

Background: Chronic exposure to Cr(VI) causes cell transformation.
Results: Cr(VI)-transformed cells exhibit activated EGFR, reduced ROS generation, and development of apoptosis resistance.
Conclusion: Constitutive activation of EGFR promotes tumorigenesis of Cr(VI)-transformed cells.
Significance: The findings provide a new understanding on carcinogenic mechanisms of not only Cr(VI) but also other metals, such as arsenic and nickel. It also can be used to formulate cancer prevention strategies.
Keywords: Antioxidant, Apoptosis, Carcinogenesis, Epidermal Growth Factor Receptor (EGFR), Reactive Oxygen Species (ROS), Cr(VI)
Abstract
Hexavalent chromium (Cr(VI)) compounds are well-established lung carcinogens. Epidermal growth factor receptor (EGFR) is a tyrosine kinase transmembrane receptor that regulates cell survival, tumor invasion, and angiogenesis. Our results show that chronic exposure of human bronchial epithelial (BEAS-2B) cells to Cr(VI) is able to cause malignant cell transformation. These transformed cells exhibit apoptosis resistance with reduced poly ADP-ribose polymerase cleavage (C-PARP) and Bax expression and enhanced expressions of Bcl-2 and Bcl-xL. These transformed cells also exhibit reduced capacity of reactive oxygen species (ROS) generation along with elevated expression of antioxidant manganese superoxide dismutase 2 (SOD2). The expression of this antioxidant was also elevated in lung tumor tissue from a worker exposed to Cr(VI) for 19 years. EGFR was activated in Cr(VI)-transformed BEAS-2B cells, lung tissue from animals exposed to Cr(VI) particles, and human lung tumor tissue. Further study indicates that constitutive activation of EGFR in Cr(VI)-transformed cells was due to increased binding to its ligand amphiregulin (AREG). Inhibition of EGFR or AREG increased Bax expression and reduced Bcl-2 expression, resulting in reduced apoptosis resistance. Furthermore, inhibition of AREG or EGFR restored capacity of ROS generation and decreased SOD2 expression. PI3K/AKT was activated, which depended on EGFR in Cr(VI)-transformed BEAS-2B cells. Inhibition of PI3K/AKT increased ROS generation and reduced SOD2 expression, resulting in reduced apoptosis resistance with commitment increase in Bax expression and reduction of Bcl-2 expression. Xenograft mouse tumor study further demonstrates the essential role of EGFR in tumorigenesis of Cr(VI)-transformed cells. In summary, the present study suggests that ligand-dependent constitutive activation of EGFR causes reduced ROS generation and increased antioxidant expression, leading to development of apoptosis resistance, contributing to Cr(VI)-induced tumorigenesis.
Introduction
Hexavalent chromium compounds (Cr(VI)),2 widely used in the industry, are well-established human lung carcinogens (1). Epidemiological studies have shown that occupational and environmental exposure to Cr(VI) is associated with a high rate of lung cancer (2–4). The International Agency for Research on Cancer (IARC) has classified Cr(VI) as Group 1 human carcinogens (2). Although Cr(VI) has been identified as a human carcinogen, the molecular mechanisms of its carcinogenesis are still poorly understood.
The tyrosine-kinase epidermal growth factor receptor (EGFR, ERBB1), one of the most versatile signaling units in biology, regulates key processes of cell biology, such as proliferation, survival, and differentiation during development, tissue homeostasis, and tumorigenesis (5). EGFR signaling is activated by binding to its ligands, resulting in homodimerization of EGFR molecules or heterodimerization with other related receptors (5). EGFR ligands include epidermal growth factor (EGF), transforming growth factor-α (TGFA), amphiregulin (AREG), diphtheria toxin receptor/heparin-binding EGF-like growth factor (DTR), epiregulin (EREG), betacellulin (BTC), and epigen (EPGN) (5). In cancer, subsequent administration of EGFR ligands caused EGFR activation of a downstream cascade, leading to un-controlled tumor proliferation (5). Downstream signaling pathways that are activated via the EGFR include phosphorylation of PI3K/AKT (5, 6). The PI3K/AKT pathway is an intracellular signaling pathway important in apoptosis and cancer (7). Activation of PI3K/AKT phosphorylates and inhibits the pro-apoptotic Bcl-2 family members Bad, Bax, and caspase-9 (8).
EGFR transactivation can be induced by many stimuli through different pathways, including ligands and mutations (5, 9). Another mechanism of EGFR transactivation is related to reactive oxygen species (ROS) (10). ROS regulate numerous intracellular signal transduction pathways as well as the activities of various transcription factors. Previous studies have demonstrated that ROS induce EGFR transactivation via transient inhibition of SHP-2 in cardiac fibroblasts (11, 12).
EGFR is commonly overexpressed or mutated in non-small cell lung cancer (7, 13). This protein is overexpressed in 40–80% of non-small cell lung cancer (NSCLC), and a subgroup of patients with a specific mutation in its gene have a marked clinical response to EGFR tyrosine kinase inhibitors. Although it has been reported that chromium-treated cells exhibited an increased EGFR expression (14, 15), the role of EGFR in Cr(VI)-induced tumorigenesis remains to be investigated. The present study has shown that chronic-exposure of Cr(VI) induced EGFR signaling and caused cell transformation. In transformed cells, constitutively activated EGFR suppressed ROS generation and elevated antioxidant SOD2 expression, leading to apoptosis resistance, which in turn contributes to tumorigenesis.
EXPERIMENTAL PROCEDURES
Chemicals and Laboratory Wares
Sodium dichromate dehydrate (Na2Cr2O7), annexin V/propidium iodide (PI), and 5-fluorouracil (5-FU: F6627) were from Sigma. Insoluble zinc chromate particle (ZnCrO4 4Zn(OH)2) was from Alfa Aesar (Ward Hill, MA). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), gentamicin, and l-glutamine were from Invitrogen (Carlsbad, CA). shRNAs of EGFR and SOD2 were from Origene (Rockville, MD). AREG siRNA was from Santa Cruz Biotechnology (Santa Cruz, CA). RNeasy Mini kit and plasmid prep kit were from Qiagen (Valencia, CA). M-MLV reverse transcriptase was from Promega (Madison, WI). Oligo (dT)20, AccuPrime TaqDNA Polymerase High Fidelity, and pGEM-T easy cloning vector were from Invitrogen. Luciferase Assay System was from Promega (Fitchburg, WI). AG1478 and LY294002 were obtained from Calbiochem (Darmstadt, Germany). 5-(and -6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) was from Molecular Probes (Eugene, OR). Antibodies against AREG, BTC, and control human IgG (1-001-A) were from R&D Systems (Minneapolis, MN). Antibodies against GAPDH and β-actin were from Santa Cruz Biotechnology. Antibody against SOD2 was from Millipore (Billerica, MA). Antibodies against AKT, pAKTSer473, Bax, Bcl2, EGFR, pEGFR1068, PI3K, pPI3K458, catalase, cleaved PARP, and wortmanin were from Cell Signaling Tech (Danvers, MA). Antibodies against Alexa Fluor® 488 goat anti-mouse IgG1 (γ1) and Alexa Fluor® 568 goat anti-rabbit IgG (H+L) were from Invitrogen.
Cr(VI)-induced Cell Transformation
Human bronchial epithelial cells (BEAS-2B) were purchased from the American Type Culture Collection. The cells were maintained in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin in 10-cm dishes. After 90% of confluence, cells were treated with 0.25 μm Cr(VI). The fresh medium was added for every 3 days. After 24 weeks, 1 × 104 cells were suspended in 2 ml of culture medium containing 0.35% agar and seeded into 6-well plates with 0.5% agar base layer, and maintained in an incubator for 3 weeks. The Cr(VI)-transformed cells (BEAS-2B-Cr) from anchorage-independent colonies were picked up and continued to grow in DMEM. Passage-matched cells without Cr(VI) treatment were used as control.
Chronic Exposure of Animals to Cr(VI) Particle
6–8 weeks old, female BALB/c mice were purchased Jackson Laboratory (Bar Harbor, MN). Endotoxin-free basic zinc chromate particle was crushed using mortar and pestle and then washed with distill water and acetone to make a size of 4.7 μm and a purity of 99–100%. Cr(VI) particle was suspended in sterile 0.9% sodium chloride solution at a concentration of 0.6 mg/ml and prepared as previously described (16). Animals under a light anesthesia (isoflurane) were intranasally exposed to a 50 μl dose of chromate or saline once a week up to 12 weeks. Lungs of animals were isolated and subject to freshly frozen and 10% formalin fixation for immunoblotting and immunohistochemistry analysis, respectively.
