

Abstract
Aims
To examine the role of physiological Akt signalling in pathological hypertrophy through analysis of PHLPP1 (PH domain leucine-rich repeat protein phosphatase) knock-out (KO) mice.
Methods and results
To investigate the in vivo requirement for ‘physiological’ control of Akt activation in cardiac growth, we examined the effect of deleting the Akt phosphatase, PHLPP, on the induction of cardiac hypertrophy. Basal Akt phosphorylation increased nearly two-fold in the cardiomyocytes from PHLPP1 KO mice and physiological hypertrophy induced by swimming exercise was accentuated as assessed by increased heart size and myocyte cell area. In contrast, the development of pathophysiological hypertrophy induced by pressure overload and assessed by increases in heart size, myocyte cell area, and hypertrophic gene expression was attenuated. This attenuation coincided with decreased fibrosis and cell death in the KO mice. Cast moulding revealed increased capillary density basally in the KO hearts, which was further elevated relative to wild-type mouse hearts in response to pressure overload. In vitro studies with isolated myocytes in co-culture also demonstrated that PHLPP1 deletion in cardiomyocytes can enhance endothelial tube formation. Expression of the pro-angiogenic factor VEGF was also elevated basally and accentuated in response to transverse aortic constriction in hearts from KO mice.
Conclusion
Our data suggest that enhancing Akt activity by inhibiting its PHLPP1-mediated dephosphorylation promotes processes associated with physiological hypertrophy that may be beneficial in attenuating the development of pathological hypertrophy.
Keywords: PH domain leucine-rich repeat protein phosphatase, PHLPP, Akt, Hypertrophy, Mouse model
1. Introduction
In response to diverse stressors, the adult heart undergoes hypertrophic enlargement, that is, a compensatory process that attempts to maintain or augment pump function. Normal hypertrophy observed in trained athletes or following postnatal development is referred to as ‘physiological’ hypertrophy.1–4 In contrast, cardiac hypertrophy observed under conditions such as hypertension or myocardial infarction is referred to as ‘pathological’ hypertrophy since it is associated with cardiac dysfunction that ultimately transitions to heart failure.5,6 Pathological growth is characterized by re-expression of a foetal gene programme, including β-myosin heavy chain (β-MHC) and α-skeletal actin (α-skActin), as well as by up-regulation of the natriuretic peptides, atrial natriuretic peptide, and brain natriuretic peptide (BNP).7 Endocrine, paracrine, and autocrine regulatory factors directly influence cardiac hypertrophy and apoptosis through their actions on membrane receptors, including G-protein-coupled receptors, which transduce signals involving enzymes such as mitogen-activated protein kinases (MAPKs), protein kinase C (PKC), calcineurin-NFAT, and Akt.6,8–10
Numerous studies have implicated Akt in regulating cardiac growth,11–13 contractile function,11,14–16 and angiogenesis.17–19 Dysregulation of Akt activity has been associated with cancer, diabetes, and cardiovascular disease.8,20,21 There are three isoforms of Akt (Akt1, Akt2, and Akt3) encoded by different genes and expressed in a tissue-specific manner in mammalian cells. The Akt isoforms share similar structures, but their physiological functions in vivo are non-redundant as indicated by distinct phenotypes in isoform-specific gene-targeted mice.22–24 Recently, a novel protein phosphatase PHLPP [PH domain leucine-rich repeat (LRR) protein phosphatase] was discovered, which dephosphorylates Akt on its hydrophobic motif, contributing to termination of Akt signalling. The PHLPP family of phosphatases is comprised of three members, PHLPP1α and PHLPP1β (splice variants of the same gene) and PHLPP2. The PHLPP isoforms have an identical domain structure in which a PH domain is followed by a region of LRRs, a PP2C phosphatase domain, and a C-terminal PDZ ligand domain.25,26 In addition, PHLPP1β and PHLPP2 contain a Ras-association domain (RA domain) preceding the PH domain.26,27 Cellular localization studies reveal that PHLPP1 and PHLPP2 are present throughout the cell25,26 and both are expressed in cardiomyocytes.28 PHLPP has also been found to dephosphorylate the hydrophobic motif of conventional and novel PKC isoforms in vitro, affecting PKC stability and thus the level of expression.29 However, studies in which PHLPP was removed in cardiomyocytes and the heart demonstrated that the expression of PKC was unaffected.28 Recently, other substrates with hydrophobic motifs have been identified as substrates for dephosphorylation by PHLPP. One such protein, S6K, is involved in protein translation and its activity is reduced by dephosphorylation by PHLPP.30 Also, Mst1, which is a pro-apoptotic kinase, is a direct target of PHLPP through dephosphorylation of its inhibitory site Thr387. However, since Akt down-regulates Mst1 by phosphorylating its inhibitory site to cause protection, PHLPP therefore can regulate Mst1 indirectly by limiting Akt activity.31 Overall, PHLPP limits second messenger signalling and opposes cell growth and survival.
Physiological hypertrophy is initiated by various stimuli including mechanical stretch, endocrine, and hormonal factors.2,3,32 Importantly, during physiological hypertrophy, there is no significant interstitial fibrosis or cardiac dysfunction nor is there up-regulation of foetal gene expression.3 Akt activation is required for physiological growth of the heart,12 and cardiac-specific overexpression of a constitutively active Akt mutant increases angiogenesis and cardiac mass and has been shown to be cardioprotective following pressure overload.33,34 However, prolonged expression of activated Akt, which is membrane targeted, has negative inotropic effects and leads to decompensation and heart failure.35 The location of constitutively activated Akt accounts for the development of pathophysiology since nuclear or SR targeted Akt has no deleterious effect on cell size in the heart and improves cardiac function and reduces apoptosis under pathological stress.36–38 The level of Akt activity in these transgenic model systems is supraphysiological thus sustained high levels of kinase activity and its location could account for the development of deleterious effects. The effects of more physiological spatiotemporal increases in Akt signalling on cardiovascular growth and function have not been directly examined. Global deletion of PHLPP1 elevates basal and transiently accentuates agonist-induced Akt activation in the heart.28 Using this genetic model, we demonstrate that a modest increase in Akt activity in the heart does not affect basal cardiomyocyte growth. In the face of stress induced by pressure overload, however, hypertrophy is diminished while in contrast the physiological hypertrophic response to swimming is accentuated. We suggest that removing the normal break on Akt activation imposed by the PHLPP1 phosphatase accentuates physiological Akt activation, which exerts salutary effects in protecting the heart against pathological stress.
