

Abstract
Previous studies by our group, using an experimental autoimmune thyroiditis (EAT) model in Strain 13 inbred guinea pigs, resulted in T cell-mediated delayed hypersensitivity; however, autoantibodies proved not to be cytotoxic to thyroid epithelial cells in the presence or absence of complement proteins. Albeit, T cell-mediated lymphocyte cytotoxicity began to diminish sharply concomitantly with increasing titers of circulating autoantibodies, indicating a skewing of the self-reactive response and amelioration of the EAT. Furthermore, immunization of guinea pigs with thyroglobulin in incomplete Freund’s adjuvant (IFA) generated a high titer of antithyroglobulin antibodies and proved to inhibit thyroiditis. These observations indicated that the shift in the immune response from Th1 to Th2 and the production of antibodies were likely responsible for ameliorating EAT. Based upon these results, we extrapolated our studies to design a multivalent vaccine, which shows promise in preventing/reversing T1D in NOD mice. A small pilot study was conducted in which a total of 34 mice, 20 non-immunized controls and 14 immunized with syngeneic islet lysate, were monitored for mean day to diabetes for a total of 28 weeks. Immunization of NOD animals with syngeneic islet lysates resulted in a significant delay in diabetes onset (P < 0.001) as compared to non-immunized controls. To further assess the vaccine’s efficacy, robustness, and delay of disease, a large-scale experiment was conducted and monitored for 32 weeks using 106 mice, 64 non-immunized controls and 42 immunized with syngeneic islet lysate. At the end of the study, 90% of the non-immunized group developed diabetes, while less than 25% of the immunized group became diabetic (P < 0.0001). The protective effect, as a result of vaccination, correlated with an increase in the levels of IL-10 and IL-4 cytokines as well as a skewing to Th2-dependent isotype antibodies in serum. Strikingly, adoptive transfer of spleen cells from immunized animals into NOD.scid recipients provided protection against transfer of diabetes by diabetogenic spleen cells. The results of this study provide evidence that vaccination with islet lysate leads to a Th2-dependent skewing of the immune response to islet beta cells as a possible mechanism of protection. This strategy may be implemented as a possible vaccination protocol for arresting and/or preventing T1D in patients.
Keywords: NOD mice, Type 1 diabetes, Autoimmunity, Vaccine, Th2
Introduction
The clinical approach for the treatment of type 1 diabetes mellitus (T1D) began with insulin therapy in an effort to maintain acceptable glycemic control with minimal complications. Even with the most meticulous regimen of insulin therapy and the most compliant patients, euglycemic control for a prolonged period is difficult to accomplish. Acute complications, such as hypoglycemia and ketoacidosis, and long-term complications, such as peripheral neuropathy and microvascular/macrovascular problems, continue to persist [1].
T1DM is a chronic autoimmune disease [2] in which T cells play a key role in the chain of events leading to β cell destruction [2]. In early efforts to block the autoimmune process and preserve β cell functions in newly diagnosed T1DM patients, immunosuppressive agents, such as azathioprine, cyclophosphamide, and cyclosporine, were introduced, but not without undesirable side effects. Direct toxicity of the agents on the kidneys, for example, or over-immunosuppression resulting in systemic infections and malignancy development due to the loss of immune surveillance, constitutes some of the deleterious side effects [3–6]. Another drawback is the transience of immunosuppressive effects, leading to the recurrence of the disease rapidly after the cessation of treatment. Therefore, it is imperative to search for an effective, safe, and long-lasting strategy to enhance the regulation of a diabetogenic immune system with limited toxicity and without global immunosuppression.
In our previous studies on the mechanism of experimental autoimmune thyroiditis (EAT) in Strain 13 inbred guinea pigs, it was demonstrated that EAT is mediated by sensitized lymphocytes. The circulating autoantibodies were not cytotoxic to thyroid epithelial cells, either in the presence or in the absence of complement [7, 8]. Of importance is the observation that both the delayed hypersensitivity and lymphocyte cytotoxicity began to decline sharply when the titers of circulating autoantibodies increased to a maximum. More interestingly, when the guinea pigs were immunized with thyroglobulin in incomplete Freund’s adjuvant (IFA), the animals did not develop thyroiditis, but instead a high titer of antithyroglobulin antibodies were detected in the circulation [7, 8]. The protective mechanism of autoimmunization in the guinea pig model at that time was not clear; however, we speculate that the protection was conveyed by either the induction of regulatory cells or skewing of the T cell repertoire to a more TH2 phenotype.