Patient Lung Sample Collection
Lung tumor tissue and adjacent normal tissue from a chromate worker at age of 62 were obtained during surgery at Tokushima University hospital, Tokushima, Japan. This nonsmoking worker had been exposed to a chromate (Na2Cr2O7, K2Cr2O7, CrO3, and Cr2O3) producing industry at Hokkaido, Japan for 19 years. The worker was diagnosed as stage 1 (T1N0M0) squamous lung carcinoma.
Immunoblot Analysis
Cell lysates were prepared in RIPA buffer. The protein concentration was measured using Bradford Protein Assay Reagent (Bio-Rad) and 30 μg of protein was separated by SDS-PAGE, and incubated with primary antibodies. The blots were then re-probed with second antibodies conjugated to horseradish peroxidase. Immunoreactive bands were detected by the enhanced chemiluminescence reagent (Amersham Biosciences).
Fluorescence Immunohistochemistry Analysis
For the cell immunohistochemistry analysis, both BEAS-2B and BEAS-2B-Cr cells were grown on chamber slides, washed with phosphate-buffered saline solution (PBS), and fixed with a 4% paraformaldehyde (PFA) solution. Then, the cells were incubated with 1% Triton X-100 followed by washing with 0.02% Tween 20. After permeabilization, cells were incubated 1 h with 0.1% Triton X-100 and 10% normal serum. Primary antibodies were added and incubated at 4 °C for overnight. The cells were incubated with secondary antibody for 1 h. The slides were mounted with Vectashield mounting medium. Finally, cells were visualized using Olympus BX53 fluorescence microscope (Center valley, PA).
For the formalin-fixed lung tissue immunohistochemistry analysis, air-dried tissue sections were rinsed, blocked, and incubated primary antibody overnight. Second antibody was added for 1 h. 300 μl of the diluted DAPI solution was added and incubated 2–5 min at room temperature. DAPI binds to DNA and is a convenient nuclear counterstain. The sections were mounted with an anti-fade mounting media. Olympus BX53 fluorescence microscope was used for visualization.
Hematoxylin and Eosin (HE) Staining
Mouse and patient lung tissue sections were deparaffinized, rehydrated, and rinsed, then hematoxylin was added. After rinsing and decolorization, the sections were counterstained in eosin and mounted with cytoseal. The sections were visualized using Olympus BX53 fluorescence microscope.
Sequencing of the EGFR Gene
RNA was isolated from BEAS-2B and BEAS-2B-Cr cells using the RNeasy mini kit according to the manufacturer's instruction. RNA concentration was determined by spectrophotometer and then adjusted to a concentration of 200 ng/ml. 2 μg of RNA was reverse transcribed by M-MLV reverse transcriptase with 0.5 μg oligo (dT)20. The reaction mixture was incubated at 42 °C for 60 min and then at 72 °C for 15 min. 1 μl of each cDNA was used for polymerase chain reaction (PCR). The PCR reactions were performed using AccuPrime TaqDNA Polymerase High Fidelity in a 50-μl reaction volume. The primer sequences for EGFR gene were as follows: the forward primer, 5-TGTAAAACGACGGCCAGTCCCTGGGGATCGGCCTCTTCATGCGA-3 and the reverse primer, 5-CAGGAAACAGCTATGACCATTCCAATGCCATCCACTTGAT-3. The cycling condition was followed: initial denaturation at 94 °C for 2 min, followed by 40 cycles at 94 °C for 30 s, 59 °C for 30 s, 68 °C for 1 min. Amplified DNAs were separated on 1% agarose gel, and the bands were visualized by ethidium bromide and photographed under ultraviolet transillumination. PCR products were subcloned into the pGEM-T easy cloning vector and sequenced. Data were analyzed using BLAST and chromatograms by manual review.
Plasmid Transfection and Stable Transfection Cells
Transfection was performed using LipofectamineTM 2000 (Invitrogen) according to the manufacturer's protocol. Briefly, cells were seeded in 6-well culture plates and transfected with 4 μg plasmid at ∼70% confluence. Expression of transfected protein was measured by immunoblotting at 48 h of post-transfection. To make stable transfection cells with shRNA EGFR, the cells were cultured in the DMEM containing 2 μg/ml puromycin. After 3 months, EGFR expression was confirmed using immunoblotting.
Real-time PCR
RNA was extracted and purified using the RNeasy mini kit. 0.5 μg of RNA was reverse transcribed using qScript cDNA synthesis kit (Quanta Biosciences). Connexin primers were designed using Primer-Blast yielding the following sense and antisense sequences: hAR, 5′-GGC CAT TAT GCT GCT GGA T- 3′ and 5′-TGT GGT CCC CAG AAA ATG GT-3′; hBTC, 5′-GGG AGA TGC CGC TTC GT- 3′ and 5′-TGC TCC AAT GTA GCC TTC ATC A-3′. Values were normalized to those of the PPIA reference gene: cyclophillin A (PPIA), 5′-AGA CAA GGT CCC AAA GAC-3′ and 5′ACC ACC CTG ACA CAT AAA-3′. All primers were tested using standard curves with 10-fold serial dilutions. The qPCR was performed in the CFX96 Real-time PCR Detection System (Bio-Rad) using Perfecta Sybr Green Fastmix (Quanta Biosciences), and the data analyzed with CFX Manager software (Bio-Rad).
Apoptosis Analysis
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) double staining was used to measure percentile of apoptosis according to manufacturer's protocol. Briefly, cells were cultured with DMEM medium in 10-cm dishes. After 80–90% confluence, the cells were treated with 20 μm of Cr(VI) for 24 h. The cells were washed with PBS and digested with 0.25% trypsin/EDTA followed by re-suspension in 500 μl of 1× binding buffer and addition of Annexin V-FITC/PI. The apoptotic cells were measured using flow cytometry.
Luciferase Assay
AREG luciferase activity was measured using luciferase reporter assay. Both BEAS-2B and BEAS-2B-Cr cells were seeded on 12-well plates (5 × 105 cells/well). The cells were transfected with 2 μg of plasmid using Lipofectamine 2000. After 24 h of transfection, the cells were harvested for luciferase assay using Luciferase Assay System in a Glomax luminometer (Promega). Data were normalized to total protein determined by the Bradford assay.
Detection of ROS
The cells were cultured in 96-well plates with 5 × 104 cells/well. The cells were treated with 5 μm of Cr(VI) for 24 h and then incubated with DCFDA (final concentration, 10 μm) for 20 min at 37 °C. The cells were washed twice with PBS. Fluorescence intensity was measured using a Gemini XPS fluorescence microplate spectrofluorometer (Molecular Devices).
Neutralization of EGFR
Chromium-transformed cells were incubated in fresh DMEM medium containing antibody against AREG (5 μg/ml) or BTC (5 μg/ml), or control IgG (1 μg/ml) for 2 h at 37 °C. Whole cell lysate was harvested for immunoblotting. Expression of p-EGFR and total EGFR was examined.
Xenograft Tumor Model
6-week-old female athymic nude mice were purchased from Charles River Laboratories (Wilmington, MA). The mice were housed in sterilized filter-topped cages and maintained in a pathogen-free animal facility at the Chandler Medical Center, University of Kentucky. All animals were handled according to the Institutional Animal Care and Use Committee (IACUC) guidelines. BEAS-2B, BEAS-2B-Cr, and EGFR knockdown BEAS-2B-Cr cells (1 × 106cells) in 100 μl of a mixture of 1× DMEM and Matrigel (BD Biosciences) were subcutaneously (s.c.) injected on the left flank of each mouse. Tumors were measured using an external caliper and volume was calculated using the formula: (length × width2)/2. After 21 days, mice were euthanized, and the tumors were harvested for Western blot analysis and immunofluorescence.
Statistical Analysis
Data were expressed as the mean ± S.D. Statistical significance of differences among treatment groups were determined by Student's t test. A p < 0.05 was considered as statistically significance.
