

Summary
Lower dietary vitamin D, or vitamin D receptor inactivation, compromises intestinal Lgr5+ stem cell functions and ability to initiate tumors. Mouse experiments routinely exceed vitamin D exposure levels in human populations, probably overestimating contribution of Lgr5+ cells to human intestinal homeostasis and tumor development.
Abstract
Lgr5+ intestinal crypt base columnar cells function as stem cells whose progeny populate the villi, and Lgr5+ cells in which Apc is inactivated can give rise to tumors. Surprisingly, these Lgr5+ stem cell properties were abrogated by the lower dietary vitamin D and calcium in a semi-purified diet that promotes both genetically initiated and sporadic intestinal tumors. Inactivation of the vitamin D receptor in Lgr5+ cells established that compromise of Lgr5 stem cell function was a rapid, cell autonomous effect of signaling through the vitamin D receptor. The loss of Lgr5 stem cell function was associated with presence of Ki67 negative Lgr5+ cells at the crypt base. Therefore, vitamin D, a common nutrient and inducer of intestinal cell maturation, is an environmental factor that is a determinant of Lgr5+ stem cell functions in vivo. Since diets used in reports that establish and dissect mouse Lgr5+ stem cell activity likely provided vitamin D levels well above the range documented for human populations, the contribution of Lgr5+ cells to intestinal homeostasis and tumor formation in humans may be significantly more limited, and variable in the population, then suggested by published rodent studies.
Introduction
Lgr5+ crypt base columnar (CBC) stem cells of the mouse intestine can give rise to all cell lineages in the villi, and to tumors upon inactivation of the Apc gene in these cells (1–3). Other intestinal stem cell populations such as Bmi1+, Lrig1+ and Dclk1+ marked cells share these properties, but Lgr5+ stem cells regularly divide, while these other populations are relatively quiescent except under stress conditions (4–6). Therefore, under standard conditions, Lgr5+ cells are the principal cells in the mouse that maintain mucosal homeostasis and that are targeted by mutation to give rise to tumors (1,2). In contrast, the more quiescent stem cells have been considered reserve populations that can be recruited back into the cell cycle by radiation induced injury to the mucosa, or chemically induced damage and inflammation to directly give rise to intestinal epithelial lineages or to repopulate the Lgr5+ cell compartment (4,7,8). Here we report a major effect of nutritional factors, and specifically vitamin D3 and its signaling through the vitamin D receptor (VDR), in determining stem cell associated properties and the ability to give rise to tumors of Lgr5+ cells in the mucosa.
NWD1, a rodent diet based on control semi-purified AIN76A diet (American Institute of Nutrition 76A), was formulated on the principal of nutrient density to adjust mouse consumption of key nutrients that are dietary risk factors for colon cancer to levels consumed by significant segments of the population with a high incidence of the disease (9,10). NWD1 is higher in fat than AIN76A, and lower in vitamin D3, calcium, fiber, folate and methionine, factors all associated with elevated risk for colon cancer (Table I). Together, these nutritional factors establish a highly protumorigenic state in the intestinal and colonic mucosa. This protumorigenic state is demonstrated by the acceleration and amplification of tumor phenotype in mice that inherit a mutant Apc allele as well as in other mouse genetic models of intestinal tumorigenesis investigated, regardless of etiology or aggressiveness (11–14). Further, as reported by three groups, the NWD1 has the unique property of causing sporadic colon and small intestinal tumors when fed to wild-type C57BL6 mice for 1–2 years with a lag, incidence, multiplicity and ratio of adenomas to carcinomas similar to that of sporadic human colon cancer; i.e. after two-thirds of their lifespan, 20% of the mice will develop one to two tumors, of which 10% are carcinoma (9,15,16). Such sporadic tumors represent the vast majority (~80%) of colon tumors in the human population. These sporadic tumors arise only after five to six decades of life, have no clear genetic risk factors, though many poorly understood loci may contribute to their probability of development, and their incidence is predominantly determined by long-term dietary patterns (17,18). Although there are data that individual nutrients that are altered in the NWD1 can amplify tumorigenesis initiated by genetic factors or carcinogens (e.g. higher fat, lower vitamin D), changing the consumption level of any factor by itself in wild-type mice does not generate tumors upon long-term feeding. Thus, mice fed NWD1 are an important model for dissecting altered homeostasis in the intestinal mucosa, including effects on stem cell biology that may contribute to higher probability of developing sporadic tumors.
Table I.