2. Methods
2.1. Animals
All procedures were performed in accordance with NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at the University of California San Diego. Generation of global homozygous Black Swiss/SV129 PHLPP1 knock-out (KO) mice has been previously described.28,39 All experiments were performed on age-matched male wild-type (WT) and KO littermates.
2.2. Hypertrophy studies
Pathological hypertrophy was induced by transverse aortic constriction (TAC) in male 8- to 10-week-old WT and KO mice as previously described.40,41 Mice were anaesthetized under 2% isoflurane, intubated, and ventilated. Mice were euthanized by cervical dislocation and analysed following 1 day or 2 weeks TAC. For functional assessment following pressure overload, non-invasive echocardiography was performed to calculate fractional shortening and pressure gradients as described previously.40,41 Echocardiographic assessment of function following TAC was performed in a blinded fashion. Swimming for 20 days as a model of exercise-induced hypertrophy was also performed.41 Swimming mice were monitored for equal exertion. Upon completion of the study, mice were euthanized by cervical dislocation and cardiac phenotype analysed.
2.3. Immunoblotting
Hearts were obtained from WT and KO mice at baseline or subjected to TAC or exercise for various times. Hearts were removed directly and immediately frozen in liquid nitrogen till further processing. Adult mouse ventricular myocytes (AMVMs) were isolated from WT and KO mice as previously described.28 Tissue or cells were homogenized in lysis buffer28 and protein concentrations determined by Bradford assay. Total and phosphorylated proteins were determined by western blot as previously described.28 The PHLPP1 and PHLPP2 antibodies used were from Bethyl Laboratories (Montgomery, TX, USA). Total Akt, phosphorylated (phopsho)-Akt (Ser473 and Thr308), phospho-MDM2, phospho-GSK3α/β, phospho-ERK1/2, phospho-Mst1(Thr183), phospho-p70S6kinase, cleaved caspase 9, cleaved caspase 3, VEGF-a, actinin, and GAPDH antibodies were obtained from Cell Signaling Technology (Boston, MA, USA). Total pan PKC, PKCα, and PKCδ were from Santa Cruz Biotechnology (Dallas, TX, USA). Fold changes were determined by densitometry, normalized to accompanying GAPDH or actinin blots, and changes were expressed as relative values compared with WT samples.
2.4. Akt kinase assay
Extracts (150 μg) from WT and KO hearts were incubated with an antibody to Akt1 overnight. Akt catalytic activity was assessed using a non-radioactive Akt kinase assay kit following the manufacturer's protocol (Cell Signaling Technology).28 The change in catalytic activity of Akt was determined by densitometry of a phosphorylated GSK-3 substrate and normalized to total immunoprecipitated Akt. The activity of Akt was expressed as fold change relative to WT or control samples.
2.5. Immunohistochemistry
Hearts were collected at the indicated times as previously described,41 fixed in 3.5% paraformaldehyde, and embedded in paraffin. Serial 5 μm sections were cut and stained with haematoxylin and eosin (H&E) to look at gross morphology or Masson's trichrome for collagen deposition. For assessment of cross-sectional area, tissue was stained with TRITC-labelled wheat germ agglutinin (WGA; Sigma, St Louis, MO, USA) and nuclei with DAPI (Vector Laboratories, Burlingame, CA, USA). Co-staining for capillaries was performed with anti-CD31 (EMD Millipore, Billerica, MA, USA). For cellular death associated with TAC, apoptosis was detected using a TRITC deoxynucleotidyltransferase-mediated dUTP nick-end labelling (TUNEL) protocol (Roche, San Francisco, CA, USA) and co-stained with DAPI for nuclear visualization.39 In brief, an operator blinded to the treatment set counted cell death, capillary-to-myocyte ratio, and measured and quantified fibrosis and the cellular area across cells with central nuclei using the ImageJ software (version 1.40 g, NIH, Bethesda, MD, USA).
2.6. RNA isolation and qPCR
Total RNA was isolated using a micro-RNA isolation kit (Invitrogen, Carlsbad, CA, USA) from WT and KO hearts following TAC. cDNA was synthesized using the Verso cDNA synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol. Amplification of genes involved in cardiac hypertrophy and angiogenesis [atrial natriuretic factor (ANF), BNP, α-skActin, β-MHC, collagen 1a (Col1a), collagen 3a (Col3a), VEGF-a, angiopoietin 2 (Angp2), and GAPDH] was analysed using specific probe sets from Applied Biosystems (Invitrogen). The fold change was calculated as previously described42 and data were normalized to WT Sham.
2.7. Microfil injection and coronary cast moulding
Corrosion casting was used to evaluate the intact three-dimensional vascular network in the heart. The microfil injection was based on the previously described protocol.43 The microfilm resin was prepared according to the protocol in Batson's no. 17 plastic replica and corrosion kit (Polysciences, Inc., Warrington, PA, USA). The hearts were visualized on a dissecting microscope with a CCD camera (×2 magnifications).
2.8. Endothelial tube formation assay
Neonatal rat ventricular myocytes (NRVMs) were isolated from 1-day-old rat pups that were euthanized by CO2 followed by decapitation as previously described.28 Isolated cells (0.15 × 106/well) were plated on Matrigel pre-coated 24-well plates. Following 24 h of plating, cardiomyocytes were transfected with either scrambled control or a pre-designed rat PHLPP1 ON-TARGETplus siRNA as previously described.28,44 After overnight incubation, cells were washed and cultured for another 24 h in serum-free media. Bovine aortic endothelial cells (BAECs; Lonza, Walkersville, MD, USA) were plated on top of NRVMs (0.75 × 105/well) in the presence of 5% foetal bovine serum and 3 ng/mL of basic fibroblast growth factor (bFGF). Tube formation was visualized 6 h later. An operator blinded to the treatment counted six fields photographed at random on an Olympus light microscope (×10 magnification) and the number of tubes/field was calculated.
2.9. Statistical analysis
Researchers were blinded to the treatment group during analyses. Data are represented as mean ± SEM. Differences are considered statistically significant (P < 0.05) assessed using unpaired Student's t-test (for two groups), one-way (for multiple comparisons), or two-way ANOVAs (for multiple comparisons involving two variables) with post hoc Tukey analysis using the GraphPad Prism software (GraphPad, La Jolla, CA, USA).