In a similar manner to EAT, T1D is a cell-mediated autoimmune disease. T cells infiltrating pancreatic islets have been identified [9], and T cell clones specific for islet-related autoantigens have been established [10]. Autoantibodies to various pancreatic islet antigens are also present in the circulation of not only diabetic patients and NOD mice but also normal individuals. Interestingly, an inverse relationship between T cell proliferation and circulating antibody titers was also found in NOD mice sensitized with ICA69 or GAD65 [11, 12]. These studies confirmed our previous observations of an inverse relationship between the antithyroglobulin antibodies and lymphocyte cytotoxicity in the EAT model [7, 8]. We, therefore, tested this autoimmunization strategy in NOD mice, using syngeneic islets in IFA, to determine whether we could induce regulation and/or protection against T1D. For this endeavor, the following experiments were conducted.
Research design and methods
Experimental animals
Female NOD mice aged 3–4 weeks were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed in the animal facility at Children’s Hospital of Pittsburgh in accordance with the National Institute of Health regulations under specific pathogen-free conditions. Some of the mice were bred and maintained in our pathogen-free facility. All experiments were conducted with the approval of the committee on animal research of the University of Pittsburgh.
Blood glucose measurement
A pentype glucometer (Precision, QID; MedisenseBedfold, MA) was used to measure blood glucose. The blood (5 μl) for glucose measurement was obtained by making a small incision at the tail of the mouse. Mice were considered diabetic when a level of 300 mg/dl or above was reached for two consecutive readings, 24 h apart.
Isolation of islet of Langerhans and preparation of islet lysates
Isolation of pancreatic islets from NOD mice was performed using a modified collagenase digestion procedure [13, 14]. Briefly, 3 ml of cold Hank’s balanced salt solution containing 1.75 mg/ml collagenase (Sigma; St. Louis, MO) was injected into the pancreatic duct. After pancreatectomy, the islets were purified from the digested pancreatic tissue by density gradient cell separation (Ficoll, 1.108, 1.096, 1.069, and 1.037 g/ml; Sigma). To prepare islet lysates, the islet suspensions were frozen and thawed three times, then sonicated at 4°C for 5 s for three cycles.
Radioimmunoassay for anti-insulin antibodies
Radioimmunoassay for anti-insulin antibodies was performed as previously described [15]. Briefly, sera to be assayed for insulin-specific antibodies were stored at −70°C until use. All assays were run in 96-well plates that were coated with 10 μCi/ml of 125I-insulin (Boehringer Mannheim, Indianapolis, IN) in triplicate at 4°C overnight and blocked at room temperature with 5% BSA in PBS for 2 h, along with standard positive and negative control serum samples (kindly supplied by Dr. Eisenbarth at the Barbara Davis Diabetes Center, Denver). Two low-positive controls and one negative control were used for monitoring each assay. The two low positives have to have an index of 0.011–0.050 and 0.020–0.080, respectively, and the negative has to be <0.01. Proficiency is monitored through the International Diabetes Autoantibody Standard Program (DASP).
Measurement of anti-islets antibodies by ELISA
To measure anti-islet antibody titers, 96-well high-binding microplates (DuoSet ELISA kits, R&D system) were used. Each well was coated with 5ug/ml of β-membrane protein in 100 μl of saline at room temperature overnight or at 4°C for 2 days. The plates were washed three times with PBST and then blocked with 300 μl of blocking buffer at room temperature for 2 h. Plates were again washed three times with PBST, and 100 μl of properly diluted antisera was added to each well and incubated at room temperature for 2 h. Triplicates were measured for each sample. The plates were again washed three times with PBST, followed by the addition of 100 μl TMB per well and a 10-min incubation in the dark at room temperature. Stop solution was added and the plates were read within 30 min on a Spectromax M2 microplate reader (Molecular Devices), and the data were analyzed using Softmax Pro v5.0.19 (Molecular Device).
Cytokine production from normal control and vaccinated female NOD mice
Samples were analyzed at different time points (0, 3, 6 and 13 weeks) for the presence of IL-4 and IL-10 by Beadlyte mouse multi-cytokine flex kit (Upstate Biotechnology, Lake Placid, NY), following the manufacturer’s protocol. Samples were read using the Luminex 100 (Upstate Biotechnology).
Immunization protocols
Four- to 6-week-old female NOD mice were primed with either 5,000 islets or 40 μg of islet lysates in appropriate adjuvant at the rear footpads. They were subsequently boosted every 2 weeks for three times with half of the priming dosage at the base of the tail. Preparation of immunization inocula with different adjuvants was carried out as following: Imject alum (Sigma) was mixed with an equal volume of islet suspension or islet lysates at room temperature for 30 min. The mix was then incubated at 4°C for 18 h. For complete (CFA) and incomplete (IFA) Freund’s adjuvant, the adjuvant was thoroughly emulsified in saline (1:1) at room temperature and then mixed with the same volume of islets or islet lysates used in Imject alum inoculum right before immunization. Each mouse received an inoculum of 50 μl.