Comparative levels of key nutrients in chow diet 5808 and semi-purified diets AIN76A, NWD1 and NWD2
| Mouse Diets | ||||
|---|---|---|---|---|
| chow | AIN-76A | Western diet | +Ca/vitD | |
| (5058)a | NWD1b | NWD2b | ||
| Fat (corn oil,%) | 9% | 5% | 20% | 20% |
| Calcium, mg/g | 8.1 | 5 | 0.5 | 7.0 |
| Vitamin D, IU/g | 3.3 | 1 | 0.11 | 2.3 |
| Phosphorous, mg/g | 6.1 | 4 | 3.6 | 3.6 |
| Folic acid, ug/g | 2.9 | 2 | 0.23 | 0.23 |
| DL methionine, % | 0.67 | 0.3 | — | — |
| Choline bitartrate, % | 0.22 | 0.2 | 0.12 | 0.12 |
| Fiber (cellulose), % | 2.2 | 5 | 2 | 2 |
| L-cysteine, % | — | — | 0.3 | 0.3 |
| kcal/g (approximate) | 3.7 | 3.6 | 4.5 | 4.5 |
aLabDiets (St Louis, MO)
bNewmark,N.L. et al., (1990) Colonic hyperplasia and hyperproliferation induced by a nutritional stress diet diet with four components of the western-style diet. JNCI, 82, 491-496; Newmark et al., (9).
Feeding the NWD1 increased relative utilization of fatty acids as an energy source compared to carbohydrates and induced glucose intolerance (19), and was accompanied by extensive changes in expression in cells of the mucosa of genes involved in energy metabolism (15). However, the NWD1 does not cause obesity; NWD1 fed mice appear grossly normal for up to 2 years, and the intestinal and colonic mucosa appear normal until one to two sporadic tumors arise in about 20% of the mice. Although NWD1 lowers mouse serum 25(OH)D levels, serum calcium remains normal. Further, there is no loss of bone mineral density nor significant change in parathyroid hormone levels, demonstrating the mice do not exhibit common effects of vitamin D or calcium deficiency (19). There is, however, altered cell maturation in the mucosa of NWD1 fed mice long before tumors develop. This includes: elevation and expansion of Wnt signaling throughout small intestinal villi and colonic crypts; altered gene expression profiles of epithelial cells in crypts and villi of the large and small intestine; increased absorptive and decreased secretory cell marker expression; and a prominent increase in ectopic expression of Paneth cell markers in intestinal villi and colonic crypt cells (15,20,21). These changes, as well as the protumorigenic effects of NWD1, are prevented by elevating vitamin D3 and calcium in the diet (i.e. NWD2 = NWD1, but with higher vitamin D3 and calcium, Table I).
To investigate further the perturbation in cell maturation caused by these diets, we determined effects on Lgr5+ stem cells caused by feeding the control AIN76A, NWD1 and NWD2 diets, and the effect of conditional inactivation of the VDR receptor specifically in Lgr5+ cells. These data demonstrate that canonical stem cell functions of Lgr5+ intestinal CBC cells in lineage tracing and as tumor initiating cells are dependent on high dietary vitamin D levels and intact VDR signaling. Thus, the biology of intestinal Lgr5+ stem cells and their role in tumor development cannot be separated from the diet fed to experimental mice. The data suggest that for significant segments of the population, and particularly for individuals with lower vitamin D levels who are at higher risk for colon tumor development, Lgr5+ stem cells may not play the same major role in intestinal homeostasis and tumor development reported for mice maintained under standard dietary conditions in which they are exposed to levels of vitamin D far higher than the range documented for human populations.
Materials and methods
Mice: C57BL6/J wild-type mice, C57BL6/J.129P2-Lgr5 tm1(cre/ERT2)Cle/J mice (3) (Lgr5GFP+), C57BL/6J-ApcMin/J mice (Apc Min/+) (22) and C57BL6;129S6-Gt(Rosa)26Sor tm14(CAG-tdTomato)Hze mice (Rosa26 RFP) were from Jackson Laboratories (Bar Harbor, Maine). Villin-cre mice were from S. Robine (23). VDR flox/flox mice were from G. Carmeliet (24). Apc 580S mice (Apc flox/+) were reported (25). Mice were maintained and bred in the Barrier Facility of the Albert Einstein College of Medicine. All experiments and procedures were approved by the Einstein IACUC. For breeding and strain maintenance, mice were fed 5058 diet (LabDiet, St Louis, MO). Mice were weaned to purified diets, as specified, for all experiments. The formulation and composition of the AIN76A, NWD1 and NWD2 diets were as described in detail (9) (Research Diets, New Brunswick, NJ). Dietary levels of key nutrients are compared in Table I.
