

Background:The mitochondrial production of formate is critical to the generation of one-carbon groups.
Results: We have shown that the in vivo production of formate is markedly reduced in folate deficiency.
Conclusion: Folate deficiency reduces both the production and utilization of one-carbon groups.
Significance: Folate status affects the production and utilization of one-carbon groups and the role of choline metabolites as precursors of these groups.
Keywords: Amino Acid, Methionine, Mitochondria, Serine, Vitamin, Choline, One-carbon Metabolism, Tetrahydrofolate
Abstract
It is now established that the mitochondrial production of formate is a major process in the endogenous generation of folate-linked one-carbon groups. We have developed an in vivo approach involving the constant infusion of [13C]formate until isotopic steady state is attained to measure the rate of endogenous formate production in rats fed on either a folate-replete or folate-deficient diet. Formate was produced at a rate of 76 μmol·h−1·100 g of body weight−1 in the folate-replete rats, and this was decreased by 44% in folate-deficient rats. This decreased formate production was confirmed in isolated rat liver mitochondria where formate production from serine, the principal precursor of one-carbon groups, was decreased by 85%, although formate production from sarcosine and dimethylglycine (choline metabolites) was significantly increased. We attribute this unexpected result to the demonstrated production of formaldehyde by sarcosine dehydrogenase and dimethylglycine dehydrogenase from their respective substrates in the absence of tetrahydrofolate and subsequent formation of formate by formaldehyde dehydrogenase. Comparison of formate production with the ingestion of dietary formate precursors (serine, glycine, tryptophan, histidine, methionine, and choline) showed that ∼75% of these precursors were converted to formate, indicating that formate is a significant, although underappreciated end product of choline and amino acid oxidation. Ingestion of a high protein diet did not result in increased production of formate, suggesting a regulation of the conversion of these precursors at the mitochondrial level to formate.
Introduction
Interest in formate metabolism has increased greatly in recent years, particularly as its role in providing one-carbon groups to the folate pool has become apparent. Formate is produced as a result of the mitochondrial metabolism of a number of metabolic substrates (serine, glycine, dimethylglycine, and sarcosine) with tetrahydrofolate (THF)3 to yield 5,10-methylene-THF, which in turn is oxidized to 10-formyl-THF. The mitochondrial isoform of 10-formyl-THF synthetase then produces formate from 10-formyl-THF. Mitochondrial formate is released to the cytosol where it may be incorporated into 10-formyl-THF, which is used for purine synthesis (1). 10-Formyl-THF may be reduced further such that it may be used for thymidine synthesis and for methylation reactions (2). In addition to the mitochondrial pathway, formate is produced in the cytoplasm in a number of other ways, including methanol oxidation, cholesterol synthesis, phytanic acid oxidation, and the catabolism of tryptophan and histidine (3). An outline of formate metabolism is provided in Fig. 1.
FIGURE 1.

An outline of one-carbon metabolism with particular reference to formate. A schematic diagram of the subcellular compartmentalization of formate metabolism. SHMT, serine hydroxymethyltransferase; DMGD, dimethylglycine (DMG) dehydrogenase; SDH, sarcosine dehydrogenase; GCS, glycine cleavage enzyme complex; FTD, 10-formyl-THF dehydrogenase; 10f-THF, 10-formyl-THF; Hcy, homocysteine; MTHFD2/2L, mitochondrial bifunctional methylene THF dehydrogenase; MTHFD1, cytosolic trifunctional methylene-THF dehydrogenase; MTHFR, methylene-THF reductase; MS, methionine synthase; BHMT, betaine:homocysteine methyltransferase; MAT, methionine adenosyltransferase; SAHH, S-adenosylhomocysteine (SAH) hydrolase; 5,10-CH2-THF, 5,10-methylene-THF; 5-CH3-THF, 5-methyl-THF; 10f-THF, 10-formyl-THF; SAM, S-adenosylmethionine.
The critical role of mitochondrial formate metabolism has been highlighted by three recent studies. Work from Stover and co-workers (4) has shown that knock-out of cytoplasmic serine hydroxymethyltransferase results in viable mice, suggesting that the mitochondrial generation of formate may be capable of providing much of the body's one-carbon pool. Monofunctional 10-formyl-THF synthetase (MTHFD1L) is the mitochondrial enzyme responsible for the production of formate. Appling et al. (5) have shown that knock-out of Mthfd1l is embryonically lethal, with embryos showing both growth defects and aberrant neural tube closure. Maternal supplementation of the drinking water with formate decreased the incidence of neural tube defects and partially rescued the growth defect (5). These experiments provide strong evidence for a critical role for mitochondrially derived formate in mammalian development. Narisawa et al. (6) have shown that mutations in genes that encode for subunits of the glycine cleavage system predispose to neural tube defects in both humans and mice. The glycine cleavage system is a mitochondrial enzyme complex that links the oxidation of glycine to the generation of 5,10-methylene-THF, from which the mitochondria produce formate.