Adoptive transfer
The adoptive transfer was performed as previously described [16]. The recipients were 20-week-old female NOD.scid mice. In the experimental group, each mouse received 107 splenocytes retro-ocularly from islet lysate–vaccinated NOD female mice under the immunization protocol described above, whereas in the control group, each mouse received the same number of splenocytes from non-immunized, non-diabetic NOD mice via the same route. One week after the priming, all mice were challenged with 8 × 106 splenocytes from diabetic female NOD mice.
Statistical analysis
GraphPad Prism version 4.0 (GraphPad Software, San Diego, CA) was used to analyze the data. Kaplan–Meier log-rank analysis was used for survival data, and unpaired ANOVA or Student’s t test was applied where appropriate. A P-value of less than 0.05 was considered statistically significant.
Results
Induction of immune response to isogenic pancreatic islet generates autoantibodies to Schwann cells and insulin in female NOD mice
The optimal dose to induce high anti-insulin and antimantle (Schwann) antibody titers was determined to be 20–40 μg islets/mouse. This immunization strategy not only produced high titers of anti-insulin antibodies (Fig. 1a), it also generated high titers of anti-peri-islet Schwann cell as shown by indirect immunofluorescence (Fig. 1b). The response to other islet-associated antigens, such as GAD-65, was less remarkable.
Fig. 1.

a Anti-insulin antibody titers 2 weeks after second booster. Each group consisted of 3 mice, and each mouse was primed with an inoculum consisting of 40 μg of sonicated islet lysates in various adjuvants. They were subsequently boosted with 2 μg of lysates with the same adjuvant used for primary immunization at 2-week intervals. b Serum generated from mice immunized with islet lysate was used to stain islet sections (left panel); right panel was a control anti-insulin antibody. Represented results show the presence of anti-β-cell and anti-Schwann cell antibodies in the sera of immunized NOD mice
Efficacy of negative vaccination in the prevention of T1D in female NOD mice
Four-week-old female NOD mice were immunized with 40 μg of sonicated pancreatic islet lysates in rear footpads in the volume of 50 μl. The vaccine group animals were boosted at 4 and 6 weeks after the primary immunization with half of the priming dose at the base of the tail. In the preliminary experiment, 34 female NOD mice were used. By the end of 24 weeks, 90% of the control group developed diabetes, while none of the vaccinated group became diabetic (P < 0.001) (Fig. 2a). In order to confirm the pilot study as well as increase the duration of monitoring after treatment, a larger-scale experiment was conducted, using a total of 106 NOD female mice, 42 in the negative vaccination group and 64 in the control. Thirty-two weeks after vaccination, 90% of the non-immunized group developed diabetes, yet less than 25% of the immunized group became diabetic (P < 0.0001) (Fig. 2b). These results (pilot and large scale) demonstrate that the negative vaccination prevents or delays the onset of T1D in NOD mice.
Fig. 2.

Pilot and large-scale negative vaccination study. a The effect of negative vaccination was determined on the incidence of T1D in female NOD mice. Thirty-four female NOD mice at the age of 4–5 weeks were used. Fourteen mice were immunized with islet lysates in IFA (solid line), and 20 of them were not immunized as controls (dashed line). Mean day to diabetes was assessed; data were analyzed by Kaplan–Meier life plot (P < 0.001). b Large-scale study with increased number of animals as well as increased duration of monitoring (4–5 weeks, 106 NOD females) were segregated into negative vaccinated and control non-vaccinated groups. The results show a significant protection (P < 0.0001) from diabetes in vaccinated NOD mice (n = 42) as compared to the control group (n = 64)
Adoptive transfer of mixed spleen cells from negatively vaccinated and diabetic NOD mice protects NOD.scid mice from T1D
In order to determine whether the protection observed in the negatively vaccinated animals was transferable, female NOD.scid mice received 107 splenocytes retro-ocularly from female NOD mice that were primed and boosted with islet lysate in IFA, as previously described. Control animals received 107 normal splenocytes from non-immunized, non-diabetic, age-matched female NOD mice. One week later, both groups of mice were injected with 107 splenocytes from diabetic female NOD mice retro-ocularly. Both groups of mice were observed for 32 weeks. All 5 mice in the control group developed diabetes between 5 and 7 weeks post-transfer. Mice that received splenocytes from the negatively vaccinated NOD became diabetic between 17 and 28 weeks (Fig. 3), which demonstrated a delay in diabetic onset (P < 0.0011) and indicated the presence of regulatory cell functions.