3. Results
3.1. Deletion of PHLPP1 increases Akt activity, but does not affect cardiac size or contractile function
Heart extracts from WT, heterozygous, and PHLPP1 KO mice were analysed for PHLPP expression. PHLPP1 protein was decreased by 25% in heterozygous mice and undetectable in the hearts of null mice (Figure 1A). No compensatory change in PHLPP2 expression was evident (Figure 1A). Removal of PHLPP1 did not affect total Akt expression; however, phosphorylation of Akt at Ser473 was increased nearly two-fold in AMVMs from the PHLPP1 KO (Figure 1B) without a change in Thr308 phosphorylation as shown previously.28 Removal of PHLPP1 did not alter basal cardiomyocyte size. Additionally, analysis of 1-year-old PHLPP1 KO mice showed no significant difference in cardiac function (Figure 1C) and no overt change in overall heart weight (Figure 1D and see Supplementary material online, Table S1) compared with WT.
Figure 1.

Analysis of cardiac changes in PHLPP1 KO mice. (A) Heart extracts (25 µg) from WT, heterozygous PHLPP1 (HET), and PHLPP1 KO mice were analysed for PHLPP isoform expression and Akt phosphorylation (n = 3). (B) Isolation of AMVMs and quantitation of Ser473 phosphorylation on Akt from WT and KO mice. (The graph represents n = 4 independent isolations WT/KO; *P <0.05, ×32 magnification.) (C) Percent fractional shortening (FS) measurement by non-invasive echocardiography of WT and KO mice at 12 months of age (n = 9). (D) Gravimetric analysis of heart weight-to-tibia length (HW/TL) ratio of WT, HET, and KO mice at 12 months of age (n = 9). n.s.: non-significant.
3.2. PHLPP1 KO mice have an accentuated response to exercise-induced hypertrophy
Since Akt activation is widely recognized as a mediator of physiological growth of the heart, as occurs during exercise and in response to hormones such as IGF-1,12,32,45,46 we investigated the effect of PHLPP1 removal on physiological hypertrophy. WT and KO mice were subjected to forced swimming for 20 days and heart size assessed (Figure 2A). The ability of exercise to induce hypertrophy was found to be significantly accentuated (39 vs. 23% increase) in PHLPP KO vs. WT mice, respectively (Figure 2B). Histological assessment of cardiomyocyte size confirmed greater physiological hypertrophy in KO vs. WT mouse hearts following exercise (Figure 2C). PHLPP KO mice also showed increased phosphorylation of Akt as well as several downstream targets (i.e. MDM2, p70S6Kinase, and ERK) relative to WT mice after 3 days swim (Figure 2D, left panel), which were normalized at 20 days swim (Figure 2D, right panel).
Figure 2.
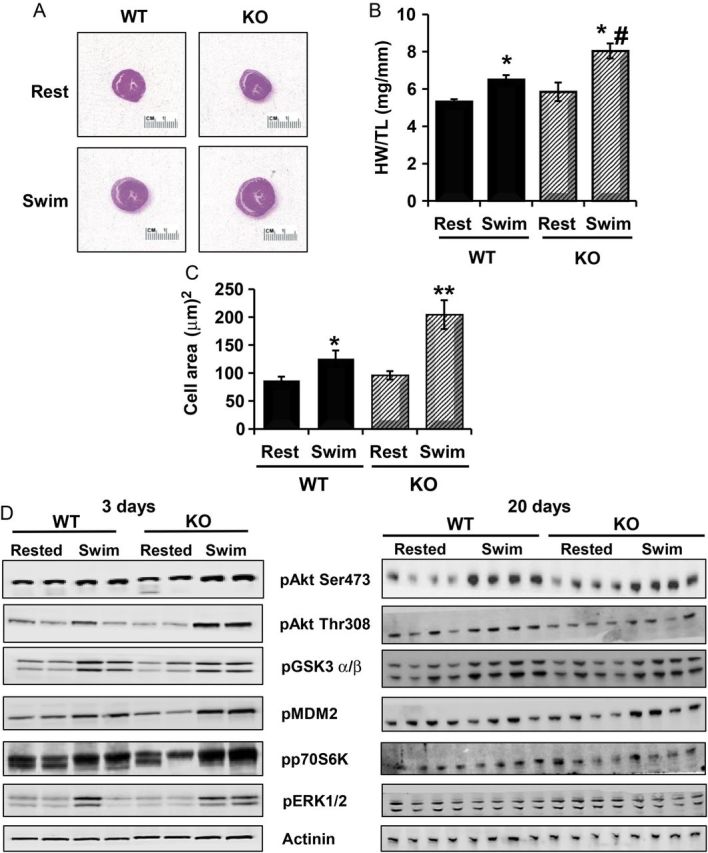
Removal of PHLPP1 increases the physiological response of the heart to exercise. WT and KO mice (12 weeks of age) were subjected to forced swimming for 20 days. (A) Representative H&E staining of WT and KO hearts at rest and following Swim. (B) Heart weight (HW) to tibia length (TL) of WT and KO mice at rest or following swim were analysed. (The graph represents n = 9 independent samples WT/KO; *P<0.05 compared with WT rest, #P<0.05 compared with WT swim.) (C) Measurement of myocyte cross-sectional area from ventricles of WT and KO mice at rest and following 20 days swim. (500 cells were counted, n = 4 WT/KO per group; *P<0.05 or **P<0.01 compared with WT rest.) (D) Protein analysis of Akt phosphorylation and downstream targets by western blotting in WT and KO heart extracts (15 µg) following 3 days (n = 2) and 20 days swim (n = 4). Actinin was blotted as a loading control.
3.3. Attenuation of pressure overload-induced hypertrophy in PHLPP1 gene-targeted mice
In contrast to the documented role of Akt in physiological hypertrophy, the role of Akt in the development of pathological hypertrophy is controversial. To determine how PHLPP1 removal and subsequent changes in Akt phosphorylation would affect the development of pathological hypertrophy, WT and KO mice were subjected to pressure overload by TAC. The pressure gradients across the aortic constriction were shown to be equivalent and no significant differences in contractile function assessed by echocardiography were observed for either group following 2 weeks TAC (see Supplementary material online, Table S2). PHLPP1 KO mice did, however, show a moderate but significant attenuation of hypertrophy, as demonstrated by a smaller increase in heart weight/body weight ratio than in WT mice (Figure 3A, 29 vs. 46% increase, respectively). Histological examination of the hearts also revealed a significantly reduced increase in cardiomyocyte cell size following TAC in PHLPP1 KO hearts compared with WT (Figure 3B). Finally, up-regulation of hypertrophic genes (α-skActin, β-MHC, ANF, and BNP) by TAC was reduced in the KO mouse heart (Figure 3C).