Formate can be rapidly catabolized by the perfused rat liver (7). Pathways of formate catabolism include its oxidation to CO2 by catalase as well as a folate-requiring mechanism, probably by means of the combined actions of 10-formyl-THF synthetase and 10-formyl-THF dehydrogenase (3). Some formate is also excreted in the urine (3). We have recently shown that both plasma and urinary formate are markedly elevated in rats made deficient in either folate or vitamin B12. We have attributed this to impairment in the cytoplasmic incorporation of formate into the folate one-carbon pool either due to frank folate deficiency (folate-deficient rats) or to the methyl folate trap (vitamin B12-deficient rats) (8). Because formate is not elevated in vitamin B6-deficient rats, we have suggested that formate levels may provide a means of discriminating between hyperhomocysteinemia brought about by defects in homocysteine remethylation (folate or vitamin B12 deficiency) and that brought about by a defect in homocysteine transsulfuration (vitamin B6 deficiency).
Despite this interest in formate, very little is known about whole-body formate metabolism. As far as we are aware, there is only one study that examined formate whole-body kinetics. This study (9) reported rates of formate appearance of ∼2–4 mg/kg/h (∼45–90 μmol/kg/h) in sheep. There appears to be no study of whole-body formate kinetics in situations where plasma formate levels are elevated. We report here a study of whole-body de novo formate synthesis in rats fed either a folate-replete or a folate-deficient diet. The results confirm impaired formate removal during folate deficiency and also indicate impairment in formate production. The impairment in formate synthesis was confirmed with isolated mitochondria. We also report an improved enzymatic assay for plasma formate.
EXPERIMENTAL PROCEDURES
Animals
Male, Sprague-Dawley rats, ∼60–80 g, were provided by Memorial University's Division of Animal Care and were housed individually in a temperature and humidity-controlled facility with a 12-h light-dark cycle (7.00–19.00 light). All animal procedures were approved by the Animal Care Committee at Memorial University of Newfoundland and followed the Guidelines of the Canadian Council on Animal Care.
Folate-deficient Diets
For our first experiment (isotopic measurement of endogenous formate synthesis) the rats were placed on either folate-replete or folate-deficient diets containing 10 g of succinylsulfathiazole/kg of diet for 15 days. These were commercial, amino acid-based diets obtained from Dyets Inc., Bethlehem, PA (#517777 and #517804 for the deficient and replete diets, respectively). These diets have been described by Bills et al. (10). For the subsequent experiments we employed the AIN-93G diet (11), prepared with high nitrogen, low vitamin (MP-Biomedical) casein and contained 10 g/kg of succinylsulfathiazole. The only difference between the deficient and replete diets was the vitamin mix. We found that after 18 days on these diets the deficient animals were comparably folate-deficient, as judged by folate, formate, and homocysteine levels as they would be on the amino acid-based diets.
High Protein Diets
AIN-93G-based diets were prepared containing either 12% or 50% casein. To accommodate the increase in protein in the 50% protein diet, each of the carbohydrate components were proportionally decreased. The decreases were as follows (in g/kg diet): cornstarch from 447 to 207; dextrinized cornstarch from 149 to 69; sucrose from 112 to 52. These diets were fed to rats for 1 week.
Plasma and Liver Folate
Plasma and liver folate were measured using the Lactobacillus casei microbiological assay (12). Liver folate content was normalized to total protein, which was determined by the modified Lowry assay (13).
In Vivo Kinetics
Rats were anesthetized with isoflurane (4% in oxygen for induction; 2% in oxygen for maintenance), and catheters (4 cm of PE-50 tubing, heat adhered to 10-cm PE-10 tubing) were surgically implanted into a femoral artery and the contralateral femoral vein. Sodium [13C]formate in saline (150 mm sodium [13C]formate diluted with 0.9% saline to a formate concentration of 12.4 mm and filtered through a 0.2-μm syringe filter) was infused without a priming dose through the venous catheter at ∼700 μl/h, a rate calculated to replace fluid losses due to sampling. Blood samples (200 μl taken at 0 time and at 20-min intervals for 1.5 h) were taken through the arterial catheter. After each sampling the arterial catheter was flushed with heparinized saline (100 units/ml). Plasma was obtained by centrifugation and derivatized, and the tracer-to-tracee ratio (TTR) (see Ref. 15 for terminology) was determined as described (14). The TTR of the derivatized zero time sample was subtracted to correct for natural abundance of heavier isotopes, as described by Wolfe (15). At steady state, the TTR can be used to calculate the endogenous rate of formate appearance (Ra) by the equation, Ra = F/Ep, where F is the rate of infusion of [13C]formate (μmol/h) and Ep is the plasma enrichment (TTR) (16).
Mitochondrial Studies
Rat liver mitochondria were isolated by differential centrifugation in a medium consisting of 0.26 m sucrose, 3.4 mm Tris, 1.0 mm EGTA, adjusted to pH 7.4. Mitochondrial quality was monitored by measuring the respiratory control ratio (oxygen consumption in State 3/oxygen consumption in State 4). With β-hydroxybutyrate as substrate these ratios were 5.4 and 4.5, respectively, from folate-replete and folate-deficient rats. The comparable ratios with succinate as substrate were 4.3 and 4.1. Mitochondria were incubated with constant stirring for 10 min at 30 °C at 2.5 mg of mitochondrial protein/ml in 2 ml of an incubation medium consisting of 140 mm KCl, 5 mm HEPES (pH 7.4), 4 mm K2HPO4, 2.5 mm MgCl2, 1.5 mm EDTA, 1.6 mm ADP, and various substrates. Using data on the hepatic content of serine and glycine in rat liver (17) and the intracellular volume of rat liver (18), we calculated the physiological intracellular concentrations of serine and glycine to be ∼1 and 3 mm, respectively. We, therefore, incubated the mitochondria with either 3 mm glycine or 1 mm serine. There is little information available on the hepatic content of sarcosine and dimethylglycine, so we employed concentrations of 1 mm for each of these to ensure that the mitochondria were adequately provided with substrates. Preliminary experiments showed that the rate of formate production was linear with mitochondrial protein under our experimental conditions. Formate was measured by GC-MS (14).