In summary, the western diets are based on AIN76A control diet (American Institute of Nutrition76A). NWD1 was adjusted to provide higher fat, and lower vitamin D3, calcium donors to the single carbon pool and fiber that produces mouse consumption levels similar to those common in segments of western societies with high incidence of colorectal cancer. NWD2 was identical to NWD1, but was repleted with higher vitamin D3 and calcium. Mice were provided food and water ad libitum. Tamoxifen was injected intraperitoneally (1mg TAM in 100μl sterile corn oil).
Histopathology.
Upon killing, intestines were rapidly dissected, separated into duodenum, jejunum, ileum and colon, and each region prepared as a swiss-roll fixed in formalin and paraffin embedded or frozen in OCT. Histopathology was evaluated on formalin and paraffin embedded sections stained with hematoxylin—eosin. Frozen sections were DAPI stained, analyzed for green, red and blue fluorescence and images overlayed using Cell Sense (Olympus). FACS analysis of Lgr5+ cells was based on their endogenous green fluorescence (BD FACS Aria, Becton-Dickinson).
Results
FACS analysis of single cell suspensions of isolated crypt cells from Lgr5 GFP+ mice fed NWD1 for 3 months showed a significantly decreased percentage of GFP positive crypt cells compared to mice fed control AIN76A, which was prevented by elevating dietary vitamin D3 and calcium (i.e. feeding NWD2) (Figure 1A, *P < 0.03). Lgr5 GFP+, Rosa26 RFP+ mice were then randomized to diets at weaning and received an injection of tamoxifen (TAM) to activate RFP expression in Lgr5+ cells and their progeny. In 3 month AIN76A fed mice, one day post-TAM, green (GFP+) and yellow (GFP+, RFP+) fluorescent cells were at the crypt bottom (Figure 1B, i). By 3 days post-TAM, Lgr5+ progeny had moved out of the crypts, populating the lower third of the villi (Figure 1B, ii), and by 5 days cells had reached the villus tip (Figure 1B, iii). This replicates lineage tracing of Lgr5+ cells that has been reported for mice in which diet was not specified, and was likely therefore a standard chow diet (3). In contrast, in 3 month NWD1 fed mice, at one day post TAM, there were fewer GFP+ cells at the bottom of the crypt (Figure 1B, iv) than in AIN76A fed mice, consistent with reduced number of Lgr-GFP+ cells (Figure 1A). At 3 days post-TAM (Figure 1B, v), RFP labeled progeny cells were still confined to the crypt. By 5 days, many fewer red-marked Lgr5+ progeny had moved into the villi of NWD1 fed mice (Figure 1B, vi) compared to mice fed AIN76A. This phenotype was also present in mice, fed the diets for 1 year (Figure 1C, compare x to xi). The restricted appearance of Lgr5+ stem cell progeny in the villi in NWD1 fed mice was not present in parallel cohorts of genetically identical mice fed NWD2 from weaning for either 3 months or 1 year (Figure 1B, vii–ix; Figure 1C, xii).
Fig. 1.

Dietary effects on Lgr5+ cells in the small intestine. (A) Lgr5 GFP+ mice, four per dietary group were fed AIN76A, NWD1 or NWD2 diets from weaning for 3 months. Single suspensions of epithelial cells were prepared from isolated intestinal crypts and percent of these cells exhibiting green fluorescence determined by FACS analysis (mean ± std; *P < 0.03, analysis of variance); (B) Lgr5 GFP+ , Rosa26 RFP+ mice, nine per dietary group were fed diets from weaning for 3 months, then given a single injection of tamoxifen and three mice in each group killed at 1, 3 or 5 days later; (C) Lgr5 GFP+ , Rosa26 RFP+ mice were fed diets from weaning for 1 year, then given a single injection of tamoxifen and killed 3 days later. In (B) and (C), frozen sections of the intestine were stained with DAPI.
NWD2 elevates vitamin D3 and calcium levels in NWD1, but leaves levels of all other components unchanged (Table 1). Feeding of NWD2 prevents all of the effects of the NWD1 on intestinal cell maturation in vivo, the increased tumor phenotype in genetic models and the eventual tumor development in wild-type mice (15,21). Further, although NWD1 lowers 25(OH)D serum levels, serum calcium levels are maintained with no significant change in serum parathyroid hormone and no loss of bone mineral density (19), suggesting it is the lower vitamin D3 level that is an important contributer to tumor development and pretumor alterations caused by feeding NWD1. The VDR, through which vitamin D signals, is expressed at similar levels in Lgr5+ cells regardless of diet (Figure 2A). Therefore, population of the villi by progeny of Lgr5+ cells was investigated in Lgr5 GFP+ , Rosa26 RFP+ , VDR flox/flox or +/+ mice fed AIN76A control diet for 3 months, in which TAM injection simultaneously activated RFP expression and inactivated the VDR flox alleles in Lgr5+ cells. At 3 days post-TAM, Lgr5+ progeny remained in the crypt compartment in mice in which the VDR was inactivated in Lgr5+ cells (Figure 2B, v) compared to Lgr5 GFP+ , Rosa26 RFP+ , VDR +/+ mice that received TAM in which the VDR gene was not inactivated (Figure 2B, ii). At 5 days post-TAM, fewer Lgr5+ cell progeny in which VDR had been inactivated had moved into the villi (Figure 2B, vi compared to Figure2B, iii). This recapitulates the effect of feeding NWD1, which are dependent on the levels of vitamin D and calcium in the diet. Thus, the effect on Lgr5+ stem cell function is a rapid, cell autonomous effect mediated by the vitamin D receptor.
