

Background: The spindle checkpoint protein BubR1 inhibits the anaphase-promoting complex through binding to Cdc20.
Results: We identify a new Cdc20-binding motif within BubR1 termed the Phe box.
Conclusion: The Phe box maintains steady-state levels of BubR1-containing checkpoint complexes in human cells.
Significance: Our study provides key insights into the homeostatic mechanisms of a key spindle checkpoint complex.
Keywords: Cell Cycle, Kinetochore, Mitosis, Protein-Protein Interaction, Ubiquitylation (Ubiquitination), APC/C, Mitotic Checkpoint Complex, Spindle Checkpoint
Abstract
The spindle checkpoint ensures accurate chromosome segregation by monitoring kinetochore-microtubule attachment. Unattached or tensionless kinetochores activate the checkpoint and enhance the production of the mitotic checkpoint complex (MCC) consisting of BubR1, Bub3, Mad2, and Cdc20. MCC is a critical checkpoint inhibitor of the anaphase-promoting complex/cyclosome, a ubiquitin ligase required for anaphase onset. The N-terminal region of BubR1 binds to both Cdc20 and Mad2, thus nucleating MCC formation. The middle region of human BubR1 (BubR1M) also interacts with Cdc20, but the nature and function of this interaction are not understood. Here we identify two critical motifs within BubR1M that contribute to Cdc20 binding and anaphase-promoting complex/cyclosome inhibition: a destruction box (D box) and a phenylalanine-containing motif termed the Phe box. A BubR1 mutant lacking these motifs is defective in MCC maintenance in mitotic human cells but is capable of supporting spindle-checkpoint function. Thus, the BubR1M-Cdc20 interaction indirectly contributes to MCC homeostasis. Its apparent dispensability in the spindle checkpoint might be due to functional duality or redundant, competing mechanisms.
Introduction
Accurate chromosome segregation relies on the correct attachment of chromosomes to the mitotic spindle. During mitosis, the two opposing kinetochores of a pair of sister chromatids must attach to microtubules emanating from the two opposite spindle poles. This type of bipolar attachment, termed bi-orientation, ensures that the separated sister chromatids are segregated evenly to daughter cells (1–3). Errors in chromosome attachment can lead to chromosome missegregation and aneuploidy (4).
The spindle checkpoint monitors and promotes proper chromosome attachment during mitosis (3, 5, 6). Unattached or improperly attached kinetochores generate an inhibitory signal that prevents progression into anaphase by inhibiting the anaphase-promoting complex/cyclosome (APC/C)5 (6, 7). APC/C is a multisubunit E3 ubiquitin ligase whose activity is required for anaphase onset and mitotic exit (8). When bound to its mitotic activator Cdc20 (9), APC/C ubiquitinates multiple substrates, including securin and cyclin B1, targeting them for degradation by the proteasome. Degradation of securin and cyclin B1 triggers sister-chromatid separation and mitotic exit. By inhibiting APC/CCdc20 in response to inappropriately attached kinetochores, the spindle checkpoint prevents chromosome segregation and mitotic exit until all chromosomes achieve bi-orientation.
The core components of the spindle checkpoint, including Mps1, Mad1, Mad2, BubR1 (Mad3 in yeasts), Bub1, and Bub3, are conserved from yeast to man (3, 5, 6). These proteins localize to unattached kinetochores and promote the generation of an APC/C inhibitory complex consisting of BubR1, Bub3, Mad2, and Cdc20, termed the mitotic checkpoint complex (MCC). Although MCC subcomplexes of Mad2-Cdc20 and BubR1-Bub3-Cdc20 can inhibit APC/C to some degree (10, 11), the intact MCC is a more potent APC/C inhibitor (12, 13).
MCC is thought to inhibit APC/C by at least two mechanisms (6, 14). First, through acting as a pseudo-substrate, it competitively blocks substrate recruitment by APC/C. APC/C substrates are recognized through multiple, short degradation motifs called degrons, such as the destruction box (D box) and the KEN box (8). The KEN box is recognized by Cdc20 or its homolog Cdh1 (15–17), whereas the D box binds at the interface between Cdc20 (or Cdh1) and the core APC/C subunit APC10 (18, 19). The MCC component BubR1 (or its yeast ortholog Mad3) contains two KEN boxes, which are required for checkpoint function and compete with KEN box-containing substrates for Cdc20 binding (20–23). Second, MCC alters the mode of Cdc20 binding to APC/C: Cdc20 on its own and Cdc20 as a part of MCC bind to different locations on APC/C (17, 24, 25), suggesting that MCC might anchor Cdc20 to a site that is not compatible for catalysis.
Biochemical and structural studies of MCC have revealed intricate interactions among Mad2, Cdc20, and BubR1. Using a seat belt-like structural element, Mad2 in its active, closed conformation traps the Mad2-interacting motif of Cdc20 in a topological embrace (26–28). The N-terminal region of BubR1 (BubR1N) contains two KEN boxes and interacts directly with Cdc20 through the first KEN box (KEN1) (16, 23). Cdc20-bound Mad2 also makes direct contact with Mad3 (and quite possibly BubR1N) (17, 29), buttressing the weak Cdc20-KEN1 interaction. Thus, through simultaneously engaging both Cdc20 and Mad2, BubR1N nucleates MCC assembly, with KEN1 playing a pivotal role in this process. The second KEN box of BubR1 (KEN2) is also required for spindle checkpoint function in human cells and blocks substrate binding by APC/C (23), although the mechanism by which KEN2 achieves this task has not been established. Even more mysteriously, the middle region of BubR1 (BubR1M) has been shown to bind to Cdc20 in the absence of Mad2 (11, 30). The function of the BubR1M-Cdc20 interaction in the spindle checkpoint is unknown.
To probe the function of Cdc20 binding by BubR1M in the spindle checkpoint, we first defined the mechanism by which BubR1M interacted with Cdc20. We found that human BubR1M contains two Cdc20-binding motifs: a D box and a novel Cdc20-binding motif that we termed the Phe box. The Phe box is conserved in vertebrate BubR1 proteins and is also present in Bub1 and cyclin A2. The D and Phe boxes of BubR1M bind cooperatively to Cdc20 and inhibit APC/CCdc20 in vitro. The D box of BubR1M binds to both human Cdc20 and Cdh1. In contrast, the Phe box selectively binds to human Cdc20, but not to Cdh1, thus explaining the inability of BubR1 to inhibit APC/CCdh1 in vitro. Strikingly, a BubR1 mutant with the Phe and D boxes deleted is deficient in MCC maintenance in human cells, indicating a contribution for BubR1M in MCC homeostasis. Consistent with previous reports (30, 31), this mutant is functional in supporting the spindle checkpoint in human cells. In fact, it is slightly more active in delaying mitotic exit when cells are challenged with low concentrations of taxol. The fact that BubR1M is not required for the spindle checkpoint suggests a possible functional duality of the BubR1M-Cdc20 interaction or the existence of compensatory APC/C inhibitory mechanisms or both.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The full-length Cdc20, Cdc20 WD40 (residues 161–477), and Mad2 L13A proteins were purified as previously described (16, 32). The coding regions of various fragments of the human BubR1 middle region (BubR1M) were cloned into the pGEX-6p vector (GE Healthcare). The coding sequence of miniBubR1 gene was commercially synthesized (Genescript) and cloned into the pGEX-6p vector. The amino acid sequence of miniBubR1 is: MGDEWELSKENVQPLRQGRIMSTLQGALAQESGGASTAELSKPTVQPWIAPPMPRAKENELQAGPWNTGRSGGGGGSVPFSIFDEFLLSEKKNKSPPADPPRVLAQRRPLAVLKTSESITSNEDVSPDVCDEFTGIEPLSEDAIITGFRNVTICPNPEDTCDFARAARFVSTPFHELE. All constructs were verified by DNA sequencing. The pGEX-6p-BubR1 plasmids were transformed into the Escherichia coli strain BL21. These GST-BubR1 proteins that included a PreScission protease cleavage site between GST and BubR1 were purified with glutathione-Sepharose 4B beads (GE Healthcare) and used in GST pulldown assays. For other assays, the fusion proteins were cleaved with the PreScission protease to remove the GST moiety. The cleaved BubR1 proteins were further purified with a Superdex 200 gel filtration column (GE Healthcare) in a buffer containing 25 mm Tris (pH 8.0), 150 mm NaCl, and 1 mm DTT. The Phe box peptide of BubR1 (with the sequence of GPSVPFSIFDEFLLSEKKNKSPPA) was chemically synthesized.