Materials
All chemicals were of the highest purity available. Sodium [13C]formate (99%) was obtained from Cambridge Isotope Laboratories (Andover, MA). Formate dehydrogenase from Candida boidinii was obtained from Sigma.
Data Presentation and Analysis
All data are provided as the means ± S.D. Differences between two means were tested for statistical significance by means of Student's t test with p < 0.05 taken to indicate a significant difference. In the isotope infusion experiments the plateaux of TTRs were verified by linear regression analysis.
RESULTS
Folate Status
The two folate-deficient diets produced a comparable degree of folate deficiency. Plasma levels of folate in the rats on the Dyets folate-deficient diet were 42.2 ± 5.1 fmol/μl (n = 4) compared with 126.6 ± 21 fmol/μl (n = 4) for the Dyets folate-replete diet (p < 0.001). The comparable folate values in liver were 6.7 ± 3.0 fmol/μg of protein (n = 4) for the folate-deficient diet compared with 46.6 ± 10.8 (n = 4) for the folate-replete diet (p < 0.001). For the AIN-93G diets, folate in plasma was 32.2 ± 4.9 fmol/μl (n = 4) and 150.5 ± 17.8 fmol/μl (n = 4), respectively, for the deficient and replete diets (p < 0.001). The liver values were 8.4 ± 0.6 and 53.3 ± 6.9 fmol/μg of protein (n = 4), respectively, for the deficient and replete diets (p < 0.001).
Enzymatic Assay for Formate
An enzymatic assay for formate, employing formate dehydrogenase and diaphorase, has been described (19). The oxidation of formate to CO2 by formate dehydrogenase is coupled to the reduction of NAD+ to NADH. The NADH is then used by diaphorase to reduce iodonitrotetrazolium to its formazan whose absorbance was read at 500 nm. Quantitation was achieved by an internal standard, i.e. plasma samples were spiked with a known amount of formate. The formate dehydrogenase assay is an endpoint assay, i.e. the increase in absorbance should eventually plateau, permitting the determination of the difference between the initial and final absorbance. However, we encountered an absorbance creep in that the absorbance continued to increase for hours. This was eventually traced to the presence of dithiothreitol (DTT) in the commercial enzyme preparation. When DTT was removed by passing the enzyme through a PD-10 size-exclusion column, the assay improved markedly, coming to a definite endpoint. Using this improved assay we compared plasma formate levels from a variety of rats (fed vitamin-replete diets or diets deficient in either folate or vitamin B12) by both the improved formate assay or by the GC-MS method of Lamarre et al. (14). There was excellent agreement between the two assays (r2 = 0.94). The enzymatic assay was routinely employed to measure plasma formate levels.
Isotopic Determination of Rates of Formate Appearance In Vivo
Fig. 2 shows a typical experiment in which [13C]formate was infused into a 198-g rat fed the folate-deficient diet until the tracer to tracee ratio attained a plateau level of ∼9% above natural abundance. The plasma formate concentration remained steady at ∼60 μm. Table 1 shows comparable data for two groups of 6 rats fed either the folate-replete or folate-deficient diet for 15 days. It is evident that the rate of formate appearance was significantly decreased in the rats fed the folate-deficient diet (43.5 μmol·h−1·100 g body wt−1) compared with rats fed the folate-replete diet (76.2 μmol·h−1·100 g body wt−1). Fig. 3 shows plots of the rate of formate appearance, measured isotopically, for each of the two groups of rats as a function of their plasma formate concentration. It is evident that the two groups of rats behave quite differently. In the folate-replete rats increased rates of formate appearance correlated with increased plasma formate concentration. In the rats fed the folate-deficient diets, however, those with the highest plasma formate concentrations displayed the lowest rates of formate appearance.
FIGURE 2.
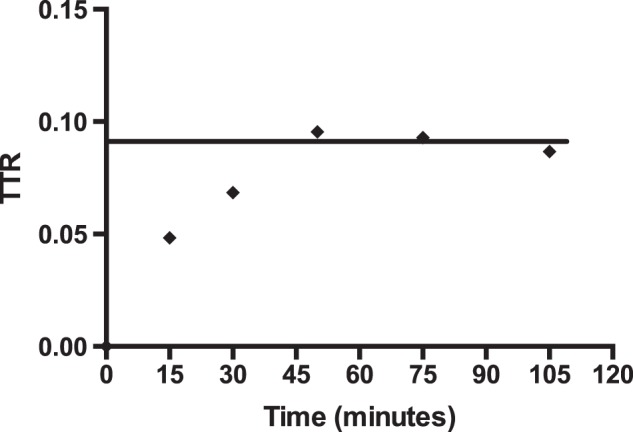
Formate enrichment (TTR) in plasma from a rat infused with [13C]formate until isotopic steady state is attained. Further details are provided in “Experimental Procedures.”
TABLE 1.
