

Background: Copper-zinc superoxide dismutase is a rare example of an intracellular protein with a disulfide bond.
Results: Disulfide mutant C57S SOD1 has 10% of the enzymatic activity of wild type.
Conclusion: The disulfide bond in SOD1 is not required for correct metal binding and enzymatic activity.
Significance: The disulfide bond in SOD1 may play a role in SOD1-linked amyotrophic lateral sclerosis.
Keywords: Amyotrophic Lateral Sclerosis (ALS) (Lou Gehrig Disease), Copper, Disulfide, Metalloenzyme, Oxidative Stress, Superoxide Dismutase (SOD), Superoxide Ion, Zinc
Abstract
The functional and structural significance of the intrasubunit disulfide bond in copper-zinc superoxide dismutase (SOD1) was studied by characterizing mutant forms of human SOD1 (hSOD) and yeast SOD1 lacking the disulfide bond. We determined x-ray crystal structures of metal-bound and metal-deficient hC57S SOD1. C57S hSOD1 isolated from yeast contained four zinc ions per protein dimer and was structurally very similar to wild type. The addition of copper to this four-zinc protein gave properly reconstituted 2Cu,2Zn C57S hSOD, and its spectroscopic properties indicated that the coordination geometry of the copper was remarkably similar to that of holo wild type hSOD1. In contrast, the addition of copper and zinc ions to apo C57S human SOD1 failed to give proper reconstitution. Using pulse radiolysis, we determined SOD activities of yeast and human SOD1s lacking disulfide bonds and found that they were enzymatically active at ∼10% of the wild type rate. These results are contrary to earlier reports that the intrasubunit disulfide bonds in SOD1 are essential for SOD activity. Kinetic studies revealed further that the yeast mutant SOD1 had less ionic attraction for superoxide, possibly explaining the lower rates. Saccharomyces cerevisiae cells lacking the sod1 gene do not grow aerobically in the absence of lysine, but expression of C57S SOD1 increased growth to 30–50% of the growth of cells expressing wild type SOD1, supporting that C57S SOD1 retained a significant amount of activity.
Introduction
Eukaryotic copper-zinc superoxide dismutase (CuZnSOD, SOD1) is an antioxidant enzyme that catalyzes the disproportionation of superoxide anion (O2⨪) to give dioxygen and hydrogen peroxide (2O2⨪ + 2H+ → O2 + H2O2) (1). An intracellular protein, CuZnSOD resides predominantly in the cytosol and in the intermembrane space of mitochondria (2), where it exists as extremely stable homodimers, each subunit of which contains a copper and a zinc ion as well as an intrasubunit disulfide bond. The maturation of CuZnSOD is complex and incompletely understood (3). It requires selective binding of copper and zinc ions, formation of the intrasubunit disulfide bond between Cys-57 and Cys-146, and dimerization of two subunits. The copper chaperone for Sod1 (CCS)3 plays a critical role in copper insertion and disulfide oxidation in vivo (4, 5), although a CCS-independent pathway has also been discovered (6, 7). It is made even more complex by the fact that the entire maturation process takes place independently in its two principal intracellular locations. SOD1 synthesized in the cytosol can mature there, but the portion of it destined for the mitochondrion must be inserted into the intermembrane space prior to maturation as the disulfide-reduced metal-free SOD1 polypeptide (2). Formation of the disulfide bond, but not metallation, is then required to trap it within the intermembrane space (2), where it matures.
CuZnSOD is also a rare example of a predominantly cytosolic protein with a stable disulfide bond. Such stable disulfide bonds are highly unusual in cytosolic proteins because, to be stable, they must be resistant to the highly reducing medium of the cytosol (8). The intrasubunit disulfide bonds in CuZnSOD are also unusual in being very tightly linked to subunit-subunit affinities in the metal-free dimeric protein. Apo human SOD1 (hSOD1) exists as a dimer when the disulfide bond is present, but, when the disulfide is reduced, it exists as marginally stable folded monomers (9).
The disulfide bond of SOD1 has also been implicated in the mechanism that causes mutant forms of SOD1 to become neurotoxic. More than 150 inherited mutations in the SOD1 gene have been linked to the familial form of the neurodegenerative disease amyotrophic lateral sclerosis (ALS) (10). These mutations convert normal WT SOD1 protein into a neurotoxic material by a mechanism likely to involve misfolding and oligomerization to form an amyloid-like substance (11, 12). The disulfide bond in ALS mutant SOD1 proteins is more readily reduced than that of WT SOD1 (13), and immature, disulfide-reduced ALS mutant SOD1 proteins have been implicated in the disease mechanism (14). Disulfide-reduced apo SOD1 has also been shown to be more prone to form amyloid fibrils than the disulfide-intact form (15), and small amounts of disulfide-reduced apo SOD1 will cause initiation of fibrilization of disulfide-intact apo SOD1 under physiologically relevant conditions (16).
In an interesting recent study in asthma patients, disulfide-reduced hSOD1 was elevated in human asthmatic erythrocyte and airway epithelial cells, leading to decreased SOD activity in asthma samples (17). The larger fraction of disulfide-reduced hSOD1 in asthma samples was evidenced by the oxidative modification of a disulfide cysteine (Cys-146) upon treatment with H2O2. Because the GSH/GSSG ratio is up-regulated in response to the oxidative inflammation that characterizes asthma, the authors observed that the greater reducing environment may reduce the disulfide bond in SOD1. Then a burst of oxidative stress could oxidatively modify Cys-146, blocking formation of the disulfide bond and decreasing total SOD1 activity.
Although studies of SOD1 with one or more of cysteines removed have been reported, detailed biophysical characterization of those mutants is unavailable. To learn more about the properties and function of the disulfide bonds in WT SOD1, we studied several mutant forms of human and yeast SOD1 proteins that are incapable of forming intramolecular Cys-57–Cys-146 disulfide bonds. By isolating and characterizing C57S mutant hSOD1 and comparing its properties to those of disulfide-reduced WT SOD1, we have established that the mutant protein is a good model for disulfide-reduced SOD1, allowing us to proceed without the heavy doses of reducing agents necessary to keep the WT protein reduced. Our comparisons of the properties of C57S mutant hSOD1 with those of disulfide-intact WT SOD1 have allowed us to estimate the degrees to which the disulfide bond influences several of the properties of the WT enzyme. We also found, in contrast to some earlier studies (2, 5), that the disulfide-free C57S mutant hSOD1 fully metallated with copper and zinc is enzymatically active, with a catalytic rate that is 10% of that of wild type hSOD1, and that the effects of this SOD activity are apparent in vivo when the C57S mutant hSOD1 gene is expressed in yeast strains lacking the normal WT SOD1 gene.
MATERIALS AND METHODS
Protein Expression and Purification
Yeast strains transformed to express human wild type or hC57S mutant SOD1 were kindly provided by Jorge Rodriguez and Peter Doucette (18). Proteins were purified using a published procedure (19) with the following minor modifications. The lysis buffer used was 250 mm Tris buffer at pH 8.0, 150 mm NaCl, 0.1 mm EDTA, and 0.1 mm DTT. After lysis, the centrifugation step was 18,600 × g for 30 min at 4 °C. The salting out step used ammonium sulfate to 65% saturation. The following centrifuge step was at 9820 × g for 40 min. Following the phenyl-Sepharose column and the pooling of Sod1 fractions, the buffer was changed to 2.25 mm potassium phosphate at pH 7.0. The sample was then loaded onto DEAE-Cellulose (U.S. Biologicals) pre-equilibrated with the same 2.25 mm potassium phosphate buffer and washed and eluted using an increasing salt gradient. The fractions containing SOD1 were pooled, concentrated, and subjected to gel filtration on a Sephadex G-75 superfine (Sigma) column. As isolated, the wild type hSOD1 contained 0.4 equivalents of copper and 2.8 equivalents of zinc per dimer, whereas the C57S hSOD1 contained no copper and 4.0 equivalents of zinc. As-isolated C146R mutant SOD1 protein, containing 0.19 equivalents of copper and 2.76 equivalents of zinc per dimer, was obtained from Peter Doucette (20). apo SOD1 proteins were prepared as described previously (21). In short, as-isolated proteins were dialyzed against 100 mm acetate buffer, pH 3.8, in the presence of 10 mm EDTA and then dialyzed against 100 mm acetate buffer, pH 3.8, containing 100 mm NaCl three times. At the second dialysis step, protein-bound EDTA was removed. Finally, proteins were dialyzed against 100 mm acetate buffer at pH 5.5 three times. Buffers that do not contain EDTA were passed through Chelex 100 resin (Bio-Rad) to remove trace metals.
Yeast C57S,C146S SOD1 was produced by site-directed mutagenesis and purified from Escherichia coli BL21(DE3). Cells were lysed by sonication in 100 mm phosphate buffer, pH 7.4, with 1 mm EDTA and PMSF. The supernatant was dialyzed overnight to yield protein in 5 mm phosphate buffer, pH 7.4. Protein was loaded onto a DE52 anion exchange column pre-equilibrated with 5 mm phosphate buffer and eluted with a potassium phosphate salt gradient. Fractions containing SOD1 were identified by SDS-PAGE, pooled, and loaded on a G75 size exclusion column pre-equilibrated with 20 mm sodium phosphate, 80 mm NaCl. Fractions containing SOD1 were similarly identified and run on a DEAE Sephadex A-50 (Pharmacia) column. Concentrations were determined by UV-visible spectrophotometry.