RESULTS
Activations of EGFR in Cr(VI)-transformed Cells and Cr(VI)-exposed Animals
To investigate whether chronic exposure of Cr(VI) activates EGFR in vitro and in vivo, both BEAS-2B cells and BALB/C mice were exposed to low doses of Cr(VI) for 6 months and 3 months, respectively, as described under “Experimental Procedures.” Fig. 1A shows that EGFR began to be activated from 2 months of Cr(VI) exposure in BEAS-2B cells. Phosphorylation of EGFR at tyrosine1068 was much higher at the exposure duration of 4 months and 6 months than that of 2 months and 3 months. At this stage of chronic Cr(VI) exposure (6 months), the cells were malignant transformed as examined by anchorage-independent cell growth (soft agar) assay (data not shown), similar to those observation in our previous study (17). Phosphorylation of EGFR at Tyr-1068 was highest in both two clones of transformed cells isolated from the cells exposed to Cr(VI) for 6 months compared with various durations of Cr(VI) exposure (Fig. 1B). To explore whether Cr(VI) exposure causes activation of EGFR in vivo, BALB/c mice were nasally exposed to insoluble Cr(VI) particle (0.6 mg/ml and 1.2 mg/ml) up to 3 months. The results show that chronic chromium exposure at both 0.6 mg/ml and 1.2 mg/ml induced phosphorylation of EGFR in mouse lung tissue (Fig. 1C). Lung tissue from control group without Cr(VI) exposure displayed normal ciliated columnar bronchial epithelial cells with 1–2 cell layers (Fig. 1D). In contrast, lung tissues from Cr(VI) exposure animals exhibited reactive bronchial epithelial hyperplasia (up to 6 and 9–10 cell layers at 0.6 mg/ml and 1.2 mg/ml of Cr(VI) treatment, respectively) (Fig. 1D). At 1.2 mg/ml Cr(VI) exposure, the ciliated columnar bronchial epithelial cells underwent squamous cells, indicating reactive process ongoing in the Cr(VI)-exposed lung. We have also examined p-EGFR expression in lung tumor and adjacent normal tissues from a patient occupationally exposed to Cr(VI) for 19 years with diagnosis of stage 1 squamous lung carcinoma. The results show that p-EGFR was highly expressed (green fluorescence) in parenchyma of tumor lung tissue, but not in the adjacent normal tissue (Fig. 1E). HE staining of these tissues shows normal bronchial vessel (artery) surrounded with normal pulmonary parenchymal tissue and alveolar macrophages (Fig. 1E, upper right) and a poorly differentiated non-small cell carcinoma (squamous cell carcinoma) with central tumor necrosis and cellular debris (Fig. 1E, bottom right). The results suggest that EGFR activation may reflect an early biomarker for Cr(VI)-induced lung damage and be involved in this process.
FIGURE 1.

Activation of EGFR in Cr(VI)-transformed cells and Cr(VI)-exposed animals and increased EGFR ligands AREG and BTC expression in Cr(VI)-transformed cells. A, BEAS-2B cells were incubated with PBS or 0.25 μm of Cr(VI) for 6 months. Cells were collected for immunoblotting analysis every month of Cr(VI) exposure. B, at 6 months of Cr(VI) exposure, the cells were subjected to cell transformation (soft agar) assay. Two clones from single colony were picked up and continued to grow. These cells are considered as transformed cells (BEAS-2B-Cr). EGFR phosphorylation and total EGFR were detected by immunoblotting. C and D, female BALB/c mice were treated intranasally with either insoluble Cr (VI) (0.6 mg/ml and 1.2 mg/ml) or PBS (vehicle for the control group), once a day, 5 days/week. After 3 months, mice were euthanized using CO2 and lung was isolated for further examination. EGFR phosphorylation and total EGFR were detected by immunoblotting (C). Hematoxylin/eosin staining was also performed to examine general pathological analysis (D). E, formalin-fixed lung tissue slides from tumor and normal adjacency obtained from a worked exposed to Cr(VI) were used to fluorescence immunostaining with p-EGFR (green) and DAPI (nuclear control, blue). F, RNA was isolated from BEAS-2B and BEAS-2B-Cr cells. EGFR was amplified using cDNA as templates. PCR product was subcloned into the pGEM-T easy cloning vector and sequenced. Sequences of exons 18–21 were analyzed by BLAST and chromatograms by manual review. G, mRNA level of EGFR-ligand AREG or BTC was detected by real-time RT-PCR in BEAS-2B and BEAS-2B-Cr. Data are normalized to those of GAPDH. *, p < 0.05, as compared with BEAS-2B cells. H, Expressions of AREG and BTC in BEAS-2B and BEAS-2B-Cr were analyzed by immunoblotting. I, neutralization of EGFR by its ligands AREG and BTC. BEAS-2B-Cr cells were incubated with anti-AREG or anti-BTC antibody. Expressions of p-EGFR and EGFR were examined using immunoblotting. J, BEAS-2B and BEAS-2B-Cr cells were transient transfected with the pGL3 control vector (pGL3) or AREG expression vector (pGL3 AREG). Cells were lysed, and luciferase activity assay was performed. Data were normalized to protein values. *, p < 0.05 compared with BEAS-2B cells.
AREG Ligand-dependent Activation of EGFR in Cr(VI)-transformed Cells
EGFR mutations have been reported in the patients of lung cancer, more frequently in Asian women with adenocarcinoma who never smoke (18). Those mutations cause activation of EGFR. To examine whether activation of EGFR is caused by its mutation in Cr(VI)-transformed cells, we have performed DNA sequence analysis in the exons from 18 to 21 in which its mutations have been reported in lung cancer patients. No mutation was observed in Cr(VI)-transformed cells (Fig. 1F), indicating that activation of EGFR was caused by mechanisms other than mutation. One possibility is an increase of binding to its ligands. In mammals, canonical EGFR activation involves binding of this protein to one of seven peptide growth factors: EGF, transforming growth factor-a (TGFA), heparin-binding EGF-like growth factor (HBEGF), AREG, BTC, EREG, and EPGN (19). We have examined the mRNA level of these ligands. The results show that mRNA levels of AREG and BTC were increased 190-fold and 90-fold in Cr(VI)-transformed cells compared with these in non-transformed ones (Fig. 1G), respectively. mRNA levels of other ligands including TGFA, HBEGF, EREG, and EPGN in Cr(VI)-transformed cells were similar to those in normal parent ones (data not shown). To test whether increased mRNA levels of AREG and BTC in Cr(VI)-transformed cells lead to increased expressions of these two ligands at translational level, immunoblotting analysis was performed. The results show that expression of AREG at translational level was dramatically increased in Cr(VI)-transformed cells compared with that in normal parent ones, while BTC expression in Cr(VI)-transformed cells also exhibited an observable elevation (Fig. 1H). Next, to investigate whether increased expressions of AREG and BTC are responsible for EGFR activation in Cr(VI)-transformed cells, a neutralization analysis using anti-AREG and anti-BTC antibody was performed. The result shows that incubation with anti-AREG antibody in Cr(VI)-transformed cells completely abolished phosphorylation of EGFR, while incubation with anti-BTC antibody did not alter phosphorylation of EGFR (Fig. 1I), indicating that activation of EGFR depends on AREG in Cr(VI)-transformed cells. Thus, we anticipate that promoter activity of AREG is also increased in Cr(VI)-transformed cells. As expected, the results from luciferase assay show that AREG promoter activity is markedly elevated in Cr(VI)-transformed cells compared with that in non-transformed one (Fig. 1J). Taking together, these results indicate that increased binding of AREG to EGFR is crucial for constitutive activation of EGFR.