In vivo kinetics of formate in folate-replete and folate-deficient rats
TTR ([13C]formate:[12C]formate), plasma formate concentration, and rate of endogenous formate production (per 100 g rat body weight) in folate-replete and folate-deficient rats after a constant infusion of sodium [13C]formate to isotopic steady state. Values are given as the mean ± S.D. for each group of rats. (n = 6).
| Kinetic Parameters | Folate-replete | Folate-deficient |
|---|---|---|
| Plateau TTR | 0.057 ± 0.017 | 0.099 ± 0.012a |
| Plasma formate concentration (μm) | 71 ± 28 | 271 ± 119a |
| Rate of endogenous formate production (μmol/h/100g) | 76.2 ± 15.8 | 43.5 ± 4.0a |
a A significant difference (p < 0.05) from the folate-replete value.
FIGURE 3.
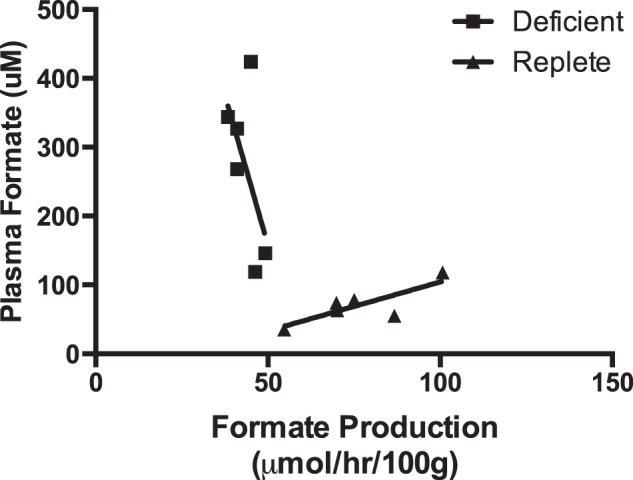
Plasma formate as a function of the formate production rate in rats fed a folate-replete or folate-deficient diet.
To gain further insights into formate metabolism, we compared rates of formate appearance with the maximal possible rate of production of one-carbon groups from dietary constituents. We could do this because we knew the composition of these chemically defined diets and we measured food consumption. From the quantity of each amino acid ingested we subtracted the amount required for net protein synthesis. We did this from knowledge of the growth rate of the rats (over the last 3 days of the experiment), the protein content of adult rats (20), and the amino acid profile of rat whole-body protein (21). Subtracting the amino acids required for net protein synthesis from those ingested gave us the quantities of each amino acid available for catabolism. We accounted for the fact that each histidine, tryptophan, methionine, and glycine catabolized could yield a maximum of one one-carbon group, serine could yield two, and each choline could potentially yield a maximum of four. Table 2 shows how the maximum potential dietary one-carbon groups were calculated. Fig. 4 shows that formate production, measured isotopically, was equal to 75% of ingested one-carbon precursors not required for net protein synthesis in the rats fed the folate-replete diet and 43% in those fed the folate-deficient diet.
TABLE 2.
Typical calculation of formate synthesis as a percentage of total dietary one-carbon potential, corrected for amino acids used for protein synthesis
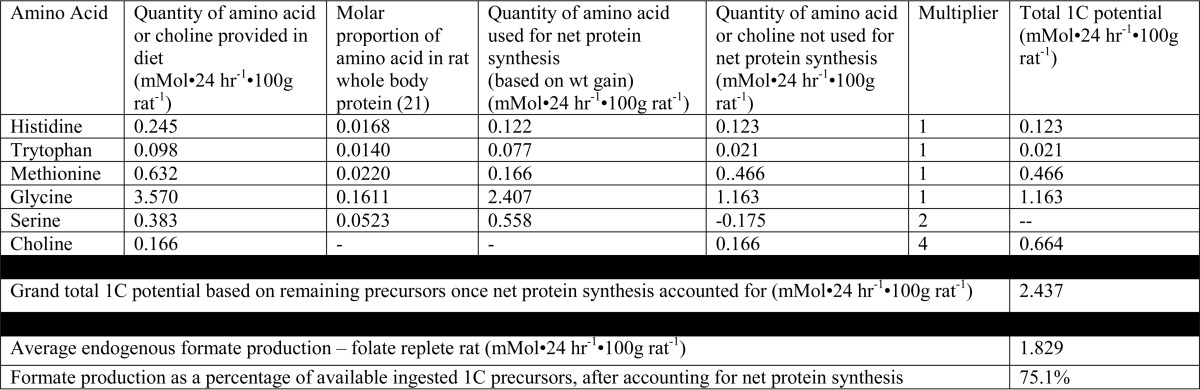
FIGURE 4.
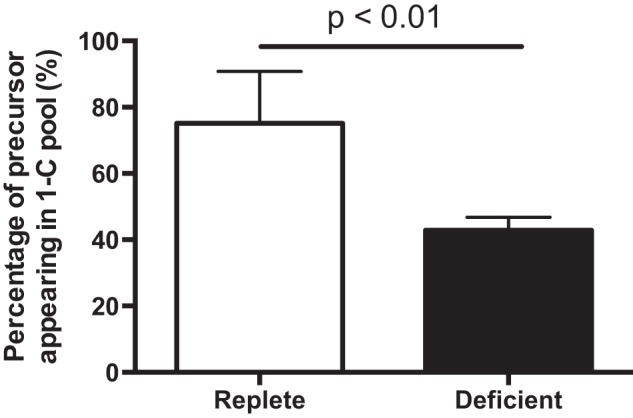
The percentage of dietary one-carbon groups that appears in formate. The maximum production of formate from dietary precursors was calculated from the dietary intake of serine, glycine, methionine, tryptophan, histidine, and choline, measured over the last 2 days of the experiment. These estimates were corrected by subtracting the quantity of each amino acid required for net protein synthesis (full details are provided under “Results”). They took into account the fact that a maximum of four one-carbon groups could be supplied by the catabolism of choline, two from serine, and one each from glycine, histidine, methionine, and tryptophan. The rate of endogenous formate production, measured isotopically, was then expressed as a percentage of the maximum rate of production of formate from these dietary precursors.