For crystallization, hC57S was purified from yeast according to previously published methods (19), with the exception that some preparations were further purified by anion exchange chromatography on a DEAE Sephadex A-50 (Pharmacia) column. apo SOD1 proteins were prepared as described above.
Crystallization, Data Collection, and Refinement
All crystals were grown by the hanging drop vapor diffusion method. Metal-bound C57S at 15 mg/ml in 2.25 mm potassium phosphate, pH 7.0, and 160 mm NaCl was mixed with an equal volume of reservoir solution containing 0.2 m ammonium sulfate, 0.1 m bis-tris, pH 5.5, and 25% polyethylene glycol 3350. apo C57S at a concentration of 19 mg/ml in 100 mm potassium phosphate, pH 5.5, was mixed with an equal volume of reservoir solution containing 3.2 m ammonium sulfate and 0.1 m MES, pH 6.0. Crystals grew in 2 weeks at room temperature. Crystals of metal-bound C57S and apo C57S were soaked in a cryoprotectant solution containing 15% ethylene glycol or glycerol in the appropriate mother liquor before flash cooling in the cryostream. X-ray data including copper and zinc anomalous edge data of metal-bound C57S were collected at Beamline 19BM at the Advanced Photon Source (Argonne, IL). X-ray data from crystals of apo C57S were collected at our home source Rigaku FR-D rotating copper anode x-ray generator equipped with R-AXIS HTC imaging plate systems. The data were indexed and scaled using HKL2000 (22). Initial phases were generated by the molecular replacement method using SOD1 mutant G37R (PDB entry 1AZV) (23) as the search model with MOLREP (24). The asymmetric unit of metal-bound C57S contains six SOD dimers, whereas apo C57S contained a single dimer. The structures were refined using PHENIX (25), and manual model building was carried out using COOT (26). The application of noncrystallographic symmetry restraints was used only in early stages of refinement for metal-bound and apo C57S. Noncrystallographic symmetry restraints were applied throughout the refinement process for apo C57S. Final coordinates and structure factors have been deposited in the Protein Data Bank (27) under accession codes 4MCM (C57S) and 4MCN (apo C57S).
Mass Spectrometry
The purity and molecular weight of proteins were determined by electrospray ionization mass spectrometry (PerkinElmer Life Sciences/Sciex API III equipped with an ion spray source), as well as SDS-PAGE. Samples (∼20 pmol/μl) were dissolved in 50:50:0.1 water/acetonitrile/formic acid (v/v/v) and injected into the electrospray source. Data collection conditions were as follows: m/z range, 400–2300; mass step, 0.3; dwell time, 1.0 ms; scan speed, 5.84 s/scan; flow rate, 10 μl/min; and orifice voltage, 90 V. Collected positive ion mass spectra were deconvoluted using BioMulti View version 1.3.1 (PerkinElmer Life Sciences/Sciex).
Analysis of Metal Content
Metal ion concentrations were quantified by either inductively coupled plasma atomic emission spectroscopy (ICP-AES) or inductively coupled plasma mass spectrometry (ICP-MS). ICP-AES used a Thermo Jarrel Ash (Thermo Electron) IRIS 1000 instrument. Samples were prepared as previously described (28), except samples were dissolved in 5.0% HNO3, and three readings were taken per wavelength and averaged. For ICP-MS, samples were prepared by mixing aqueous protein samples with an equal volume of concentrated OPTIMA grade nitric acid (Fisher) and then digested at 95 °C for 2 h in open vessels in a dust-free environment. The digested contents of the tubes were then extracted with 500 μl of 2% OPTIMA grade nitric acid. The individual extracts were diluted to a total volume of 2.00 ml with 2% nitric acid. 20 μl of a 5-ppm aqueous germanium ion solution (ICP-MS grade) was then added to each tube as an internal standard, and the contents were vortexed vigorously. ICP-MS measurements were then conducted on an Agilent 7500ce ICP-MS in both helium and H2 collision gas modes. Reported measurements of copper and zinc in each sample represent the averages of three measurements. The individual measurements are typically obtained with a relative standard deviation of ∼3% or less per sample. In both methods, metal contents were calculated to obtain the number of metal ions bound per SOD1 dimer.
Differential Scanning Calorimetry (DSC)
The thermal stabilities of proteins were measured as previously reported (29). DSC experiments on disulfide-reduced apo proteins were carried out in 100 mm phosphate buffer containing 100 mm DTT, pH 7.4. Buffer plus 100 mm DTT was used in the reference cell. Experiments on disulfide-reduced as-isolated proteins were performed in 100 mm acetate buffer with 40 mm Tris (2-carboxyethyl)phosphine) (TCEP), pH 5.5. CpCalc software (Calorimetry Sciences Corp.) was used to determine molar heat capacity values, and Origin software (Microcal) was used to fit the data.
Metal Ion Titrations and Remetallation of Enzymes
Metal ion titrations were performed in 100 mm acetate buffer at pH 5.5, unless otherwise specified, in accordance with previous procedures (30). Briefly, titrations of the apoproteins with solutions of cupric and cobaltous sulfate solutions were monitored by UV-visible spectroscopy using Shimadzu UV-2501 PC spectrophotometer. A typical enzyme solution of 150 μl of ∼0.3 mm (as dimeric enzyme concentration, equaling 0.6 mm concentration of monomers) was titrated with the appropriate 10 mm metal sulfate solutions. In all titrations, the total volume of metal solution was no more than 10% of the total volume of apoprotein titrated. Protein samples were centrifuged before spectra were taken to avoid turbidity. End points were reached when the spectra of successive metal additions overlapped. To avoid oxidative damage to the protein used in the pulse radiolysis experiments, the copper titration into as-isolated human C57S SOD1 was performed anaerobically. Lack of oxidative damage to this copper-titrated, as-isolated human C57S SOD1 was verified by mass spectrometry. One-zinc C57S and wild type and two-zinc wild type were prepared by titrating the appropriate amount of metal into the apoproteins. Metal content was verified by ICP-AES. The two-zinc C57S SOD1 was prepared by adding excess amounts of Zn2+ to apo human C57S SOD1 and incubating for 1 h at 37 °C. After washing with metal-free buffer, metal contents were measured by ICP-AES.
Electrospray MS and Hydrogen/Deuterium Exchange (HDX)
Global H/D exchange experiments were carried out as previously reported (18). Site-specific H/D exchange experiments were conducted at 4 °C as described previously (31).
Electron Paramagnetic Resonance
Protein samples for EPR were prepared by loading 150 μl of SOD1 variants containing copper concentrations between 0.4 and 1.5 mm, into 4-mm EPR tubes (Wilmad Glass) and rapidly freezing using ethanol/dry ice. Solutions were buffered with 100 mm phosphate at pH 7.4. Spectra were recorded on a Bruker X-band spectrometer. Samples were held at 97 K during the data collection using a variable temperature nitrogen gas setup with a Wilmad quartz Dewar insert. Instrumental parameters were as follows: center field, 3000 G; sweep width, 1600 G; frequency, 9.44 GH; microwave power, 20 milliwatt; modulation amplitude, 5 G; modulation frequency, 100 kHz; receiver gain, 5.02 × 104; and time constant, 81.92 ms. Each spectrum is representative of an average of eight scans.
Yeast Cell Growth and Native Gel Activity Staining
The yeast strain for yeast growth studies and native activity gels was EG118 (MAT α, leu2-3, 112, his3Δ1, trp1-289, ura3-52; Sod1::URA3) (32) transformed to express the wild type or the C57S mutant SOD1 on the plasmid pRS424 (2μ, trp marker) (33). Yeast were plated onto synthetic dextrose media plates supplemented with amino acids except for tryptophan (SD−Trp). Overnight startup cultures were grown from a fresh single colony in liquid SD−Trp medium. Cells were inoculated at A600 of 0.05 in liquid SD−Trp−Lys and incubated at 30 °C with shaking for a maximum of 24 h. Total cell growth was monitored by measuring optical density at 600 nm. SOD activity by native gel used nondenaturing gel electrophoresis and staining with nitro blue tetrazolium (34). The SOD activity appeared as a clear band in contrast to the purple background.
Activity Measurement and Pulse Radiolysis
SOD activity in purified protein was measured directly by pulse radiolysis using the 2-MeV Van de Graaff accelerator at Brookhaven National Laboratory using previously published procedures (35), with the exception that experiments using yeast SOD1 were carried out in 12 mm phosphate, 12 mm formate, and 10 μm (for C57S,C146S) or 30 μm (for wild type) EDTA. The rate constant of hSOD1 was determined at pH 5.5 in the presence and absence of EDTA. The amount of copper bound to the enzyme was determined by subtracting free copper concentration from total copper concentration determined by ICP-AES (for hSOD1) or ICP-MS (for ySOD1). The free copper concentration was determined by plotting the measured rate constant of the buffer solution alone into a standard curve (rate constant versus free copper concentration) or by dividing the difference in rate with and without EDTA by the rate constant for the dismutation of O2⨪ by free copper (36). The reported data were averages of five measurements for every pH data point. To calculate specific activities, the observed rates were divided by the concentration of enzyme-bound copper. Ionic strength was varied by addition of NaCl.
RESULTS
Metal Binding Properties of Disulfide-free C57S Mutant Human SOD1
Recombinant WT and C57S mutant hSOD1, when expressed and purified under identical conditions, contained 0.4 equivalent copper and 2.8 equivalent zinc per dimer and 0.0 equivalents copper and 4.0 equivalents zinc per dimer, respectively (20). To gain insight into the metal binding process in apo disulfide-reduced hSOD1, we prepared the apoproteins of both WT hSOD1 and C57S hSOD1 and performed in vitro remetallation studies. The results of all titrations are summarized in Table 1 and described in detail below.