Apoptosis Resistance of Cr(VI)-transformed Cells
Resistance to apoptotic cell death and increased cell survival in response to genotoxic insults are key characteristics of cancer cells (20). To test whether Cr(VI)-transformed cells develop apoptosis resistance, apoptosis assay was conducted in both non-transformed parent and Cr(VI)-transformed BEAS-2B cells. A relative high dose (20 μm) of Cr(VI) treatment (24 h) causes 80 and 40% of cell apoptosis in normal non-transformed cells and Cr(VI)-transformed cells (Fig. 2A), respectively. Further study shows that the levels of cleaved PARP and Bax were reduced in Cr(VI)-transformed cells compared with those in non-transformed ones (Fig. 2B). In contrast, expressions of anti-apoptotic proteins Bcl-2 and Bcl-xL were elevated, indicating that both apoptotic and anti-apoptotic proteins contribute to apoptosis resistance in Cr(VI)-transformed cells. It has been reported that short-term exposure to Cr(VI) generates reactive oxygen species (ROS), which is very important in Cr(VI)-induced carcinogenesis (17, 21–23). Although ROS generated by Cr(VI) are responsible for Cr(VI)-induced cell transformation (16), these species are also capable of causing apoptosis (24). To explore the mechanisms of development of apoptosis resistance in Cr(VI)-transformed cells, we have examined ROS generation in both Cr(VI)-transformed cells and non-transformed parent cells. The capability of ROS generation of Cr(VI)-transformed cells with or without Cr(VI) treatment was much lower than that of non-transformed cells (Fig. 2C). Cr(VI) treatment caused 30% increase of ROS generation in normal BEAS-2B cells compared with the cells without treatment, 13% increase of ROS generation was observed in Cr(VI)-transformed cells (Fig. 2C), indicating the reduced capacity of ROS generation in Cr(VI)-transformed cells. To examine that whether apoptosis resistance in Cr(VI)-transformed cells is Cr(VI) treatment specific, 5-FU, a common used chemotherapeutic drug to treat NSCLC in clinic, was used to treat both Cr(VI)-transformed cells and non-transformed ones, apoptosis was measured. The result shows that 5-FU caused apoptosis in both Cr(VI)-transformed cells and non-transformed ones in a dose-dependent manner (Fig. 2F). The percentile of apoptosis of Cr(VI)-transformed cells with or without 5-FU treatment was much lower than that of non-transformed cells (Fig. 2F). Percentile of apoptosis induced by 100 μm and 200 μm of 5-FU in non-transformed BEAS-2B cells was 81.1% and 39.6%, respectively (Fig. 2F). It was 19.2% and 36.3% in Cr(VI)-transformed cells (Fig. 2F), indicating that Cr(VI)-transformed cells is much more resistance to apoptosis compared with non-transformed ones. ROS generation in Cr(VI)-transformed cells was significantly lower than that in non-transformed cells (Fig. 2G). Antioxidants are responsible for scavenging ROS once they are produced. Among those antioxidants, superoxide dismutase (SOD) is a key one to scavenge ROS. Our result shows that expression of SOD2, a member of SOD family, was markedly increased in Cr(VI)-transformed cells (Fig. 2D). Similar to in vitro study, results from a fluorescence immunostaining of lung tissues from a worker occupationally exposed to Cr(VI) show that SOD2 (red fluorescence) was highly expressed in the parenchyma of tumor tissue, but not in the adjacent normal tissues (Fig. 2E). To test whether increased expression of SOD2 is responsible for apoptosis resistance of Cr(VI)-transformed cells, SOD2 was knockdown by its shRNA (Fig. 2H). Knockdown of SOD2 substantially increased ROS generation and apoptosis compared with scramble cells (Fig. 2, I and J), suggesting that SOD2 may be a key player for the reduced ROS generation in Cr(VI)-transformed cells and reduced ROS generation is important in apoptosis resistance of those transformed cells.
FIGURE 2.

Resistance to apoptosis and reduced capacity of ROS generation in Cr(VI)-transformed cells. A, both BEAS-2B and BEAS-2B-Cr were exposed to 20 μm of Cr(VI) for 24 h. Annexin V-FITC/PI assay was used for apoptosis analysis. B and D, BEAS-2B and BEAS-2B-Cr cells were cultured in 10-cm cell culture dishes. After 90% of confluence, the cells were harvested and cell lysates were prepared for immunoblotting. Expressions of apoptotic proteins C-PARP and Bax, anti-apoptotic proteins Bcl-2 and Bcl-xL, and antioxidant enzyme SOD2 were detected using specific antibody. C, spectrofluorometric measurement of DCFDA fluorescence in BEAS-2B and BEAS-2B-Cr cells. Cells were treated with Cr(VI) (5 μm) for 24 h. E, formalin-fixed lung tissue slides from tumor and normal adjacency obtained from a worked exposed to Cr(VI) were used to fluorescence immunostaining with SOD2 (red) and DAPI (nuclear control, blue). F and G, Both BEAS-2B and BEAS-2B-Cr cells were treated with 100 and 200 μm of 5-FU for 24 h. Apoptosis (F) and ROS generation (G) were analyzed as described in A and C. H, I, and J, BEAS-2B-Cr cells were transiently transfected with scramble and SOD2 shRNA for 48 h. SOD2 expression, ROS generation, and apoptosis were analyzed using immunoblotting, DCFDA fluorescence, and Annexin V-FITC/PI assay, respectively. A, C, F, and G, * and #, p < 0.05 compared with control without treatment and BEAS-2B cells with treatment, respectively. I and J, *, p < 0.05 compared with scramble cells.
Inhibition of EGFR or Its Ligand Restores Apoptosis and ROS Generation in Cr(VI)-transformed Cells
EGFR binding to its ligand results in receptor dimerization and autophosphorylation, triggering several signal transduction cascades (5). Our results have demonstrated that activation of EGFR is dependent on its ligand AREG. To determine whether activations of EGFR and AREG contribute to apoptosis resistance and reduced ROS production in Cr(VI)-transformed cells, expression of EGFR or AREG was inhibited by transfection of its shRNA to the cells. The results show that either knockdown of AREG or EGFR increased apoptosis (Figs. 3B and 4C) in Cr(VI)-transformed cells with increased expression of apoptotic protein Bax and reduced expression of anti-apoptotic protein Bcl-2 in response to Cr(VI) treatment (Figs. 3A and 4A). As expected, by knockdown of either EGFR or AREG, Cr(VI)-transformed cells generated more ROS upon Cr(VI) stimulation (Figs. 3C and 4E) with a decreased expression of SOD2 (Figs. 3A and 4A). We also treated Cr(VI)-transformed cells with EGFR tyrosine kinase inhibitor AG1478 to examine whether blockage of EGFR phosphorylation is able to increase apoptosis in Cr(VI)-transformed cells. The similar results (Fig. 4, B, D, and F) were observed as those using knockdown of EGFR and AREG. The above results suggest that an important role of EGFR and its ligand in apoptosis resistance of Cr(VI)-transformed cells and that ROS are downstream targets of EGFR.
FIGURE 3.
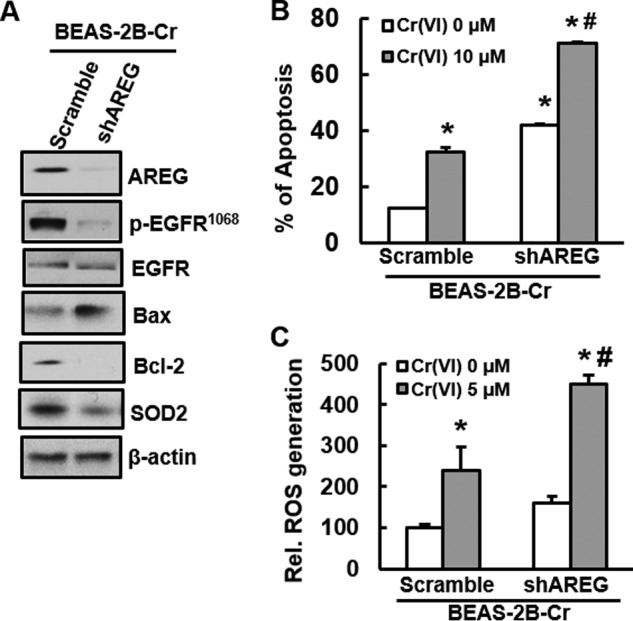
Knockdown of AREG induces apoptosis and ROS generation in Cr(VI)-transformed cells. BEAS-2B-Cr cells were transfected with scramble shRNA and AREG-shRNA for 48 h and treated with or without 5 μm of Cr (VI) as described under “Experimental Procedures.” A, immunoblotting. Expression of AREG, EGFR, Bax, Bcl-2, or SOD2 was examined in BEAS-2B-Cr cells with or without AREG knockdown. B, apoptosis analysis. BEAS-2B-Cr cells with or without AREG knockdown were treated with 10 μm of Cr(VI) for 24 h. Annexin V-FITC/PI assay was used for apoptosis analysis. C, ROS generation. DCFDA fluorescence intensity was measured in BEAS-2B-Cr with or without knockdown of AREG in the presence or absence of 5 μm Cr(VI). * and #, p < 0.05 compared with its control cells without Cr(VI) treatment and scramble cells with 5 μm Cr(VI) treatment.