Fig. 2.
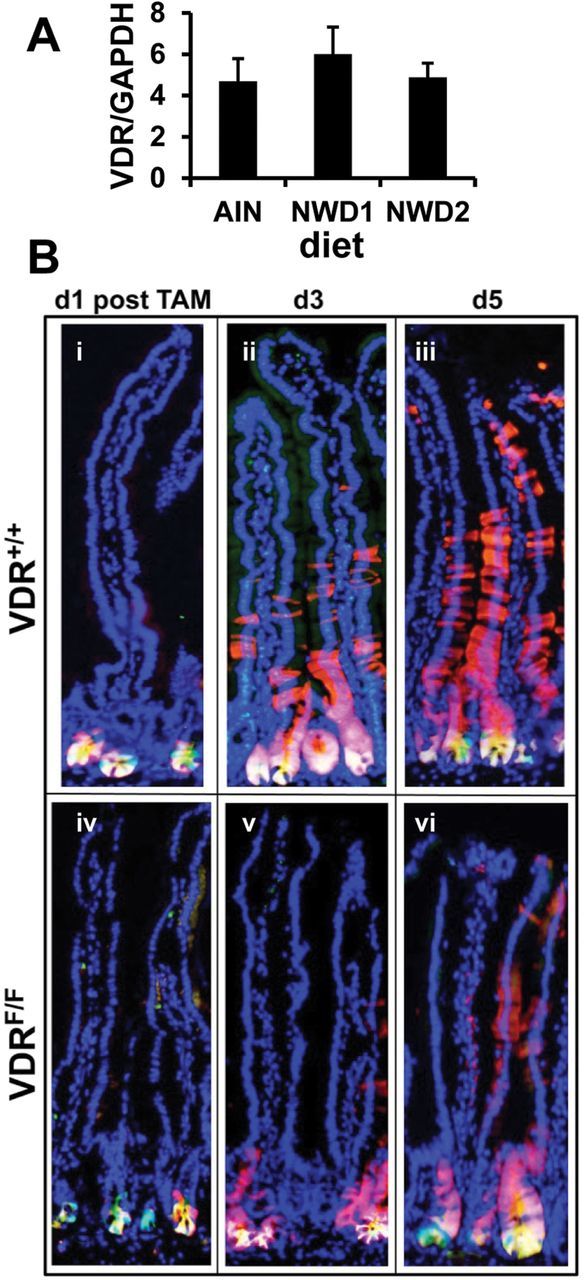
The VDR in intestinal stem cell function. (A) GFP marked cells were isolated by FACS from single cell suspensions of purified intestinal crypts of Lgr5 GFP+ mice fed AIN76A, NWD1 or NWD2 diet from weaning for 6 months. mRNA levels for VDR and for GAPDH were determined by quantitative real-time PCR for the Lgr5 GFP+ cells of three mice in each dietary group (mean ± std) with each sample assayed in quadruplicate; (B) Lgr5 GFP+ , Rosa26 RFP+ , VDR flox/flox or +/+ mice, nine mice per group were fed AIN76A diet for 3 months, given a single tamoxifen injection and three mice of each group killed 1, 3 or 5 days thereafter. Frozen sections of the intestine were stained with DAPI.
Colonic mucosa was also investigated in these mice. Supplementary Figure 1A, available at Carcinogenesis online, illustrates that in mice fed AIN76A control diet, at 3 days post-TAM injection, the progeny of Lgr5+ cells had populated approximately two-thirds of the crypt length. However, the cells remained confined to the bottom of the crypt in mice fed NWD1 (Supplementary Figure 1B, available at Carcinogenesis online), or in mice in which the VDR was inactivated (Supplementary Figure 1C, available at Carcinogenesis online). Therefore, the ability of Lgr5+ cells to give rise to cells that populate the colonic crypts was similarly dependent on dietary vitamin D3 intake and the VDR.
