rate ∼8-fold relative to NSF alone (Table 4). In the presence of αSNAP3·SC, NSF ATPase activity is stimulated further relative to αSNAP3, and is 13-fold higher than that of NSF alone (Table 4).
TABLE 4.
αSNAP3 activation of NSF-mediated ATP hydrolysis
NSF ATP hydrolysis rates were measured as a function of increasing ATP concentration (0–600 μm) and fit to the Michaelis-Menten equation using nonlinear regression with [NSF hexamer] = 4–8 nm, [αSNAP] = 4.5 μm, [αSNAP3] = 1.5 μm, and [αSNAP3·SC] = 2 μm. At these concentrations, which were limited by protein solubility, αSNAP and αSNAP3 in the absence of SC do not fully saturate NSF. Specifically, there is predicted to be 50% free NSF and 50% NSF·αSNAP in the experiment for NSF·αSNAP and 19% free NSF and 81% NSF·αSNAP3 in the experiment for NSF·αSNAP3 (calculated using Km for αSNAP and αSNAP3 stimulation of ATPase activity; Fig. 2, D and E, respectively), and the kcat values have been corrected for these fractions bound using the ATP hydrolysis rate for NSF alone for the αSNAP-free fraction in this table.
| NSF | NSF·αSNAP | NSF·αSNAP3 | NSF·αSNAP3·SC | |
|---|---|---|---|---|
| kcatATP (min−1) | 5.7 ± 0.21 | 10.3 ± 0.25; 14.9 (corrected) | 40.0 ± 3.6; 48.1 (corrected) | 77.2 ± 3.9 |
| KmATP (μm) | 20 ± 3.6 | 46.6 ± 4.6 (uncorrected) | 56.9 ± 20 (uncorrected) | 52.9 ± 9.0 |
| kcatATP/KmATP (μm−1 min−1) | 0.29 ± 0.05 | 0.22 ± 0.02 | 0.70 ± 0.26 | 1.46 ± 0.26 |
Direct Measurement of ATP Utilization in SNARE Complex Disassembly
To determine directly the amount of ATP needed to disassemble the neuronal SNARE complex, we sought to measure SC disassembly under pre-steady-state conditions (i.e. with NSF in excess over its αSNAP·SC substrate). Under these conditions, the observed rate is that of the actual complex disassembly independent of enzyme concentration. By comparing this rate with the rate of ATP hydrolysis in the 20 S complex under the same conditions, the number of ATPs consumed in a single disassembly reaction can be obtained.
NSF is well behaved up to concentrations of about 5 μm in the buffers used in our assays. To have NSF in sufficient excess for pre-steady-state measurements, we chose to have the αSNAP·SC substrate at concentrations in the range 10–100 nm. A low concentration of SC (<1 μm) is also desirable to avoid reassociation of SNAREs during the experiment. However, the affinity of αSNAP for the SC (Kd = 450 nm) indicated that it would be difficult to prepare a significant amount of αSNAP·SC (i.e. the NSF substrate) at these concentrations. We therefore exploited αSNAP3, which binds sufficiently strongly to carry out pre-steady-state measurements of SC disassembly. Moreover, given the apparent need for more than one αSNAP in SC disassembly, using the unimolecular αSNAP3 (Fig. 3E) avoids the potential complication of having only one or two αSNAP molecules bound in the NSF·αSNAP·SC complex. SC was combined with a limiting concentration of αSNAP3 and an excess of NSF (Fig. 5A). The reaction was monitored for SNARE disassembly and for ATP hydrolysis, and the initial rates were determined by a linear fit to the early time points in the SC disassembly reaction (Fig. 5, B and C).
FIGURE 5.