FIGURE 4.

Inhibition of EGFR promotes apoptosis and increases ROS generation in Cr(VI)-transformed cells. A, C, and E, Cr(VI)-transformed cells were stably transfected with shRNA EGFR. A, BEAS-2B-Cr and its stable knockdown of EGFR (shEGFR) were cultured in 10-cm dishes. After 90% confluence, the cells were harvested and subjected to immunoblotting to examine expression of EGFR, apoptotic proteins, or SOD2 using specific antibody. C, Apoptosis analysis. BEAS-2B-Cr with or without EGFR knockdown were treated with 10 μm of Cr(VI) for 24 h. Annexin V-FITC/PI assay was used for apoptosis analysis. E, ROS generation. DCF fluorescence intensity was measured in BEAS-2B-Cr cells with or without knockdown of EGFR in the presence or absence of 5 μm Cr(VI). B, BEAS-2B-Cr cells were treated with different doses of EGFR tyrosine kinase inhibitor AG1478 (2.5 and 5.0 μm). After 24 h, the cells were collected for examination of EGFR, Bcl-2, Bax, and SOD2 expression by immunoblotting. D and F, BEAS-2B-Cr cells were pre-treated with different doses of AG1478 (2.5, 5.0, and 10.0 μm) 1 h prior to Cr(VI) treatment (10 μm). After 24 h, the cells were harvested for measurement of apoptosis (D) and ROS generation (F) using flow cytometry and spectrofluorometer, respectively. * and #, p < 0.05 compared with its control cells without Cr(VI) treatment and scramble cells with Cr(VI) treatment.
EGFR-dependent Activation of PI3K/AKT Signaling and Its Role in Apoptosis Resistance in Cr(VI)-transformed Cells
EGFR activation targets its downstream PI3K/AKT pathway (25, 26). The present study has investigated whether Cr(VI)-transformed BEAS-2B cells displays an activation of PI3K/AKT signaling, whether the activation of PI3K/AKT is dependent on EGFR, and whether activation of this pathway is important in apoptosis resistance of Cr(VI)-transformed cells. Fig. 5A shows that both phosphorylations of PI3K at Tyr-458 and AKT at Ser-473 were elevated in Cr(VI)-transformed cells compared with those in non-transformed parent cells. We have also examined the phosphorylation of PI3K and AKT at other sites. The results show that there were no observable difference between Cr(VI)-transformed cells and their passage-matched non-transformed cells (data not shown). Inhibition of EGFR by either its shRNA or tyrosine inhibitor AG1478 in Cr(VI)-transformed cells abolished phosphorylation of PI3K or AKT (Fig. 5A), indicating that activation of PI3K/AKT in Cr(VI)-transformed cells is EGFR dependent. To determine whether PI3K/AKT signaling regulates apoptosis resistance and reduced capacity of ROS generation in Cr(VI)-transformed cells, we pre-treated the cells with PI3K/AKT inhibitors wortmannin and LY294002. The results show that pre-treatment of the cells with either LY294002 or wortmannin inhibited expression of phosphorylation AKT, but not at protein level (Fig. 5B). Inhibition of AKT by those two inhibitors significantly increased percentile of apoptosis upon Cr(VI) stimulation in Cr(VI)-transformed cells (Fig. 5C). Further study shows that pre-treatment with LY294002 or wortmannin increased expression of apoptotic protein Bax and reduced anti-apoptotic protein Bcl-2 (Fig. 5B). To explore whether those two PI3K inhibitors are able to restore reduced ROS generating capacity in Cr(VI)-transformed cells, DCF fluorescence intensity, reflecting H2O2 generation, was measured. The results show that pre-treatment of the cells with LY294002 or wortmannin increased ROS generation with or without Cr(VI) treatment (Fig. 5D). In addition, SOD2 expression was markedly reduced after pre-treatment of the cells with LY294002 or wortmannin (Fig. 5B), indicating that inhibition of PI3K/AKT signaling reduces apoptosis resistance of Cr(VI)-transformed cells. These results correlate with restoration of capacity of ROS generation in Cr(VI)-transformed cells.
FIGURE 5.

EGFR-dependent activation of PI3K/AKT and its role in apoptosis resistance in Cr(VI)-transformed cells. A, BEAS-2B-Cr cells were either transfected with EGFR shRNA for 48 h (left) or treated with AG1478 for 24 h (right). The cells were harvested for immunoblotting to examine expression of EGFR, PI3K, and AKT. B, BEAS-2B-Cr cells were treated with LY294002 (10 and 50 μm) or wortmannin (100 and 500 nm) for 24 h. The cells were harvested for examination of AKT, Bcl-2, Bax, and SOD2 expression. C and D, BEAS-2B-Cr cells were pre-treated with LY294002 (10 and 50 μm) or wortmannin (100 and 500 nm) for 1 h prior to Cr(VI) treatment (10 μm). After 24 h, the cells were harvested for measurement of apoptosis (C) and ROS generation (D) using flow cytometry and spectrofluorometer, respectively. * and #, p < 0.05 compared with its control cells without Cr(VI) treatment and scramble cells with 10 μm Cr(VI) treatment.
Inhibition of EGFR Retards Tumor Growth in Vivo
To investigate the role of EGFR in tumorigenesis, non-transformed BEAS-2B cells, Cr(VI)-transformed cells, and Cr(VI)-transformed cells with EGFR knockdown were injected subcutaneously into nude mice. The results show that none of the mice injected with non-transformed normal BEAS-2B cells grew any tumor and that 4 of 4 mice injected with Cr(VI)-transformed cells grew tumor (Fig. 6A). Silence of EGFR reduced both tumor size and frequency (3 out of 4 mice) (Fig. 6A). Consistent with the in vitro studies, phosphorylation of EGFR, PI3K, or AKT was reduced in the EGFR silencing tumor tissues compared with that in the tumor tissues from Cr(VI)-transformed cells as evidence by both immunoblotting (Fig. 6B) and immunofluorescence staining (Fig. 6C). In addition, EGFR silencing tumor tissues expressed less SOD2 than that of Cr(VI)-transformed cells (Fig. 6B).
FIGURE 6.

Silencing of EGFR inhibits in vivo xenograft tumor growth. 1 × 106 cells of either BEAS-2B, or BEAS-2B-Cr, or BEAS-2B-Cr with EGFR knockdown (BEAS-2B-Cr-shEGFR) were injected subcutaneously into nude mice (4 mice/group). After 3 weeks, the mice were euthanized using CO2 and tumors were isolated. A, upper, representative photograph of excised tumors. Lower, tumor volume were quantified from those mice and presented as mean ± S.D. * and #, p < 0.05 compared with BEAS-2B cells and BEAS-2B-Cr cells, respectively. B, whole protein was extracted from fresh frozen tumor tissues. Expression of EGFR, PI3K, AKT, or SOD2 was examined by immunoblotting. C, tissue sections from formalin-fixed tumor tissues were subjected to fluorescence immunochemistry analysis.
DISCUSSION
It has been reported that more than 60% of non-small lung carcinomas (NSCLCs) have elevated expression EGFR (9). EGFR tyrosine kinase inhibitor (TKI) is widely used and is thought to be one of most effective chemotherapeutic drugs in the clinic for lung cancer patients. Mutations in tyrosine kinase (TK) domain of the EGFR gene in NSCLCs were discovered and presence of these mutations corrected with sensitivity to the EGFR inhibitor (9). It has been reported that EGFR mRNA level was increased in BEAS-2B cells exposed to 1 μm of Cr(VI) up to 8 culture passages (14). Our previous study has demonstrated that chronic exposure of BEAS-2B cells to Cr(VI) was able to induce malignant cell transformation (17). The present study has found that EGFR was constitutively activated in Cr(VI)-transformed BEAS-2B cells. Phosphorylation of EGFR was not present in the non-transformed passage-matched mock-treated BEAS-2B cells. Most importantly, a marked activation of EGFR in lung tumor tissue obtained from a worker occupationally exposed to Cr(VI) was observed, confirming our in vitro observation. EGFR expression was elevated starting at two months of 0.25 μm Cr(VI) treatment and elevated expression of EGFR displayed a time-dependent manner up to six months, indicating that EGFR may be an early biomarker for the cells exposed to Cr(VI).