To determine whether altered maturation of Lgr5+ progeny altered the efficiency by which Lgr5+ cells can give rise to tumors, another important characteristic of stem cells, Lgr5 GFP+ , Apc flox/+ mice were randomized to diets at weaning. After 3 months, each mouse received a single TAM injection to inactivate the Apc flox allele in Lgr5 cells, each continued on its specific diet for 6 more months and was then killed. Neoplastic intestinal growths, ranging from dysplasias to adenomas, were detected in mice fed AIN76A or NWD2 (Figure 3A and C), but no abnormal histopathology was identified in mice fed NWD1 (Figure 3A). The difference in tumors in mice fed AIN76A compared to NWD1 was statistically significant (Figure 3B, *P < 0.03). Thus, compromise of Lgr5+ cell movement out of the crypts (Figure 1B and C) was associated with decreased development of intestinal neoplasia and tumors.
Fig. 3.

Effect of diet or vitamin D receptor inactivation on Apc initiated tumor development. (A–C) Lgr5 GFP , Apc flox/+ mice, five mice per dietary group were fed diets from weaning for 3 months, received a single injection of tamoxifen and then continued on their respective diets for 6 more months before killing and evaluation of histopathology. (A) tumor incidence and histopathology for each of the five mice in each dietary group; (B) mean tumor incidence per dietary group ± std (*P < 0.03, analysis of variance); (C) examples of histopathology of tumors from mice fed AIN76A or NWD2; (D) Apc Min/+, VDR flox/flox mice that were cre(-) or villin-cre+ were fed AIN76A or NWD1 for 4 months, killed, sections from formalin fixed swiss rolls of the small intestine were stained with hematoxylin/eosin and tumor multiplicity determined.
To determine whether inactivation of the VDR in intestinal epithelial cells altered intestinal tumorigenesis in general, Apc Min/+ , VDR flox/flox , villin-cre+ mice were bred in which the VDR was constitutively inactivated in most intestinal epithelial cells. When fed control AIN76A diet, there was no effect on small intestinal tumor multiplicity in these mice compared to Apc Min/+ mice with an intact VDR gene (i.e. cre(-), Figure 3D), confirming prior reports that VDR inactivation did not alter Apc Min/+ mouse tumor multiplicity (26,27). Tumor multiplicity increased by ~53% in villin-cre(-) mice fed NWD1 (no VDR inactivation) compared to the genetically same mice fed AIN76A (Figure 3D). This was not significant, but was similar to the previously reported 55% increase in tumor multiplicity stimulated by a related western-diet in Apc 1638N/+ mice (28). The important conclusion from these data was that although feeding the NWD1 abrogated intestinal tumor development when an Apc allele was mutated specifically in Lgr5+ cells, this was not the case in Apc Min/+ mice in which mutation of an Apc allele is present in all cells, and inactivation of the VDR in all epithelial cells of Apc Min/+ mice did not alter tumor frequency.
A characteristic of Lgr5+ CBC cells is that they proliferate regularly. Therefore, intestinal sections of the Lgr5 GFP+ , Rosa26 RFP mice that had been fed different diets from weaning for 3 months, and killed 3 days after injection with TAM, were stained for Ki67, a marker of cell cycling that is expressed, though differentially localized subcellularly, in all phases of the cell cycle. Figure 4 illustrates that in mice fed control AIN76A or NWD2, in which Lgr5+ cells function as canonical stem cells, all Lgr5+ cells and their immediate progeny stained positive for Ki67 (Figure 4, top and bottom rows). However, in mice fed NWD1, crypts were identified in which Lgr5+ cells were negative for Ki67 (Figure 4, middle row). Note that these negative cells were always Lgr5+ cells at the very bottom of the crypt.
Fig. 4.

Ki67 postive cells at the crypt base in mice fed different diets. Lgr5 GFP+ , Rosa26 RFP+ mice were from the experiment shown in Figure 1B: mice were fed AIN76A, NWD1 or NWD2 diets from weaning, given a single injection of tamoxifen at 3 months and then killed 3 days later, three mice per dietary group. Frozen sections of the intestine were stained with antibody to Ki67 (rabbit anti-mouse, Novus Biologicals, 1:200) detected with a secondary goat anti-rabbit Ab conjugated to Alexa Fluor 350 (InVitrogen, 1:200). The crypts were photographed separately for green, red and blue emission, representing the GFP and RFP expressed in Lgr5+ cells and their progeny, and the expression of Ki67, respectively. Images at the three different wavelengths for each crypt shown were overlayed; shown next to each of these crypts is the isolated blue fluorescence, indicating expression of Ki67.