Pre-steady-state analysis of NSF ATP hydrolysis and SNARE disassembly. A, NSF hydrolyzes ATP in the NSF·αSNAP·SC (20 S) complex, leading to SC disassembly (i), and outside the 20 S complex (ii and iii). B, SC disassembly in the presence (filled circles) and absence (open circles) of Mg2+ (n = 3; error bars show S.E.). C, ATP hydrolysis in the presence (filled circles) and absence (open circles) of αSNAP3. C and D, a representative pre-steady-state experiment ([NSF] = 1.96 μm, [αSNAP3] = 100 nm, [SC] = 750 nm) (n = 3; error bars show S.E.) used to obtain reaction rates. D, pre-steady-state disassembly and ATP hydrolysis experiments were performed at different SC concentrations (38–900 nm), and the rate constant for ATP hydrolysis versus SC concentration is plotted with the full data from Table 4. The percentage of αSNAP3 in the αSNAP3·SC complex in each experiment (dashed line, right y axis) was calculated from experimental protein concentrations and the independently determined dissociation constant of KdαSNAP·SC = 100 nm (see “Experimental Procedures”) (error bars show propagated uncertainty in the linear regression of ATP hydrolysis and SC disassembly measurements in B and C above). E, the number of ATPs hydrolyzed per SC disassembly are plotted versus SC concentration of each experiment (data points, left y axis). The percentage of αSNAP3·SC fits the data points well (dashed line, right y axis). Error bars, propagated uncertainty in the calculation of ATP per SC.
With NSF in excess, three different NSF-containing species contribute to the observed ATP hydrolysis (Fig. 5A): NSF·αSNAP3·SC (20 S complex; Fig. 5A, i); free NSF (νNSFATP; Fig. 5A, ii); and NSF·αSNAP3 (νNSF·αSNAPATP; Fig. 5A, iii). Because NSF is present in excess (∼20-fold) the amount of NSF involved in disassembly represents a small fraction of the total enzyme present. The hydrolysis mediated by the 20 S complex is of interest, because only this species leads to SNARE complex disassembly. To eliminate the contribution of NSF·αSNAP3 to the observed ATP hydrolysis, ATPase activity was determined as a function of increasing concentrations of SC, thereby saturating and removing free αSNAP3. At nearly saturating SNARE complex concentrations (Table 5 and Fig. 5, D and E), the observed ATP hydrolysis from the remaining ∼5% NSF·αSNAP3 is negligible, because the ATPase activity of this species is only about half of the NSF·αSNAP3·SC complex (Table 4). The contribution of free NSF to ATP hydrolysis was determined in parallel control reactions in which αSNAP3 was omitted (Fig. 5C); the SNARE complex does not interact with NSF directly and therefore does not affect hydrolysis activity. The rate of ATP hydrolysis in the 20 S complex was then obtained by subtracting the rate of ATP hydrolysis of free NSF (the αSNAP-free control; Fig. 5C) from the rate of ATP hydrolysis in the SC disassembly reaction measured at nearly saturating SC concentrations (Table 5); note that the amount of NSF involved in disassembly is negligible compared with total enzyme concentration (Table 5), so it can be omitted in the background subtraction. The ratio of this value and the SC disassembly rate gives the number of ATPs hydrolyzed per SC disassembly.
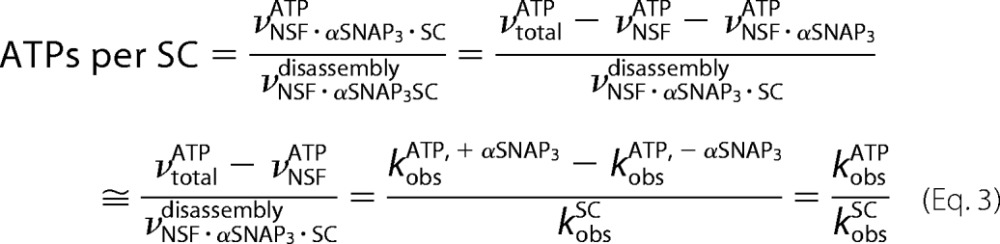 |
A mean value of 10.2 ± 1.1 ATPs hydrolyzed per SC disassembly is obtained by averaging the four points at the plateau of Fig. 5. D and E (Table 5).
TABLE 5.