The BubR1-Bub3 complexes used in the APC/C ubiquitination assays were expressed in Sf9 insect cells as a Strep-His6-BubR1 fusion in complex with His6-Bub3. Cells were lysed in the lysis buffer (50 mm Tris-HCl, pH 8.0, 250 mm NaCl, 3 mm MgCl2, 5 mm NaF, 10 mm β-glycerophosphate, 1.5 mm DTT, 5% glycerol, and protease inhibitors). The complexes were affinity-purified using the Strep-Tactin Superflow Plus resin (Qiagen) and eluted with buffer containing 15 mm d-desthiobiotin (Sigma). Proteins were concentrated and stored in the storage buffer (25 mm Tris-HCl, pH 7.7, 100 mm NaCl, 1 mm MgCl2, 1 mm DTT, and 5% glycerol).
Protein Binding Assays
For binding between human BubR1M and Cdc20 (or Cdh1), purified GST-BubR1M proteins were bound to glutathione-Sepharose 4B beads (GE Healthcare). Full-length Myc-Cdc20, truncation mutants of Myc-Cdc20, and full-length Myc-Cdh1 were in vitro translated in reticulocyte lysate containing [35S]methionine and incubated with the GST-BubR1M beads. The beads were washed four times with TBS containing 0.05% Tween. The proteins bound to beads were eluted by boiling in SDS loading buffer and separated on SDS-PAGE. The 35S signal was detected and quantified using a Fuji phosphorimager. Glutathione-Sepharose beads bound to GST were used as controls. The relative binding intensity was defined as the binding intensity of each sample (subtracting background binding by GST beads) divided by 20% input.
Microscale Thermophoresis
Cdc20 WD40 was labeled with a fluorophore on cysteines using the blue-maleimide protein labeling kit (Nanotemper, Munich, Germany), following the manufacturer's instructions. The labeled protein was exchanged into the experimental buffer containing 20 mm Tris (pH 9.0) and 150 mm NaCl. Serial dilutions of BubR1506–583 and the Phe box peptide were made by 15 successive 1:1 dilutions of the highest titrant concentration (1 mm and 200 μm, respectively) into the experimental buffer. These solutions were each diluted 1:1 with a solution of 400 nm labeled Cdc20 WD40. Thus, the final concentration of the labeled Cdc20 WD40 was 200 nm in all samples, and the highest concentrations of BubR1506–583 and the Phe box peptide were 100 and 500 μm, respectively. The samples were placed in glass capillary tubes for ∼30 min before the final measurements were taken in a Monolith NT.115 device (Nanotemper). For the Cdc20 WD40-Phe box peptide titration, the LED (illumination) power of the instrument was set to 90%, and the IR laser power was set to 80%. For the Cdc20 WD40-BubR1506–583 titration, these values were set to 50 and 80%, respectively. Once the LED was turned on, fluorescence was monitored as a function of time. There was a 5-s waiting period before the IR laser was ignited. The IR laser remained on for 30 s, followed by a 5-s monitoring of the recovery period. The resulting fluorescence time traces were analyzed to generate the isotherms. The mean fluorescence from a 0.5-s time span just after the “temperature jump” phase of the traces (F̄c) was compared with that from a 2-s span just prior to the extinguishment of the IR laser (F̄h) as follows.
 |
The isotherm is thus a comparison of the concentration of the ligand versus these Fn values. Subsequent analysis was performed as previously described (33). Figures from the thermophoretic experiments were generated in GUSSI.
Ubiquitination Assays
APC/C ubiquitination assays were performed as previously described (34).
Mammalian Cell Culture and Transfection
HeLa TetON cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% bovine serum (Invitrogen). Plasmid and siRNA transfections were performed using Effectene (Qiagen) and Lipofectamine RNAiMAX (Invitrogen), respectively, according to instructions from the manufacturers. The siRNAs used in this study were synthesized by Dharmacon and were used at a final concentration of 5 nm. The siRNAs had the following sequences: siBubR1, CAAGAUGGCUGUAUUGUUU; and siLuc, UCAUUCCGGAUACUGCGAU.
Antibodies, Immunoblotting, and Immunoprecipitation (IP)
Antibodies against Mad2, BubR1, and Bub3 were described previously (11). The commercial antibodies used were anti-Cdc20 (Santa Cruz; sc-5296), anti-Myc (Roche), anti-tubulin (Sigma), MPM2 (Millipore, 1:400 dilution), and CREST serum (ImmunoVision). The secondary antibodies used were anti-mouse IgG (H+L) (Dylight 680 conjugates), anti-rabbit IgG (H+L) (Dylight 800 conjugates) (Cell Signaling), and anti-mouse Alexa 488 secondary antibody (Invitrogen).
For anti-Myc IP, cells were lysed in the lysis buffer (40 mm Tris-HCl, pH 7.5, 105 mm KCl, 1 mm MgCl2, 0.05% Nonidet P-40) freshly supplemented with 1× SigmaFast protease inhibitors (Sigma), 0.5 μm okadaic acid (LC Labs), 10 μm cytochalasin B (Sigma), 10 units/ml TurboNuclease (Accelagen), and 1.5 mm DTT. Lysates were incubated with anti-Myc beads at 4 °C for at least 2 h. The beads were washed with the high salt buffer (lysis buffer with 200 mm KCl and 0.2% Nonidet P-40) twice and with the lysis buffer three times. Bound proteins were eluted with the SDS loading buffer. For total cell lysates, cells were directly lysed with SDS loading buffer. Lysates and IP were separated on SDS-PAGE, transferred to nitrocellulose membranes (Bio-Rad), and blotted with the desired antibodies. The blots were scanned with an Odyssey Infrared Imaging System (LI-COR).
Flow Cytometry
HeLa cells were transfected with the indicated plasmids. At 8 h after plasmid transfection, they were transfected with siRNAs. At 24 h after siRNA transfection, taxol or nocodazole were added at the indicated concentrations, and samples were collected another 15 h later. Cells were washed with PBS and fixed in cold 70% ethanol. After fixation, cells were washed with PBS, incubated for at least 3 h with the MPM2 antibody (1:400 dilution in PBS containing 0.2% Triton X-100 and 3% BSA). Cells were washed with PBS and incubated for 30 min with the anti-mouse Alexa 488 secondary antibody (1:200 dilution in PBS containing 0.2% Triton X-100 and 3% BSA). Cells were washed again and resuspended in PBS containing 200 μg/ml DNase-free RNase A (Qiagen) and 2 μg/ml propidium iodide (Sigma). Samples were scanned with a FACScalibur flow cytometer (BD Biosciences), and the data were processed with the FlowJo software.
Live Cell Microscopy
HeLa cells were transfected with the BubR1 plasmids together with a plasmid encoding eGFP in 8-well chambered coverslips (LabTek) and were transfected again with siRNAs at 8 h after plasmid transfection. At 40 h after siRNA transfection, taxol was added to a final concentration of 30 nm. Cells were then imaged with a DeltaVision system (GE Healthcare) fitted with a humidified environmental chamber at 37 °C and 5% CO2. Differential interference contrast and eGFP images were acquired with a 40× objective at 10-min intervals for 30–36 h. The images were manually analyzed to determine the mitotic durations. Only cells expressing eGFP were included in the quantification. The t50 is defined as the time at which 50% of the mitotic cells have undergone adaptation or apoptosis.
Immunofluorescence
HeLa cells were cultured and transfected with the desired siRNAs in 4-well chambered slides. At 42 h after transfection, nocodazole (40 nm) was added for 30 min to depolymerize microtubules. Cells were then prefixed with ice-cold 0.5% formaldehyde in PBS for 10 min on ice. The cells were briefly washed with PBS and pre-extracted in ice-cold 0.2% Triton X-100 in PBS for 5 min. Cells were postfixed with ice-cold 4% formaldehyde in PBS for 15 min. After being washed twice with PBS, cells were permeabilized with 0.2% Triton X-100 in PBS for 20 min at room temperature and incubated with mouse anti-Cdc20 (1:100), rabbit anti-BubR1 (1 μg/ml), and CREST antibodies (1:1000) diluted in PBS containing 3% BSA and 0.2% Triton X-100 for 3 h at room temperature. Cells were then washed three times with 0.2% Triton X-100 in PBS and incubated with the appropriate secondary antibodies diluted in PBS with 3% BSA and 0.2% Triton X-100 for 3 h at room temperature. Cells were washed three times with 0.2% Triton X-100 in PBS and once with PBS. The slides were mounted using ProLong Gold with DAPI (Invitrogen). Images were acquired with a DeltaVision system (GE Healthcare), fitted with a 100× objective. The images were deconvolved using the provided algorithm with the “conservative” setting, and projected by sum projection. Projections were quantified in ImageJ. For each cell, a mask was created using the CREST signal. This mask was then applied to the Cdc20 and BubR1 channels to measure the total intensity. Normalized intensities were defined as the ratios between the Cdc20 (or BubR1) intensities and those of CREST.