TABLE 1.
Summary of metal titrations
Separate experiments titrating cobalt, copper, and zinc to apo human C57S SOD1 all show a phantom subunit in which one subunit of the dimer does not bind metals (see text and figures cited below for detail).
| Species | Original metal | Metal added | pH | Figure/reference | Analytical technique | Result |
|---|---|---|---|---|---|---|
| hC57S | None | Co2+ | 5.5 | Fig. 1B | UV-visible | 1 Co2+ per dimer (phantom subunit) |
| hWT | None | Co2+ | 5.5 | Fig. 1A | UV-visible | 2 Co2+ per dimer (normal binding) |
| hC57S | None | Co2+ | 7.4 | Fig. 1D | UV-visible | 2 Co2+ per dimer (probable phantom subunit) |
| hWT | None | Co2+ | 7.4 | Ref. 38 | UV-visible | 4 Co2+ per dimer |
| hC57S | None | Cu2+ | 5.5 | Fig. 3B | UV-visible | One copper in copper site, one copper in zinc site (probable phantom subunit) |
| hWT | None | Cu2+ | 5.5 | Fig. 3A | UV-visible | Both coppers in copper sites (normal binding) |
| hC57S | None | Zn2+ | 5.5 | Fig. 2 | Temperature-dependent H/D exchange | Heterodimer (one phantom, one metal subunit) |
| hWT | None | Zn2+ | 5.5 | Fig. 2 | Temperature-dependent H/D exchange | Homodimer |
| hC57S | Four zincs | Cu2+ | 5.5 | Fig. 3C | UV-visible | Normal binding to both subunits |
Cobalt Binding to Apoproteins
Cobalt is commonly used as a zinc substitute in studying SOD1 metal binding properties. Zn2+ is spectroscopically silent, and Co2+, in contrast, gives three intense absorption bands in the visible range. In previous studies (30) and in this work (Fig. 1A), remetallation of apo WT hSOD1 by stepwise titrations of one-quarter equivalents of Co2+ at pH 5.5 revealed an end point at two equivalents Co2+ per dimer. In the case of apo C57S hSOD1, stepwise addition of the first equivalent of Co2+ resulted in an electronic spectrum (Fig. 1B) superimposable upon that of WT hSOD1 titrated with one equivalent of Co2+ (Fig. 1A), indicating that the two proteins bind cobalt ions to their zinc sites with comparable coordination geometries. Calculations using the extinction coefficient previously determined for Co2+ in the zinc site of hSOD1 (370 m−1 cm−1 (37)) confirmed that this end point was reached at one equivalent of cobalt per SOD1 dimer. Washing with a metal-free buffer followed by ICP-AES measurements further confirmed that C57S hSOD1 contained only one Co2+ per dimer. However, in contrast to WT hSOD1, further stepwise additions of Co2+ to one-cobalt C57S hSOD1 yielded no significant increase in the absorbance (Fig. 1B). Moreover, fast addition of excess cobalt to apo C57S hSOD1 yielded the same end point at one equivalent of cobalt bound per dimer (Fig. 1C).
FIGURE 1.

Titration of Co2+ into wild type and C57S hSOD1, monitored by UV-visible absorption spectroscopy at 300–800 nm. A, stepwise titrations into wild type SOD1 at pH 5.5 show an end point at two equivalents Co2+ per dimer. Spectrum a, 1 equivalent Co2+ per dimer; spectrum b, 1.25 equivalents Co2+ per dimer; spectrum c, 1.5 equivalents Co2+ per dimer; spectrum d, 1.75 equivalents Co2+ per dimer; spectrum e, 2 equivalents Co2+ per dimer. B, stepwise titrations into C57S SOD1 at pH 5.5 show an end point at one equivalent Co2+ per dimer. Spectrum a, 1 equivalent Co2+ per dimer; spectrum b, 1.25 equivalents Co2+ per dimer; spectrum c, 1.5 equivalents Co2+ per dimer. C, addition of excess Co2+ to C57S SOD1 yields one equivalent per dimer after washing. Six equivalent Co2+ per dimer were added to C57S SOD1 at once. After washing, spectrum was taken again. Spectrum a, the end point of stepwise Co2+ titration (spectrum c from Fig. 1B); spectrum b, excess Co2+ addition before washing; spectrum c, excess Co2+ addition after washing. D, stepwise titrations into C57S SOD1 at pH 7.4 show an end point at two equivalents Co2+ per dimer. Spectrum a, apo; spectrum b, 1 equivalent Co2+ per dimer; spectrum c, 2 equivalent Co2+ per dimer; spectrum d, difference b − a. Squares and diamonds symbolize the zinc-binding and copper-binding sites, respectively.
To mimic physiological conditions more closely, we titrated cobalt ions into C57S hSOD1 at pH 7.4 and found that approximately two equivalents of Co2+ were bound per dimer (Fig. 1D). By contrast, apo WT human SOD1 and apo bovine SOD1 are known to bind four Co2+ per dimer under the same conditions (38), with Co2+ occupying all zinc and copper sites. The asymmetry of the difference spectrum for human C57S in Fig. 1D relative to the one- and two-cobalt spectra shows that the second cobalt binds to a site different from the first cobalt. The difference spectrum also resembles what was reported in an analogous study of cobalt binding to apo yeast SOD1 at pH 5.5 (37). In that study, it was determined that the first Co2+ bound to one of the zinc sites, and the second bound to the copper site of the same subunit, leaving a metal-free subunit that has thus been termed a “phantom subunit.” We conclude, therefore, that two Co2+ ions bind to one subunit of the human apo C57S dimer at pH 7.4, leaving empty a phantom subunit.
Zinc Binding to Apoproteins
WT hSOD1 containing two zinc ions is known to be dimeric with one zinc bound to the zinc site in each of the two subunits. C57S hSOD1 containing two zinc is also known to be dimeric (19), but the exact locations of the two zinc ions were unknown. To address whether two-zinc C57S hSOD1 dimer is a Zn1,Zn1-hSOD1 homodimer, with one zinc in each subunit, or a Zn2,E2-hSOD1 heterodimer, with an apo subunit and two zinc ions in the other subunit, we titrated Cu2+ ions into two-zinc C57S hSOD1. Copper binding caused the appearance of a 670-nm band, which is characteristic of copper in the copper site (37), and the zinc contents of the sample decreased from two equivalents per subunit to one as determined by ICP-AES. It appears, therefore, that the displaced zinc ion had been bound in a copper site, leading to the conclusion that two-zinc C57S SOD1 is a heterodimer.
We also used HDX to determine the location of the zinc binding sites. For these experiments, we compared five species: one- and two-zinc per dimer human wild type SOD1 and apo and one- and two-zinc per dimer human C57S SOD1. The protein was first incubated for 2 h in 10 mm phosphate buffer (90% D2O) at ambient temperature, pH 7.4, to exchange all rapidly exchanging amide protons in SOD1 with deuterons. The masses of the protein subunits were then monitored while the temperature of the solution was increased stepwise. Because temperature increases caused the subunits to unfold, additional exchange, evidenced by an increase in mass, could be observed.
For apo C57S hSOD1, a mass increase started at 26 °C when the fully deuterium-exchanged form began to appear (mass 16,030 Da), suggesting that the protein had begun to unfold (Fig. 2). By 32 °C, the protein was fully exchanged, as evidenced by the disappearance of the partially exchanged species (mass, 16,005 Da). Because the mass/temperature profile is a function of the metal binding to each subunit, two identical apo subunits would be expected to unfold at the same temperature, resulting in a single unfolding event, as was observed. For one-zinc C57S hSOD1, however, two unfolding events were observed. The first took place at 47 °C (Fig. 2) followed by a second at 53 °C. These two mass increases correspond to the unfolding of the metal-free and the one-zinc subunit, respectively. Similarly, one-zinc WT hSOD1 also has two pronounced mass increases (Fig. 2), at temperatures ∼10 °C higher than the corresponding increases in C57S hSOD1.
FIGURE 2.

Temperature-dependent H/D exchange kinetics for apo C57S SOD1, one-zinc, and two-zinc derivatives of human WT and C57S SOD1. Thermal scans of unfolding of apo, one-zinc, and two-zinc derivatives of human WT and C57S SOD1 were monitored by H/D exchange and electrospray ionization-MS. In the two-dimensional charts, the mass distribution versus temperature is shown. The colors indicate the intensity of the mass signal, which, in the case of identical polypeptides, is roughly proportional to the concentration of the protein at each mass and temperature.
As expected, two-zinc WT hSOD1 shows a single mass increase at 65 °C (Fig. 2), consistent with its assignment as a homodimer containing one zinc ion per monomer. In the case of two-zinc C57S hSOD1, however, two mass increases were observed, at 53 and 63 °C (Fig. 2), indicating that one subunit is metallated with two zinc ions and thus unfolds at the higher temperature, and the other is metal-free and unfolds at the lower temperature. This is consistent with our findings in the copper titration into the two-zinc protein described above.