Pre-steady-state measurement of SC disassembly and ATP hydrolysis by NSF
These rate constants of ATP hydrolysis and SC disassembly were used to determine the number of ATP hydrolyzed per SC ([NSF] = 2 μm, [αSNAP3] = 100 nm, [ATP] = 500 μm). The fraction of αSNAP3 bound to SC was determined using the protein concentrations in each experiment in conjunction with the quadratic equation for binding (see “Experimental Procedures”).
| [SC] | kobsATP, +αSNAP3 | kobsATP, −αSNAP3 | kobsATP, +αSNAP3 − kobsATP, −αSNAP3 | kobsSC | αSNAP3 bound to SC |
|---|---|---|---|---|---|
| nm | min−1 | min−1 | min−1 | min−1 | % |
| 37.5 | 1.47 ± 0.06 | 0.68 ± 0.11 | 0.80 ± 0.13 | 0.015 ± 0.001 | 30 |
| 75 | 2.17 ± 0.17 | 0.63 ± 0.12 | 1.54 ± 0.21 | 0.039 ± 0.001 | 54 |
| 150 | 2.81 ± 0.10 | 0.69 ± 0.10 | 2.12 ± 0.14 | 0.089 ± 0.002 | 79 |
| 300 | 3.40 ± 0.05 | 0.64 ± 0.10 | 2.76 ± 0.11 | 0.174 ± 0.006 | 92 |
| 450 | 3.44 ± 0.09 | 0.65 ± 0.13 | 2.79 ± 0.16 | 0.215 ± 0.012 | 95 |
| 600 | 3.86 ± 0.27 | 0.62 ± 0.12 | 3.24 ± 0.30 | 0.243 ± 0.017 | 97 |
| 750 | 3.94 ± 0.40 | 0.70 ± 0.11 | 3.24 ± 0.41 | 0.305 ± 0.019 | 97 |
| 900 | 3.53 ± 0.28 | 0.60 ± 0.15 | 2.93 ± 0.32 | 0.299 ± 0.026 | 98 |
DISCUSSION
The SNARE complex that is disassembled by NSF and αSNAP is a parallel four-helix bundle composed of four distinct helices. Recent biophysical analysis has shown that αSNAP binds to purified, non-membrane-anchored SNARE complexes in a 1:1 stoichiometry (15). Our αSNAP·SC binding data (Fig. 2B) are consistent with these findings while not ruling out the presence of much weaker binding sites. Indeed, all published structural analyses, including chemical cross-linking, mass spectrometry, and electron microscopy, suggest that three copies of αSNAP are present in the 20 S complex (17–21). It is possible that, although there is only one detectable binding site for αSNAP on the SC, there are much weaker binding sites that are not detected in the assays used here or used previously (15). In any case, the kinetic titration analyses presented here (Figs. 2E and 3E) represent the first functional analysis of αSNAP stoichiometry in NSF-mediated SNARE complex disassembly and suggest that multiple αSNAP molecules are required (Fig. 2E). Consistent with this analysis, a trimeric αSNAP acts unimolecularly in disassembly (Fig. 3F).
The observation that only a single αSNAP appears to bind to the SNARE complex in solution suggests that there is one preferred binding site (Fig. 6). It is possible that other binding sites are present but too weak to be detected in solution state binding assays. αSNAP binds to different ternary SNARE complexes as well as binary t-SNARE complexes, which may indicate that the preferred site is a surface created by the association of the t-SNARE components (15). Although cryo-EM analysis of the 20 S complex led to a model in which each of the three αSNAP molecules contacts the SNARE complex, the analysis required 3-fold averaging of the images and therefore cannot discern the inherent asymmetry of the SNARE complex or determine potential differences in its interactions with αSNAP (21).
FIGURE 6.