Discussion
The data establish that the diet consumed by mice profoundly influences stem cell functions of Lgr5+ CBC cells. In the purified diets used, the levels of vitamin D3 and calcium are the determining nutrients of these effects. Serum 25(OH)D level is reduced significantly in mice fed the NWD1 compared to those fed AIN76A or NWD2 (19); the latter differing from NWD1 only by higher levels of vitamin D3 and calcium (Table I). However, serum calcium levels do not differ among mice feed these three diets (19). Moreover, inactivation of the vitamin D receptor in Lgr5+ cells rapidly recapitulates the loss of Lgr5+ stem cell function, strongly suggesting that vitamin D is the key nutrient in this pretumorigenic effect of the NWD1. Therefore, vitamin D exposure, similar to the stress of radiation and chemically induced injury with subsequent inflammation, is a common environmental variable that determines how Lgr5+ CBC cells function as stem cells in the intestinal mucosa. The effects of lower vitamin D level and inactivation of the VDR is consistent with an extensive literature that vitamin D is a potent regulator of growth and/or maturation of intestinal tumor cells in vitro and in vivo, possibly due to down-regulat ion of Wnt signaling (e.g. ref. 29 and ref. 30) or Notch signaling (31). However, since vitamin D regulates cellular calcium uptake (e.g. ref. 32), a contributing down-stream effect on intestinal intracellular calcium levels cannot be ruled out in determining whether Lgr5+ cell progeny populate the villi and can serve as tumor initiating cells.
The compromise of Lgr5+ stem cell function in mice fed lower levels of vitamin D and calcium was associated with appearance of a subset of Lgr5+ CBC cells in the intestinal crypt that did not stain for Ki67. In regard to this, it is interesting that although all Lgr5+ cells cycle, only a subset has the property of self-renewal and therefore serve as stem cells (33). Moreover, we reported that there are rare cells at the crypt base that express the cyclin dependent kinase inhibitor p27Kip1 that regulates Rb phosphorylation and hence cell cycling (34), and that genetic inactivation of p27 Kip1 can cause intestinal tumors (12,35).
The sensitivity of Lgr5+ stem cell functions to vitamin D levels is important in the debate regarding which intestinal stem cells maintain intestinal homeostasis and serve as tumor initiating cells. As measured by serum 25(OH)D levels, the range of vitamin D exposure in the US population from all sources (e.g. sunlight and dietary exposure) has been established by the Centers for Disease Control and Prevention, National Health and Nutrition Examination Survey (NHANES) (http://www.cdc.gov/nchs/nhanes/nhanes_products.htm). Figure 5 illustrates this range for males and females, age 31–50, and also shows that feeding AIN76A to mice, which is formulated with 1IU of vitamin D3/g, establishes a 25(OH)D serum level of ~100 nM/l (19) at the very upper limit of the range and thus characterizes at most a few percent of individuals in the US population. Almost all publications that have established Lgr5+ stem cell functions in the mouse do not specify dietary conditions. Thus, the mice were likely fed standard chow diets that are convenient, relatively inexpensive, and therefore, routinely supplied by animal facilities. However, the level of vitamin D3 in standard chow diets is about 3-fold higher than in AIN76A (e.g. 3.3 IU/g for chow 5058 from LabDiet, St Louis, MO), and chow diets establish 25(OH)D levels even higher than those established by AIN76A (~125 nM/l in the mouse (36,37), well beyond the range documented for humans (Figure 5). Since the data presented demonstrate that stem cell and tumor initiating functions of Lgr5+ cells are only seen at the higher levels of vitamin D3 exposure, the contribution of Lgr5+ cells to intestinal homeostasis and tumor development in human populations may be much more variable and less important than assumed from the published mouse data. This is especially true in individuals at risk for development of colon cancer, who generally have lower vitamin D levels (38,39). The data emphasize the fundamental importance of diet in generating mouse models that accurately reflect mechanisms of pathogenesis and that identify cellular, biochemical or molecular targets for prevention or treatment that will translate well to human populations (40).
Fig. 5.

Serum 25(OH)D levels in the US population and in mice exposed to different diets. The cumulative percent of the US population of males or females, age 31–50, showing different levels of serum 25(OH)D was plotted from the NHANES data established by the Center for Disease Control and Prevention (http://www.cdc.gov/nchs/nhanes/nhanes_products.htm). Dashed lines indicate the mouse serum levels of 25(OH)D established by feeding control AIN76A diet or a standard chow diet (5058 from LabDiets), as previously reported.