Fig. 3.

Twenty-week-old female NOD.scid mice received 107 splenocytes retro-ocularly from islet lysate–vaccinated (n = 6) or control treated animals (n = 5). One week after the priming, all mice were challenged with 8 × 106 splenocytes from diabetic female NOD mice. Animals were monitored for mean day to hyperglycemia. A statistically significant delay in T1DM onset was measured in mice receiving the islet lysate–vaccinated splenocytes (P < 0.0011)
Kinetic studies on cytokine production from normal control and vaccinated female NOD splenocytes demonstrate a vaccine-dependent TH1 to TH2 skewing
In this experiment, a total of forty-eight 4-week-old female NOD mice were used: 16 of them were immunized with islet lysates, 16 immunized with IFA alone, and 16 with PBS. In our control studies, it was shown that female NOD mice began to develop diabetes at the age of 12 weeks (Fig. 2a, b). Since our primary interest is the pathogenic mechanisms in the pre-diabetic stage, the mice were bled at 0, 3, 6 and 13 weeks after priming. The lymphoid cells from peripheral lymph nodes and splenocytes were removed from normal and immunized NOD mice and used to determine the effect of vaccination on cytokine milieu. It was apparent that over time, the islet lysate–vaccinated mice showed a skewing to the TH2-type profile. IL-4 and IL-10 levels peaked at the 13-week mark in the islet lysate–vaccinated group, while the IFA and control treated animals demonstrated little to no change in cytokine levels (Fig. 4a–c). Interestingly, the control unimmunized animals started out with high levels of IL-10 at time 0 that fell sharply after 3 weeks, while IL-10 levels in the islet lysate–vaccinated animals rose over time. This drop in IL-10 in the controls seems to echo the loss of regulation and the progression to T cell-mediated β cell destruction, as these animals started to go diabetic at ~13 weeks. Furthermore, assessment of the antibody isotypes also revealed that the switch to the TH2 cytokine profile demonstrated in the islet lysate–treated animals led to a switch from the TH1-dependent IgG2b isotype to IgG1 (Fig. 4d). This further demonstrates that the islet lysate–immunized mice were likely protected by a skewing of the T cell response.
Fig. 4.

a–c The rate of cytokine synthesis was assessed kinetically (0, 3, 6 and 13 weeks after immunization) from islet lysate, IFA-only or control vaccinated mice. Samples from pooled serum obtained from the various groups were assessed by Beadlyte mouse multi-cytokine flex kit. d Serum samples from islet lysate, IFA-only or control vaccinated mice were assayed for immunoglobulin isotype kinetically at 3 and 13 weeks
Discussion
T1D is an autoimmune disease that results from a failure or breakdown of the mechanism normally responsible for maintaining unresponsiveness (tolerance) to self. The breakdown of tolerance, either central or peripheral, is caused by complex processes likely from the summation of multiple interacting factors, including immunological abnormalities affecting lymphocytes and APC (antigen-presenting cells), genes that predispose to autoimmunity, and/or environmental factors, such as microbial infections and tissue injuries [17]; thus, it is almost impossible to control all the factors in such a synchronized way that autoimmune diabetes can be avoided. No matter the complexity involved in the progression to T1D, it is clear that it is a T cell-mediated autoimmune disease that leads to β cell destruction. In animal models, it has been well documented that both CD4+ and CD8+ T cells are responsible and necessary for the progression of the disease [18]. The CD4+ TH2 compartment, however, has been ascribed with a protective role due to its ability to cause a cytokine shift away from the TH1 cytokines like IFN-γ and TNF-α that have been reported to be instrumental in the disease progression [19–23]. The polarization of T helper subsets has been heavily implicated in both progression (TH1) and protection (TH2) of T cell-mediated β cell destruction [24]. Because of this, many studies have been published describing strategies that lead to the TH2 T cell switch for protection against TH1 [25]. For example, it has been reported that protection from diabetes was afforded by infecting NOD mice with various helminthes, which lead to a functional polarization of the T helper subsets to the protective TH2 phenotype and away from the pathogenic TH1 [25]. Others have used the delivery of apoptotic α cells in vivo to induce suppression of the anti-α-cell response as well as to skew the T cell response to the TH2 phenotype, leading to the production of IL-10-producing regulatory cells [26]. It is clear that various immune strategies have been employed to protect diabetes-prone animals from developing diabetes [24], and although they vary in approach, the primary protocols implicate that a skewing of the T cell compartment from the TH1 pathogenic subset to the TH2 phenotype provides protection.