Model for SNARE complex disassembly by NSF. αSNAP molecules (blue cylinders) associated with the membrane using hydrophobic loops (red circles) bind the SC (multicolored cylinder) and NSF to form the 20 S complex (i). The primary binding site on the SC for αSNAP is shown as a yellow patch, and the gray bar indicates the membrane. Outward and downward movement of the N-domains upon ATP hydrolysis (power stroke) may release αSNAP transiently (ii), and rotation of the NSF hexamer positions a different αSNAP at the primary binding site (iii). ATP hydrolysis and rotation steps are repeated (iv and v), and disassembled SNARE proteins and αSNAP remain membrane-associated (vi). Displacement of ADP for ATP resets the N-domains into an “up” conformation, where a new αSNAP·SC complex can be disassembled (vii). If αSNAP were always bound to both the NSF N-domain and to the SC, then as the SC is unwound and NSF translocates, the NSF N-domain would tilt more and more with respect to the SC, shortening the lever arm and potentially reducing the force that can be applied to the complex. See “Discussion” for details.
Our design of the trimerized αSNAP3 was inspired by the observation that a 20-fold lower αSNAP concentration was needed to support the NSF-mediated disassembly of liposome-anchored SNARE complexes versus soluble complexes, such as those used in this paper (23). In that work, it was suggested that the enhanced effect of lipid-associated αSNAP arises from an increase in local concentration due to restriction to the two-dimensional bilayer, or perhaps that the lipid association produces a conformational change in αSNAP. This observation raises the possibility that fewer molecules of αSNAP might be needed for SC disassembly on membranes (i.e. that the functional stoichiometry of αSNAP in SC disassembly differs from that found here). Nevertheless, as noted above, the 14-fold lower KmαSNAP for disassembly mediated by αSNAP3 than by monomeric αSNAP strongly argues that more than one αSNAP molecule is utilized in disassembly. If the reduced concentration of αSNAP required to disassemble lipid-anchored SC were to arise from a change in stoichiometry, then trimerized αSNAP3 would be predicted to produce only a 3-fold change in KmαSNAP. Moreover, the similarity in kcatdisassembly between αSNAP and αSNAP3 (Table 3) suggests that the reaction is proceeding through the same rate-limiting step. Given that the soluble NSF enzyme disassembles both membrane-bound and soluble SNARE complexes, the most parsimonious interpretation of these data is that the effect of the interaction of αSNAP with the lipid bilayer is to increase local concentration rather than to fundamentally alter the mechanism of NSF-mediated disassembly of the 20 S complex, in particular the stoichiometry of αSNAP in this reaction.
αSNAP interacts with NSF independent of the SNARE complex, as shown by the enhancement of ATP hydrolysis activity (Table 4) (12–14). ATP hydrolysis is further enhanced when the SNARE complex is present, giving a 13-fold increase in hydrolysis versus free NSF. If multiple αSNAP molecules are required to bind to and stabilize a conformation of NSF that efficiently hydrolyzes ATP, the SNARE complex may help to organize αSNAP and NSF molecules into a productive arrangement, given the suggestion from the cryo-EM data that the N-terminal portion of the SNARE complex interacts with the NSF N-domains in the 20 S complex as well as with αSNAP. The enhanced SNARE disassembly and ATPase efficiency of the unimolecular αSNAP3 may be due to the reduced probability of forming non-productive NSF·αSNAP complexes (i.e. those with only one or two αSNAPs bound) versus monomeric αSNAP. An alternative model is that αSNAP serves as a processivity factor that must remain associated with NSF and the SNARE complex during each round of ATP hydrolysis.
The nature of the force transduction needed to disassemble the SNARE complex and the role of multiple αSNAP molecules in this process are not known. The αSNAP C-terminal domain binds to the NSF N-domain (19, 22). The cryo-EM structure of the ADP·AlFx-bound 20 S complex indicates that the N-terminal region of αSNAP interacts with the C-terminal (membrane-proximal) surface of the SNARE complex, and the αSNAP C-terminal domain binds to the NSF N-domain, which in turn contacts the SNARE complex (21) (Fig. 6). Structural analysis of isolated NSF reveals that in the ATP-bound state, the NSF N-domains lie above the plane of the D1 ring, where they interact with the αSNAP·SNARE complex (21, 33), whereas in the ADP-bound state, the N-domains lie near the periphery of the D1 ring (Fig. 6, vi). The nucleotide-dependent downward movement of the NSF N-domains may represent the power stroke that pulls αSNAP and one or more components of the SNARE complex as part of the disassembly process (Fig. 6, i to ii, iii to iv, and v to vi). We can envision at least two scenarios for the role of multiple αSNAP molecules in this process. As illustrated in Fig. 6, the processive unwinding of the SNARE complex might require NSF to rotate relative to the SNARE complex as part of the power stroke, with engagement of the principal binding site (yellow patch) now mediated by a different αSNAP molecule (Fig. 6, iv and v). Alternatively, although there is one principal binding site on the SNARE complex, weaker sites might provide separate attachments to the different NSF subunits, which can move with respect to one another during the ATPase cycle and thereby generate force needed to separate the SNARE proteins.