Figure 3.

Attenuation of pathological hypertrophy induced by pressure overload in PHLPP1 KO mice. WT (black) and KO (striped) mice (8 weeks old) were subjected to pressure overload by TAC for 2 weeks. (A) Representative H&E staining of WT and PHLPP1 KO hearts following TAC and graph of heart weight-to-body weight ratio. (The graph represents n = 15 independent samples WT/KO; *P<0.05 compared with WT TAC.) (B) Measurement of myocyte cross-sectional area from ventricles of WT (black) and KO mice (striped). Representative WGA staining for WT and KO hearts following TAC and the quantified graph represents cell area analysis. (500 cells were counted, n = 3 WT/KO per group; **P<0.01 compared with WT TAC.) (C) Isolation of RNA from WT (solid) and KO (striped) hearts and the relative mRNA fold changes in hypertrophic gene expression. (The graphs represent n = 8 independent samples; *P<0.05 compared with WT TAC.) ANF: atrial natriuretic factor; BNP: brain natriuretic peptide; skActin: α-skeletal muscle actin; β-MHC: beta myosin heavy chain.
3.4. Akt activation in response to pressure overload
As previously reported, PHLPP1 KO hearts have increased basal Akt Ser473 phosphorylation (Figure 4A and C) and Akt1 activity (Figure 4B).28 TAC significantly increased Akt Ser473 phosphorylation between 1 and 14 days TAC in PHLPP1 KO, but not in WT, mice (Figure 4A). Akt1 kinase activity also remained significantly greater in PHLPP1 KO vs. WT animals subjected to TAC (Figure 4B). All Akt targets examined (i.e. GSK3α/β, MDM2, p70S6Kinase, and ERK1/2) had enhanced phosphorylation in the PHLPP1 KO vs. WT hearts at 1 day TAC (Figure 4C, left panel), which were equalized by 14 days (Figure 4C, right panel).
Figure 4.

Akt activation and downstream targets following pressure overload in the PHLPP1 KO mice. WT and KO mice (12 weeks old) were subjected to TAC. (A) Quantification of Akt phosphorylation at Ser473 in WT and KO hearts following 1 and 14 days TAC. (The graph represents n = 5 hearts WT/KO; *P<0.05 and **P<0.01 compared with WT Sham and #P<0.05 compared with 1 day TAC.) (B) Fold change in Akt1 catalytic activity in WT and KO cardiac extracts following 1 and 14 days TAC. (The graphs represents n = 4 WT/KO, *P<0.05 compared with WT Sham.) (C) Phosphorylation of Akt and its downstream targets in heart extracts (15 μg) from WT and KO mice following 1 day (n = 2) and 14 days TAC (n = 3). Actinin was blotted as a loading control.
3.5. PHLPP1 KO mice have decreased fibrosis and cell death following TAC
Since removal of PHLPP1 decreased pathological hypertrophy and activation of foetal genes, we examined cellular fibrosis and cell survival in WT and KO hearts following 2 weeks TAC. Remarkably, PHLPP1 KO hearts showed little fibrosis (Figure 5A) as demonstrated by Mason's trichrome staining and changes in Col1a or Col3a gene expression (Figure 5B). There was also significantly less cell death following 2 weeks TAC in the PHLPP1 KO mice as assessed by TUNEL staining (Figure 5C), and levels of cleaved caspase 9 and caspase 3 were significantly reduced (Figure 5D). In addition, activation of Mst1, a pro-apoptotic protein, which is inhibited by Akt activity, was reduced in the KO hearts relative to WT mice following TAC (Figure 5D).
Figure 5.
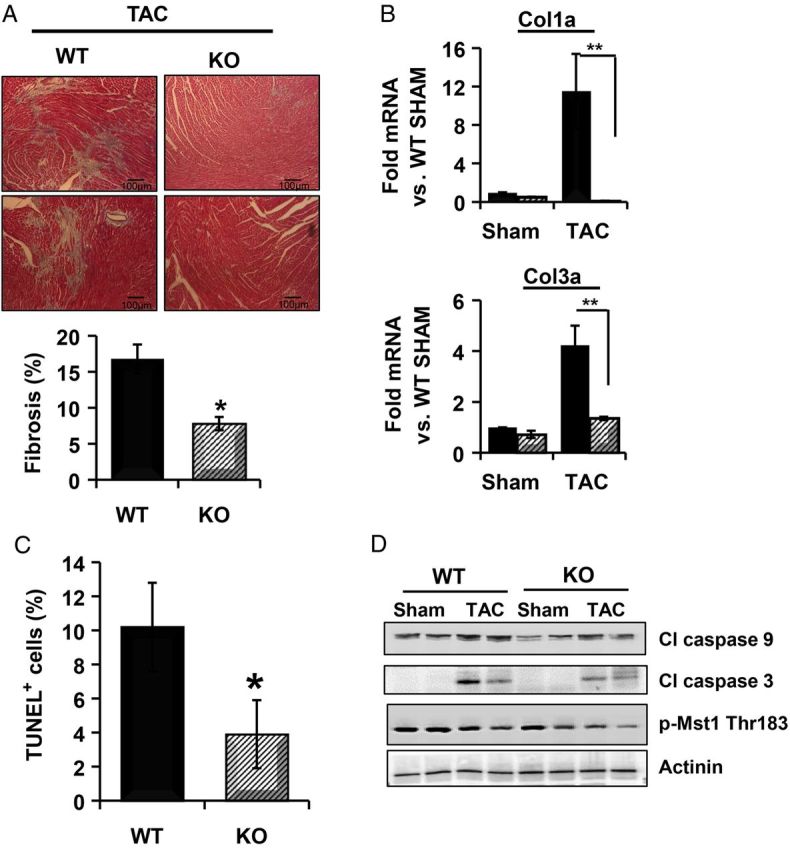
PHLPP1 KO mice have reduced fibrosis and cell death following TAC. (A) Representative Masson's trichrome-stained sections of WT and KO mice following 14 days TAC (×10 magnification). Percent fibrosis was calculated in WT and KO hearts from trichrome-stained sections using the Image J software. (The graph represents n = 5 hearts WT/KO; *P<0.05 compared with WT TAC.) (B) Isolation of RNA from WT (solid) and KO (striped) hearts subjected to 2 weeks TAC and the relative mRNA fold changes in collagen gene expression. (The graphs represent n = 8 independent samples; **P<0.01 compared with WT TAC.) Col1a: collagen 1a and Col3a: Col3a. (C) Cell death following 2 weeks TAC was determined in WT and KO hearts by TUNEL staining. The graph represents four hearts and six areas analysed (*P<0.05 compared with WT TAC). (D) Western blot analysis of cleaved caspase 3, 9, and Mst1 phosphorylation at Thr183 from WT and KO hearts (n = 2) following TAC. Actinin was blotted as a loading control.