Activation of EGFR signaling might induce uncontrolled cell growth and a malignant phenotype. The two major classes of EGFR mutations are an L858R point mutation and small, in-frame deletions in exon 19; both types of mutation enhance the activity and oncogenicity of EGFR (27). These mutations induced lung adenocarcinoma in a mouse model and confer hypersensitivity to EGFR-tyrosine kinase inhibitors (EGFR-TKIs) in humans (28, 29). To investigate whether activation of EGFR is due to its mutations in Cr(VI)-transformed cells, we have examined the mutations in exons 18 and 19 by a PCR-restriction fragment length polymorphism assay, no mutation was found. Apart from EGFR mutations, overexpression of EGFR or its ligands, or co-expression of ligands and receptor might lead to an abnormal EGFR-mediated signal transduction. In the present study, we have measured all seven EGFR ligands. Only elevated expressions of AREG and BTC at transcriptional and translational levels were observed in Cr(VI)-transformed cells. Increased expression of AREG is responsible for activation of EGFR, but not BTC. Knockdown of EGFR in Cr(VI)-transformed cells reduced both the incidence of tumorigenesis and volume of tumor, indicating a key role of EGFR in tumorigenicity of Cr(VI)-transformed cells.
Redox homeostasis and signaling are associated with protein kinases and growth factor receptor activation (30). Previous study has documented that several agents such as radiation, oxidants, and alkylating agents induce ligand-independent activation of the EGFR (31). ROS play an essential role in EGFR signaling as an intracellular signal transducer (10). Several studies have reported that NADPH oxidase 1 (NOX1) enhances the expression of EGFR signaling components and confers autocrine growth (32, 33). Early hydrogen peroxide-derived NOX1 activation is required for the phosphorylation of several EGFR signaling pathways, such as c-Src/ERKs and p38/AKT (34). The activation of EGFR components is required as a mechanism for NOX-1 to regulate its own expression and the expression of an EGFR ligand (TGF-α) (32). In leiomyoma smooth muscle cells, the inhibition of NOX impaired MAPK signaling activation and decreased the cell proliferation response to EGF (33). Our previous study has reported that Cr(VI) is able to activate NOX (17). It appears that that Cr(VI)-induced NOX activation may be one of the mechanisms for EGFR activation.
Acquired resistance to apoptosis is a critical cellular event during carcinogenesis, and disruption of apoptosis has been shown to play a major role in tumor formation and malignant progression (35–37). When cells acquire apoptosis resistance, these cells continue to proliferate, leading to carcinogenesis. The present study has observed that Cr(VI)-transformed cells exhibited resistance to apoptosis in response to Cr(VI) with decreased levels of C-PARP and Bax and elevated expressions of anti-apoptotic proteins Bcl-2 and Bcl-xL. To show that the apoptosis resistance in Cr(VI)-transformed cells is not specific for Cr(VI) treatment, 5-FU, a chemotherapeutic drug used in clinic, was used to treat Cr(VI)-transformed cells. The result is consistent with that treatment with Cr(VI), indicating development of apoptosis resistance of Cr(VI)-transformed cells. It has been reported that Cr(VI)-transformed cells displayed a high level of Bcl-2 with an enhanced angiogenesis and a decreased apoptosis (38). Bcl-2 knockdown reduced the tumorigenesis of Cr(VI)-transformed cells (37). Bcl-2 and Bcl-xL are oncogenes that inhibit apoptosis (39). To understand the role of EGFR in resistance to apoptosis in Cr(VI)-transformed cells, we analyzed the levels of anti-apoptotic protein Bcl-2 and apoptotic protein Bax in Cr(VI)-transformed cells with knockdown of either AREG or EGFR or with tyrosine inhibitor AG1478. Inhibition of EGFR restored apoptosis in Cr(VI)-transformed cells with commitment to increase of Bax expression and reduction of Bcl-2 expression. This observation is supported by the previous report that treatment the HCC827 cells with EGFR TKIs induced apoptosis via decrease of Mcl-1 expression, an anti-apoptotic protein of Bcl-2 family (40).
Excessive amounts of ROS can cause irreversible oxidative damage to biomacromolecules, apoptosis, and cell death (12). Therefore, maintaining ROS homeostasis at proper levels is crucial for normal cell survival, while a moderate enhancement of ROS is associated with abnormal cancer cell growth and disruption of redox homeostasis (12). Cancer cells are usually characterized by increased antioxidant capacity (41). Cells that survive intrinsic oxidative stress mobilize a set of adaptive mechanisms, which not only activate ROS-scavenging systems to cope with the oxidative stress, but also inhibit apoptosis (42). It has been shown that ROS are responsible for Cr(VI)-induced apoptosis in normal non-transformed cells (24, 43). The present study shows that, upon the chronic exposure of Cr(VI) at low dose (0.5 μm), survived BEAS-2B cells obtained adaptive machinery which is believed to booster antioxidant enzymes against elevated ROS. Therefore, in both Cr(VI)-transformed cells and lung tumor tissue obtained from a worker exposed to Cr(VI) for 19 years expression of antioxidant enzyme SOD2 was increased and capacity of those cells to generate ROS was reduced. Inhibition of SOD2 by its shRNA increased ROS generation, resulting in an increase of apoptosis. These observations indicate that decreased ROS together with increased Bcl-2 and decreased Bax in Cr(VI)-transformed cells is likely to be an important factor for apoptosis resistance.
Previous studies have shown that oxidative stress led to EGFR phosphorylation, which conferred protection against oxidative stress-induced apoptosis (44–47). The present study hypothesizes that Cr(VI)-transformed cells increase EGFR signaling to protect against oxidative stress induced cell death. If EGFR signaling is involved in the protection of Cr(VI)-transformed cells from oxidative stress, then inhibition of EGFR activation would be expected to increase oxidative stress. As expected, either knockdown of EGFR or tyrosine kinase inhibitor AG1478 or ligand AREG restored apoptosis in Cr(VI)-transformed cells with a commitment increase in ROS generation and reduction of antioxidant enzyme SOD2 expression, indicating that increased oxidative stress contributes to cell death induced by inhibition of EGFR in Cr(VI)-transformed cells.
Activation of EGFR signaling pathway promotes EGFR-mediated prosurvival and antiapoptotic signals through downstream targets, such as PI3K-AKT, ERK, and STAT (7, 48, 49). The PI3K/AKT pathway is switched on through activation of membrane receptors, including tyrosine-kinase receptors (RTKs), such as EGFR, HER2, IGFR-1, VEGFR, and PDGFR (50). Activated receptor tyrosine kinases recruit to the cell membrane a complex including PI3Ks. PI3Ks phosphorylate the phosphatidylinositol (PtdIns) lipid substrates in the plasma membrane, generating PtdIns(3,4,5)P3, PtdIns (3,4)P2 and PtdIns3P, which interact with multiple effector proteins and transduce the signaling from the membrane to the cytoplasm (50). Short term exposure of Cr(VI) to BEAS-2B cells activates PI3K-AKT/MAPK (15), but whether Cr(VI) utilizes EGFR to activate these pathways has not been studied. The present study has found that activation of PI3K/AKT in Cr(VI)-transformed cells was dependent on EGFR. Inhibition of PI3K/AKT by its specific inhibitors restored apoptosis in Cr(VI)-transformed cells. Bcl-2 is a known downstream target gene of PI3K/AKT (51). Our further study shows that PI3K inhibitor LY294002 decreased SOD2, increased ROS, elevated expression of apoptotic protein Bax, and ameliorated expression of anti-apoptotic protein Bcl-2 in Cr(VI)-transformed cells. Treatment of Cr(VI)-transformed cells with both inhibitors increased ROS generation and reduced SOD2 expression, indicating dependence of PI3K/AKT on oxidative stress. Our previous study has shown that Cr(VI) is able to cause ROS generation, leading to cell transformation, and inhibition of ROS generation by overexpressing antioxidants catalase and SOD reduced cell transformation induced by Cr(VI) (17), suggesting that ROS generation by Cr(VI) exposure is responsible for Cr(VI)-induced cells transformation, which is considered as the first stage of Cr(VI) carcinogenesis. The present study shows that after cell transformation or in the second stage of Cr(VI) carcinogenesis, however, these cells show elevated level of antioxidant enzyme and reduced ROS generation, leading to expression of Bcl-2 and Bcl-xL and apoptosis resistance of the Cr(VI)-transformed cells. This process promotes cell proliferation and tumorigenesis. Thus in the first stage of Cr(VI) carcinogenesis or cell transformation, the excess amount of ROS generated by Cr(VI) exposure is oncogenic by causing cell transformation. In the second stage of Cr(VI) carcinogenesis, once the cells were malignant transformed, the decreased capacity of ROS generation in those transformed cells is also carcinogenic by causing apoptosis resistance. The oncogenic role of ROS is further illustrated in Fig. 7.