The available structural data complicate any model in which αSNAP serves as a lever arm in the force transduction needed to disassemble SNARE complex. If αSNAP is bound by the SNARE complex at one end and by the NSF N-domain at the other, then as the enzyme traverses the SNARE complex, the rigid αSNAP molecule would have to be held at successively larger angles relative to the 6-fold axis of NSF and the long axis of the SNARE coiled-coil, with the N-domain positioned increasingly away from the axis (Fig. 6). There may be sufficient flexibility in the radial position of the N-domain to allow this, but if downward motion of NSF-N constitutes the power stroke, a starting point farther from the axis would diminish the force applied to the SNARE bundle as it shortens (Fig. 6, compare i, iii, and v). A possible model to reconcile this is that SNARE disassembly is cooperative; more energy is required initially to unwind the bundle than when most of it is disassembled. On the other hand, it is possible that αSNAP has no direct role in the force transduction between NSF and the SNARE complex but rather stabilizes a conformation of the enzyme in which hydrolysis and N-domain movements are more strongly coupled, and/or enhances processivity.
Single molecule force measurements suggest that the free energy needed to disassemble a single SNARE complex is about 39 kcal mol−1 (4). We find that 10 ATPs are needed to disassemble a SNARE complex in the presence of the trimeric αSNAP3, considerably lower than the value of 50 ATPs reported to be needed when nearly saturating monomeric αSNAP is used (32). The 14-fold diminution of Km for αSNAP3 in SNARE complex disassembly suggests that the trimerized molecule does more than simply providing more copies of the molecule. The foldon domain may help to restrict the orientation of the multiple αSNAPs needed for disassembly and thereby favor the correct orientation for binding to the SNARE complex and to NSF (Fig. 3A). In vivo, the SNARE complex includes transmembrane anchors (2), and αSNAP contains a membrane-interacting loop near its N terminus (23). The restriction of these components to the two-dimensional plane of the membrane probably increases the probability of forming the αSNAP·SNARE complex, so it is conceivable that these differences lead to in vivo disassembly kinetics and energetics different from those measured with the soluble components. Nonetheless, the more efficient SNARE complex disassembly observed with αSNAP3 versus monomeric αSNAP may due to αSNAP3 acting as a mimic of membrane-associated αSNAPs, and should prove to be a useful tool for further mechanistic studies of NSF activity and SNARE disassembly.
Acknowledgment
We thank Axel Brunger for comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM64798 (to D. H.) and R01 MH58570 (to W. I. W.).
- NSF
- N-ethylmaleimide-sensitive factor
- AAA+
- ATPase associated with various cellular activities
- SC
- SNARE complex
- αSNAP
- soluble NSF attachment protein α
- αSNAP3
- αSNAP fused to bacteriophage T4 fibritin “foldon” domain (trimeric αSNAP).