3.6. PHLPP1 KO mice have increased capillary density
Since it has been demonstrated that increased Akt activity in the heart can increase angiogenesis33,47 and recent evidence suggests that angiogenesis can contribute to hypertrophy development,35,48 we explored the possibility that changes in angiogenesis might contribute to the altered cardiac growth response observed in the PHLPP1 KO mouse heart. To this end, cast moulding was used to look for possible changes in heart vessel structure. At baseline, the KO mice had an evident increase in capillary bed density compared with WT mice (Figure 6A). To determine the cause of the increase in capillary density, the expression of VEGFa, a protein that can stimulate angiogenesis, was examined. VEGFa protein was significantly elevated in PHLPP1 KO compared with WT mouse hearts at baseline (Figure 6B). We then examined the effect of TAC on angiogenesis in WT vs. KO mice as assessed by the ratio of capillaries to myocytes in the heart. KO mice had a significant increase in the ratio of capillary to myocyte at baseline compared with WT mice and following 2 weeks TAC displayed a significantly greater increase than that of their WT counterparts (Figure 6C). Furthermore, the expression of mRNA for Angp2 and VEGFa, markers of angiogenesis, were increased in KO, but not in WT, mice following 2 weeks TAC (Figure 6D).
Figure 6.

Increased angiogenesis in PHLPP1 KO mice. WT and KO mice (8 weeks of age) were analysed by (A) microfil polymer injection and corrosion casting of hearts for vasculature changes [posterior (a and c) and left ventricle (LV) view (b and d), ×2 magnification]. (B) Quantification and representative western blot of VEGFa levels at baseline in WT and PHLPP1 KO hearts. GAPDH was blotted for normalization (n = 4 hearts WT/KO; *P<0.05 compared with WT baseline). (C) Quantification of capillary density between WT (solid) and KO (striped) mice following 2 week TAC. (The graph represents n = 3 independent hearts WT/KO; *P<0.05 compared with WT Sham; #P<0.05 compared with WT TAC.) (D) Isolation of RNA from WT (solid) and KO (striped) hearts subjected to 2 weeks TAC. The relative mRNA fold changes in angiogenesis markers. (The graphs represent n = 8 independent samples; *P<0.05 and **P<0.01 compared with WT Sham.) Angp2: angiopoietin 2; VEGFa: vascular endothelial growth factor a.
To determine whether paracrine signalling from cardiomyocytes in PHLPP KO mice could affect capillary density, we carried out in vitro capillary tube formation experiments. Neonatal rat cardiomyocytes treated with scrambled or PHLPP1 siRNA were co-cultured with BAECs. siRNA-mediated knockdown of PHLPP1 in cardiomyocytes increased the number of tubes formed following serum stimulation compared with scrambled siRNA-treated cells (Figure 7A). Without BAECs, siRNA-mediated knockdown of PHLPP1 increased VEGFa mRNA expression compared with siRNA-scrambled control in the presence of serum (Figure 7B), suggesting that increased Akt in cardiomyocytes stimulates the production of angiogenic factors leading to increased angiogenesis.
Figure 7.
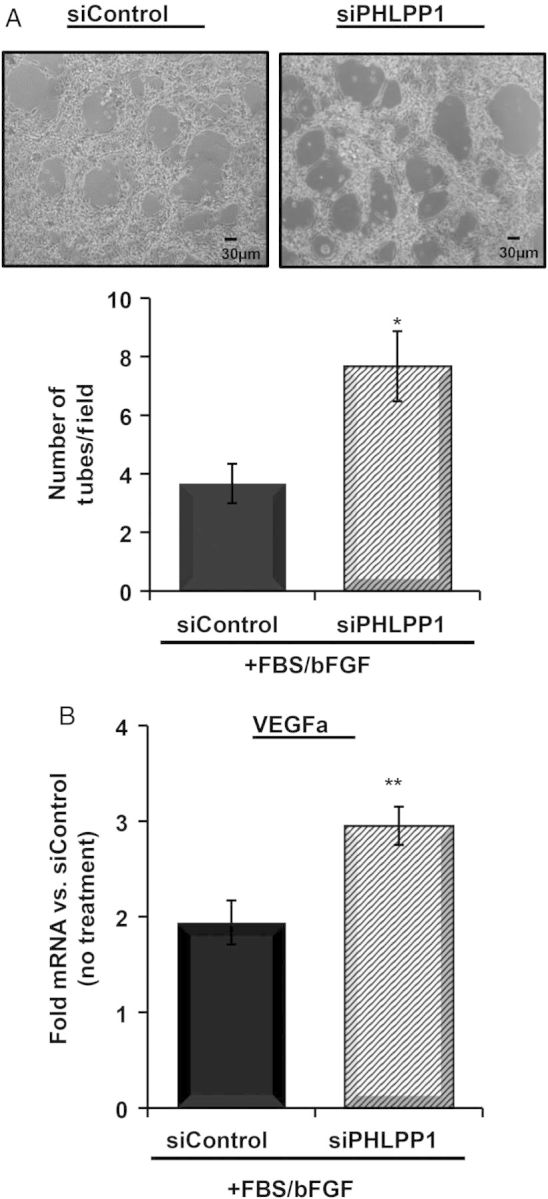
Loss of PHLPP1 in cardiomyocytes increases endothelial cell tube formation. NRVMs were transfected with scrambled control or PHLPP1 siRNA. Following 48 h, (A) BAECs were co-cultured with the NRVMs in the presence of serum (5%) and bFGF (3 ng/mL) for 6 h. Bright field images were taken at ×10 magnification. Quantification of tubes per field was determined. (The graph represents n = 3 independent experiments, six fields per experiment; *P<0.05 compared with siControl.) (B) Isolation of RNA from NRVMs with scrambled control or PHLPP1 siRNA with or without serum (5%) and bFGF (3 ng/mL) for 6 h (no BAEC). The relative mRNA fold changes in VEGFa were determined compared with siControl (no treatment). [The graph represents n = 6 independent experiments; **P<0.01 compared with siControl (treated).]