Formate Production by Isolated Rat Liver Mitochondria
To confirm the finding that de novo formate production was decreased in folate deficiency, we examined formate production by isolated liver mitochondria from rats fed either a folate-replete or folate-deficient diet. Table 3 shows a substantial rate of formate production from serine in mitochondria from the folate-replete rats, but the rates from the other substrates were very low. Formate production from serine was very much reduced in mitochondria from the folate-deficient rats, and formate production from glycine remained low. Unexpectedly, in these mitochondria, formate production from sarcosine and dimethylglycine was increased ∼4-fold.
TABLE 3.
Formate production by liver mitochondria from folate-replete and folate-deficient rats
Rate of formate production from different precursors (nmol/mg of protein/10 min) by isolated rat liver mitochondria from animals fed either a folate-replete or folate-deficient diet. Values are given as the mean (S.D.), n = 5, in each group.
| Substrate | Folate-replete | Folate-deficient |
|---|---|---|
| Serine (1 mm) | 6.81 ± 1.24 | 1.03 ± 0.64a |
| Glycine (3 mm) | 0.96 ± 0.69 | 0.64 ± 0.76 |
| Dimethylglycine (1 mm) | 1.11 ± 0.75 | 3.98 ± 2.83a |
| Sarcosine (1 mm) | 1.03 ± 0.77 | 4.57 ± 2.55a |
a A significant difference (p < 0.05) from the folate-replete value.
In Vivo Formate Production in Rats Fed a High Protein Diet
Because a high protein diet would provide a greater quantity of potential one-carbon precursors, we examined whether such a diet would increase the rate of endogenous formate production. Rats were fed AIN-93G-based diets containing either 12% or 50% casein for 7 days and then infused with [13C]formate as described above. Table 4 shows that there was no significant difference in plasma formate levels, TTR, or rates of endogenous formate production. This could be explained by the occurrence of a regulatory mechanism, which balances the flux of one-carbon groups to formate relative to the flux to direct oxidation depending on the quantity of potential one-carbon groups available. Formate production amounted to 37% of the available dietary intake of potential one-carbon precursors in the rats fed the high protein diet and, remarkably, 176% in the rats fed the low-protein diet. This surprising result is discussed below.
TABLE 4.
In vivo kinetics of formate in rats fed high-protein or low-protein diets
Rats were fed AIN-based diets, containing either 12% or 50% casein for 7 days. Data are given as the mean ± S.D. (n = 5).
| Kinetic Parameters | High protein | Low protein |
|---|---|---|
| Plateau TTR | 0.046 ± 0.006 | 0.049 ± 0.011 |
| Plasma formate (μm) | 43.2 ± 4.6 | 38.3 ± 5.7 |
| Rate of endogenous formate production (μmol/h/100g) | 103.4 ± 21.3 | 98.2 ± 24.3 |
DISCUSSION
As far as we are aware there has been only one previous study that examined formate production in an animal model; in 1962, Annison and White (9) used [14C]formate to estimate endogenous formate production in sheep. They reported a value of 2–4 mg·kg−1·h−1, which amounts to ∼40–80 μmol·kg−1·h−1, approximately of the rates that occur in rats. Undoubtedly, this reflects to some degree the lower metabolic rates that occur in larger animals. A major finding of the present study is the relatively high rate of de novo formate production in the rat. There are many sources of formate. The metabolism of serine, glycine, dimethylglycine, and sarcosine in mitochondria can give rise to 5,10-methylene-THF, which may be converted to formate, as outlined in Fig. 1. There is good evidence that serine is the major source of one-carbon groups for the provision of methyl groups for methyltransferase reactions (22). There is also good evidence that mitochondrial serine hydroxymethyltransferase plays a critical role in the donation of one-carbon groups. This conclusion has been drawn from experiments with [2,3,3-2H3]serine (23). Metabolism of this labeled serine through serine hydroxymethyltransferase 1 in the cytosol yields 5,10-methylene-THF, which contains two atoms of 2H that, when reduced to 5-methyl-THF, yields methionine molecules that contain two atoms of 2H (M+2). Metabolism of [2,3,3-2H3]serine via the mitochondrial serine hydroxymethyltransferase 2 also yields 5,10-methylene-THF that is (M+2). However, it retains only one of these deuterons on conversion to 10-formyl-THF, with the result that M+1 formate is produced. When incorporated into the cytoplasmic THF pool this becomes (M+1) 5-methyl-THF and produces (M+1) methionine when used by methionine synthase. The ratio of (M+1) to (M+2) methionine produced from [2,3,3-2H3]serine is, therefore, a measure of the relative production of one-carbon groups from serine by the mitochondrial and cytoplasmic pathways (23). A variety of studies have shown that the mitochondrial pathway is the dominant route for the production of one-carbon units (22, 24, 25).