We, therefore, set our strategy to treat this disease by preventing the generation and activation of existing autoreactive T cells, instead of dealing with the complex genetic and environmental factors. Based on this premise, our previous studies of EAT, as well as reports defining the role that both autoantibodies [27] and B cells play in the overall progression of T1D pathogenesis [28–32], we decided to immunize NOD mice with isogenic islet lysates in IFA to determine whether autoimmunization could skew the immune system to the more protective TH2-type response, leading to the prevention of T1D, as well as to induce TH2-isotype-dependent, anti-insulin or anti-islet antibodies indicative of an immunological switch in phenotype.
We found that vaccination of NOD mice with islet lysate resulted in the generation of insulin as well as antimantle (Schwann) cell antibodies when the lysate was emulsified in incomplete Freund’s adjuvant (IFA) while Alum + lysate alone failed to generate a strong response (Fig. 1a, b). The unique anti-mantle cell response was highest in the islet lysate group, the same group that demonstrated maximum protection from progression to spontaneous diabetes. Interestingly, these data demonstrate that the elevated antibody response did not correlate with an accelerated progression to diabetes.
In our preliminary vaccine experiments, the incidence of T1D was remarkably reduced to 10% in the immunized NOD mice as compared to that of controls at 90% (Fig. 2a). Furthermore, a larger-scale experiment was conducted in order to assess the efficacy of the negative vaccine treatment in a larger cohort over a longer period of time. The total number of animals used in this study was 106 NOD mice, 42 in the negative vaccination group and 64 in the control. In this larger-scale study, 90% of the control animals went diabetic, while only 25% of the vaccinated animals went diabetic. The number of animals used in these studies clearly proved that the results are highly robust, and the two studies, pilot and larger scale, demonstrate strong reproducibility, warranting further study and design for use in more clinically relevant settings. Moreover, these findings confirm our previous studies in another autoimmune model, experimental autoimmune thyroiditis (EAT), displaying that this type of negative vaccination prevents cell-mediated autoimmune disease [7].
The results of adoptive cotransfer of diabetic spleen cells with cells isolated from the negatively vaccinated NOD mice, shown in Fig. 3, confirm that the negatively vaccinated NOD mice produced a regulatory lymphocyte population that could disable diabetic spleen cells so that they too could not induce diabetes. This process of “infectious” tolerance may explain why the transfer of these T cells from the negatively vaccinated NOD mice provided the protection. This tolerance was despite the fact that the number of CD25+ CD4+ Foxp3-positive cells was not increased in the splenocytes of the vaccinated NOD mice (data not shown), indicating that the protection was not mediated by the induction of conventional regulatory cells, but instead another mechanism was afforded.
The characteristics of this regulation was further investigated by assessing the cytokine profiles of T cells from negatively vaccinated mice as compared to the control unimmunized, IFA-only immunized, and the control immunized animals. As previously stated, in T1D, both CD4 and CD8 T cells comprise the cell-mediated arm of the immune response [33–36] and are responsible for the production of IL-2 and IFN-γ. CD4+ TH2 cells produce IL-4 and IL-10: IL-4 is essential in triggering and sustaining a humoral immune response, such as protective antibodies, while IL-10, an anti-inflammatory cytokine [37]. Thus, a shift in the phenotype of a TH1-rich cytokine environment toward a TH2-polarized environment could prevent or ameliorate the development of cell-mediated T1D [38]. As shown in Fig. 5, when NOD mice were immunized with pancreatic islet lysates in IFA, synthesis of TH2 cytokines, such as IL-4 and IL-10, was enhanced, peaking at the 13-week time point, while the control treated animals showed marginal cytokine responses after immunization (Fig. 5). Interestingly, in the islet cell–vaccinated group, there was an increase in the TH1-dependent cytokine IFN-γ, which also peaked at the 13-week time point. Recent work has demonstrated the need for IFN-γ in order for regulatory T cells to exert their functions [39, 40]. Therefore, the production of IFN-γ in the negatively vaccinated mice may play an important role in facilitating the formation of immune tolerance through controlling regulatory T cell function.
Fig. 5.