4. Discussion
Despite intensive research into the role of Akt activation in the heart, its importance in regulating pathophysiological hypertrophy remains controversial. Modulation of PHLPP, which can directly dephosphorylate and inactivate Akt,25,26,28 provides a mechanism for fine tuning this pathway and testing the physiological role of Akt in cardiac growth. We demonstrate that gene-targeted removal of PHLPP1 results in a very modest basal Akt activation (∼25% increase)28 without affecting cell size and accentuates the response to physiological stimulation of Akt as occurs with exercise training or IGF32 (Figures 1, 2, and 4B). Based on several in vivo findings and as discussed in Introduction, the intensity and duration of Akt activation is a critical determinant of its role in physiological and pathological hypertrophy.33–35,49–51 Hormones such as insulin and IGF-1, which are highly effective activators of the Akt signalling pathway,45,46 enhance physiological growth of the heart in both rodents and humans. Conversely, removal of Akt1 in the heart inhibits exercise-induced cardiac hypertrophy.12 Our finding that removal of PHLPP1 accentuates physiological hypertrophy induced by swimming (Figure 2) suggests that inhibition of PHLPP1 could be beneficial in augmenting physiological adaption of the heart following exercise.
Supraphysiological levels and localization of Akt in the heart lead to dysregulation of the signalling pathway and its targets, which ultimately culminates in pathological hypertrophy and failure.33–35,49,50 The Akt signalling pathway is not, however, typically considered as an intrinsic maladaptive regulator of pathological hypertrophy. Since the magnitude and duration of Akt activity is clearly relevant to its effect on cardiomyocyte growth following pressure overload, we reasoned that more subtle changes in Akt activation kinetics might actually have salutary effects on pathological hypertrophy development. In support of this possibility, the hypertrophic response to TAC was blunted in PHLPP1 KO compared with WT mice (Figure 3A). Removal of PHLPP1 significantly blunted the increase in cardiomyocyte size (Figure 3B), attenuated the re-expression of hypertrophic markers (Figure 3C), and increased Akt Ser473 phosphorylation. Akt enzyme activity assays using Akt1 and Akt2 antibodies demonstrated that Akt1 activity was increased in PHLPP 1 KO mice at baseline and following pressure overload (Figure 4A and B), whereas Akt 2 was not (data not shown).
The finding that removal of PHLPP1 accentuates Akt activation and decreases cell death after TAC is consistent with the well-documented anti-apoptotic effect of Akt on cardiomyocytes both in vitro28,52 and in vivo.16,22,33,53 PHLPP1 KO mice challenged with TAC have increased phosphorylation of Akt downstream targets that are important for cell survival, and maintenance of cardiac function (Figure 4C) as well as decreased TUNEL-positive cells (Figure 5C) and phosphorylation of pro-apoptotic targets (Figure 5D). In contrast, WT and PHLPP1 KO mice showed no significant differences in PKC isoform expression (see Supplementary material online, Figure S1) or Raf-1 phosphorylation (data not shown), both of which have been shown to be direct PHLPP targets in other systems.54,55 There was also no difference in dephosphorylated NFAT between WT and KO mice following 1 day TAC (data not shown), suggesting no underlying defect in the calcineurin signalling pathway10 in PHLPP1 KO mice.
Blunted hypertrophic growth in the presence of increased Akt activity would not, however, be expected to result from increased cardiomyocyte survival. We determined that removal of PHLPP1 increased the baseline capillary density in the heart compared with WT mice (Figure 6A and C). The observation that the KO heart had increased VEGFa mRNA (Figure 6D) and protein expression (Figure 6B) at baseline is of particular interest as Akt activation within cardiomyocytes has been found to enhance expression of VEGFa,56,57 which has in turn been demonstrated to increase coronary angiogenesis.35,58 Thus, an increase in Akt activity in cardiomyocytes from PHLPP1 KO is likely to contribute to the increased capillary density found in the heart. Since removal of PHLPP1 is global in our mouse, our finding that knockdown of PHLPP1 specifically in cardiomyocytes in vitro increases their ability to cause endothelial cell tube formation in a myocyte/endothelial cell co-culture system supports our notion (Figure 7A and B).
Under hypertrophic stress, the heart increases capillary density to meet the demand of oxygen required for growth. WT mice increase their capillary density in response to TAC-induced pressure overload; however, the KO further increases the capillary-to-myocyte ratio compared with their WT counterparts (Figure 6C) and angiogenic marker expression (Figure 6D). Through co-culture experiments, we have established the possibility that increases in capillary density may in part be due to paracrine signalling from cardiomyocytes lacking PHLPP1 to surrounding endothelial cells within the heart (Figure 7A and B). We hypothesize that the increased capillary density provides more oxygen to meet cellular demands during hypertrophic growth, which would ultimately allow for reduced overall cell enlargement. In support of our findings, it has been recently demonstrated that inhibition of apoptosis during pressure overload preserves cardiac function through decreased fibrosis and is associated with increased overall angiogenesis and decreased hypertrophy.48 Our previous finding that ex vivo perfused hearts from PHLPP1 KO mice were protected from oxidative damage could be explained by increased capillary density as well as by the protection of cardiomyocytes by increased Akt activity.28
Our work is the first to show that removal of the serine/threonine phosphatase PHLPP1, which increases Akt activity in the heart to physiological levels without affecting cell size, increases basal and TAC-induced angiogenesis and ultimately blunts the overall development of pathological hypertrophy in response to pressure overload-induced stress. These findings are consistent with the data of Shiojima et al.,16 demonstrating that activation of inducible expression of Akt following long-term TAC improves cardiac function, and the suggestion that this occurs through inhibition of cell death. Whether removal of PHLPP1, which activates Akt and inhibits cell death, would also rescue the development of heart failure following long-term TAC or myocardial infarction remains to be determined. Future studies using cardiac-specific removal of PHLPP1 will be necessary to determine whether increasing Akt activity in cardiomyocytes is sufficient to affect angiogenesis or whether endothelial or other cells in which PHLPP1 is deleted are also involved. In either case, our work raises the possibility that PHLPP1 inhibition would have therapeutic potential for protecting the heart against stress.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by grants from the National Institute of Health (HL114949 to N.H.P., HL087023 to A.B.G., and HL085577 to N.H.P. and A.B.G.) and also from the Tobacco-Related Disease Research Program of the University of California 20KT-0048 (N.H.P. and A.E.T.).
Acknowledgements
The authors thank Joan Heller Brown for her initial support and insightful review of the manuscript and thank Shigeki Miyamoto for helpful discussion and Katherine Huang and Jeffery M. Smith for technical assistance.