Copper Binding to Apoproteins
UV-visible spectra of apo WT hSOD1 and apo C57S hSOD1 titrated with Cu2+ are shown in Fig. 3. The addition of one equivalent Cu2+ to either protein yielded an absorption peak near 670 nm, suggesting that the copper site geometry in the mutant protein is similar to that of the native WT SOD1 (39). In the case of WT hSOD1, the addition of the second equivalent of Cu2+ per dimer increased the intensity of 670 nm peak by 2-fold (Fig. 3A), consistent with the second Cu2+ binding to the copper site of the second subunit (40). By contrast, the addition of the second equivalent Cu2+ to C57S hSOD1 generated a new visible band at 810 nm rather than increasing the magnitude of the 670-nm peak (Fig. 3B). The d-d transition centered at 810 nm has previously been assigned to copper in the zinc site of SOD1 (39, 40). Thus, we conclude that the first equivalent Cu2+ binds to the copper site and the second equivalent to the zinc site of C57S hSOD1. After intensive washing, the electronic absorption spectrum of two-copper C57S hSOD1 was unchanged, and ICP-AES results confirmed that two Cu2+ bound per dimer. This behavior was also observed in studies of copper binding to the yeast WT SOD1 (37) and is consistent with a phantom subunit in which the two Cu2+ bind to one subunit only.
FIGURE 3.
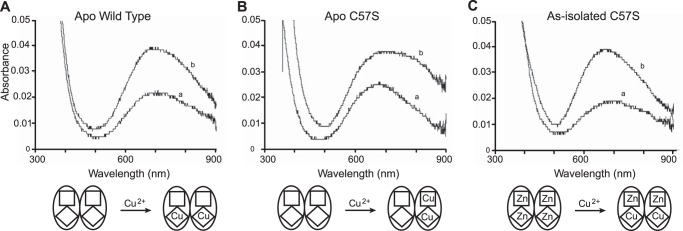
Comparison of spectra from copper titration into wild type and C57S SOD1. A–C, apo human WT SOD1 (A), apo C57S SOD1 (B), and as-isolated C57S SOD (C) (0.0 copper and 4.0 equivalents zinc), all in 100 mm acetate buffer at pH 5.5. Spectrum a, 1 equivalent Cu2+ per dimer; spectrum b, 2 equivalents Cu2+ per dimer. Squares and diamonds symbolize the zinc-binding and the copper-binding sites, respectively.
Copper Binding to Four-zinc Human C57S SOD1
ICP-AES measurements of as-isolated C57S hSOD1 demonstrated that each protein dimer contained negligible copper and four zinc ions, even after extensive washing, and the x-ray structure of as-isolated C57S hSOD1 confirmed that zinc ions occupy all four metal binding sites. Although titrations of copper and zinc into the C57S hSOD1 apoprotein failed to give proper reconstitution of the protein, titrating the as-isolated four-zinc C57S hSOD1 with copper ions readily reconstituted the “native” metal content. The UV-visible spectrum of this two-copper, two-zinc C57S hSOD1 displayed a d-d transition centered at 670 nm (Fig. 3C), similar to that observed for WT hSOD1 (39), indicating that the added copper replaced the zinc ions bound to the copper sites. Extensive washing with a metal-free buffer did not change the spectra. ICP-AES results confirmed the binding of two copper and two zinc ions per dimer.
Thermal Stability of Disulfide-free C57S Mutant Human SOD1
Apoproteins
A previous DSC study demonstrated that the melting temperature of apo WT hSOD1 was reduced from 52 to 42 °C, when DTT was added to reduce the disulfide bond (18) and that apo C57S hSOD1 melts at 42 °C. Here, DSC experiments on apo C57S hSOD1 in phosphate buffer at pH 7.4 in the presence or absence of DTT each yielded one endotherm with a Tm value of 42 °C (Fig. 4A), confirming that the presence of DTT had no effect on the stability of hSOD1 other than in its role as a reducing agent.
FIGURE 4.
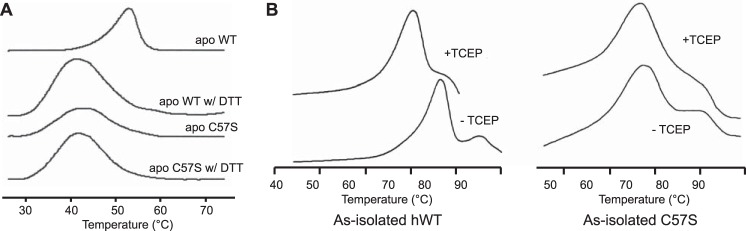
Thermal stability of wild type and C57S SOD1. A, DSC profiles of apo WT SOD1 and apo C57S SOD1 in 100 mm phosphate buffer at pH 7.4 with or without 100 mm DTT. The intrasubunit disulfide bond reduction of WT apo SOD1 lowers the melting temperature to the same value as apo C57S SOD1. B, DSC profiles of as-isolated WT and C57S SOD1 in 100 mm acetate at pH 5.5 with or without 40 mm TCEP. The entire thermal profile of as-isolated hWT under reducing conditions was shifted to lower temperatures without destroying the multiple overlapping endothermic transitions. The reduction of intrasubunit disulfide bond of as-isolated WT SOD1 lowers the melting temperature to the similar value as as-isolated C57S SOD1. As-isolated C57S SOD1 looks nearly identical in both conditions, with or without TCEP.
Metallated Proteins
Using DSC, we compared thermal stability of as-isolated WT hSOD1 (0.4 equivalents copper and 2.8 equivalents zinc) and as-isolated C57S hSOD1 (0.0 copper and 4.0 equivalents zinc) with or without the reducing reagent TCEP. The multiple overlapping endothermic transitions shown in all scans represent the unfolding of differently metallated SOD1 species (29, 41).
Reduction of the disulfide bond by TCEP decreased the melting temperature of as-isolated WT hSOD1 by ∼8 °C (Fig. 4B). By contrast, the addition of TCEP did not change the scanned trace of C57S hSOD1 (Fig. 4B). Because both proteins are dimers, the lack of change in the thermostability of the dimeric mutant protein is due to the absence of the disulfide bond. Thus, the presence of the disulfide bond appears to impart stability in metallated dimeric hSOD1.
Dynamic Behavior of Disulfide-free C57S Mutant Human SOD1
To assess further the effect of disulfide bond formation on the dynamics of hSOD1, we performed both global and site-specific HDX measurements. First the overall dynamics of disulfide-oxidized and disulfide-reduced apo hSOD1 can be compared by the number of unexchanged amide backbone hydrogens as a function of time. As seen in Fig. 5, apo disulfide-oxidized WT hSOD1 at 10 °C retained ∼50 unexchanged protons following an 80-min incubation in 90% D2O, whereas disulfide-reduced WT protein retained ∼35. The disulfide-reduced wild type protein also had faster HDX kinetics, as determined by biexponential curve fitting of the global HDX curves (42). These results suggest that the apoprotein has a more rigid structure when the disulfide bonds are present than when they are reduced. In addition, the disulfide-reduced apo WT hSOD1, apo C57S hSOD1, and apo C146R hSOD1 had nearly identical numbers of unexchanged protons over time (Fig. 5). The similarity between the three suggests that it is the absence of the disulfide bond that causes the faster exchange rather than some other property that has been altered by one of the disulfide bond mutations.
FIGURE 5.
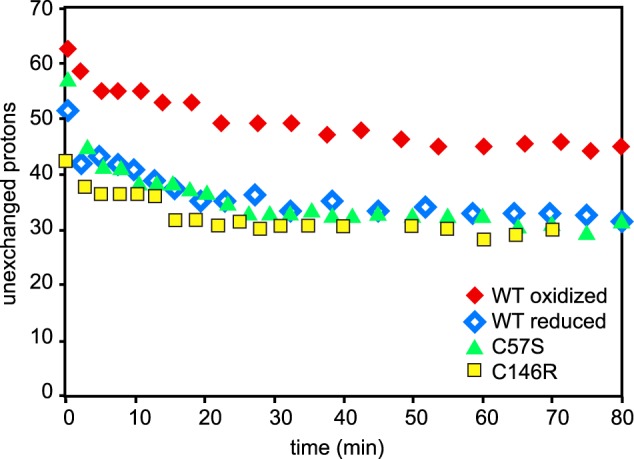
Global H/D exchange kinetics of human wild type apo disulfide-oxidized SOD1, disulfide-reduced SOD1, apo C57S, and apo C146R SOD1. Apo C57S SOD1 shows the identical behavior to reduced apo hWT SOD1 in global H/D exchange experiments. apo C146R SOD1 also exhibits comparable behavior to wild type disulfide-reduced hWT SOD1 and C57S SOD1.
Finally, we compared apo WT hSOD1 to apo C57S hSOD1 by site-specific HDX. These experiments showed that the peptides in the two proteins exhibited the same HDX behavior in all of the detectable peptides, except the one covering amino acids 104–116 (Fig. 6) and a slight difference in a small section of the β-barrel (residues 46–53). That small change in amino acids 104–116 is considered to result from monomer-dimer equilibrium or from mutation of the cysteine to serine, and a slight loosening of structure near residue 57 is consistent with breaking the disulfide bond. Therefore, it appears that the local dynamics of the two proteins are not profoundly altered by the removal of the disulfide bond, when the metal cofactors are not present.
FIGURE 6.

Site specific H/D exchange kinetics of WT apo SOD1 and apo C57S SOD1. Site-specific H/D exchange kinetics of apo SOD1 and apo C57S SOD1 are shown for each proteolytic fragment that was separated by reverse phase chromatography. apo C57S SOD1 possesses a local structure similar to WT apo SOD1. Squares, WT apo SOD1; diamonds, apo C57S SOD1.