FIGURE 7.
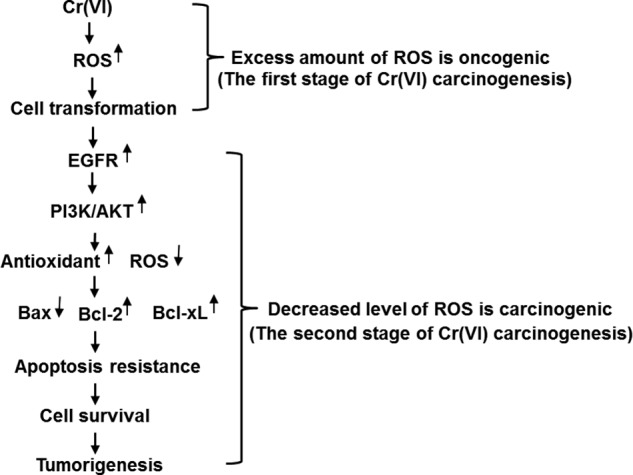
The role of ROS in Cr(VI) carcinogenesis. Our previous study has demonstrated that chronic exposure of cells to Cr(VI) causes ROS generation which is responsible for malignant cell transformation. Thus, at the first stage of Cr(VI)-carcinogenesis (cell transformation), excess amount of ROS is oncogenic. The present study has found that Cr(VI)-transformed cells exhibit activated EGFR, reduced ROS generation, elevated antioxidant expression, leading to apoptosis resistance and therefore tumorigenesis. At the second stage of Cr(VI)-carcinogenesis (after cell transformation), decreased level of ROS is carcinogenic.
In summary, the present study shows that chronic exposure of BEAS-2B cells to Cr(VI) is able to cause cell malignant transformation. Cr(VI)-transformed cells exhibit apoptosis resistance. Ligand-dependent constitutive activation of EGFR was observed in Cr(VI)-transformed cells. PI3K/AKT was downstream target of EGFR signaling. The activated EGFR/PI3K/AKT signal pathway up-regulated antioxidant enzyme, SOD and reduced cellular ROS level and increased Bcl-2 and Bcl-xL, leading to apoptosis resistance of Cr(VI)-transformed cells. The activated EGFR/PI3K/AKT signal is important in tumorigenesis of Cr(VI)-transformed cells.
This study was supported, in whole or in part, by National Institutes of Health/NIEHS R01ES018883 and National Institutes of Health/NCI R03CA171604 (to Z. Z.).
- Cr(VI)
- chromium compound VI
- AREG
- amphiregulin
- BTC
- betacellulin
- DCFDA
- 5-(and -6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate
- DTR
- diphtheria toxin receptor/heparin-binding EGF-like growth factor
- EGF
- epidermal growth factor
- EGFR
- epidermal growth factor receptor
- EPGN
- epigen
- EREG
- epiregulin
- FITC
- fluorescein isothiocyanate
- NOX1
- NADPH oxidase 1
- NSCLC
- non-small cell lung cancer
- PI
- propidium iodide
- ROS
- reactive oxygen species
- SOD2
- superoxide dismutase 2
- TGFA
- transforming growth factor-α
- TK
- tyrosine kinase
- TKI
- tyrosine kinase inhibitor.
REFERENCES
- 1. Hill R., Leidal A. M., Madureira P. A., Gillis L. D., Cochrane H. K., Waisman D. M., Chiu A., Lee P. W. (2008) Hypersensitivity to chromium-induced DNA damage correlates with constitutive deregulation of upstream p53 kinases in p21-/- HCT116 colon cancer cells. DNA Repair 7, 239–252 [DOI] [PubMed] [Google Scholar]
- 2. International Agency for Research on Cancer (IARC) (1990) Chromium, nickel and welding. IARC Monogr. Eval. Carcinog Risks Hum. 49, 1–648 [PMC free article] [PubMed] [Google Scholar]
- 3. Langård S. (1990) One hundred years of chromium and cancer: a review of epidemiological evidence and selected case reports. Am. J. Ind. Med. 17, 189–215 [DOI] [PubMed] [Google Scholar]
- 4. Pellerin C., Booker S. M. (2000) Reflection on hexavalent chromium: health hazards of an industrial heavy weigh. Environ. Health Perspect. 108, A402–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schneider M. R., Wolf E. (2009) The epidermal growth factor receptor ligands at a glance. J. Cell. Physiol. 218, 460–466 [DOI] [PubMed] [Google Scholar]
- 6. Engelman J. A., Luo J., Cantley L. C. (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619 [DOI] [PubMed] [Google Scholar]
- 7. Scagliotti G. V., Selvaggi G., Novello S., Hirsch F. R. (2004) The biology of epidermal growth factor receptor in lung cancer. Clin. Cancer Res. 10, 4227s–4232s [DOI] [PubMed] [Google Scholar]
- 8. Marinov M., Ziogas A., Pardo O. E., Tan L. T., Dhillon T., Mauri F. A., Lane H. A., Lemoine N. R., Zangemeister-Wittke U., Seckl M. J., Arcaro A. (2009) AKT/mTOR pathway activation and BCL-2 family proteins modulate the sensitivity of human small cell lung cancer cells to RAD001. Clin. Cancer Res. 15, 1277–1287 [DOI] [PubMed] [Google Scholar]
- 9. Santos G., Shepherd F. A., Tsao M. S. (2011) EGFR mutation and lung cancer. Annu. Rev. Pathol. Mech. Dis. 6, 49–69 [DOI] [PubMed] [Google Scholar]
- 10. Chiarugi P., Buricchi F. (2007) Protein tyrosine phosphorylation and reversible oxidation: two cross-talking posttranslation modifications. Antioxid. Redox Signal 9, 1–24 [DOI] [PubMed] [Google Scholar]
- 11. Chen C. H., Cheng T. H., Lin H., Shih N. L., Chen Y. L., Chen Y. S., Cheng C. F., Lian W. S., Meng T. C., Chiu W. T., Chen J. J. (2006) Reactive oxygen species generation is involved in epidermal growth factor receptor transactivation through the transient oxidization of Src homology 2-containing tyrosine phosphatase in endothelin-1 signaling pathway in rat cardiac fibroblasts. Mol. Pharmacol. 69, 1347–1355 [DOI] [PubMed] [Google Scholar]
- 12. Trachootham D., Alexandre J., Huang P. (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 8, 579–591 [DOI] [PubMed] [Google Scholar]
- 13. Irmer D., Funk J. O., Blaukat A. (2007) EGFR kinase domain mutations-functional impact and relevance for lung cancer therapy. Oncogene 26, 5693–5701 [DOI] [PubMed] [Google Scholar]
- 14. Rodrigues C. F., Rodrigues C. F., Urbano A. M., Matoso E., Carreira I., Almeida A., Santos P., Botelho F., Carvalho L., Alves M., Monteiro C., Costa A. N., Moreno V., Alpoim M. C. (2009) Human bronchial epithelial cells malignantly transformed by hexavalent chromium exhibit an aneuploid phenotype but no microsatellite instability. Mutat. Res. 670, 42–52 [DOI] [PubMed] [Google Scholar]
- 15. Wang B. J., Sheu H. M., Guo Y. L., Lee Y. H., Lai C. S., Pan M. H., Wang Y. I. (2010) Hexavalent chromium induced ROS formation, Akt, NF-κB, and MAPK activation, and TNF-α and IL-1α production in keratinocytes. Toxicol. Lett. 198, 216–224 [DOI] [PubMed] [Google Scholar]
- 16. Beaver L. M., Stemmy E. J., Schwartz A. M., Damsker J. M., Constant S. L., Ceryak S. M., Patierno S. R. (2010) Lung Inflammation, Injury, and Proliferative Response after Repetitive Particulate Hexavalent Chromium Exposure. Environ. Health Perspec. 117, 1898–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X., Son Y. O., Chang Q., Sun L., Hitron J. A., Budhraja A., Zhang Z., Ke Z., Chen F., Luo J., Shi X. (2011) NADPH oxidase activation is required in reactive oxygen species generation and cell transformation induced by hexavalent chromium. Toxicol. Sci. 123, 399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun Y., Ren Y., Fang Z., Li C., Fang R., Gao B., Han X., Tian W., Pao W., Chen H., Ji H. (2010) Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J. Clin. Oncol. 28, 4616–4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harris R. C., Chung E., Coffey R. J. (2003) EGF receptor ligands. Exp. Cell Res. 284, 2–13 [DOI] [PubMed] [Google Scholar]
- 20. Portt L., Norman G., Clapp C., Greenwood M., Greenwood M. T. (2011) Anti-apoptosis and cell survival: a review. Biochim. Biophys. Acta 1813, 238–259 [DOI] [PubMed] [Google Scholar]