RESULTS
Human BubR1M Contains Two Cdc20-binding Motifs
Initial studies on the BubR1-Cdc20 interaction suggested the existence of multiple, independent Cdc20-binding regions (11). In addition to the N-terminal region, the middle region of BubR1 (BubR1M) containing residues 526–700 was implicated in Cdc20 binding. This middle Cdc20-binding region of BubR1 was later further narrowed down to a fragment containing residues 490–560 (30). We set out to further define the Cdc20-binding elements in this region. We purified various BubR1M fragments as GST fusion proteins and tested their binding to in vitro translated 35S-labeled Cdc20. As expected, a BubR1M fragment containing residues 526–613 (BubR1526–613) bound to Cdc20 (Fig. 1A). It also bound to Cdc20 lacking the N-terminal 60 or 157 residues (Cdc20 ΔN60 or ΔN157). A smaller, 21-residue BubR1 fragment (BubR1526–546) also bound Cdc20 but less efficiently than BubR1526–613 did. These results suggest that at least two motifs of BubR1M might cooperatively bind to the C-terminal WD40 domain of Cdc20.
FIGURE 1.

Identification of a conserved Phe box in BubR1 that interacts with Cdc20. A, autoradiograph (top panel) and Coomassie staining (bottom panel) of the input and bound proteins of the binding reactions between GST or GST-BubR1 fragments (bound to beads) and in vitro translated 35S-labeled full-length (FL) or truncated Cdc20. The relative binding intensities are quantified and indicated below each lane. B, sequence alignment of the Phe and D boxes in human, mouse, and Xenopus BubR1 proteins, with high or low similarity residues shaded in black or gray, respectively. C, sequence alignment the Phe boxes of human BubR1, Bub1, and cyclin A2. D, list of Phe and D box mutants (termed mPhe and mD) used in this study. E, autoradiograph (top panel) and Coomassie staining (bottom panel) of the input and bound proteins of the binding reactions between GST or GST-BubR1526–613 WT, mPhe, or mD and 35S-labeled full-length or truncated Cdc20. All samples were run in the same gel. Certain lanes were removed for clarity, and the remaining lanes were spliced together. The relative binding intensities are indicated below each lane. F, microscale thermophoresis analysis of binding of BubR1506–583 (left panel) or a synthetic BubR1 Phe box peptide (right panel) to the Cdc20 WD40 domain. The dissociation constant (Kd) of each binding reaction is indicated. fluo., fluorescence.
Sequence alignment of BubR1 proteins from different vertebrate species revealed two conserved motifs (Fig. 1B). The first motif has the consensus of FXIFDE (where X indicates any residue) and will be hereafter referred to as the Phe box because it contains two phenylalanines. The second motif is a D box, with the well established consensus RXXL. Intriguingly, two other Cdc20-binding proteins, Bub1 and cyclin A2, each contain a motif that conforms to the Phe box consensus, suggesting that the Phe box might be a widespread Cdc20-binding motif (Fig. 1C).
To test whether the Phe and D boxes mediated the binding of BubR1526–613 to Cdc20, we generated point mutants targeting key residues in their consensus sequences (Fig. 1D). Mutation of the Phe box (mPhe) markedly reduced the binding of BubR1526–613 to either the full-length Cdc20 or Cdc20 ΔN157 (Fig. 1E). Mutation of the D box (mD) also weakened the binding between BubR1 and Cdc20, albeit to a lesser degree. These results confirm the involvement of both the Phe and D boxes in mediating BubR1M binding to the WD40 repeat domain of Cdc20.
We next measured the binding affinity between BubR1506–583 (which had fewer proteolytic fragments) and the WD40 domain of Cdc20 (Cdc20 WD40; residues 161–477), using microscale thermophoresis (Fig. 1F). BubR1506–583 (which contained both the Phe and D boxes) bound to Cdc20 WD40 with a dissociation constant (Kd) of 0.49 μm. A synthetic peptide containing the Phe box alone (BubR1523–546) exhibited weaker binding to Cdc20 WD40 (Kd = 1.20 μm). Taken together, our results indicate that the Phe and D boxes in BubR1M bind cooperatively to the WD40 domain of Cdc20, with the Phe box contributing the bulk of the binding energy.
Both Phe and D Boxes of BubR1 Contribute to APC/CCdc20 Inhibition in Vitro
Having established the involvement of Phe and D boxes in mediating the binding of BubR1M to Cdc20, we next asked whether these motifs were required for BubR1-mediated inhibition of APC/CCdc20. Full-length BubR1-Bub3 complexes containing wild type BubR1 or the indicated mutants were assayed for their ability to inhibit APC/CCdc20 in vitro. As expected, wild type BubR1-Bub3 inhibited the APC/CCdc20 activity toward cyclin B1 (Fig. 2A). Mutations of either the KEN1 or KEN2 box did not affect this inhibition. In contrast, a BubR1 mutant with residues 525–561 (which encompass both the Phe and D boxes) deleted, termed ΔPheD, reduced the ability of BubR1 to inhibit APC/CCdc20. These results are consistent with previous observations that, in the absence of Mad2, the middle region of BubR1 is critical for its APC/C inhibitory activity in vitro (11). The dispensability of the KEN boxes in BubR1-mediated inhibition of APC/C (in the absence of Mad2) is consistent with the fact that APC/CCdc20-catalyzed ubiquitination of cyclin B1 relies on the D box of cyclin B1. The D box of BubR1M likely directly competes with the D box of cyclin B1 for Cdc20 binding, whereas the Phe box provides the necessary affinity.
FIGURE 2.

The Phe and D boxes of BubR1 contribute to APC/CCdc20 inhibition in vitro in the absence of Mad2. A, anti-Myc blot of the in vitro ubiquitination reactions of APC/CCdc20 with cyclin B11–97-Myc as the substrate, in the presence of the indicated BubR1-Bub3 proteins at 200 nm. B, schematic drawing of miniBubR1, a miniaturized BubR1 protein containing only four Cdc20-binding motifs: KEN1, KEN2, Phe, and D boxes. C, anti-Myc blot of the in vitro ubiquitination reactions of APC/CCdc20 with cyclin B11–97-Myc as the substrate, in the presence of increasing concentrations of BubR1-Bub3 or miniBubR1. D, anti-Myc blot of the in vitro ubiquitination reactions of APC/CCdc20 with cyclin B11–97-Myc as the substrate, in the presence of the indicated miniBubR1 proteins at 200 nm. E, anti-Myc blot of the in vitro ubiquitination reactions of APC/CCdc20 with cyclin B11–97-Myc as the substrate, in the presence of the indicated BubR1-Bub3 proteins at 85 nm with or without 1.25 μm Mad2 L13A protein.
To simplify and extend the analysis, we constructed a miniaturized version of BubR1, termed miniBubR1, which contained only the four identified Cdc20-binding motifs of BubR1: KEN1, KEN2, Phe, and D boxes (Fig. 2B). As expected, the full-length BubR1-Bub3 complex inhibited the activity of APC/CCdc20 toward cyclin B1 (Fig. 2B). Remarkably, miniBubR1 on its own was capable of inhibiting APC/CCdc20, albeit with lower potency (Fig. 2C). For example, miniBubR1 at 400 nm achieved a level of APC/C inhibition comparable to that of the full-length BubR1-Bub3 at 50 nm. Thus, miniBubR1 has substantial APC/C inhibitory activity but cannot fully recapitulate the APC/C inhibition by BubR1-Bub3. Other BubR1-Bub3 domains or elements contribute to APC/CCdc20 inhibition, either by providing additional interaction surfaces or by correctly positioning the multiple Cdc20-binding motifs.
We then introduced mutations into each of the four Cdc20-binding motifs in miniBubR1 and assayed the ability of these mutants to inhibit APC/CCdc20. To better reveal the potential involvement of these motifs, we used a concentration of miniBubR1 (200 nm) that produced only partial APC/C inhibition. Consistent with the results observed using full-length BubR1-Bub3, mutations of either the Phe or the D box reduced the APC/C inhibitory activity of miniBubR1 (Fig. 2D). In contrast, mutations of either the KEN1 or the KEN2 box did not affect the ability of miniBubR1 to inhibit APC/CCdc20. Therefore, the Phe and D boxes are critical for APC/C inhibition by BubR1 in vitro.
Because the MCC contains also Mad2, we next tested whether the Phe and D boxes are required for APC/CCdc20 activity in the presence of all MCC components. Addition of BubR1-Bub3 and Mad2 individually at low concentrations was not sufficient to inhibit APC/CCdc20, but addition of BubR1-Bub3 and Mad2 together resulted in synergistic APC/CCdc20 inhibition (Fig. 2E). In contrast to the results observed when BubR1-Bub3 was added alone, mutation of the KEN1 or KEN2 box decreased the synergistic APC/C inhibition in the presence of Mad2. Deletion of the Phe and D boxes did not decrease the ability of BubR1-Bub3 to synergize with Mad2. These results indicate that the BubR1M region has the ability to inhibit APC/CCdc20 in the absence of Mad2. However, when Mad2 is present, APC/CCdc20 inhibition by BubR1 relies more on the KEN1 and KEN2 boxes. These results suggest the intriguing possibility that BubR1 binds and inhibits APC/CCdc20 using different modes, depending on whether Mad2 is present or not.