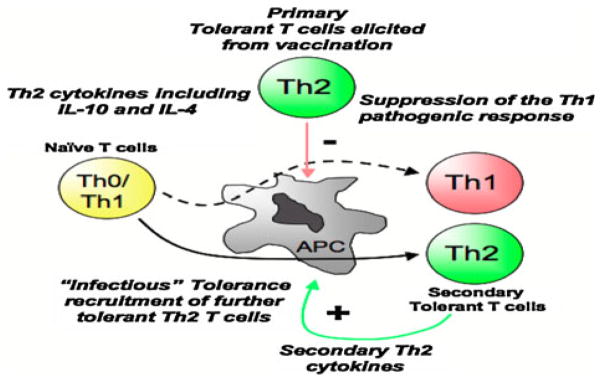
Model depicting Th2-mediated deviation and regulation of Th1 pathogenic T cells in T1D. Negative vaccination leads to the generation of T cells that impart regulatory function on pathogenic autoreactive T cells either by deviation or by regulation mechanisms. This mechanism affords endogenous protection of the vaccinated host, and adoptively transferred splenocytes protect naïve recipients from diabetogenic T cells, possibly through an infectious tolerance mechanism
Finally, the interrogation of the serum for the humoral response demonstrated that the islet lysate–vaccinated animals produced increased IgG1 antibody in the serum, peaking at the same time point as the TH2-type cytokines, as compared to the IFA-only immunized animals. The IFA treatment produced elevated levels of the TH1-dependent isotype IgG2b. These results provide supporting evidence that the protective effect of the islet lysate–negative vaccination strategy elicited an immune response that led to a skewing of the T cell repertoire to the TH2 phenotype, which protected against spontaneous diabetes and adoptive transfer of diabetes.
Overall, our autoimmunization strategy leads to the generation of T cells that impart regulatory functions on pathogenic autoreactive T cells by either deviation or regulation mechanisms. This approach affords endogenous protection of the vaccinated host, and adoptively transferred splenocytes protect naïve recipients from diabetogenic T cells, possibly through an infectious tolerance mechanism (Fig. 5).
Acknowledgments
The authors wish to express their appreciation to Professor Dr. Philip Fireman for critical review of this manuscript.
Contributor Information
Ming S. Lin, Immunogenetics/Pulmonary Medicine, Allergy and Immunology, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA
Hubert M. Tse, Immunogenetics, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA
Meghan M. Delmastro, Immunogenetics, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA
Suzanne Bertera, Immunogenetics/Immunology, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA.
Caterina T. Wong, Immunogenetics/Immunology, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA
Robert Lakomy, Immunogenetics/Immunology, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA.
Jing He, Immunogenetics/Immunology, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA.
Martha M. Sklavos, Immunogenetics/Immunology, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA
Gina M. Coudriet, Immunogenetics/Immunology, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA
Massimo Pietropaolo, Immunogenetics/Immunology, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA.
Massimo M. Trucco, Immunogenetics/Immunology, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA
Jon D. Piganelli, Email: jdp51@pitt.edu, Immunogenetics/Immunology, Children’s Hospital of Pittsburgh and University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh 15224, USA
References
- 1.Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med. 1986;314:1360–8. doi: 10.1056/NEJM198605223142106. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–9. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 3.Haller MJ, Gottlieb PA, Schatz DA. Type 1 diabetes intervention trials 2007: where are we and where are we going? Curr Opin Endocrinol Diabetes Obes. 2007;14:283–7. doi: 10.1097/MED.0b013e32825a673b. [DOI] [PubMed] [Google Scholar]
- 4.Like AA, Dirodi V, Thomas S, Guberski DL, Rossini AA. Prevention of diabetes mellitus in the BB/W rat with Cyclosporin-A. Am J Pathol. 1984;117:92–7. [PMC free article] [PubMed] [Google Scholar]
- 5.Salama AD, Remuzzi G, Harmon WE, Sayegh MH. Challenges to achieving clinical transplantation tolerance. J Clin Invest. 2001;108:943–8. doi: 10.1172/JCI14142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stiller CR, Dupre J, Gent M, Jenner MR, Keown PA, Laupacis A, Martell R, Rodger NW, von Graffenried B, Wolfe BM. Effects of cyclosporine immunosuppression in insulin-dependent diabetes mellitus of recent onset. Science. 1984;223:1362–7. doi: 10.1126/science.6367043. [DOI] [PubMed] [Google Scholar]