- 21. Shi X., Dalal N. S. (1989) Chromium (V) and hydroxyl radical formation during the glutathione reductase-catalyzed reduction of chromium (VI). Biochem. Biophys. Res. Commun. 163, 627–634 [DOI] [PubMed] [Google Scholar]
- 22. Yao H., Guo L., Jiang B., Luo J., Zhang Z., Shi X. (2008) Oxidative stress and chromium carcinogenesis. J. Environ. Pathol. Toxicol. Oncol. 27, 77–88 [DOI] [PubMed] [Google Scholar]
- 23. Leonard S. S., Harris G. K., Shi X. (2004) Metal-induced oxidative stress and signal transduction. Free Radic. Biol. Med. 37, 1921–1942 [DOI] [PubMed] [Google Scholar]
- 24. Ye J., Wang S., Leonard S. S., Sun Y., Butterworth L., Antonini J., Ding M., Rojanasakul Y., Vallyathan V., Castranova V., Shi X. (1999) Role of reactive oxygen species and p53 in chromium (VI)-induced apoptosis. J. Biol. Chem. 274, 34974–34980 [DOI] [PubMed] [Google Scholar]
- 25. Lee D. H., Szczepanski M. J., Lee Y. J. (2009) Magnolol induces apoptosis via inhibiting the EGFR/PI3K/Akt signaling pathway in human prostate cancer cells. J. Cell. Biochem. 106, 1113–1122 [DOI] [PubMed] [Google Scholar]
- 26. Zhou C., Qiu L., Sun Y., Healey S., Wanebo H., Kouttab N., Di W., Yan B., Wan Y. (2006) Inhibition of EGFR/PI3K/AKT cell survival pathway promotes TSA's effect on cell death and migration in human ovarian cancer cells. Int. J. Oncol. 29, 269–278 [PubMed] [Google Scholar]
- 27. Greulich H., Chen T. H., Feng W., Jänne P. A., Alvarez J. V., Zappaterra M., Bulmer S. E., Frank D. A., Hahn W. C., Sellers W. R., Meyerson M. (2005) Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLOS Med. 2, e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yun C. H., Boggon T. J., Li Y., Woo M. S., Greulich H., Meyerson M., Eck M. J. (2007) Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 11, 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou B. B., Peyton M., He B., Liu C., Girard L., Caudler E., Lo Y., Baribaud F., Mikami I., Reguart N., Yang G., Li Y., Yao W., Vaddi K., Gazdar A. F., Friedman S. M., Jablons D. M., Newton R. C., Fridman J. S., Minna J. D., Scherle P. A. (2006) Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell 10, 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Truong T. H., Carroll K. S. (2013) Redox regulation of protein kinases. Crit. Rev. Biochem. Mol. Biol. 48, 332–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Knebel A., Rahmsdorf H. J., Ullrich A., Herrlich P. (1996) Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J. 15, 5314–5325 [PMC free article] [PubMed] [Google Scholar]
- 32. Sancho P., Fabregat I. (2010) NADPH oxidase NOX1 controls autocrine growth of liver tumor cells through up-regulation of the epidermal growth factor receptor pathway. J. Biol. Chem. 285, 24815–24824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mesquita F. S., Dyer S. N., Heinrich D. A., Bulun S. E., Marsh E. E., Nowak R. A. (2010) Reactive oxygen species mediate mitogenic growth factor signaling pathways in human leiomyoma smooth muscle cells. Biol. Reprod. 82, 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paletta-Silva R., Rocco-Machado N., Meyer-Fernandes J. R. (2013) NADPH oxidase biology and the regulation of tyrosine kinase receptor signaling and cancer drug cytotoxicity. Int. J. Mol. Sci. 14, 3683–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ellis R. E., Yuan J., Horvitz H. R. (1991) Mechanisms and functions of cell death. Annu. Rev. Cell Biol. 7, 663–698 [DOI] [PubMed] [Google Scholar]
- 36. Hanahan D., Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- 37. Wyllie A. H., Bellamy C. O., Bubb V. J., Clarke A. R., Corbet S., Curtis L., Harrison D. J., Hooper M. L., Toft N., Webb S., Bird C. C. (1999) Apoptosis and carcinogenesis. Br. J. Cancer 80, 34–37 [PubMed] [Google Scholar]
- 38. Medan D., Luanpitpong S., Azad N., Wang L., Jiang B., Davis M. E., Barnett J. B., Guo L., Rojanasakul Y. (2012) Multifunctional Role of Bcl-2 in Malignant Transformation and Tumorigenesis of Cr(VI)-Transformed Lung Cells. PLOS One 7, e37045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zinkel S., Gross A., Yang E. (2006) BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 13, 1351–1359 [DOI] [PubMed] [Google Scholar]
- 40. Faber A. C., Li D., Song Y., Liang M. C., Yeap B. Y., Bronson R. T., Lifshits E., Chen Z., Maira S. M., García-Echeverría C., Wong K. K., Engelman J. A. (2009) Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc. Natl. Acad. Sci. U.S.A. 106, 19503–19508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kagan V., Bakalova R., Karakashev P. (1990) Lipid peroxidation in tumor cells and tissues of tumor-bearing animals, in: “Membrane Lipid Oxidation”, Vigo-Pelfrey C. (ed), CRC Press, Boca-Raton, Florida: 3, 192–208 [Google Scholar]
- 42. Ivanova D., Bakalova R., Lazarova D., Gadjeva V., Zhelev Z. (2013) The impact of reactive oxygen species on anticancer therapeutic strategies. Adv. Clin. Exp. Med. 22, 899–908 [PubMed] [Google Scholar]
- 43. Chen F., Vallyathan V., Castranova V., Shi X. (2001) Cell apoptosis induced by carcinogenic metals. Mol. Cell Biochem. 222, 183–188 [PubMed] [Google Scholar]
- 44. Wang X., McCullough K. D., Franke T. F., Holbrook N. J. (2000) Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J. Biol. Chem. 275, 14624–14631 [DOI] [PubMed] [Google Scholar]
- 45. Fischer O. M., Hart S., Gschwind A., Prenzel N., Ullrich A. (2004) Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor. Mol. Cell. Biol. 24, 5172–5183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peus D., Vasa R. A., Meves A., Beyerle A., Pittelkow M. R. (2000) UVB-induced epidermal growth factor receptor phosphorylation is critical for downstream signaling and keratinocyte survival. Photochem. Photobiol. 72, 135–140 [DOI] [PubMed] [Google Scholar]
- 47. Orcutt K. P., Parsons A. D., Sibenaller Z. A., Scarbrough P. M., Zhu Y., Sobhakumari A., Wilke W. W., Kalen A. L., Goswami P., Miller F. J., Jr., Spitz D. R., Simons A. L. (2011) Erlotinib-mediated inhibition of EGFR signaling induces metabolic oxidative stress through NOX4. Cancer Res. 71, 3932–3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yarden Y., Sliwkowski M. X. (2001) Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137 [DOI] [PubMed] [Google Scholar]
- 49. Sordella R., Bell D. W., Haber D. A., Settleman J. (2004) Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 305, 1163–1167 [DOI] [PubMed] [Google Scholar]
- 50. Fumarola C., Bonelli M. A., Petronini P. G., Alfieri R. R. (2014) Targeting PI3K/AKT/mTOR pathway in non-small cell lung cancer. Biochem. Pharmacol. 90, 197–207 [DOI] [PubMed] [Google Scholar]
- 51. Pugazhenthi S., Nesterova A., Sable C., Heidenreich K. A., Boxer L. M., Heasley L. E., Reusch J. E. (2000) Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J. Biol. Chem. 275, 10761–10766 [DOI] [PubMed] [Google Scholar]