The Phe Box Does Not Interact with Human Cdh1
Both APC/C activators, Cdc20 and Cdh1, contain highly homologous C-terminal WD40 repeat domains that form similar β propeller structures to directly interact with KEN and D boxes. Surprisingly, in the absence of Mad2, human BubR1 alone is capable of inhibiting APC/CCdc20 in vitro but does not inhibit APC/CCdh1 efficiently (11). We wondered whether the Phe box had binding specificity toward Cdc20. Consistent with data in Fig. 1, the BubR1526–583 and BubR1526–613 fragments that contained both Phe and D boxes bound to Cdc20 efficiently (Fig. 3A). Mutation of either box reduced Cdc20 binding. The smaller BubR1526–546 fragment containing only the Phe box retained substantial Cdc20 binding. In contrast, this Phe box-containing fragment failed to interact with Cdh1 (Fig. 3B). The BubR1526–583 and BubR1526–613 fragments containing both Phe and D boxes also bound less efficiently to Cdh1 than to Cdc20. This weaker Cdh1 binding by BubR1M was completely dependent on its D box, as mutation of the D box abolished the interaction between BubR1M and Cdh1 (Fig. 3B). Taken together, these results indicate that the Phe box in BubR1 preferably binds to human Cdc20. The cooperative binding of both Phe and D boxes thus enables BubR1 to bind to Cdc20 with higher affinity. Cdh1, on the other hand, can only engage the D box of BubR1 and thus binds with weaker affinity.
FIGURE 3.

The Phe box of BubR1 binds to human Cdc20, but not to human Cdh1. A and B, binding between the indicated GST-BubR1 fragments and in vitro translated 35S-labeled full-length human Cdc20 (A) or Cdh1 (B). The relative binding intensities are indicated below each lane. C, cartoon drawing of the superimposed structures of Saccharomyces cerevisiae (Sc) Cdh1-Acm1 (gray) and human (Hs) Cdc20-BubR1-KEN1 (magenta). The KEN, D, and Phe boxes from Acm1 are colored yellow, with their side chains shown as sticks. D, a close-up view of the Phe box-binding site, with HsCdc20 residues corresponding to the Phe box-binding residues of ScCdh1 shown as magenta sticks. The amino acid types of the corresponding HsCdh1 residues are listed in parentheses. E, binding between GST-BubR1526–546 and in vitro translated 35S-labeled full-length human Cdc20 WT or mutants. The relative binding intensities are indicated below each lane.
Structural studies of Cdc20 and Cdh1 have revealed similar APC/C degron-binding surfaces in these activators (15–17). They each contain a single, highly conserved KEN box-binding site on the top face of the β propeller formed by their WD40 domain and a D box-binding site at the side of the β propeller (Fig. 3C). Despite extensive efforts, we could not determine the crystal structure of human Cdc20 bound to the Phe box of BubR1. The electron density maps of crystals grown with Cdc20 bound to the Phe box peptide did not contain interpretable density belonging to the peptide.
The structure of the budding yeast Cdh1 bound to its inhibitor Acm1 revealed the details of how Cdh1 interacts with Acm1 (15). Interestingly, the N-terminal region of the Cdh1-binding region of Acm1 (residues 61–66) has the sequence of FMLYEE, which conforms to the consensus of the Phe box. This motif binds to the bottom face of WD40 propeller of yeast Cdh1 (Fig. 3, C and D). This region in Acm1 was named the A motif, although the previous analysis focused on the two glutamates, and the potential importance of the aromatic and hydrophobic residues preceding them was not shown (35). We will henceforth refer to the FMLYEE motif in Acm1 as the Phe box. While this manuscript was under review, a study identified a motif similar to the A motif and Phe box in yeast Clb5, termed the ABBA motif, and showed that mutation of this motif delays Clb5 degradation (36). Thus, the Phe box is present in both APC/C inhibitors and substrates.
Superposition of the yeast Cdh1 and human Cdc20 structures underscores the overall similarity between their β propellers (Fig. 3C). A closer inspection of the Phe box-binding site revealed that many residues in yeast Cdh1 critical for Phe box binding are conserved in human Cdc20 (Fig. 3D). For example, the two hydrophobic residues Phe-61 and Leu-63 in the Phe box form extensive hydrophobic interactions with Ile-311, His-316, His-355, and Val-371 of yeast Cdh1. These residues correspond to Ile-235, Tyr-240, Tyr-279, and Val-295 in human Cdc20, respectively, which are compatible for hydrophobic interactions with Phe-528 and Ile-530 of the BubR1 Phe box. Surprisingly, these yeast Cdh1 residues are not conserved in human Cdh1 and correspond to Ser-238, Leu-243, Glu-282, and Thr-300 in human Cdh1. Similarly, the two acidic residues Glu-65 and Glu-66 of the Phe box in Acm1 exhibit favorable electrostatic interactions with Lys-333, Lys-335, and Arg-338 in yeast Cdh1, which correspond to Gln-257, Lys-259, and Arg-262 in human Cdc20. Two of these three basic or polar residues are again not conserved in human Cdh1.
Our structural modeling thus suggests the intriguing possibility that the BubR1 Phe box might bind to human Cdc20 in a manner similar to the binding between yeast Cdh1 and the Phe box/A motif of Acm1. This binding mode provides a straightforward explanation for the inability of human Cdh1 to recognize the Phe box, because it lacks many of the putative Phe box-binding residues. Consistent with this hypothesis, three of the five mutations targeting predicted Phe box-binding residues, including K259E, R262S, and Y279E, greatly reduced Cdc20 binding to the Phe box of BubR1 (Fig. 3E).
BubR1 Phe and D Boxes Contribute to MCC Homeostasis in Mitotic Human Cells
Our results so far established the roles for the Phe and D boxes of BubR1 in Cdc20 binding in vitro. We next tested whether these motifs also mediated the binding of BubR1 to Cdc20 in human cells. We constructed a BubR1 mutant with residues 525–561 (which encompass both the Phe and D boxes) deleted, termed ΔPheD. As important controls, we also included the BubR1 mutants with its KEN boxes mutated (mKEN1 and mKEN2) in our experiments. HeLa cells were transfected with Myc-BubR1 WT, mKEN1, mKEN2, mD, mPhe, or ΔPheD and arrested in mitosis with the microtubule-depolymerizing drug nocodazole. Lysates of mitotic cells collected through shake-off were subjected to immunoprecipitation with anti-Myc antibody beads. The lysates and the anti-Myc immunoprecipitates were analyzed with quantitative Western blot. Myc-BubR1 WT efficiently pulled down Bub3, Cdc20, and Mad2, consistent with MCC formation (Fig. 4, A and B). Myc-BubR1 mKEN1 completely lost its binding to Cdc20 or Mad2, whereas mKEN2 still retained some binding (Fig. 4A). These results were consistent with published reports showing that the KEN1 box, but not the KEN2 box, of BubR1 was required for MCC formation in human cells (23). Strikingly, despite retaining proper Bub3 binding, Myc-BubR1 ΔPheD bound to Cdc20 and Mad2 much less efficiently, at 70 and 30% of the WT levels (Fig. 4A). Mutations of either the Phe or D box also reduced Cdc30 and Mad2 binding, indicating that both motifs are involved in maintaining MCC levels.
FIGURE 4.

The BubR1 Phe and D boxes maintain MCC levels in human cells. A, anti-Myc, anti-Bub3, anti-Cdc20, and anti-Mad2 blots of the lysates (Input) and anti-Myc immunoprecipitates (IP) from mitotic HeLa cells transfected with the indicated Myc-BubR1 plasmids and treated with nocodazole. The relative band intensities of Cdc20 or Mad2 normalized to the intensities of BubR1 in each sample are indicated below each lane. B, HeLa cells transfected with siLuc or siBubR1 for 42 h were treated with 40 nm nocodazole for 30 min to depolymerize microtubules, then fixed, and stained with DAPI and the indicated antibodies. Scale bars, 5 μm. C, quantification of the normalized staining intensities of BubR1 and Cdc20 of cells in B. Each dot represents an individual cell. Error bars, S.D.
One possibility is that BubR1 recruits Cdc20 to kinetochores through the Phe and D boxes. In fact, a role for the BubR1 middle region in Cdc20 kinetochore targeting was recently reported (37). We first tested whether BubR1 was required for the kinetochore localization of Cdc20. The kinetochore localization of Cdc20 was slightly elevated in cells depleted of BubR1 (Fig. 4, B and C). Thus, BubR1 is not required for the kinetochore targeting of Cdc20 in our system. Whether this discrepancy is due to technical differences in experimental design or reflects the existence of multiple Cdc20 targeting mechanisms differentially favored in specific cell lines remains to be determined.