Crystal Structures of As-isolated and apo C57S
Here we report the x-ray crystal structure of metal-bound (as-isolated; PDB entry 4MCM) and metal-deficient (apo; PDB entry 4MCN) human C57S SOD1s, refined to 2.15 Å and 2.6 Å, respectively (Table 2).
TABLE 2.
Data collection and refinement statistics
| C57S |
||||
|---|---|---|---|---|
| apo | As isolated |
|||
| Copper edge | Zinc edge | |||
| Data collection | ||||
| Space group | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | ||||
| a, b, c (Å) | 39.9, 56.0, 105.5 | 74.5, 163.9, 174.1 | 74.5, 164.0, 174.4 | 74.5, 164.1, 174.3 |
| Wavelength | 1.5418 | 1.0332 | 1.3779 | 1.2828 |
| Resolution (Å) | 50-2.6 | 30-2.2 | 50-2.8 | 50-2.8 |
| Rsyma | 0.119 (0.576) | 0.064 (0.482) | 0.119 (0.510) | 0.120 (0.512) |
| I/σI | 17.0 (3.6) | 25.6 (2.9) | 17.9 (3.9) | 13.4 (3.5) |
| Completeness (%) | 100 (100) | 99.3 (95.4) | 100 (100) | 100 (100) |
| Redundancy | 7.0 (7.0) | 6.1 (4.4) | 5.7 (5.6) | 5.5 (5.1) |
| Wilson B value | 33.9 | 32.9 | 30.8 | 31.7 |
| Refinement | ||||
| Resolution (Å) | 38.4-2.6 | 29.8-2.2 | ||
| No. reflections | 7,738 | 106,830 | ||
| Rwork/Rfree | 0.189/0.245 | 0.211/0.262 | ||
| No. atoms | ||||
| Protein | 1,831 | 13,315 | ||
| Metals | - | 24 Zn2+ | ||
| Ligand | 5 (1 SO42 −) | 70 (14 SO42 −) | ||
| Solvent | 42 | 1094 | ||
| B-factors | ||||
| Protein | 29.4 | 31.6 | ||
| Metals | 29.6 (24 Zn2+) | |||
| Ligand | 44.9 (1 SO42 −) | 35.9 (14 SO42 −) | ||
| Solvent | 29.0 | 28.9 | ||
| Root mean square deviations | ||||
| Bond lengths (Å) | 0.010 | 0.008 | ||
| Bond angles (°) | 1.219 | 1.154 | ||
a The values in parentheses are for the highest resolution shell.
As-isolated, Metal-bound C57S
The overall fold observed for wild type SOD (43, 44) is largely preserved in the as-isolated, metal-bound C57S mutant. The structure contains six SOD1 dimers in the crystallographic asymmetric unit, designated as A/B, C/D, E/F, G/H, I/J, and K/L, where the slash (/) represents the naturally occurring homodimer interface. The electron density allowed the positioning of 153 residues in all of the monomers except N-terminal residue 1 in two monomers. Ninety-nine percent of the amino acid residues fall in the allowed regions of a Ramachandran plot.
Copper and Zinc Binding Sites
In the as-isolated, metal-bound C57S structure, all the metal ligands are clearly visible in the electron density, and no disorder is observed compared with wild type SOD1. Both the copper and zinc sites are occupied. Analysis of x-ray diffraction data collected at the copper and zinc absorption edges reveal that both copper and zinc sites are occupied mainly by zinc ions (Table 2 and Fig. 7A). In addition, the sulfate ion coordinated to the copper site (Fig. 7A) was previously found only when zinc occupies the copper site (45).
FIGURE 7.
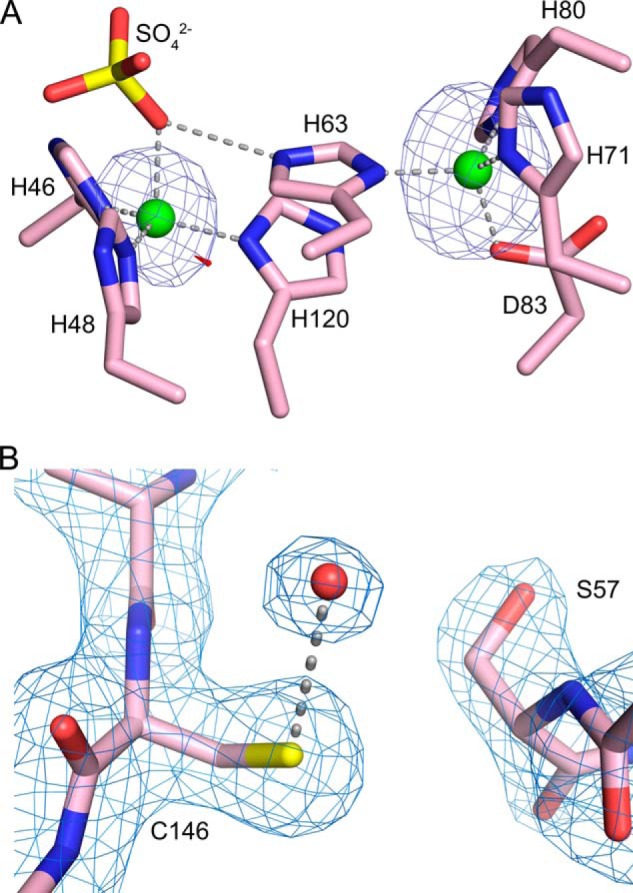
A, identification of zinc ions in the as-isolated, metal-bound C57S hSOD1 structure (PDB entry 4MCM) using x-rays tuned to the copper and zinc absorption edges. Subunit A is shown, which is representative of all 12 SOD1 subunits in the asymmetric unit. The copper-binding site is on the left, and the zinc-binding site is on the right. Zinc ions are shown as green spheres, and a sulfate ion coordinated to the zinc is shown as yellow and red sticks. X-rays tuned to the copper edge do not promote anomalous scattering by zinc and thereby act as the discriminator between the two metal ions. The small amount of electron density shown in red, near the metal in the copper site, comes from the anomalous scattering by copper calculated using data from the copper edge. The electron density shown in blue in both metal-binding sites comes from the anomalous scattering by zinc calculated using data from the zinc edge. The copper edge and zinc edge anomalous difference Fourier electron density maps are both contoured at 5 σ. B, apo C57S hSOD1 (PDB entry 4MCN) lacks the intrasubunit disulfide bond. σ-A weighed electron density with coefficients 2mFo − DFc (63) superimposed on the refined structure of C57S SOD1 in subunit A. The electron density map is contoured at 1.3 σ. The rotation of Ser-57 away from Cys-146 is clearly shown; the distance between the sulfur of Cys-146 and the oxygen of Ser-57 is 5 Å. The red sphere coordinated to Cys-146 is a water molecule.
Metal-deficient apoC57S
This structure has one dimer in the crystallographic asymmetric unit with subunit designated as A/B. Both the zinc and electrostatic loops are disordered in each monomer, and there is no bond between Ser-57 and Cys-146 (Fig. 7B).
Rescue of sod1Δ Phenotypes by Disulfide Mutant SOD1s
Saccharomyces cerevisiae from which the sod1 gene has been deleted (sod1Δ) requires lysine to grow under aerobic conditions (46). Monitoring the growth of sod1Δ cells expressing mutant SOD1 in media lacking lysine is an indirect but very sensitive method for determining whether or not a mutant SOD1 has some SOD activity in vivo. As seen in Fig. 8A, in media lacking lysine, the growth of sod1Δ yeast cells expressing either human or yeast C57S SOD1 was 30–50% of the growth of cells expressing WT yeast SOD1. This ability of C57S SOD1 to “rescue” or alleviate the lysine auxotrophy strongly suggests that C57S SOD1 retains a significant amount of SOD activity in vivo despite the absence of intrasubunit disulfide bonds. This result is in contradiction to earlier studies that concluded that intact disulfide bonds are essential for SOD activity (2, 5).
FIGURE 8.

Disulfide mutant yeast and human SOD1s are active in vitro and in vivo. A, yeast growth trials. The indicated yeast or human C57S sod1 genes, on overexpression plasmid pRS424 or vector control (VC), were transformed into EG118 sod1Δ yeast. Cultures were inoculated, in complete media or minus-lysine media, to A600 = 0.05 and grown for 24 h at 30 °C with 220 rpm shaking. The results indicate that both yeast and human disulfide mutant SOD1 partially rescue yeast from the sod1Δ lysine auxotrophy. B, activity by native activity gel. The presence of white bands indicates both human wild type and disulfide mutant (C57S) SOD1 are enzymatically active in cell lysates. C, activity by pulse radiolysis. Rate constants are shown for dismutation of superoxide as a function of pH for human and yeast wild type and disulfide mutant SOD1. Rates were normalized for the copper content of each enzyme. D, ionic strength trials. Using pulse radiolysis, results indicate that disulfide mutant SOD1 is less sensitive (lower slope) to increased ionic strength than is wild type SOD1.
Further characterization of the SOD activity of the disulfide mutant enzyme was carried out using native gel SOD activity assays. In these assays, SOD activity produces a clear band as it lowers O2⨪ concentrations and therefore stops reduction of nitro blue tetrazolium to the blue precipitate formazan. In Fig. 8B, white bands indicate the presence of active SOD1 in yeast cell lysates containing either WT or C57S hSOD1. The band for human C57S disappeared in a separate experiment where 1 mm EDTA was added to all the reagents (47). Western blotting revealed this disappearance was not due to different expression levels of WT and C57S hSOD1.