Conflict of interest: none declared.
References
- 1.Kemi OJ, Ceci M, Wisloff U, Grimaldi S, Gallo P, Smith GL, Condorelli G, Ellingsen O. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J Cell Physiol. 2008;214:316–321. doi: 10.1002/jcp.21197. [DOI] [PubMed] [Google Scholar]
- 2.Catalucci D, Latronico MV, Ellingsen O, Condorelli G. Physiological myocardial hypertrophy: how and why? Front Biosci. 2008;13:312–324. doi: 10.2741/2681. [DOI] [PubMed] [Google Scholar]
- 3.Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14:38–48. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dorn GW., II The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–970. doi: 10.1161/HYPERTENSIONAHA.106.079426. [DOI] [PubMed] [Google Scholar]
- 5.Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122:2727–2735. doi: 10.1161/CIRCULATIONAHA.110.942268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–571. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- 8.Sussman MA, Volkers M, Fischer K, Bailey B, Cottage CT, Din S, Gude N, Avitabile D, Alvarez R, Sundararaman B, Quijada P, Mason M, Konstandin MH, Malhowski A, Cheng Z, Khan M, McGregor M. Myocardial AKT: the omnipresent nexus. Physiol Rev. 2011;91:1023–1070. doi: 10.1152/physrev.00024.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorn GW, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 11.Condorelli G, Drusco A, Stassi G, Bellacosa A, Roncarati R, Iaccarino G, Russo MA, Gu Y, Dalton N, Chung C, Latronico MV, Napoli C, Sadoshima J, Croce CM, Ross J., Jr Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci USA. 2002;99:12333–12338. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ. Akt1 is required for physiological cardiac growth. Circulation. 2006;113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 13.Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catalucci D, Latronico MV, Ceci M, Rusconi F, Young HS, Gallo P, Santonastasi M, Bellacosa A, Brown JH, Condorelli G. Akt increases sarcoplasmic reticulum Ca2+ cycling by direct phosphorylation of phospholamban at Thr17. J Biol Chem. 2009;284:28180–28187. doi: 10.1074/jbc.M109.036566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Latronico MVG, Costinean S, Lavitrano ML, Peschle C, Condorelli G. Regulation of cell size and contractile function by AKT in cardiomyocytes. Ann N Y Acad Sci. 2004;1015:250–260. doi: 10.1196/annals.1302.021. [DOI] [PubMed] [Google Scholar]
- 16.Shiojima I, Schiekofer S, Schneider JG, Belisle K, Sato K, Andrassy M, Galasso G, Walsh K. Short-term Akt activation in cardiac muscle cells improves contractile function in failing hearts. Am J Path. 2012;181:1969–1976. doi: 10.1016/j.ajpath.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schiekofer S, Belisle K, Galasso G, Schneider JG, Boehm BO, Burster T, Schmitz G, Walsh K. Angiogenic-regulatory network revealed by molecular profiling heart tissue following Akt1 induction in endothelial cells. Angiogenesis. 2008;11:289–299. doi: 10.1007/s10456-008-9112-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitamura T, Asai N, Enomoto A, Maeda K, Kato T, Ishida M, Jiang P, Watanabe T, Usukura J, Kondo T, Costantini F, Murohara T, Takahashi M. Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol. 2008;10:329–337. doi: 10.1038/ncb1695. [DOI] [PubMed] [Google Scholar]
- 19.Karar J, Maity A. PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci. 2011;4:51. doi: 10.3389/fnmol.2011.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newton AC, Trotman LC. Turning Off AKT: PHLPP as a drug target. Annu Rev Pharmacol Toxicol. 2014;54:537–558. doi: 10.1146/annurev-pharmtox-011112-140338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mullonkal CJ, Toledo-Pereyra LH. Akt in ischemia and reperfusion. J Invest Surg. 2007;20:195–203. doi: 10.1080/08941930701366471. [DOI] [PubMed] [Google Scholar]
- 22.Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, Kadowaki T, Hay N. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, Sundararajan D, Chen WS, Crawford SE, Coleman KG, Hay N. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 26.Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25:917–931. doi: 10.1016/j.molcel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu K, Okada M, Nagai K, Fukada Y. Suprachiasmatic nucleus circadian oscillatory protein, a novel binding partner of K-Ras in the membrane rafts, negatively regulates MAPK pathway. J Biol Chem. 2003;278:14920–14925. doi: 10.1074/jbc.M213214200. [DOI] [PubMed] [Google Scholar]
- 28.Miyamoto S, Purcell NH, Smith JM, Gao T, Whittaker R, Huang K, Castillo R, Glembotski CC, Sussman MA, Newton AC, Brown JH. PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ Res. 2010;107:476–484. doi: 10.1161/CIRCRESAHA.109.215020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao T, Brognard J, Newton AC. The phosphatase PHLPP controls the cellular levels of protein kinase C. J Biol Chem. 2008;283:6300–6311. doi: 10.1074/jbc.M707319200. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Stevens PD, Li X, Schmidt MD, Gao T. PHLPP-mediated dephosphorylation of S6K1 inhibits protein translation and cell growth. Mol Cell Biol. 2011;31:4917–4927. doi: 10.1128/MCB.05799-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiao M, Wang Y, Xu X, Lu J, Dong Y, Tao W, Stein J, Stein GS, Iglehart JD, Shi Q, Pardee AB. Mst1 is an interacting protein that mediates PHLPPs’ induced apoptosis. Mol Cell. 2010;38:512–523. doi: 10.1016/j.molcel.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 32.McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, Schinke M, Kong S, Sherwood MC, Brown J, Riggi L, Kang PM, Izumo S. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J Biol Chem. 2004;279:4782–4793. doi: 10.1074/jbc.M310405200. [DOI] [PubMed] [Google Scholar]
- 33.Ceci M, Gallo P, Santonastasi M, Grimaldi S, Latronico MV, Pitisci A, Missol-Kolka E, Scimia MC, Catalucci D, Hilfiker-Kleiner D, Condorelli G. Cardiac-specific overexpression of E40K active Akt prevents pressure overload-induced heart failure in mice by increasing angiogenesis and reducing apoptosis. Cell Death Differ. 2007;14:1060–1062. doi: 10.1038/sj.cdd.4402095. [DOI] [PubMed] [Google Scholar]