To further probe the mechanism by which BubR1M contributes to MCC homeostasis, we transfected HeLa cells with various Myc-BubR1 plasmids, arrested them in mitosis with nocodazole or the microtubule-stabilizing drug taxol, and treated the cells with the proteasome inhibitor MG132 for 1 h before collecting the mitotic cells through shake-off. The lysates and the anti-Myc immunoprecipitates were again analyzed with quantitative Western blot. Cdc20 degradation by the proteasome is an important pathway for MCC disassembly (38–40). MG132 blocks this pathway and hampers MCC disassembly. Under these conditions, mutation on BubR1 KEN1 still abolished Mad2 binding and greatly reduced Cdc20 binding to BubR1 (Fig. 5), consistent with the notion that KEN1 is directly required for MCC formation. In contrast, the amounts of Cdc20 and Mad2 bound to BubR1 ΔPheD were similar to those bound to BubR1 WT in cells treated with nocodazole and MG132 and were slightly reduced in cells arrested in taxol and MG132. These results indicate that the Phe and D boxes of BubR1 are not strictly required for MCC formation. They might contribute to MCC homeostasis through interfering with proteasome-dependent MCC disassembly.
FIGURE 5.
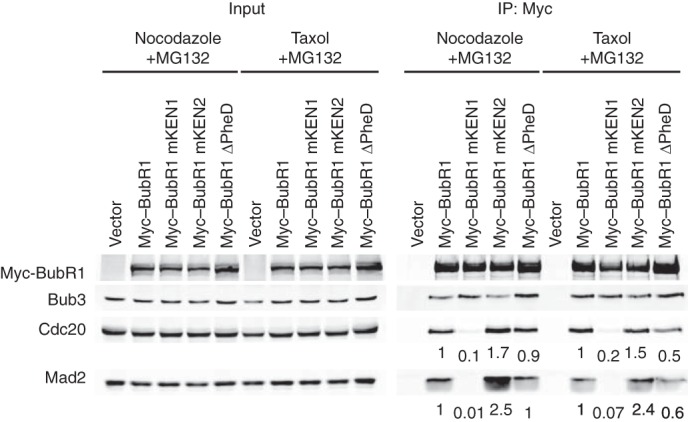
The BubR1 Phe and D boxes contribute to MCC homeostasis in human cells. Anti-Myc, anti-Bub3, anti-Cdc20, and anti-Mad2 blots of the lysates (Input) and anti-Myc immunoprecipitates (IP) from mitotic HeLa cells transfected with the indicated Myc-BubR1 plasmids and treated with nocodazole or taxol and MG132. The relative Cdc20 and Mad2 intensities of the IP samples were quantified and are indicated below each lane.
The Phe and D Boxes of BubR1 Are Dispensable for the Spindle Checkpoint in Human Cells
The Phe and D boxes of BubR1 are required for the ability of BubR1 to inhibit APC/CCdc20 in vitro, in the absence of Mad2. They are involved in maintaining the normal levels of MCC in mitotic HeLa cells. We next tested whether these motifs were required for the spindle checkpoint in human cells. HeLa cells were depleted of the endogenous BubR1 by transfection with a BubR1 siRNA (siBubR1) and were simultaneously transfected with siBubR1-resistant transgenes encoding Myc-BubR1 WT or mutants targeting various motifs. The cells were incubated with high concentrations of nocodazole (500 nm) or taxol (200 nm) for 15 h, stained with the MPM2 antibody (which recognizes mitotic phospho-epitopes), and analyzed with flow cytometry. As expected, depletion of BubR1 greatly reduced the mitotic index of cells treated with nocodazole or taxol (Fig. 6, A and B). Ectopic expression of Myc-BubR1 WT, but not mKEN1 or mKEN2, restored the mitotic arrest in cells depleted of the endogenous BubR1, consistent with the known requirement for both KEN boxes in the spindle checkpoint. Myc-BubR1 mPhe, mD, and ΔPheD were fully functional in restoring nocodazole- or taxol-triggered mitotic arrest. All Myc-BubR1 proteins were expressed at levels comparable with that of the endogenous BubR1 (Fig. 6C). Thus, although the Phe and D boxes are important for Cdc20 binding and APC/CCdc20 inhibition in vitro and for MCC homeostasis in human cells, they are apparently dispensable for the mitotic arrest triggered by high concentrations of spindle poisons. Our observation is in fact consistent with previous reports showing that deletions of the middle region of BubR1 do not produce strong checkpoint defects (30, 31).
FIGURE 6.

The BubR1 Phe and D boxes attenuate checkpoint strength in HeLa cells with low grade spindle damage. A and B, quantification of the mitotic index (defined as the percentage of MPM2+, 4n cells) of HeLa cells transfected with the indicated siRNAs and plasmids and treated with 500 nm nocodazole (A) or 200 nm taxol (B). Error bars, S.D., n = 3 technical repeats (taxol) and n = 2 technical repeats (nocodazole). C, anti-BubR1 and anti-tubulin blot of total lysates of HeLa cells transfected with the indicated plasmids and siRNAs and treated with taxol. D, HeLa cells transfected with the indicated plasmids and siRNAs were incubated with 30 nm taxol for 15 h. Samples were stained with the MPM2 antibody and propidium iodide and subjected to FACS analysis. The mitotic index was quantified as in A and B. Error bars, S.D., n = 3 technical repeats. The p values (compared with WT) are determined by t test: *, p = 0.01; **, p < 0.01. E, representative FACS graphs of samples in D are shown. The mitotic index of each sample (defined as MPM2+, 4n cells) is indicated.
Recent studies have shown that the length of mitotic arrest and presumably the strength of the spindle checkpoint can be tuned by the number of unattached kinetochores or the severity of spindle damage (41–43). We wondered whether the Phe and D boxes of BubR1 were required for the less robust mitotic arrest triggered by low concentrations of taxol (Fig. 6, D and E). The mitotic index of control HeLa cells treated with 30 nm taxol (∼30%) was indeed lower than that of cells treated with 200 nm taxol (∼45%). Surprisingly, in the presence of 30 nm taxol, cells expressing Myc-BubR1 mPhe, mD, or ΔPheD all exhibited a small but statistically significant increase in mitotic index, as compared with cells expressing Myc-BubR1 WT (Fig. 6, D and E). This result suggests that when cells are challenged with low degrees of spindle damage, the Phe and D boxes of BubR1 might even attenuate the strength of the spindle checkpoint.
To further confirm this surprising result, we carefully measured the duration of mitotic arrest in HeLa cells expressing BubR1 WT, mD, mPhe, and ΔPheD in the presence of 30 nm taxol with live cell imaging. Under these conditions, control-depleted cells were arrested in mitosis for several hours (Fig. 7A). Most cells then underwent mitotic slippage or adaptation (Fig. 7B). This outcome was in stark contrast to cells treated with high concentrations of taxol, virtually all of which underwent apoptosis following the mitotic arrest (44). This difference was again consistent with the notion that low concentrations of taxol induce a weaker spindle checkpoint response. As expected, cells transfected with siBubR1 and a control vector quickly exited mitosis and underwent adaptation, indicative of a gross checkpoint defect (Fig. 7, A and B). Expression of Myc-BubR1 WT fully rescued the checkpoint defect caused by siBubR1. Consistent with the flow cytometry data, cells expressing Myc-BubR1 ΔPheD remained in mitosis slightly longer than cells expressing Myc-BubR1 WT did (Δt50 = 4.1 ± 2.5 h) (Fig. 7A). Myc-BubR1 mD had a smaller effect, whereas mPhe was comparable with WT. Myc-BubR1 WT and mutants were expressed at comparable levels (Fig. 7C). Thus, despite its positive contribution to MCC levels, the middle region of BubR1 is dispensable for the spindle checkpoint. It appears to attenuate the strength of the checkpoint when cells experience low grade spindle damage.
FIGURE 7.
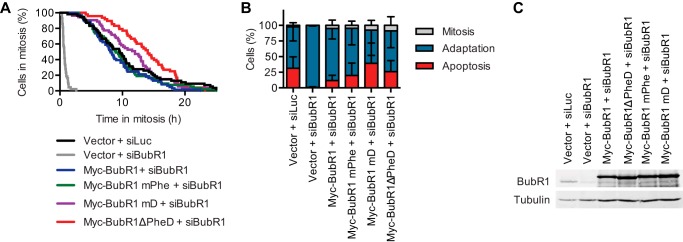
The BubR1 Phe and D boxes shorten mitotic arrest duration of HeLa cells with low grade spindle damage. A, live cell imaging analysis of HeLa cells transiently transfected with the indicated plasmids and siRNAs and treated with 30 nm taxol. Cumulative percentage of mitotic cells was plotted against their mitotic durations. Δt50(Myc-BubR1 + siBubR1 − Myc-BubR1ΔPheD + siBubR1) = 4.1 ± 2.5 h, n = 2. B, quantification of the terminal phenotypes of the mitotic cells in A. Error bars, S.D., n = 2 independent experiments. C, anti-BubR1 and anti-tubulin blots of lysates from cells treated with the same conditions as in A and handled in parallel.