SOD Activity Determined by Pulse Radiolysis
To confirm the activity of C57S-SOD1, we directly measured the activity using pulse radiolysis, a precise and quantitative method for measurement of SOD activity (35). Pulse radiolysis generates a pulse of superoxide, the rate of disappearance of which is then monitored spectrophotometrically at 260 nm. Here, experiments were performed separately on the C57S hSOD1 and on C57S,C146S ySOD1.
Human WT SOD1 reacts with superoxide with a rate constant of ∼2 × 109 m−1 s−1 (48), independent of pH in the range of 5.5–9 (48). As shown in Fig. 8C, both the human and yeast disulfide mutant SOD1 enzymes were found to be SOD active with rate constants of 1–2 × 108, i.e. 5–10% of WT SOD1, independent of pH in the range of 5.5–9. Note that the lower rate in the disulfide mutant is not due to lower copper content because rate constants were normalized for the copper content of the enzymes.
One possible explanation for the lower rate is that the ionic attraction in the active site for superoxide ion is diminished in the mutant. We therefore performed an ionic strength trial in which the catalytic activity of SOD1 was measured over a range of ionic strengths using pulse radiolysis. The catalytic activity of yeast C57S,C146S SOD1 was less affected by increased ionic strength than was that of WT yeast SOD1 (Fig. 8D), suggesting that the former has less attraction for the superoxide anion than the latter, possibly explaining a portion of the lower catalytic activity.
Active Site Geometry by EPR
Another possible explanation for the lower catalytic rate of disulfide mutant SOD1 is an altered geometry at the copper site. Such an alteration could be detectable by EPR. EPR spectra were determined for as-isolated C57S hSOD1, into which copper was titrated, and for as-isolated WT hSOD1. The C57S hSOD1 displayed only subtle differences in g⊥ and A‖ values relative to the as-isolated WT SOD1 (Fig. 9), indicating that copper inserted into C57S hSOD1 in vitro has a geometry very similar to that of biologically metallated WT hSOD1 and that the environment around the copper is highly similar despite the absence of the intrasubunit disulfide bond. Therefore, altered copper coordination geometry is not likely to be the cause of the decreased SOD activity in the disulfide mutant SOD1.
FIGURE 9.
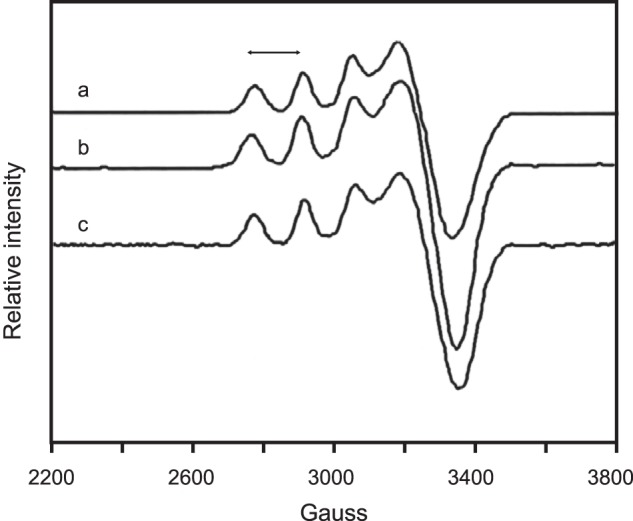
EPR shows copper binding and environment are similar in C57S and wild type human SOD1. The spectra were taken at 97 K in 100 mm phosphate buffer, pH 7.4. Spectrum a, as-isolated hWT SOD1 (g⊥, 2.078; A‖, 139); spectrum b, copper-titrated as-isolated C57S SOD1 (C57S Cu2Zn2 hSOD1) (g⊥, 2.070; A‖, 140); spectrum c, as-isolated C146R SOD1 (g⊥, 2.0758; A‖, 145). The double-headed arrow denotes A‖.
We also analyzed by EPR the as-isolated C146R hSOD1, one of FALS mutants also lacking the disulfide bond. The spectrum of C146R hSOD1 (Fig. 9) showed shifts and sharpening of the g⊥ features as gxx and gyy converge toward equivalence, suggesting that the copper site in C146R is slightly more planar in character than that of WT hSOD1. Overall, however, the copper geometry appeared not to be significantly altered compared with WT hSOD1.
DISCUSSION
apo and Metallated C57S hSOD1 Shares Many Properties with Analogous Forms of Disulfide-reduced WT hSOD1
The use of C57S SOD1 as a model for disulfide-reduced wild type SOD1 is justified only if most of their properties are similar. We therefore compared the thermal stability, metal binding, and hydrogen/deuterium exchange properties for both disulfide-reduced WT and C57S hSOD1.
We found from DSC calorimetric studies that the apo forms of hC57S SOD1 and disulfide-reduced apo WT hSOD1 both unfold at 42 °C, indicating that the two have similar thermostability (Fig. 4). Global (Fig. 5) and site-specific (Fig. 6) H/D exchange experiments on apo C57S hSOD1 also demonstrated that its behavior was nearly identical to that of WT disulfide-reduced apo hSOD1 in both experiments.
We also compared metallated C57S hSOD1 to metallated WT hSOD1 protein by EPR. Both C57S hSOD1 and C146R hSOD1 showed copper geometries that were not significantly altered compared with WT hSOD1 (Fig. 9). In addition, the crystal structure of metallated C57S SOD1 also showed metal binding geometry consistent with wild type SOD1. Unfortunately, we could not compare the thermostability and HDX behaviors of metallated C57S hSOD1 with that of disulfide-reduced metallated WT hSOD1, because the disulfide bond is extremely resistant to reduction in the fully metallated WT hSOD1. Based on the above findings, we conclude that both apo and metallated C57S hSOD1 are very good models for disulfide-reduced SOD1.
The Disulfide Bond Is Not Essential for SOD1 Activity
The disulfide bond was previously reported to be required for SOD1 activity (2, 5). However, in this study, we found that the copper-titrated, as-isolated C57S hSOD1 and C57S,C146S ySOD1 both possess 5–10% activity compared with the WT enzyme (Fig. 8C). In addition, sod1Δ yeast cells (without the ySOD1 gene) were able to grow in the absence of lysine when C57S hSOD1 was overexpressed (Fig. 8A).
Comparison of the rate constants determined for WT and C57S,C146S yeast SOD1 at various concentrations of NaCl suggests that the C57S,C146S mutant is less dependent on ionic strength than the WT enzyme. Hence, electrostatic interactions between superoxide and the active site are weakened in C57S,C146S ySOD1. Because both are disulfide-null mutants, we believe C57S hSOD1 resembles C57S,C146S ySOD1 in the weakened electrostatic interactions, leading to decreased enzymatic activity. The weakening in electrostatic interactions likely results from a changed mobility of a second sphere residue, Arg-143, which carries a positive charge and is critical for superoxide to access the active site. This idea is supported by a solution structural analysis of hSOD1 that showed that Arg-143 is less ordered in disulfide-reduced than in disulfide-oxidized wild type hSod1 (49, 50).
Disulfide-reduced SOD1 Exhibits a Phantom Subunit That Does Not Bind Metals
To gain insight into hSOD1 metal binding in the absence of a disulfide bond, we performed in vitro remetallation studies on apo WT and C57S hSOD1. In the case of bovine and human SOD1 proteins, the apoprotein readily binds metal ions to the copper and zinc sites of both subunits under appropriate conditions. By contrast, apo yeast SOD1 binds only two metal ions per SOD1 dimer, both in the same subunit (37, 51). Such metal ion binding behavior for apo SOD1 has been termed the phantom subunit phenomenon. It has also been observed for tomato SOD1 (52). In the current study, we observed similar phantom subunit behavior in remetallations of apo C57S hSOD1 with cobalt at pH 5.5 (Fig. 1, compare B and C with A) and pH 7.4 (Fig. 1D), with copper at pH 5.5 (Fig. 3), and with zinc at pH 7.4 (Fig. 2). The results of these metal titrations are summarized in Table 1.
It has already been demonstrated that the phantom subunit behavior is not due to modification or denaturation of protein (37, 52). In vitro metal titration studies of C146S hSOD1 also showed a phantom subunit, excluding the possibility that the specific mutation of C57 might be the cause (47).
It is possible to explain the phenomena of the phantom subunit from the results of computational assays by Das and Plotkin (53), which showed that disulfide-oxidized, apo WT hSOD1 possesses more internal allosteric strain relative to the disulfide-oxidized, fully metallated enzyme, and this internal strain generates high metal binding affinity in the apoprotein. The disulfide-reduced apo WT hSOD1 has less internal strain than the disulfide-oxidized apoprotein, and this greater thermostability likely results from increased unfolded entropy. Based on these findings, we speculate that apo C57S hSOD1, which does not possess a disulfide bond (Fig. 7B), resembles the disulfide-reduced apo wild type SOD1 and is less internally strained than the disulfide-oxidized apo wild type SOD1. Because the internal frustration is a prerequisite for high metal binding affinity, the metal binding ability of apo C57S hSOD1 is likely impaired. As suggested by biophysical studies, binding of one metal ion per dimer significantly stabilizes the dimeric form of WT hSOD1 (54). Therefore, when apo C57S hSOD1 binds one metal ion, the metallated subunit would stabilize and increase the rigidity of the metal-deficient subunit, which would further reduce the metal binding affinity of the latter so that it could not bind any metal ions.