Since Lgr5+ cell contribution to mucosal function and tumor development is compromised in mice fed NWD1, and yet the mice are healthy and the mucosa does not exhibit altered histology until solitary tumors form after 1–2 years, what cells maintain mucosal function and what is the cell of origin from which tumors arise? First, quiescent stem cell populations (Bmi1+, Lrig1+ or Dclk1+) may be mobilized, as they are in response to other stressors (4,7,8). Second, and not mutually exclusive, differentiated cells, such as Paneth cells may be recruited back into the cell cycle acquiring stem cell-like properties (41,42). This would be consistent with: the perturbed cell maturation of intestinal epithelial cells in mice fed the NWD1, indicated by elevated Wnt signaling throughout intestinal villi and colonic crypts (21), a common characteristic of cells that exhibit a stem cell phenotype; altered balance of lineage marker expression; and ectopic Paneth cell marker expression throughout the intestinal villi and colonic crypts in cells without typical Paneth cell morphology (21). It was reported recently that epigenetic changes in differentiated intestinal cells are consistent with this hypothesis in obesity induced changes in the intestinal mucosa (43). Support for the recruitment of differentiated cells to provide stem cell functions has also been published for the stomach (44), lung (45) and hair follicle (46), and plasticity of differentiated cells in acquiring stem cell functions has been reviewed recently (47). We suggest that it is important to determine the biological functions of potential stem cells from different intestinal cell compartments across the range of vitamin D exposures that characterize the human population.
Funding
The National Institutes of Health; United States Public Health Service (R01CA151494, R01CA135561 and RO1 CA174432 to L.H.A.) and Albert Einstein Cancer Center (P30 CA13330).
Conflict of Interest Statement: None declared.
Supplementary material
Supplementary Figure 1 can be found at http://carcin.oxfordjournals.org/
Glossary
Abbreviations:
- CBC
Lgr5+ crypt base columnar
- VDR
vitamin D receptor
- TAM
tamoxifen.
References
- 1. Clevers H., et al. (2013). SnapShot: the intestinal crypt. Cell, 152, 1198–1198.e2. [DOI] [PubMed] [Google Scholar]
- 2. Barker N., et al. (2009). Crypt stem cells as the cells-of-origin of intestinal cancer. Nature, 457, 608–611. [DOI] [PubMed] [Google Scholar]
- 3. Barker N., et al. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature, 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- 4. Powell A.E., et al. (2012). The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell, 149, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y., et al. (2013). LRIG1 is a triple threat: ERBB negative regulator, intestinal stem cell marker and tumour suppressor. Br. J. Cancer, 108, 1765–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Westphalen C.B., et al. (2014). Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Invest., 124, 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tian H., et al. (2011). A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature, 478, 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buczacki S.J., et al. (2013). Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature, 495, 65–69. [DOI] [PubMed] [Google Scholar]
- 9. Newmark H.L., et al. (2001). A Western-style diet induces benign and malignant neoplasms in the colon of normal C57Bl/6 mice. Carcinogenesis, 22, 1871–1875. [DOI] [PubMed] [Google Scholar]
- 10. Newmark H.L. (1987). Nutrient density: an important and useful tool for laboratory animal studies. Carcinogenesis, 8, 871–873. [DOI] [PubMed] [Google Scholar]
- 11. Yang W.C., et al. (2001). Targeted inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated tumor formation and the tumor-promoting activity of a Western-style high-risk diet by altering cell maturation in the intestinal mucosal. Cancer Res., 61, 565–569. [PubMed] [Google Scholar]
- 12. Yang W., et al. (2003). Targeted inactivation of p27kip1 is sufficient for large and small intestinal tumorigenesis in the mouse, which can be augmented by a Western-style high-risk diet. Cancer Res., 63, 4990–4996. [PubMed] [Google Scholar]
- 13. Yang W., et al. (2005). Inactivation of p21WAF1/cip1 enhances intestinal tumor formation in Muc2-/- mice. Am. J. Pathol., 166, 1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang K., et al. (2008). Interaction of Muc2 and Apc on Wnt signaling and in intestinal tumorigenesis: potential role of chronic inflammation. Cancer Res., 68, 7313–7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang K., et al. (2008). Dietary induction of colonic tumors in a mouse model of sporadic colon cancer. Cancer Res., 68, 7803–7810. [DOI] [PubMed] [Google Scholar]
- 16. Aslam M.N., et al. (2010). A mineral-rich red algae extract inhibits polyp formation and inflammation in the gastrointestinal tract of mice on a high-fat diet. Integr. Cancer Ther., 9, 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Willett W.C. (2001). Diet and cancer: one view at the start of the millennium. Cancer Epidemiol. Biomarkers Prev., 10, 3–8. [PubMed] [Google Scholar]
- 18. Terry P., et al. (2001). Fruit, vegetables, dietary fiber, and risk of colorectal cancer. J. Natl. Cancer Inst., 93, 525–533. [DOI] [PubMed] [Google Scholar]