There are also a number of cytoplasmic reactions whereby formate is produced. These include reactions in the catabolism of tryptophan and histidine, a number of cytochrome P450-catalyzed demethylation reactions (such as the demethylation of lanosterol during cholesterol synthesis and the α-oxidation of phytanic acid). The magnitude of these reactions in aggregate is not known. An important conclusion from our experiments is that formate production represents a significant, although underappreciated, end product of amino acid and choline metabolism. In folate-replete animals, formate metabolism amounted to ∼75% of the maximum of one-carbon groups that could be produced from dietary amino acids and choline.
In our previous study we attributed the elevated plasma formate seen in folate deficiency to impairment in the utilization of one-carbon groups (8). This is certainly true. However, our present study revealed an additional impairment, an appreciable decrease in the rate of endogenous formate production (Table 1). This can be attributed to the depletion of the mitochondrial folate pool. In the study reported herein, we observed quite high rates of formate production from serine by mitochondria from folate-replete rats (Table 3) but much lower rates from glycine, sarcosine, and dimethylglycine. This is entirely consistent with the in vivo findings of Davis et al. (22) that serine is the primary source of methyl groups for homocysteine methylation. Formate production from serine by liver mitochondria from folate-deficient rats was greatly decreased (Table 3), confirming the greatly decreased rate of de novo formate production observed in vivo (Fig. 3). A very surprising result, however, was the increased production of formate from sarcosine and dimethylglycine in liver mitochondria from folate-deficient rats. These results may be explained by findings that both purified sarcosine dehydrogenase and dimethylglycine dehydrogenase are capable of enzymatic activity in the absence of THF (26). In the presence of THF, these enzymes transfer one-carbon groups from their respective substrates to produce 5,10-methylene-THF and the corresponding products (glycine from sarcosine and sarcosine from dimethylglycine). FAD is also reduced to FADH2. When assayed in the absence of THF, these enzymatic reactions still proceed except that the one-carbon groups are transferred from the substrates to water, producing formaldehyde. We suggest that such formaldehyde can be oxidized to formate by the mitochondrial formaldehyde dehydrogenase (27), accounting for the formate production from sarcosine and dimethylglycine that occurs in mitochondria from folate-deficient rats. These results may have broad implications. Dimethylglycine arises from choline catabolism. Sarcosine arises both via choline metabolism and from the activity of glycine N-methyltransferase, which uses S-adenosylmethionine to methylate glycine to sarcosine. It is conceivable, therefore, that formate may be produced in vivo, albeit at rather lower than normal rates, by mitochondria from folate-deficient rats. In such a situation, dietary methionine, choline, and betaine may be more important than serine as one-carbon precursors.
We fed rats on a high protein diet to determine whether this would oblige increased production of formate; it did not do so. There was no difference in the rate of de novo formate synthesis between rats fed a 12% and a 50% protein diet; neither was plasma formate elevated in the rats fed the high protein diet. Remarkably, in the rats fed the low protein diet, de novo formate production amounted to 176% of the available dietary intake of one-carbon groups (dietary intake minus the quantity required for net protein synthesis). This can only be accounted for if there is a high rate of de novo synthesis of one or more of the one-carbon precursors. Such a synthesis of serine has been demonstrated in Sprague-Dawley rats by Kalhan et al. (28). They found appreciable rates of serine synthesis (95 and 60 μmol·h−1·100 g body wt−1, respectively, in rats fed diets containing 6 and 24% protein). Because serine potentially produces 2 one-carbon groups per molecule, this de novo synthesis of serine can increase the potential availability of one-carbon groups by 120–190 μmol·h−1·100 g body wt−1. Taking a mean value of 155 μmol·h−1·100 g body wt−1, this increases the potential availability of one-carbon groups to 444 and 211 μmol·h−1·100 g body wt−1, respectively, in the rats fed the high and low protein diets. Thus formate synthesis in our rats fed the high-protein diet amounted to 23% of the total available one-carbon groups (net dietary intake plus de novo synthesis) and 46% in the rats fed the low-protein diet. These calculations are entirely consistent with the suggestion of Kalhan et al. (28) that the high rates of de novo serine synthesis are related to one-carbon metabolism.