- 34.Kim YK, Kim SJ, Yatani A, Huang Y, Castelli G, Vatner DE, Liu J, Zhang Q, Diaz G, Zieba R, Thaisz J, Drusco A, Croce C, Sadoshima J, Condorelli G, Vatner SF. Mechanism of enhanced cardiac function in mice with hypertrophy induced by overexpressed Akt. J Biol Chem. 2003;278:47622–47628. doi: 10.1074/jbc.M305909200. [DOI] [PubMed] [Google Scholar]
- 35.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsujita Y, Muraski J, Shiraishi I, Kato T, Kajstura J, Anversa P, Sussman MA. Nuclear targeting of Akt antagonizes aspects of cardiomyocyte hypertrophy. Proc Natl Acad Sci USA. 2006;103:11946–11951. doi: 10.1073/pnas.0510138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rota M, Boni A, Urbanek K, Padin-Iruegas ME, Kajstura TJ, Fiore G, Kubo H, Sonnenblick EH, Musso E, Houser SR, Leri A, Sussman MA, Anversa P. Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ Res. 2005;97:1332–1341. doi: 10.1161/01.RES.0000196568.11624.ae. [DOI] [PubMed] [Google Scholar]
- 38.Catalucci D, Zhang DH, DeSantiago J, Aimond F, Barbara G, Chemin J, Bonci D, Picht E, Rusconi F, Dalton ND, Peterson KL, Richard S, Bers DM, Brown JH, Condorelli G. Akt regulates L-type Ca2+ channel activity by modulating Cavalpha1 protein stability. J Cell Biol. 2009;184:923–933. doi: 10.1083/jcb.200805063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen B, Van Winkle JA, Lyden PD, Brown JH, Purcell NH. PHLPP1 gene deletion protects the brain from ischemic injury. J Cereb Blood Flow Metab. 2013;33:196–204. doi: 10.1038/jcbfm.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maillet M, Purcell NH, Sargent MA, York AJ, Bueno OF, Molkentin JD. DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J Biol Chem. 2008;283:31246–31255. doi: 10.1074/jbc.M806085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purcell NH, Wilkins BJ, York A, Saba-El-Leil MK, Meloche S, Robbins J, Molkentin JD. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc Natl Acad Sci USA. 2007;104:14074–14079. doi: 10.1073/pnas.0610906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dusaban SS, Purcell NH, Rockenstein E, Masliah E, Cho MK, Smrcka AV, Brown JH. Phospholipase C{varepsilon} links G protein-coupled receptor activation to inflammatory astrocytic responses. Proc Natl Acad Sci USA. 2013;110:3609–3614. doi: 10.1073/pnas.1217355110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang C, Zhang X, Ramil JM, Rikka S, Kim L, Lee Y, Gude NA, Thistlethwaite PA, Sussman MA, Gottlieb RA, Gustafsson AB. Juvenile exposure to anthracyclines impairs cardiac progenitor cell function and vascularization resulting in greater usceptibility to stress-induced myocardial injury in adult mice. Circulation. 2010;121:675–683. doi: 10.1161/CIRCULATIONAHA.109.902221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen M, Pratt CP, Zeeman ME, Schultz N, Taylor BS, O'Neill A, Castillo-Martin M, Nowak DG, Naguib A, Grace DM, Murn J, Navin N, Atwal GS, Sander C, Gerald WL, Cordon-Cardo C, Newton AC, Carver BS, Trotman LC. Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell. 2011;20:173–186. doi: 10.1016/j.ccr.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, Wayment BE, Litwin SE, Holzenberger M, LeRoith D, Abel ED. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endo. 2008;22:2531–2543. doi: 10.1210/me.2008-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–3365. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- 48.Park M, Vatner SF, Yan L, Gao S, Yoon S, Lee GJ, Xie LH, Kitsis RN, Vatner DE. Novel mechanisms for caspase inhibition protecting cardiac function with chronic pressure overload. Basic Res Cardiol. 2013;108:324. doi: 10.1007/s00395-012-0324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cittadini A, Monti MG, Iaccarino G, Di Rella F, Tsichlis PN, Di Gianni A, Stromer H, Sorriento D, Peschle C, Trimarco B, Sacca L, Condorelli G. Adenoviral gene transfer of Akt enhances myocardial contractility and intracellular calcium handling. Gene Ther. 2006;13:8–19. doi: 10.1038/sj.gt.3302589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hua Y, Zhang Y, Ceylan-Isik AF, Wold LE, Nunn JM, Ren J. Chronic Akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res Cardiol. 2011;106:1173–1191. doi: 10.1007/s00395-011-0222-8. [DOI] [PubMed] [Google Scholar]
- 51.Cook SA, Matsui T, Li L, Rosenzweig A. Transcriptional effects of chronic Akt activation in the heart. J Biol Chem. 2002;277:22528–22533. doi: 10.1074/jbc.M201462200. [DOI] [PubMed] [Google Scholar]
- 52.Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008;415:333–344. doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- 53.Yamashita K, Kajstura J, Discher DJ, Wasserlauf BJ, Bishopric NH, Anversa P, Webster KA. Reperfusion-activated Akt kinase prevents apoptosis in transgenic mouse hearts overexpressing insulin-like growth factor-1. Circ Res. 2001;88:609–614. doi: 10.1161/01.res.88.6.609. [DOI] [PubMed] [Google Scholar]
- 54.Jackson TC, Verrier JD, Semple-Rowland S, Kumar A, Foster TC. PHLPP1 splice variants differentially regulate AKT and PKCalpha signaling in hippocampal neurons: characterization of PHLPP proteins in the adult hippocampus. J Neurochem. 2010;115:941–955. doi: 10.1111/j.1471-4159.2010.06984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X, Stevens PD, Liu J, Yang H, Wang W, Wang C, Zeng Z, Schmidt MD, Yang M, Lee EY, Gao T. PHLPP is a negative regulator of RAF1, which reduces colorectal cancer cell motility and prevents tumor progression in mice. Gastroenterology. 2014;146:1301–1312. doi: 10.1053/j.gastro.2014.02.003. e1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, Walsh K, Sessa WC. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J Clin Invest. 2005;115:2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giordano FJ, Gerber HP, Williams SP, VanBruggen N, Bunting S, Ruiz-Lozano P, Gu Y, Nath AK, Huang Y, Hickey R, Dalton N, Peterson KL, Ross J, Jr, Chien KR, Ferrara N. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci USA. 2001;98:5780–5785. doi: 10.1073/pnas.091415198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47:887–893. doi: 10.1161/01.HYP.0000215207.54689.31. [DOI] [PMC free article] [PubMed] [Google Scholar]