Implications for SOD1 Maturation in Vivo
In many organisms, including humans and yeast, CCS delivers copper directly to SOD1 (55). The copper incorporation is proposed to occur via formation of a heterodimer complex of CCS and SOD1 (2, 56). The delivery of copper by CCS was found to require a free cysteine(s) on SOD1 (5). A crystal structure of the SOD1-CCS heterodimer revealed an intermolecular disulfide bond between Cys-57 of SOD1 and Cys-229 of CCS, although this disulfide bond was not necessary for stable heterodimer formation (56). Thus, it was proposed that this intermolecular disulfide bond was transient but played a part in CCS function. Indeed, ySOD1 cannot be activated by Cu-yCCS if the disulfide bond of ySOD1 was already oxidized, clearly illustrating that the disulfide status plays a significant role in recognition of SOD1 by CCS.
This work provides some pieces for completing the puzzle of SOD1 maturation in vivo. C57S hSOD1 isolated for both biochemical assays and for the crystal structure contained four zinc ions per dimer but lacked copper. This supports a hypothesis that an intermolecular disulfide bond between SOD1 (Cys-57) and CCS (Cys-229) may be related to the mechanism of subsequent copper insertion into SOD1. It also supports the idea that the form of SOD1 that interacts with CCS in vivo may have zinc in both the copper and zinc sites of disulfide-reduced SOD1.
Implications for SOD1-linked Familial ALS
In a previous study, as-isolated FALS mutants were showed to be more susceptible to disulfide reduction than WT, and the disulfide-reduced FALS mutants were more easily degraded by protease K and trypsin (13). Based on these results, conformational destabilization caused by disulfide reduction was proposed to cause toxicity, consistent with hypotheses that the toxic function gained by FALS mutants results from an altered protein conformation. Dimer destabilization was also proposed to be a key part of the SOD1-linked ALS disease mechanism based on the fact that the crystal structure of a FALS mutant, A4V, with properties similar to those of WT hSOD1, showed noticeable alterations of the dimer interface despite the absence of significant changes in the individual monomer structure (57). Subsequently, it was demonstrated that dissociated monomeric hSOD1 was an intermediate in aggregation formation and also a good substrate for 20 S proteasome (58, 59). Besides the monomeric form of hSOD1, exposed Cys residues from disulfide reduction appeared to be involved in aggregation through disulfide-cross-linked multimerization (14, 60–62). Disulfide reduction was also shown to destabilize the metal-depleted form of FALS mutants significantly, leading to unfolding and oxidative aggregation (14).
Because C57S hSOD1 is a good model of disulfide-reduced WT hSOD1 based on biophysical and spectroscopic characterizations, the former can be utilized to investigate the aggregation propensity of the latter in fibrillation assays and in cell culture and animal models. These investigations could shed light on the mechanism of SOD1 aggregation and the etiology of FALS.
Acknowledgment
Radiolysis studies were carried out at the Accelerator Center for Energy Research at Brookhaven National Laboratory.
This work was supported, in whole or in part, by National Institutes of Health Grants DK46828 and GM28222 (to J. S. V.), GM08496 (to K. S.), and NS39112 (to P. J. H.). This work was also supported by Veterans Affairs Grant VA 101 BX00506 (to P. J. H.) and Contract DE-AC02-98CH10886 from the U.S. Department of Energy Division of Chemical Sciences, Geosciences, and Biosciences (to Brookhaven National Laboratory).
The atomic coordinates and structure factors (codes 4MCM and 4MCN) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CCS
- copper chaperone for Sod1
- ALS
- amyotrophic lateral sclerosis or Lou Gehrig disease
- DSC
- differential scanning calorimetry
- FALS
- familial (inherited) ALS
- H/D
- hydrogen/deuterium
- HDX
- H/D exchange
- hSOD1
- human SOD1
- ICP-AES
- inductively coupled plasma-atomic emission spectroscopy
- ICP-MS
- inductively coupled plasma-mass spectrometry
- NBT
- nitro blue tetrazolium
- TCEP
- tris(2-carboxyethyl)phosphine
- SOD1 or CuZnSOD
- copper-zinc superoxide dismutase
- hSOD
- human SOD1
- ySOD1
- yeast (S. cerevisiae) SOD1
- PDB
- Protein Data Bank.
REFERENCES
- 1. Fridovich I. (1989) Superoxide dismutases an adaptation to a paramagnetic gas. J. Biol. Chem. 264, 7761–7764 [PubMed] [Google Scholar]
- 2. Field L. S., Furukawa Y., O'Halloran T. V., Culotta V. C. (2003) Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J. Biol. Chem. 278, 28052–28059 [DOI] [PubMed] [Google Scholar]
- 3. Banci L., Barbieri L., Bertini I., Luchinat E., Secci E., Zhao Y., Aricescu A. R. (2013) Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat. Chem. Biol. 9, 297–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Culotta V. C., Klomp L. W., Strain J., Casareno R. L., Krems B., Gitlin J. D. (1997) The copper chaperone for superoxide dismutase. J. Biol. Chem. 272, 23469–23472 [DOI] [PubMed] [Google Scholar]
- 5. Furukawa Y., Torres A. S., O'Halloran T. V. (2004) Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J. 23, 2872–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carroll M. C., Girouard J. B., Ulloa J. L., Subramaniam J. R., Wong P. C., Valentine J. S., Culotta V. C. (2004) Mechanisms for activating Cu- and Zn-containing superoxide dismutase in the absence of the CCS Cu chaperone. Proc. Natl. Acad. Sci. U.S.A. 101, 5964–5969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sea K. W., Sheng Y., Lelie H. L., Kane Barnese L., Durazo A., Valentine J. S., Gralla E. B. (2013) Yeast copper, zinc superoxide dismutase can be activated in the absence of its copper chaperone. J. Biol. Inorg. Chem. 18, 985–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Le Moan N., Clement G., Le Maout S., Tacnet F., Toledano M. B. (2006) The Saccharomyces cerevisiae proteome of oxidized protein thiols: contrasted functions for the thioredoxin and glutathione pathways. J. Biol. Chem. 281, 10420–10430 [DOI] [PubMed] [Google Scholar]
- 9. Lindberg M. J., Normark J., Holmgren A., Oliveberg M. (2004) Folding of human superoxide dismutase: disulfide reduction prevents dimerization and produces marginally stable monomers. Proc. Natl. Acad. Sci. U.S.A. 101, 15893–15898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shaw B. F., Valentine J. S. (2007) How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem. Sci. 32, 78–85 [DOI] [PubMed] [Google Scholar]
- 11. Chattopadhyay M., Valentine J. (2009) Aggregation of copper-zinc superoxide dismutase in familial and sporadic ALS. Antioxid. Redox. Signal. 11, 1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sheng Y., Chattopadhyay M., Whitelegge J., Valentine J. S. (2012) SOD1 aggregation and ALS: role of metallation states and disulfide status. Curr. Top. Med. Chem. 12, 2560–2572 [DOI] [PubMed] [Google Scholar]
- 13. Tiwari A., Hayward L. (2003) Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J. Biol. Chem. 278, 5984–5992 [DOI] [PubMed] [Google Scholar]
- 14. Furukawa Y., O'Halloran T. (2005) Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 280, 17266–17274 [DOI] [PubMed] [Google Scholar]
- 15. Oztug Durer Z. A., Cohlberg J. A., Dinh P., Padua S., Ehrenclou K., Downes S., Tan J. K., Nakano Y., Bowman C. J., Hoskins J. L., Kwon C., Mason A. Z., Rodriguez J. A., Doucette P. A., Shaw B. F., Valentine J. S. (2009) Loss of metal ions, disulfide reduction and mutations related to familial ALS promote formation of amyloid-like aggregates from superoxide dismutase. PLoS One 4, e5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chattopadhyay M., Durazo A., Sohn S. H., Strong C. D., Gralla E. B., Whitelegge J. P., Valentine J. S. (2008) Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc. Natl. Acad. Sci. U.S.A. 105, 18663–18668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghosh S., Willard B., Comhair S. A., Dibello P., Xu W., Shiva S., Aulak K. S., Kinter M., Erzurum S. C. (2013) Disulfide bond as a switch for copper-zinc superoxide dismutase activity in asthma. Antioxid. Redox. Signal. 18, 412–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodriguez J. A., Shaw B. F., Durazo A., Sohn S. H., Doucette P. A., Nersissian A. M., Faull K. F., Eggers D. K., Tiwari A., Hayward L. J., Valentine J. S. (2005) Destabilization of apoprotein is insufficient to explain Cu,Zn-superoxide dismutase-linked ALS pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 102, 10516–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Doucette P. A., Whitson L. J., Cao X., Schirf V., Demeler B., Valentine J. S., Hansen J. C., Hart P. J. (2004) Dissociation of human copper-zinc superoxide dismutase dimmers suing chaotrope and reductant. J. Biol. Chem. 279, 54558–54566 [DOI] [PubMed] [Google Scholar]
- 20. Doucette P. (2004) Biophysical studies of human copper-zinc superoxide dismutase and mutants associated with the neurodegenerative disease amyotrophic lateral sclerosis. Ph.D. dissertation, University of California, Los Angeles [Google Scholar]