That formate synthesis is not a stoichiometric product of amino acid oxidation suggests its production may be adjusted, probably in proportion to the need to provide one-carbon groups. In such a situation we suggest that excess one-carbon groups may be oxidized to CO2. There are a number of mechanisms whereby the inflow of one-carbon groups to the folate cycle may be regulated. One of these, the oxidation of formate by the peroxidative action of catalase, appears to be limited by the production of hydrogen peroxide (27). Krebs et al. (29) have described an elegant regulatory system whereby elevated S-adenosylmethionine levels inhibit methylene-THF reductase, giving rise to increased levels of 5,10-methylene-THF. Because the reactions between 10-formyl-THF and 5,10-methylene-THF are reversible, the increased 5,10-methylene-THF results in increased levels of 10-formyl-THF and increased rates of oxidation of the formyl group to CO2 via the 10-formyl-THF dehydrogenase. However, we cannot invoke these cytoplasmic mechanisms to explain the increased disposition of one-carbon groups by rats fed the high protein diet as they presuppose increased mitochondrial formate production in the first instance. However, there is a mitochondrial mechanism that permits the oxidation of excess one-carbon groups without requiring the production of increased quantities of formate. Mitochondrial 10-formyl-THF is at a metabolic crossroad. It may be converted to formate and THF by the mitochondrial MTHFD1L coupled to the synthesis of ATP via a substrate-level phosphorylation; it may also be oxidized to CO2 by the mitochondrial 10-formyl-THF dehydrogenase, accompanied by the reduction of NADP+ to NADPH. Relatively little is known about the regulation of this important crossroad; however, the ratio of CO2 to formate is not invariant. The relative conversion of the 3-carbon of serine to CO2 or to formate has been studied in rat liver mitochondria by García-Martínez and Appling (30) who reported a variable ratio of formate to CO2, depending on the serine concentration and whether the mitochondria are coupled or uncoupled. Virtually no CO2 is produced by avian liver mitochondria as birds have a very large requirement for formate for uric acid synthesis (31). We suggest that the mitochondrial disposition of 10-formyl-THF may be regulated in rats fed a high protein diet such that the ratio of CO2 to formate is increased. The accompanying increased production of intramitochondrial NADPH would make electrons available to the respiratory chain for ATP synthesis. Experiments to test this hypothesis are under way.
We describe the first study of in vivo formate kinetics in folate-deficient animals. Unexpectedly, it demonstrated decreased de novo formate production in folate deficiency. This is attributed to the requirement for folate coenzymes for the mitochondrial generation of formate. However, it is likely that the choline metabolites, sarcosine and dimethylglycine, may produce formate via formaldehyde in the absence of folate coenzymes. Formate production is a significant, although unappreciated, product of oxidative metabolism, particularly of amino acids and choline. Feeding a high protein diet did not increase formate production, indicating that the rate of formate production is not simply regulated by substrate supply. We anticipate that the method we describe for measuring formate kinetics in vivo will be applied to other experimental and nutritional paradigms in humans and non-human animals.
This work was supported, in whole or in part, by grants from the Canadian Institutes of Health Research and the Research Development Corporation (to J. T. B. and M. E. B.). We also acknowledge support by graduate fellowships from the School of Graduate Studies, Memorial University of Newfoundland (to G. P. M. and to L. M.), and Canadian Institutes of Health Research and Research Development Corp. (to L. M.).
This article is dedicated to the memory of Dr. Richard W. Hanson of Case Western Reserve University School of Medicine, a friend and champion of metabolism.
- THF
- tetrahydrofolate
- MTHFD1L
- monofunctional mitochondrial 10-formyl-THF synthetase
- TTR
- tracer:tracee ratio.
REFERENCES
- 1. Barlowe C. K., Appling D. R. (1988) In vitro evidence for the involvement of mitochondrial folate metabolism in the supply of cytoplasmic one-carbon units. Biofactors 1, 171–176 [PubMed] [Google Scholar]
- 2. Tibbetts A. S., Appling D. R. (2010) Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30, 57–81 [DOI] [PubMed] [Google Scholar]
- 3. Lamarre S. G., Morrow G., Macmillan L., Brosnan M. E., Brosnan J. T. (2013) Formate: an essential metabolite, a biomarker, or more? Clin. Chem. Lab. Med. 51, 571–578 [DOI] [PubMed] [Google Scholar]
- 4. MacFarlane A. J., Liu X., Perry C. A., Flodby P., Allen R. H., Stabler S. P., Stover P. J. (2008) Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J. Biol. Chem. 283, 25846–25853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Momb J., Lewandowski J. P., Bryant J. D., Fitch R., Surman D. R., Vokes S. A., Appling D. R. (2013) Deletion of Mthfd1l causes embryonic lethality and neural tube and faciocranial defects in mice. Proc. Natl. Acad. Sci. U.S.A. 110, 549–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Narisawa A., Komatsuzaki S., Kikuchi A., Niihori T., Aoki Y., Fujiwara K., Tanemura M., Hata A., Suzuki Y., Relton C. L., Grinham J., Leung K. Y., Partridge D., Robinson A., Stone V., Gustavsson P., Stanier P., Copp A. J., Greene N. D., Tominaga T., Matsubara Y., Kure S. (2012) Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Hum. Mol. Genet. 21, 1496–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Damian P., Raabe O. G. (1996) Toxicokinetic modeling of dose-dependent formate elimination in rats: in vivo-in vitro correlations using the perfused rat liver. Toxicol. Appl. Pharmacol. 139, 22–32 [DOI] [PubMed] [Google Scholar]