- 7.Lin MS, Salvin SB. In vitro and in vivo studies on the mechanism of experimental autoimmune thyroiditis in guinea pigs. Cell Immunol. 1976;27:177–87. doi: 10.1016/0008-8749(76)90227-6. [DOI] [PubMed] [Google Scholar]
- 8.Lin MS, Salvin SB. Further studies on the mechanism of experimental autoimmune thyroiditis in guinea pigs. Properties of thyroid cytotoxic factor (TCF) and its relationship to pathogenesis of the disease. Cell Immunol. 1976;27:188–99. doi: 10.1016/0008-8749(76)90228-8. [DOI] [PubMed] [Google Scholar]
- 9.Itoh N, Hanafusa T, Miyazaki A, Miyagawa J, Yamagata K, Yamamoto K, Waguri M, Imagawa A, Tamura S, Inada M, et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993;92:2313–22. doi: 10.1172/JCI116835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peakman M, Wen L, McNab GL, Watkins PJ, Tan KC, Vergani D. T cell clones generated from patients with type 1 diabetes using interleukin-2 proliferate to human islet antigens. Autoimmunity. 1994;17:31–9. doi: 10.3109/08916939409014656. [DOI] [PubMed] [Google Scholar]
- 11.Harrison LC, Honeyman MC, DeAizpurua HJ, Schmidli RS, Colman PG, Tait BD, Cram DS. Inverse relation between humoral and cellular immunity to glutamic acid decarboxylase in subjects at risk of insulin-dependent diabetes. Lancet. 1993;341:1365–9. doi: 10.1016/0140-6736(93)90940-i. [DOI] [PubMed] [Google Scholar]
- 12.Roep BO, Duinkerken G, Schreuder GM, Kolb H, de Vries RR, Martin S. HLA-associated inverse correlation between T cell and antibody responsiveness to islet autoantigen in recent-onset insulin-dependent diabetes mellitus. Eur J Immunol. 1996;26:1285–9. doi: 10.1002/eji.1830260616. [DOI] [PubMed] [Google Scholar]
- 13.Bertera S, Crawford ML, Alexander AM, Papworth GD, Watkins SC, Robbins PD, Trucco M. Gene transfer of manganese superoxide dismutase extends islet graft function in a mouse model of autoimmune diabetes. Diabetes. 2003;52:387–93. doi: 10.2337/diabetes.52.2.387. [DOI] [PubMed] [Google Scholar]
- 14.Sklavos MM, Bertera S, Tse HM, Bottino R, He J, Beilke JN, Coulombe MG, Gill RG, Crapo JD, Trucco M, Piganelli JD. Redox modulation protects islets from transplant-related injury. Diabetes. 2010;59:1731–8. doi: 10.2337/db09-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu L, Robles DT, Abiru N, Kaur P, Rewers M, Kelemen K, Eisenbarth GS. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc Natl Acad Sci USA. 2000;97:1701–6. doi: 10.1073/pnas.040556697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piganelli JD, Flores SC, Cruz C, Koepp J, Batinic-Haberle I, Crapo J, Day B, Kachadourian R, Young R, Bradley B, Haskins K. A metalloporphyrin-based superoxide dismutase mimic inhibits adoptive transfer of autoimmune diabetes by a diabetogenic T-cell clone. Diabetes. 2002;51:347–55. doi: 10.2337/diabetes.51.2.347. [DOI] [PubMed] [Google Scholar]
- 17.van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91:79–118. doi: 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- 18.Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes. 1993;42:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- 19.Bergman B, Haskins K. Autoreactive T-cell clones from the nonobese diabetic mouse. Proc Soc Exp Biol Med. 1997;214:41–8. doi: 10.3181/00379727-214-44067. [DOI] [PubMed] [Google Scholar]
- 20.Cantor J, Haskins K. Effector function of diabetogenic CD4 Th1 T cell clones: a central role for TNF-alpha. J Immunol. 2005;175:7738–45. doi: 10.4049/jimmunol.175.11.7738. [DOI] [PubMed] [Google Scholar]
- 21.Cantor J, Haskins K. Recruitment and activation of macrophages by pathogenic CD4 T cells in type 1 diabetes: evidence for involvement of CCR8 and CCL1. J Immunol. 2007;179:5760–7. doi: 10.4049/jimmunol.179.9.5760. [DOI] [PubMed] [Google Scholar]
- 22.Haskins K, Wegmann D. Diabetogenic T-cell clones. Diabetes. 1996;45:1299–305. doi: 10.2337/diab.45.10.1299. [DOI] [PubMed] [Google Scholar]