REFERENCES
- 1. Brunger A. T. (2005) Structure and function of SNARE and SNARE-interacting proteins. Q. Rev. Biophys. 38, 1–47 [DOI] [PubMed] [Google Scholar]
- 2. Söllner T., Whiteheart S. W., Brunner M., Erdjument-Bromage H., Geromanos S., Tempst P., Rothman J. E. (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324 [DOI] [PubMed] [Google Scholar]
- 3. Jahn R., Scheller R. H. (2006) SNAREs: engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643 [DOI] [PubMed] [Google Scholar]
- 4. Gao Y., Zorman S., Gundersen G., Xi Z., Ma L., Sirinakis G., Rothman J. E., Zhang Y. (2012) Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337, 1340–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Block M. R., Glick B. S., Wilcox C. A., Wieland F. T., Rothman J. E. (1988) Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc. Natl. Acad. Sci. U.S.A. 85, 7852–7856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fleming K. G., Hohl T. M., Yu R. C., Müller S. A., Wolpensinger B., Engel A., Engelhardt H., Brünger A. T., Söllner T. H., Hanson P. I. (1998) A revised model for the oligomeric state of the N-ethylmaleimide-sensitive fusion protein, NSF. J. Biol. Chem. 273, 15675–15681 [DOI] [PubMed] [Google Scholar]
- 7. Tagaya M., Wilson D. W., Brunner M., Arango N., Rothman J. E. (1993) Domain structure of an N-ethylmaleimide-sensitive fusion protein involved in vesicular transport. J. Biol. Chem. 268, 2662–2666 [PubMed] [Google Scholar]
- 8. Whiteheart S. W., Rossnagel K., Buhrow S. A., Brunner M., Jaenicke R., Rothman J. E. (1994) N-Ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J. Cell Biol. 126, 945–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matveeva E., Whiteheart S. W. (1998) The effects of SNAP/SNARE complexes on the ATPase of NSF. FEBS Lett. 435, 211–214 [DOI] [PubMed] [Google Scholar]
- 10. White S. R., Lauring B. (2007) AAA+ ATPases: achieving diversity of fundtion with conserved machinery. Traffic 8, 1657–1667 [DOI] [PubMed] [Google Scholar]
- 11. Nagiec E. E., Bernstein A., Whiteheart S. W. (1995) Each domain of the N-ethylmaleimide-sensitive fusion protein contributes to its transport activity. J. Biol. Chem. 270, 29182–29188 [DOI] [PubMed] [Google Scholar]
- 12. Barnard R. J. O., Morgan A., Burgoyne R. D. (1997) Stimulation of NSF ATPase activity by α-SNAP is required for SNARE complex disassembly and exocytosis. J. Cell Biol. 139, 875–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morgan A., Dimaline R., Burgoyne R. D. (1994) The ATPase activity of N-ethylmaleimide-sensitive fusion protein (NSF) is regulated by soluble NSF attachment proteins. J. Biol. Chem. 269, 29347–29350 [PubMed] [Google Scholar]
- 14. Steel G. J., Morgan A. (1998) Selective stimulation of the D1 ATPase domain of N-ethylmaleimide-sensitive fusion protein (NSF) by soluble NSF attachment proteins. FEBS Lett. 423, 113–116 [DOI] [PubMed] [Google Scholar]
- 15. Vivona S., Cipriano D. J., O'Leary S., Li Y. H., Fenn T. D., Brunger A. T. (2013) Disassembly of all SNARE complexes by N-ethylmaleimide-sensitive factor (NSF) is initiated by a conserved 1:1 interaction between α-soluble NSF attachment protein (SNAP) and SNARE complex. J. Biol. Chem. 288, 24984–24991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rice L. M., Brunger A. T. (1999) Crystal structure of the vesicular transport protein Sec17: implications for SNAP function in SNARE complex assembly. Mol. Cell 4, 85–95 [DOI] [PubMed] [Google Scholar]
- 17. Marz K. E., Lauer J. M., Hanson P. I. (2003) Defining the SNARE complex binding surface of α-SNAP: implications for SNARE complex disassembly. J. Biol. Chem. 278, 27000–27008 [DOI] [PubMed] [Google Scholar]