DISCUSSION
BubR1 is an evolutionarily conserved spindle checkpoint protein and a critical component of MCC (the BubR1-Bub3-Cdc20-Mad2 complex), which is a potent checkpoint inhibitor of APC/C. Two regions of human BubR1, the N-terminal region (BubR1N) and the middle region (BubR1M), mediate Cdc20 binding and APC/C inhibition. BubR1N shares sequence similarity with its yeast homolog Mad3 and, on its own, binds to Cdc20 weakly and does not inhibit APC/C (23). In the presence of Mad2, however, BubR1N (or Mad3) simultaneously engages Cdc20 with its first KEN box and contacts the Cdc20-bound Mad2, thus nucleating the formation of MCC (17, 23, 29). Thus, the spindle checkpoint function of BubR1N is well established. It collaborates with Mad2 to bind to Cdc20 cooperatively and is critical for MCC assembly and APC/C inhibition. In contrast, BubR1M binds to Cdc20 and inhibits APC/C in the absence of Mad2 in vitro (11), but deletions of BubR1M do not produce spindle checkpoint defects in mammalian cells (30, 31). The nature and function of the BubR1M-Cdc20 interaction have thus remained murky. In this study, we show that BubR1M contains two Cdc20-binding motifs conserved in vertebrates: a D box and a Phe box. These two tandem motifs bind cooperatively to Cdc20 and are required for APC/C inhibition in vitro. They also contribute to the maintenance of MCC levels in human cells during checkpoint-mediated mitotic arrest.
Role of the D box of BubR1M in APC/C inhibition
The D box with the consensus RXXLXXXXN is a well established APC/C degron found in many APC/C substrates and inhibitors and interacts with both Cdc20 and Cdh1. The D box of cyclin B1 is critical for its ubiquitination by APC/C (45). The fact that BubR1M contains a D box required for APC/C inhibition strongly suggests that, in the absence of Mad2, BubR1M inhibits APC/C through acting as a pseudo-substrate to competitively block D box-dependent recruitment of cyclin B1. Why BubR1M is not itself ubiquitinated is unclear, but it may possibly be because the BubR1M D box lacks the conserved asparagine at the end. This type of shorter D boxes might only bind to Cdc20 but might not effectively engage the D box co-receptor APC10. Additionally, the full-length BubR1 is protected against APC/C-dependent ubiquitination by post-translational modifications, such as acetylation (46). Interestingly, the budding yeast Mad3 also contains a D box in its C-terminal region (429RKAL432), which is critical for its checkpoint function (20). Mutation of this D box reduces, but does not abolish, Cdc20 binding. It will be interesting to test whether, in the absence of Mad2, the D box of Mad3 suffices to inhibit ubiquitination of D box substrates by APC/CCdc20 in vitro.
Role of the Phe Box in Cdc20 Binding
The Phe box is related to the A motif previously identified in Acm1 (35) and is also present in other Cdc20-binding proteins, including human Bub1 and cyclin A2 (Fig. 1C). Structural modeling (with the human Cdc20 and yeast Cdh1-Acm1 structures as templates) and mutagenesis identified the Phe box-binding site in human Cdc20 (Fig. 3). Intriguingly, yeast Acm1 has been shown to bind specifically to yeast Cdh1, but not to yeast Cdc20 (15). Several of the key yeast Cdh1 residues that interact with the Phe box are not conserved in yeast Cdc20, suggesting that the Acm1 specificity for yeast Cdh1 is at least partially dependent on binding to the Phe box. As expected from the fact that yeast Cdc20 does not recognize the Phe box, Mad3 does not appear to have a Phe box. On the other hand, our results show that the BubR1 Phe box mediates binding to human Cdc20, but not to human Cdh1. Although it is unclear whether this switch in binding specificity has functional implications, this phenomenon can be explained by structural comparisons. Many of Phe box-binding residues are not conserved in human Cdh1, explaining the specificity of the Phe box in binding to human Cdc20.
It will be interesting to test whether the putative Phe boxes of Bub1 and cyclin A2 are also involved in their interaction with Cdc20. Intriguingly, the Phe box in Bub1 is located only two residues N-terminal to its first KEN box (KEN1) and ∼100 residues N-terminal to the second KEN box (KEN2) (47). The two KEN boxes of Bub1 have been previously shown to bind cooperatively to Cdc20 (48). Because Cdc20 only has one KEN box-binding site, this cooperativity remained unexplained. In light of the fact that a Phe box in Bub1 lies immediately adjacent to KEN1, it is possible that the Bub1 Phe box and KEN2 bind cooperatively to Cdc20. The effects of KEN1 mutations on Bub1 binding to Cdc20 might be due to collateral damage to the conjoined Phe box.
Different Modes of APC/C Inhibition by BubR1 in the Presence or Absence of Mad2
Despite the ability of BubR1 to directly bind Cdc20 and inhibit APC/C in vitro, it is clearly established that the full MCC is a more potent inhibitor than either Mad2 or BubR1 alone. Interestingly, our results indicate that the BubR1M region is critical for APC/C inhibition in the absence of Mad2, but this region is largely dispensable for the inhibitory synergy observed in the presence of Mad2. These in vitro observations could explain the lack of checkpoint defects in human cells expressing BubR1 ΔPheD, because these cells contain endogenous Mad2. Finally, a recent study showed that MCC could inhibit a second Cdc20 molecule already bound to APC/C (49). This trans-inhibitory activity of MCC requires the KEN2 box and a newly discovered D box adjacent to KEN2 in the N-terminal region of BubR1. Therefore, BubR1 contains five Cdc20-binding motifs: two KEN boxes, two D boxes, and a Phe box. These motifs can in theory engage three Cdc20 molecules simultaneously. How these elements collaborate to inhibit APC/CCdc20 clearly requires further investigation.
Mechanisms by Which D and Phe Boxes Contribute to MCC Homeostasis
The Phe and D boxes are not only involved in Cdc20 binding and APC/C inhibition in vitro but also contribute to maintaining high level MCC in human cells. As compared with the wild type, a BubR1 mutant with a small region encompassing the Phe and D boxes deleted is less efficient in binding Cdc20 and Mad2 in human cells arrested in mitosis by nocodazole. Interestingly, blocking the activity of the proteasome largely bypasses the need for these motifs in maintaining MCC homeostasis. Thus, the Phe and D boxes contribute to MCC homeostasis, possibly by slowing the proteasome-dependent MCC disassembly.
The structure of the fission yeast MCC suggests that the KEN1-containing, N-terminal region of Mad3 (and by analogy BubR1N) is critical for MCC formation by contacting both Cdc20 and Mad2 (17). BubR1M is unlikely to directly contact Cdc20 in the context of MCC. The role of the BubR1M-Cdc20 interaction in maintaining MCC is probably indirect.
How then can the BubR1M-Cdc20 interaction maintain MCC levels in human cells? We propose the following speculative model (Fig. 8), which provides a plausible answer to this question. In this model, two types of Cdc20-containing checkpoint complexes exist at equilibrium: the BubR1-Bub3-Cdc20 complex (BBC) and the intact MCC comprising BubR1-Bub3-Cdc20-Mad2. BBC involves the cooperative binding of the Phe and D boxes to Cdc20, whereas MCC requires the cooperative binding of Mad2 and the KEN1 of BubR1 to Cdc20. MCC is a more potent APC/C inhibitor, but Cdc20 as a subunit of MCC is more prone to undergo autoubiquitination and proteasome-dependent degradation (50), triggering complete MCC disassembly. With the help of p31comet and TRIP13 (38–40, 51–56), Mad2 can dissociate from MCC in a proteasome-independent pathway, producing BBC. Because Mad2 promotes Cdc20 autoubiquitination, Cdc20 in BBC should be more resistant to proteasome-dependent degradation. With the addition of active Mad2 (produced at unattached kinetochores), this partially disassembled MCC subcomplex can readily reassemble into the intact MCC. BubR1 lacking the Phe and D boxes cannot form BBC. The p31comet- and TRIP13-dependent Mad2 extraction causes the complete disassembly of MCC containing this BubR1 mutant, slowing its reassembly and reducing its steady-state levels.
FIGURE 8.