- 21. Valentine J., Pantoliano M. (1981) in Metal Ions in Biology (Spiro T., ed) pp. 291–358, John Wiley & Sons, Inc., New York [Google Scholar]
- 22. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 306–326 [DOI] [PubMed] [Google Scholar]
- 23. Hart P. J., Liu H., Pellegrini M., Nersissian A. M., Gralla E. B., Valentine J. S., Eisenberg D. (1998) Subunit asymmetry in the three-dimensional structure of a human CuZnSOD mutant found in familial amyotrophic lateral sclerosis. Protein Sci. 7, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vagin A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 25. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The protein data bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liba A. (2000) Characterization of tomato Sod1. Ph.D. dissertation, University of California, Los Angeles [Google Scholar]
- 29. Rodriguez J. A., Valentine J. S., Eggers D. K., Roe J. A., Tiwari A., Brown R. H., Jr., Hayward L. J. (2002) Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J. Biol. Chem. 277, 15932–15937 [DOI] [PubMed] [Google Scholar]
- 30. Lyons T. J., Nersissian A., Huang H., Yeom H., Nishida C. R., Graden J. A., Gralla E. B., Valentine J. S. (2000) The metal binding properties of the zinc site of yeast copper-zinc superoxide dismutase: implications for amyotrophic lateral sclerosis. J. Biol. Inorg. Chem. 5, 189–203 [DOI] [PubMed] [Google Scholar]
- 31. Shaw B. F., Durazo A., Nersissian A. M., Whitelegge J. P., Faull K. F., Valentine J. S. (2006) Local unfolding in a destabilized, pathogenic variant of superoxide dismutase 1 observed with H/D exchange and mass spectrometry. J. Biol. Chem. 281, 18167–18176 [DOI] [PubMed] [Google Scholar]
- 32. Gralla E. B., Valentine J. S. (1991) Null mutants of Saccharomyces cerevisiae Cu,Zn superoxide dismutase: characterization and spontaneous mutation rates. J. Bacteriol. 173, 5918–5920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sikorski R. S., Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Flohé L., Otting F. (1984) Superoxide dismutase assays. Methods Enzymol. 105, 93–104 [DOI] [PubMed] [Google Scholar]
- 35. Goto J. J., Gralla E. B., Valentine J. S., Cabelli D. E. (1998) Reactions of hydrogen peroxide with familial amyotrophic lateral sclerosis mutant human copper-zinc superoxide dismutases studied by pulse radiolysis. J. Biol. Chem. 273, 30104–30109 [DOI] [PubMed] [Google Scholar]
- 36. Von Piechowski M., Nauser T., Hoignè J., Bühler R. E. (1993) O2-decay catalyzed by Cu2+ and Cu+ ions in aqueous solutions: A pulse radiolysis study for atmospheric chemistry. Bericht. Bunsen-Gesell. 97, 762–771 [Google Scholar]
- 37. Lyons T., Nersissian A., Goto J., Zhu H., Gralla E., Valentine J. (1998) Metal ion reconstitution studies of yeast copper-zinc superoxide dismutase: the “phantom” subunit and the possible role of Lys7p. J. Biol. Inorg. Chem. 3, 650–662 [Google Scholar]
- 38. Ming L., Banci L., Luchinat C., Bertini I., Valentine J. (1988) NMR-study of cobalt(II)-substituted yeast and human copper-zinc superoxide dismutase. Inorg. Chem. 27, 728–733 [Google Scholar]
- 39. Pantoliano M. W., Valentine J. S., Nafie L. A. (1982) Spectroscopic studies of copper(II) bound at the native copper site or substituted at the native zinc site of bovine erythrocuprein (sueproxide dismutase). J. Am. Chem. Soc. 104, 6310–6317 [Google Scholar]
- 40. Goto J. J., Zhu H., Sanchez R. J., Nersissian A., Gralla E. B., Valentine J. S., Cabelli D. E. (2000) Loss of in vitro metal ion binding specificity in mutant copper-zinc superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 275, 1007–1014 [DOI] [PubMed] [Google Scholar]
- 41. Roe J. A., Butler A., Scholler D. M., Valentine J. S., Marky L., Breslauer K. J. (1988) Differential scanning calorimetry of Cu,Zn-superoxide dismutase, the apoprotein, and its zinc-substituted derivatives. Biochemistry 27, 950–958 [DOI] [PubMed] [Google Scholar]
- 42. Durazo A. (2007) Hydrogen/deuterium exchange studies on copper-zinc superoxide dismutase. Ph.D. dissertation, University of California, Los Angeles [Google Scholar]
- 43. Strange R. W., Antonyuk S., Hough M. A., Doucette P. A., Rodriguez J. A., Hart P. J., Hayward L. J., Valentine J. S., Hasnain S. S. (2003) The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J. Mol. Biol. 328, 877–891 [DOI] [PubMed] [Google Scholar]
- 44. Tainer J. A., Getzoff E. D., Richardson J. S., Richardson D. C. (1983) Structure and mechanism of copper, zinc superoxide dismutase. Nature 306, 284–287 [DOI] [PubMed] [Google Scholar]
- 45. Elam J. S., Malek K., Rodriguez J. A., Doucette P. A., Taylor A. B., Hayward L. J., Cabelli D. E., Valentine J. S., Hart P. J. (2003) An alternative mechanism of bicarbonate-mediated peroxidation by copper-zinc superoxide dismutase: rate enhanced via proposed enzyme-associated peroxycarbonate intermediate. J. Biol. Chem. 278, 21032–21039 [DOI] [PubMed] [Google Scholar]
- 46. Liu X. F., Elashvili I., Gralla E. B., Valentine J. S., Lapinskas P., Culotta V. C. (1992) Yeast lacking superoxide dismutase. J. Biol. Chem. 267, 18298–18302 [PubMed] [Google Scholar]
- 47. Sohn S. H. (2006) Redesigning of Cu,Zn-superoxide dismutase to investigate the function of the intrasubunit disulfide bond and other properties related with familial amyotrophic lateral sclerosis. Ph.D. Dissertation, University of California, Los Angeles [Google Scholar]
- 48. Bertini I., Mangani S., Viezzoli S. (1998) Structure and properties of copper-zinc superoxide dismutases. Adv. Inorg. Chem. 45, 127–250 [Google Scholar]
- 49. Banci L., Bertini I., Cantini F., D'Amelio N., Gaggelli E. (2006) Human SOD1 before harboring the catalytic metal: solution structure of copper depleted, disulfide reduced form. J. Biol. Chem. 281, 2333–2337 [DOI] [PubMed] [Google Scholar]
- 50. Ferraroni M., Rypniewski W., Wilson K. S., Viezzoli M. S., Banci L., Bertini I., Mangani S. (1999) The crystal structure of the monomeric human SOD mutant F50E/G51E/E133Q at atomic resolution. the enzyme mechanism revisited. J. Mol. Biol. 288, 413–426 [DOI] [PubMed] [Google Scholar]
- 51. Dunbar J. C., Johansen J. T. (1981) Structural and functional properties of the Cu,Zn superoxide dismutases. Bull. Eur. Physiopathol. Respir. 17, 51–61 [PubMed] [Google Scholar]
- 52. Liba A. (2003) Studies of iron homeostasis in Saccharomyces cerevisiae and biophysical characterization of tomato chloroplast superoxide dismutase. Ph.D. dissertation, University of California [Google Scholar]
- 53. Das A., Plotkin S. S. (2013) SOD1 exhibits allosteric frustration to facilitate metal binding affinity. Proc. Natl. Acad. Sci. U.S.A. 110, 3871–3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Potter S. Z., Zhu H., Shaw B. F., Rodriguez J. A., Doucette P. A., Sohn S. H., Durazo A., Faull K. F., Gralla E. B., Nersissian A. M., Valentine J. S. (2007) Binding of a single zinc ion to one subunit of copper-zinc superoxide dismutase apoprotein substantially influences the structure and stability of the entire homodimeric protein. J. Am. Chem. Soc. 129, 4575–4583 [DOI] [PubMed] [Google Scholar]
- 55. Culotta V. C., Yang M., O'Halloran T. V. (2006) Activation of superoxide dismutases: Putting the metal to the pedal. Biochim. Biophys. Acta 1763, 747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lamb A. L., Torres A. S., O'Halloran T. V., Rosenzweig A. C. (2001) Heterodimeric structure of superoxide dismutase in complex with its metallochaperone. Nat. Struct. Biol. 8, 751–755 [DOI] [PubMed] [Google Scholar]
- 57. Hough M. A., Grossmann J. G., Antonyuk S. V., Strange R. W., Doucette P. A., Rodriguez J. A., Whitson L. J., Hart P. J., Hayward L. J., Valentine J. S., Hasnain S. S. (2004) Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc. Natl. Acad. Sci. U.S.A. 101, 5976–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rakhit R., Crow J. P., Lepock J. R., Kondejewski L. H., Cashman N. R., Chakrabartty A. (2004) Monomeric Cu,Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J. Biol. Chem. 279, 15499–15504 [DOI] [PubMed] [Google Scholar]
- 59. Hart P. J. (2006) Pathogenic superoxide dismutase structure, folding, aggregation and turnover. Curr. Opin. Chem. Biol. 10, 131–138 [DOI] [PubMed] [Google Scholar]
- 60. Furukawa Y., Fu R., Deng H.-X., Siddique T., O'Halloran T. V. (2006) Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu,Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc. Natl. Acad. Sci. U.S.A. 103, 7148–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Niwa J., Yamada S., Ishigaki S., Sone J., Takahashi M., Katsuno M., Tanaka F., Doyu M., Sobue G. (2007) Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1. J. Biol. Chem. 282, 28087–28095 [DOI] [PubMed] [Google Scholar]
- 62. Wang J., Xu G., Borchelt D. R. (2006) Mapping superoxide dismutase 1 domains of non-native interaction: roles of intra- and intermolecular disulfide bonding in aggregation. J. Neurochem. 96, 1277–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Read R. (1986) Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A Found. Crystallogr. 42, 140–149 [Google Scholar]