- 23.Peterson JD, Berg R, Piganelli JD, Poulin M, Haskins K. Analysis of leukocytes recruited to the pancreas by diabetogenic T cell clones. Cell Immunol. 1998;189:92–8. doi: 10.1006/cimm.1998.1377. [DOI] [PubMed] [Google Scholar]
- 24.Sia C. Imbalance in Th cell polarization and its relevance in type 1 diabetes mellitus. Rev Diabet Stud. 2005;2:182–6. doi: 10.1900/RDS.2005.2.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saunders KA, Raine T, Cooke A, Lawrence CE. Inhibition of autoimmune type 1 diabetes by gastrointestinal helminth infection. Infect Immun. 2007;75:397–407. doi: 10.1128/IAI.00664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia CQ, Peng R, Qiu Y, Annamalai M, Gordon D, Clare-Salzler MJ. Transfusion of apoptotic beta-cells induces immune tolerance to beta-cell antigens and prevents type 1 diabetes in NOD mice. Diabetes. 2007;56:2116–23. doi: 10.2337/db06-0825. [DOI] [PubMed] [Google Scholar]
- 27.Greeley SA, Katsumata M, Yu L, Eisenbarth GS, Moore DJ, Goodarzi H, Barker CF, Naji A, Noorchashm H. Elimination of maternally transmitted autoantibodies prevents diabetes in non-obese diabetic mice. Nat Med. 2002;8:399–402. doi: 10.1038/nm0402-399. [DOI] [PubMed] [Google Scholar]
- 28.Forsgren S, Andersson A, Hillorn V, Soderstrom A, Holmberg D. Immunoglobulin-mediated prevention of autoimmune diabetes in the non-obese diabetic (NOD) mouse. Scand J Immunol. 1991;34:445–51. doi: 10.1111/j.1365-3083.1991.tb01567.x. [DOI] [PubMed] [Google Scholar]
- 29.Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes. 1997;46:941–6. doi: 10.2337/diab.46.6.941. [DOI] [PubMed] [Google Scholar]
- 30.Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, Fleming SA, Leiter EH, Shultz LD. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med. 1996;184:2049–53. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol. 1998;161:3912–8. [PubMed] [Google Scholar]
- 32.Silveira PA, Serreze DV, Grey ST. Invasion of the killer B’s in type 1 diabetes. Front Biosci. 2007;12:2183–93. doi: 10.2741/2221. [DOI] [PubMed] [Google Scholar]
- 33.Kay TW, Campbell IL, Harrison LC. Characterization of pancreatic T lymphocytes associated with beta cell destruction in the non-obese diabetic (NOD) mouse. J Autoimmun. 1991;4:263–76. doi: 10.1016/0896-8411(91)90023-6. [DOI] [PubMed] [Google Scholar]
- 34.Kay TW, Chaplin HL, Parker JL, Stephens LA, Thomas HE. CD4+ and CD8+ T lymphocytes: clarification of their pathogenic roles in diabetes in the NOD mouse. Res Immunol. 1997;148:320–7. doi: 10.1016/s0923-2494(97)87241-0. [DOI] [PubMed] [Google Scholar]
- 35.Thomas HE, Darwiche R, Corbett JA, Kay TW. Evidence that beta cell death in the nonobese diabetic mouse is Fas independent. J Immunol. 1999;163:1562–9. [PubMed] [Google Scholar]
- 36.Thomas HE, Kay TW. Beta cell destruction in the development of autoimmune diabetes in the non-obese diabetic (NOD) mouse. Diabetes Metab Res Rev. 2000;16:251–61. doi: 10.1002/1520-7560(200007/08)16:4<251::aid-dmrr126>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 37.Hussain S, Delovitch TL. Intravenous transfusion of BCR-activated B cells protects NOD mice from type 1 diabetes in an IL-10-dependent manner. J Immunol. 2007;179:7225–32. doi: 10.4049/jimmunol.179.11.7225. [DOI] [PubMed] [Google Scholar]
- 38.Suarez-Pinzon WL, Rabinovitch A. Approaches to type 1 diabetes prevention by intervention in cytokine immunoregulatory circuits. Int J Exp Diabetes Res. 2001;2:3–17. doi: 10.1155/EDR.2001.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mangada J, Pearson T, Brehm MA, Wicker LS, Peterson LB, Shultz LD, Serreze DV, Rossini AA, Greiner DL. Idd loci synergize to prolong islet allograft survival induced by costimulation blockade in NOD mice. Diabetes. 2009;58:165–73. doi: 10.2337/db08-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pearson T, Weiser P, Markees TG, Serreze DV, Wicker LS, Peterson LB, Cumisky AM, Shultz LD, Mordes JP, Rossini AA, Greiner DL. Islet allograft survival induced by costimulation blockade in NOD mice is controlled by allelic variants of Idd3. Diabetes. 2004;53:1972–8. doi: 10.2337/diabetes.53.8.1972. [DOI] [PubMed] [Google Scholar]