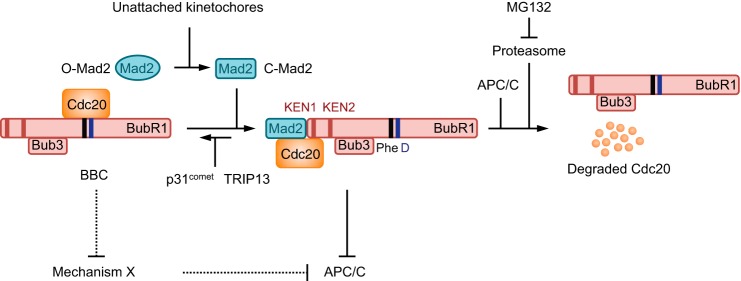
A speculative model explaining the functions of BubR1 Phe and D boxes. During checkpoint activation, the intact MCC (BubR1-Bub3-Cdc20-Mad2) and its subcomplex BBC (BubR1-Bub3-Cdc20) exist in a dynamic equilibrium. In the forward reaction, unattached kinetochores convert the inactive, open Mad2 (O-Mad2; oval) to active, closed Mad2 (C-Mad2; rectangle), which binds to BBC to promote MCC formation. The N-terminal region of BubR1 is critical for MCC formation, with the first KEN box (KEN1; indicated by a red bar) contacting Cdc20 directly. The Phe and D boxes (indicated by black and blue bars, respectively) might not directly contact Cdc20 in MCC. In the reverse reaction, p31comet and the ATPase TRIP13 extract C-Mad2 from MCC to produce BBC and O-Mad2. Cdc20 in MCC is more efficiently autoubiquitinated by APC/C and is degraded through the proteasome. Because the Phe and D boxes bind to Cdc20 in BBC, extraction of Mad2 from MCC formed by BubR1 ΔPheD will cause its complete disassembly into free BubR1-Bub3, Cdc20, and Mad2. Reassembly of these components into MCC might occur at a slower rate than that of MCC assembly through Mad2 incorporation into BBC. Rates of MCC disassembly pathways (proteasome-dependent degradation of Cdc20 and p31comet/TRIP13-mediated Mad2 extraction) are unlikely to be affected by the Phe and D box deletion. The slower rate of reassembly in the presence of constant rates of disassembly explains why the steady-state level of MCC containing BubR1 ΔPheD is lower. A potential explanation for why BubR1 ΔPheD is still functional in the checkpoint is the existence of a redundant mechanism (mechanism X) of APC/C inhibition. This mechanism might compete with the Phe and D box of BubR1 for Cdc20 binding. In cells with BubR1 ΔPheD, this mechanism might be enhanced, resulting in longer mitotic arrest under conditions of low grade spindle damage.
Mutation of KEN1 completely abolishes the BubR1-Bub3-Cdc20 interaction in nocodazole-arrested mitotic human cells (Fig. 4A), suggesting that the vast majority of Cdc20-containing complexes exist as the intact MCC. The steady-state level of BBC is very low. Nevertheless, the transient existence of this complex is critical for maintaining high levels of MCC, presumably through preventing its complete disassembly.
Functions of the Phe and D Boxes in the Spindle Checkpoint
Although deletion of the Phe and D boxes reduces MCC levels severalfold, the BubR1 ΔPheD mutant is fully functional in the spindle checkpoint. One simple explanation for this conundrum is that the residual amount of MCC is still above the threshold needed for APC/CCdc20 inhibition. This possibility, however, does not explain why BubR1 ΔPheD is slightly more effectively in maintaining mitotic arrest when the checkpoint is activated by low grade spindle damage, such as that caused by low concentrations of taxol.
To explain the apparent gain of function of BubR1 ΔPheD, we envision two additional possibilities that are not mutually exclusive. In the first possibility, the checkpoint employs an independent mechanism that collaborates with MCC to inhibit APC/C (Fig. 8). This mechanism is partially redundant to and might even compete with BubR1M for Cdc20 binding. The removal of BubR1M strengthens this independent mechanism, which sustains the checkpoint along with the decreased amount of MCC. This functional compensation might even produce longer mitotic arrest in response to low grade spindle damage, at least in HeLa cells. Along this vein, we note that Bub1 also contains a Phe box. Deletion of the Phe box in BubR1 might increase Bub1 binding to Cdc20 and enhance Bub1-mediated Cdc20 phosphorylation, which can inhibit APC/CCdc20 in the absence of MCC (57). In the second possibility, the Phe and D boxes of BubR1 have dual, opposite functions in the spindle checkpoint. Aside from their positive roles in maintaining MCC, they might also have negative roles. For example, BubR1 binds to protein phosphatase 2A through the KARD motif (residues 664–681) (58), which lies ∼100 residues C-terminal to the D box in BubR1M. The close proximity of Cdc20 bound to BubR1M and protein phosphatase 2A bound to KARD might promote Cdc20 dephosphorylation and checkpoint silencing. Thus, similar to Mad2, which inhibits APC/C as part of the MCC and at the same time promotes MCC disassembly through its interaction with p31comet, BubR1 might also have dual positive and negative functions that contribute to APC/C inhibition in the presence of an active checkpoint but promote fast MCC disassembly and APC/C activation when the checkpoint needs to be silenced. Deletion of the Phe and D boxes is expected to eliminate both their positive and putative negative functions, resulting in a largely net neutral effect on the checkpoint. Under specific conditions or in certain cell lines, elimination of the negative function by Phe and D box mutations might even outweigh the reduction in the positive function, resulting in gain of function phenotypes.
Conclusion
In summary, we have identified two functional motifs, the Phe and D boxes, in the middle region of BubR1 that mediate Cdc20 binding and APC/C inhibition. These motifs promote MCC homeostasis in human cells but are not required for the spindle checkpoint. This apparent contradiction highlights an important gap in our understanding of the spindle checkpoint and leads to several plausible and testable hypotheses, including functional duality of these motifs or the existence of redundant, competing mechanisms that inhibit APC/C.
Acknowledgment
We thank Dr. Hayden Ball for peptide synthesis.
This work was supported, in whole or in part, by National Institutes of Health Grant GM085004 (to X. L.). This work was also supported by Welch Foundation Grant I-1441 (to H. Y.) and Cancer Prevention and Research Institute of Texas Grant RP120717-P2 (to H. Y.).
- APC/C
- anaphase-promoting complex/cyclosome
- D box
- destruction box
- mD
- mutated D box
- IP
- immunoprecipitation
- KEN box
- lysine-glutamate-asparagine box
- MCC
- mitotic checkpoint complex
- Phe box
- phenylalanine box
- mPhe
- mutated Phe box
- WD40
- tryptophan-aspartate 40
- BBC
- BubR1-Bub3-Cdc20 complex.
REFERENCES
- 1. Cheeseman I. M., Desai A. (2008) Molecular architecture of the kinetochore-microtubule interface. Nat. Rev. Mol. Cell Biol. 9, 33–46 [DOI] [PubMed] [Google Scholar]
- 2. Tanaka T. U. (2010) Kinetochore-microtubule interactions: steps towards bi-orientation. EMBO J. 29, 4070–4082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foley E. A., Kapoor T. M. (2013) Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat. Rev. Mol. Cell Biol. 14, 25–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Holland A. J., Cleveland D. W. (2012) Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep. 13, 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lara-Gonzalez P., Westhorpe F. G., Taylor S. S. (2012) The spindle assembly checkpoint. Curr. Biol. 22, R966–980 [DOI] [PubMed] [Google Scholar]
- 6. Jia L., Kim S., Yu H. (2013) Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem. Sci. 38, 302–311 [DOI] [PubMed] [Google Scholar]
- 7. Kim S., Yu H. (2011) Mutual regulation between the spindle checkpoint and APC/C. Semin. Cell Dev. Biol. 22, 551–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peters J. M. (2006) The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 7, 644–656 [DOI] [PubMed] [Google Scholar]
- 9. Yu H. (2007) Cdc20: a WD40 activator for a cell cycle degradation machine. Mol. Cell 27, 3–16 [DOI] [PubMed] [Google Scholar]
- 10. Fang G., Yu H., Kirschner M. W. (1998) The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 12, 1871–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang Z., Bharadwaj R., Li B., Yu H. (2001) Mad2-independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev. Cell 1, 227–237 [DOI] [PubMed] [Google Scholar]
- 12. Sudakin V., Chan G. K., Yen T. J. (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 154, 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fang G. (2002) Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol. Biol. Cell 13, 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo X., Yu H. (2012) Mitosis: short-circuiting spindle checkpoint signaling. Curr. Biol. 22, R128–R130 [DOI] [PubMed] [Google Scholar]
- 15. He J., Chao W. C., Zhang Z., Yang J., Cronin N., Barford D. (2013) Insights into degron recognition by APC/C coactivators from the structure of an Acm1-Cdh1 complex. Mol. Cell 50, 649–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tian W., Li B., Warrington R., Tomchick D. R., Yu H., Luo X. (2012) Structural analysis of human Cdc20 supports multisite degron recognition by APC/C. Proc. Natl. Acad. Sci. U.S.A. 109, 18419–18424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chao W. C., Kulkarni K., Zhang Z., Kong E. H., Barford D. (2012) Structure of the mitotic checkpoint complex. Nature 484, 208–213 [DOI] [PubMed] [Google Scholar]