- 8. Lamarre S. G., Molloy A. M., Reinke S. N., Sykes B. D., Brosnan M. E., Brosnan J. T. (2012) Formate can differentiate between hyperhomocysteinemia due to impaired remethylation and impaired transsulfuration. Am. J. Physiol. Endocrinol. Metab. 302, E61–E67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Annison E. F., White R. R. (1962) Formate metabolism in sheep. Biochem. J. 84, 552–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bills N. D., Koury M. J., Clifford A. J., Dessypris E. N. (1992) Ineffective hematopoiesis in folate-deficient mice. Blood 79, 2273–2280 [PubMed] [Google Scholar]
- 11. Reeves P. G., Nielsen F. H., Fahey G. C., Jr (1993) AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 123, 1939–1951 [DOI] [PubMed] [Google Scholar]
- 12. Horne D. W., Patterson D. (1988) Lactobacillus casei microbiological assay of folic acid derivatives in 96-well microtiter plates. Clin. Chem. 34, 2357–2359 [PubMed] [Google Scholar]
- 13. Bensadoun A., Weinstein D. (1976) Assay of proteins in the presence of interfering materials. Anal. Biochem. 70, 241–250 [DOI] [PubMed] [Google Scholar]
- 14. Lamarre S. G., MacMillan L., Morrow G. P., Randell E., Pongnopparat T., Brosnan M. E., Brosnan J. T. (2014) An isotope-dilution, GC-MS assay for formate and its application to human and animal metabolism. Amino Acids 46, 1885–1891 [DOI] [PubMed] [Google Scholar]
- 15. Wolfe R. R. (1992) in Radioactive and Stable Isotope Tracers in Biomedicine, pp. 258–259, Wiley-Liss, New York [Google Scholar]
- 16. Wolfe R. R., Goodenough R. D., Wolfe M. H., Royle G. T., Nadel E. R. (1982) Isotopic analysis of leucine and urea metabolism in exercising humans. J. Appl. Physiol. 52, 458–466 [DOI] [PubMed] [Google Scholar]
- 17. Brosnan J. T., Man K.-C., Hall D. E., Colbourne S. A., Brosnan M. E. (1983) Interorgan metabolism of amino acids in streptozotocin-diabetic ketoacidotic rat. Am. J. Physiol. 244, E151–E158 [DOI] [PubMed] [Google Scholar]
- 18. Qian D., Brosnan J. T. (1996) Administration of Escherichia coli endotoxin to rat increases liver mass and hepatocyte volume in vivo. Biochem. J. 313, 479–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grady S., Osterloh J. (1986) Improved enzymatic assay for serum formate with colorimetric end-point. J. Anal Toxicol. 10, 1–5 [DOI] [PubMed] [Google Scholar]
- 20. Ferrell C. L., Koong K. J. (1986) Influence of plane of nutrition on body composition, organ size and energy utilization of Sprague-Dawley rats. J. Nutr. 116, 2525–2535 [DOI] [PubMed] [Google Scholar]
- 21. Rafecas I., Esteve M., Fernández-López J. A., Remesar X., Alemany M. (1994) Whole-rat protein content estimation: applicability of the N x 6.25 factor. Brit. J. Nutr. 72, 199–209 [DOI] [PubMed] [Google Scholar]
- 22. Davis S. R., Stacpoole P. W., Williamson J., Kick L. S., Quinlivan E. P., Coats B. S., Shane B., Bailey L. B., Gregory J. F., 3rd (2004) Tracer-derived total and folate-dependent homocysteine remethylation and synthesis rates in humans indicate that serine is the main one-carbon donor. Am. J. Physiol. Endocrinol. Metab. 286, E272–E279 [DOI] [PubMed] [Google Scholar]
- 23. Gregory J. F., 3rd, Cuskelly G. J., Shane B., Toth J. P., Baumgartner T. G., Stacpoole P. W. (2000) Primed, constant infusion with [2H3]serine allows in vivo kinetic measurement of serine turnover, homocysteine remethylation, and transsulfuration processes in human one-carbon metabolism. Am. J. Clin. Nutr. 72, 1535–1541 [DOI] [PubMed] [Google Scholar]
- 24. Herbig K., Chiang E. P., Lee L. R., Hills J., Shane B., Stover P. J. (2002) Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosynthesis. J. Biol. Chem. 277, 38381–38389 [DOI] [PubMed] [Google Scholar]
- 25. Pike S. T., Rajendra R., Artzt K., Appling D. R. (2010) Mitochondrial C1-tetrahydrofolate synthase (MTHFD1L) supports the flow of mitochondrial one-carbon units into the methyl cycle in embryos. J. Biol. Chem. 285, 4612–4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Porter D. H., Cook R. J., Wagner C. (1985) Enzymatic properties of dimethylglycine dehydrogenase and sarcosine dehydrogenase from rat liver. Arch. Biochem. Biophys. 243, 396–407 [DOI] [PubMed] [Google Scholar]
- 27. Waydhas C., Weigl K., Sies H. (1978) The disposition of formaldehyde and formate arising from drug N-demethylations dependent on cytochrome P-450 in hepatocytes and in perfused rat liver. Eur. J. Biochem. 89, 143–150 [DOI] [PubMed] [Google Scholar]
- 28. Kalhan S. C., Uppal S. O., Moorman J. L., Bennett C., Gruca L. L., Parimi P. S., Dasarathy S., Serre D., Hanson R. W. (2011) Metabolic and genomic response to dietary isocaloric protein restriction in the rat. J. Biol. Chem. 286, 5266–5277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krebs H. A., Hems R., Tyler B. (1976) The regulation of folate and methionine metabolism. Biochem. J. 158, 341–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. García-Martínez L. F., Appling D. R. (1993) Characterization of the folate-dependent mitochondrial oxidation of carbon 3 of serine. Biochemistry 32, 4671–4676 [DOI] [PubMed] [Google Scholar]
- 31. Sonne J. C., Buchanan J. M., Delluva A. M. (1948) Biological precursors of uric acid: I, the role of lactate, acetate, and formate in the synthesis of the ureide groups of uric acid. J. Biol. Chem. 173, 69–79 [PubMed] [Google Scholar]