- 18. Hohl T. M., Parlati F., Wimmer C., Rothman J. E., Söllner T. H., Engelhardt H. (1998) Arrangement of subunits in 20 S particles consisting of NSF, SNAPs, and SNARE complexes. Mol. Cell 2, 539–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wimmer C., Hohl T. M., Hughes C. A., Müller S. A., Söllner T. H., Engel A., Rothman J. E. (2001) Molecular mass, stoichiometry, and assembly of 20 S particles. J. Biol. Chem. 276, 29091–29097 [DOI] [PubMed] [Google Scholar]
- 20. Furst J., Sutton R. B., Chen J., Brunger A. T., Grigorieff N. (2003) Electron cryomicroscopy stucture of N-ethyl maleimide sensitive factor at 11 Å resolution. EMBO J. 22, 4365–4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chang L. F., Chen S., Liu C. C., Pan X., Jiang J., Bai X. C., Xie X., Wang H. W., Sui S. F. (2012) Structural characterization of full-length NSF and 20S particles. Nat. Struct. Mol. Biol. 19, 268–275 [DOI] [PubMed] [Google Scholar]
- 22. Matveeva E. A., May A. P., He P., Whiteheart S. W. (2002) Uncoupling the ATPase activity of the N-ethylmaleimide sensitive factor (NSF) from 20S complex disassembly. Biochemistry 41, 530–536 [DOI] [PubMed] [Google Scholar]
- 23. Winter U., Chen X., Fasshauer D. (2009) A conserved membrane attachment site in α-SNAP facilitates N-ethylmaleimide-sensitive factor (NSF)-driven SNARE complex disassembly. J. Biol. Chem. 284, 31817–31826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Güthe S., Kapinos L., Möglich A., Meier S., Grzesiek S., Kiefhaber T. (2004) Very fast folding and association of a trimerization domain from bacteriophage T4 fibritin. J. Mol. Biol. 337, 905–915 [DOI] [PubMed] [Google Scholar]
- 25. Papanikolopoulou K., Forge V., Goeltz P., Mitraki A. (2004) Formation of highly stable chimeric trimers by fusion of an adenovirus fiber shaft fragment with the foldon domain of bacteriophage t4 fibritin. J. Biol. Chem. 279, 8991–8998 [DOI] [PubMed] [Google Scholar]
- 26. Rydzanicz R., Zhao X. S., Johnson P. E. (2005) Assembly PCR oligo maker: a tool for designing oligodeoxynucleotides for constructing long DNA molecules for RNA production. Nucleic Acids Res. 33, W521–W525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stemmer W. P., Crameri A., Ha K. D., Brennan T. M., Heyneker H. L. (1995) Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164, 49–53 [DOI] [PubMed] [Google Scholar]
- 28. Fasshauer D., Antonin W., Margittai M., Pabst S., Jahn R. (1999) Mixed and non-cognate SNARE complexes. J. Biol. Chem. 274, 15440–15446 [DOI] [PubMed] [Google Scholar]
- 29. Lauer J. M., Dalal S., Marz K. E., Nonet M. L., Hanson P. I. (2006) SNARE complex zero layer residues are not critical for N-ethylmaleimide-sensitive factor-mediated disassembly. J. Biol. Chem. 281, 14823–14832 [DOI] [PubMed] [Google Scholar]
- 30. Buxbaum E. (1999) Co-operating ATP sites in the multiple drug resistance transporter Mdr1. Eur. J. Biochem. 265, 54–63 [DOI] [PubMed] [Google Scholar]
- 31. Fasshauer D., Eliason W. K., Brünger A. T., Jahn R. (1998) Identification of a minimal core of the synaptic SNARE complex sufficient for reversible assembly and disassembly. Biochemistry 37, 10354–10362 [DOI] [PubMed] [Google Scholar]
- 32. Cipriano D. J., Jung J., Vivona S., Fenn T. D., Brunger A. T., Bryant Z. (2013) Processive ATP-driven substrate disassembly by the N-ethylmaleimide-sensitive facttor (NSF) molecular machine. J. Biol. Chem. 288, 23436–23445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moeller A., Zhao C., Fried M. G., Wilson-Kubalek E. M., Carragher B., Whiteheart S. W. (2012) Nucleotide-dependent conformational changes in the N-ethylmaleimide sensitive factor (NSF) and their potential role in SNARE complex disassembly. J. Struct. Biol. 177, 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]