- 18. Buschhorn B. A., Petzold G., Galova M., Dube P., Kraft C., Herzog F., Stark H., Peters J. M. (2011) Substrate binding on the APC/C occurs between the coactivator Cdh1 and the processivity factor Doc1. Nat. Struct. Mol. Biol. 18, 6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. da Fonseca P. C., Kong E. H., Zhang Z., Schreiber A., Williams M. A., Morris E. P., Barford D. (2011) Structures of APC/CCdh1 with substrates identify Cdh1 and Apc10 as the D-box co-receptor. Nature 470, 274–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burton J. L., Solomon M. J. (2007) Mad3p, a pseudosubstrate inhibitor of APCCdc20 in the spindle assembly checkpoint. Genes Dev. 21, 655–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. King E. M., van der Sar S. J., Hardwick K. G. (2007) Mad3 KEN boxes mediate both Cdc20 and Mad3 turnover, and are critical for the spindle checkpoint. PLoS One 2, e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sczaniecka M., Feoktistova A., May K. M., Chen J. S., Blyth J., Gould K. L., Hardwick K. G. (2008) The spindle checkpoint functions of Mad3 and Mad2 depend on a Mad3 KEN box-mediated interaction with Cdc20-anaphase-promoting complex (APC/C). J. Biol. Chem. 283, 23039–23047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lara-Gonzalez P., Scott M. I., Diez M., Sen O., Taylor S. S. (2011) BubR1 blocks substrate recruitment to the APC/C in a KEN-box-dependent manner. J. Cell Sci. 124, 4332–4345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Herzog F., Primorac I., Dube P., Lenart P., Sander B., Mechtler K., Stark H., Peters J. M. (2009) Structure of the anaphase-promoting complex/cyclosome interacting with a mitotic checkpoint complex. Science 323, 1477–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Izawa D., Pines J. (2011) How APC/C-Cdc20 changes its substrate specificity in mitosis. Nat. Cell Biol. 13, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luo X., Tang Z., Rizo J., Yu H. (2002) The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol. Cell 9, 59–71 [DOI] [PubMed] [Google Scholar]
- 27. Mapelli M., Musacchio A. (2007) MAD contortions: conformational dimerization boosts spindle checkpoint signaling. Curr. Opin. Struct. Biol. 17, 716–725 [DOI] [PubMed] [Google Scholar]
- 28. Luo X., Yu H. (2008) Protein metamorphosis: the two-state behavior of Mad2. Structure 16, 1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tipton A. R., Wang K., Link L., Bellizzi J. J., Huang H., Yen T., Liu S. T. (2011) BUBR1 and closed MAD2 (C-MAD2) interact directly to assemble a functional mitotic checkpoint complex. J. Biol. Chem. 286, 21173–21179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davenport J., Harris L. D., Goorha R. (2006) Spindle checkpoint function requires Mad2-dependent Cdc20 binding to the Mad3 homology domain of BubR1. Exp. Cell Res. 312, 1831–1842 [DOI] [PubMed] [Google Scholar]
- 31. Malureanu L. A., Jeganathan K. B., Hamada M., Wasilewski L., Davenport J., van Deursen J. M. (2009) BubR1 N terminus acts as a soluble inhibitor of cyclin B degradation by APC/CCdc20 in interphase. Dev. Cell 16, 118–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang M., Li B., Liu C. J., Tomchick D. R., Machius M., Rizo J., Yu H., Luo X. (2008) Insights into Mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric Mad2 dimer. PLoS Biol. 6, e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wienken C. J., Baaske P., Rothbauer U., Braun D., Duhr S. (2010) Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100. [DOI] [PubMed] [Google Scholar]
- 34. Tang Z., Yu H. (2004) Functional analysis of the spindle-checkpoint proteins using an in vitro ubiquitination assay. Methods Mol. Biol. 281, 227–242 [DOI] [PubMed] [Google Scholar]
- 35. Burton J. L., Xiong Y., Solomon M. J. (2011) Mechanisms of pseudosubstrate inhibition of the anaphase promoting complex by Acm1. EMBO J. 30, 1818–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu D., Hsiao J. Y., Davey N. E., Van Voorhis V. A., Foster S. A., Tang C., Morgan D. O. (2014) Multiple mechanisms determine the order of APC/C substrate degradation in mitosis. J. Cell Biol. 207, 23–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Han J. S., Vitre B., Fachinetti D., Cleveland D. W. (2014) Bimodal activation of BubR1 by Bub3 sustains mitotic checkpoint signaling. Proc. Natl. Acad. Sci. U.S.A. 111, E4185–E4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reddy S. K., Rape M., Margansky W. A., Kirschner M. W. (2007) Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature 446, 921–925 [DOI] [PubMed] [Google Scholar]
- 39. Jia L., Li B., Warrington R. T., Hao X., Wang S., Yu H. (2011) Defining pathways of spindle checkpoint silencing: functional redundancy between Cdc20 ubiquitination and p31comet. Mol. Biol. Cell 22, 4227–4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Varetti G., Guida C., Santaguida S., Chiroli E., Musacchio A. (2011) Homeostatic control of mitotic arrest. Mol. Cell 44, 710–720 [DOI] [PubMed] [Google Scholar]
- 41. Collin P., Nashchekina O., Walker R., Pines J. (2013) The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat. Cell Biol. 15, 1378–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dick A. E., Gerlich D. W. (2013) Kinetic framework of spindle assembly checkpoint signalling. Nat. Cell Biol. 15, 1370–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Heinrich S., Geissen E. M., Kamenz J., Trautmann S., Widmer C., Drewe P., Knop M., Radde N., Hasenauer J., Hauf S. (2013) Determinants of robustness in spindle assembly checkpoint signalling. Nat. Cell Biol. 15, 1328–1339 [DOI] [PubMed] [Google Scholar]
- 44. Díaz-Martínez L. A., Karamysheva Z. N., Warrington R., Li B., Wei S., Xie X. J., Roth M. G., Yu H. (2014) Genome-wide siRNA screen reveals coupling between mitotic apoptosis and adaptation. EMBO J. 33, 1960–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fang G., Yu H., Kirschner M. W. (1998) Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell 2, 163–171 [DOI] [PubMed] [Google Scholar]
- 46. Choi E., Choe H., Min J., Choi J. Y., Kim J., Lee H. (2009) BubR1 acetylation at prometaphase is required for modulating APC/C activity and timing of mitosis. EMBO J. 28, 2077–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Qi W., Yu H. (2007) KEN-box-dependent degradation of the Bub1 spindle checkpoint kinase by the anaphase-promoting complex/cyclosome. J. Biol. Chem. 282, 3672–3679 [DOI] [PubMed] [Google Scholar]
- 48. Kang J., Yang M., Li B., Qi W., Zhang C., Shokat K. M., Tomchick D. R., Machius M., Yu H. (2008) Structure and substrate recruitment of the human spindle checkpoint kinase Bub1. Mol. Cell 32, 394–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Izawa D., Pines J. (2014) The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature 10.1038/nature13911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Foster S. A., Morgan D. O. (2012) The APC/C subunit Mnd2/Apc15 promotes Cdc20 autoubiquitination and spindle assembly checkpoint inactivation. Mol. Cell 47, 921–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Teichner A., Eytan E., Sitry-Shevah D., Miniowitz-Shemtov S., Dumin E., Gromis J., Hershko A. (2011) p31comet promotes disassembly of the mitotic checkpoint complex in an ATP-dependent process. Proc. Natl. Acad. Sci. U.S.A. 108, 3187–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Westhorpe F. G., Tighe A., Lara-Gonzalez P., Taylor S. S. (2011) p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J. Cell Sci. 124, 3905–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xia G., Luo X., Habu T., Rizo J., Matsumoto T., Yu H. (2004) Conformation-specific binding of p31comet antagonizes the function of Mad2 in the spindle checkpoint. EMBO J. 23, 3133–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang M., Li B., Tomchick D. R., Machius M., Rizo J., Yu H., Luo X. (2007) p31comet blocks Mad2 activation through structural mimicry. Cell 131, 744–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Eytan E., Wang K., Miniowitz-Shemtov S., Sitry-Shevah D., Kaisari S., Yen T. J., Liu S. T., Hershko A. (2014) Disassembly of mitotic checkpoint complexes by the joint action of the AAA-ATPase TRIP13 and p31comet. Proc. Natl. Acad. Sci. U.S.A. 111, 12019–12024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang K., Sturt-Gillespie B., Hittle J. C., Macdonald D., Chan G. K., Yen T. J., Liu S. T. (2014) Thyroid hormone receptor interacting protein 13 (TRIP13) AAA-ATPase is a novel mitotic checkpoint silencing protein. J. Biol. Chem. 289, 23928–23937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tang Z., Shu H., Oncel D., Chen S., Yu H. (2004) Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol. Cell 16, 387–397 [DOI] [PubMed] [Google Scholar]
- 58. Suijkerbuijk S. J., Vleugel M., Teixeira A., Kops G. J. (2012) Integration of kinase and phosphatase activities by BUBR1 ensures formation of stable kinetochore-microtubule attachments. Dev. Cell 23, 745–755 [DOI] [PubMed] [Google Scholar]