- 19. Bastie C.C., et al. (2012). Dietary cholecalciferol and calcium levels in a Western-style defined rodent diet alter energy metabolism and inflammatory responses in mice. J. Nutr., 142, 859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang K., et al. (2007). Molecular targets of calcium and vitamin D in mouse genetic models of intestinal cancer. Nutr. Rev., 65, S134–S137. [DOI] [PubMed] [Google Scholar]
- 21. Wang D., et al. (2011). Paneth cell marker expression in intestinal villi and colon crypts characterizes dietary induced risk for mouse sporadic intestinal cancer. Proc. Natl Acad. Sci. U S A, 108, 10272–10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moser A.R., et al. (1990). A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science, 247, 322–324. [DOI] [PubMed] [Google Scholar]
- 23. el Marjou F., et al. (2004). Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis, 39, 186–193. [DOI] [PubMed] [Google Scholar]
- 24. Van Cromphaut S.J., et al. (2001). Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc. Natl Acad. Sci. U S A, 98, 13324–13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shibata H., et al. (1997). Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science, 278, 120–123. [DOI] [PubMed] [Google Scholar]
- 26. Zheng W., et al. (2012). Inactivation of the vitamin D receptor in APC(min/+) mice reveals a critical role for the vitamin D receptor in intestinal tumor growth. Int. J. Cancer, 130, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Larriba M.J., et al. (2011). Vitamin D receptor deficiency enhances Wnt/β-catenin signaling and tumor burden in colon cancer. PLoS One, 6, e23524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang K., et al. (1998). Dietary modulation of carcinoma development in a mouse model for human familial adenomatous polyposis. Cancer Res., 58, 5713–5717. [PubMed] [Google Scholar]
- 29. Fleet J.C., et al. (2012). Vitamin D and cancer: a review of molecular mechanisms. Biochem. J., 441, 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Welsh J. (2006). Calcium and Vitamin D. In Heber D, et al. (eds.), Nutritional Oncology. Elsevier, Massachusettes, MA, pp. 545–558. [Google Scholar]
- 31. Kovalenko P.L., et al. (2010). 1,25 dihydroxyvitamin D-mediated orchestration of anticancer, transcript-level effects in the immortalized, non-transformed prostate epithelial cell line, RWPE1. BMC Genomics, 11, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xue Y., et al. (2009). Intestinal vitamin D receptor is required for normal calcium and bone metabolism in mice. Gastroenterology, 136, 1317–1327, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kozar S., et al. (2013). Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell, 13, 626–633. [DOI] [PubMed] [Google Scholar]
- 34. Smartt H.J., et al. (2007). p27kip1 Regulates cdk2 activity in the proliferating zone of the mouse intestinal epithelium: potential role in neoplasia. Gastroenterology, 133, 232–243. [DOI] [PubMed] [Google Scholar]
- 35. Yang W., et al. (2005). p27kip1 in intestinal tumorigenesis and chemoprevention in the mouse. Cancer Res., 65, 9363–9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bolton C., et al. (2013). Serum levels of 25-hydroxy vitamin D in normal Biozzi and C57BL/6 mice and during the course of chronic relapsing experimental autoimmune encephalomyelitis (CR EAE). Inflamm. Res., 62, 659–667. [DOI] [PubMed] [Google Scholar]
- 37. Wang Y., et al. (2012). Development of experimental autoimmune encephalomyelitis (EAE) in mice requires vitamin D and the vitamin D receptor. Proc. Natl Acad. Sci. U S A, 109, 8501–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Feldman D., et al. (2014). The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer, 14, 342–357. [DOI] [PubMed] [Google Scholar]
- 39. Gorham E.D., et al. (2007). Optimal vitamin D status for colorectal cancer prevention: a quantitative meta analysis. Am. J. Prev. Med., 32, 210–216. [DOI] [PubMed] [Google Scholar]
- 40. Perrin S. (2014). Preclinical research: Make mouse studies work. Nature, 507, 423–425. [DOI] [PubMed] [Google Scholar]
- 41. Roth S., et al. (2012). Paneth cells in intestinal homeostasis and tissue injury. PLoS One, 7, e38965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schwitalla S., et al. (2013). Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell, 152, 25–38. [DOI] [PubMed] [Google Scholar]
- 43. Li R., et al. (2014). Obesity, rather than diet, drives epigenomic alterations in colonic epithelium resembling cancer progression. Cell Metab., 19, 702–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stange D.E., et al. (2013). Differentiated Troy+ chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell, 155, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tata P.R., et al. (2013). Dedifferentiation of committed epithelial cells into stem cells in vivo . Nature, 503, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rompolas P., et al. (2013). Spatial organization within a niche as a determinant of stem-cell fate. Nature, 502, 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Blanpain C., et al. (2014). Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science, 344, 1242281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.