

Abstract
Insulin resistance (IR) increases cardiovascular morbidity and is associated with mitochondrial dysfunction. IR is now recognized to be present in type 1 diabetes; however, its relationship with mitochondrial function is unknown. We determined the relationship between IR and muscle mitochondrial function in type 1 diabetes using the hyperinsulinemic-euglycemic clamp and 31P-MRS before, during, and after near-maximal isometric calf exercise. Volunteers included 21 nonobese adolescents with type 1 diabetes and 17 nondiabetic control subjects with similar age, sex, BMI, Tanner stage, and activity levels. We found that youths with type 1 diabetes were more insulin resistant (median glucose infusion rate 10.1 vs. 18.9 mg/kglean/min; P < 0.0001) and had a longer time constant of the curve of ADP conversion to ATP (23.4 ± 5.3 vs. 18.8 ± 3.9 s, P < 0.001) and a lower rate of oxidative phosphorylation (median 0.09 vs. 0.21 mmol/L/s, P < 0.001). The ADP time constant (β = −0.36, P = 0.026) and oxidative phosphorylation (β = 0.02, P < 0.038) were related to IR but not HbA1c. Normal-weight youths with type 1 diabetes demonstrated slowed postexercise ATP resynthesis and were more insulin resistant than control subjects. The correlation between skeletal muscle mitochondrial dysfunction in type 1 diabetes and IR suggests a relationship between mitochondrial dysfunction and IR in type 1 diabetes.
Introduction
Type 1 diabetes is increasing in youth and is associated with increased cardiovascular morbidity and mortality (1). In type 2 diabetes, insulin resistance (IR) has a critical role in the development of cardiovascular disease, exercise impairment, and other features of the metabolic syndrome (2). IR typically occurs in association with the metabolic syndrome phenotype (increased hepatic, visceral, and intramyocellular lipid [IMCL]; hypertriglyceridemia; obesity; hyperinsulinemia; hyperglycemia; and low HDL cholesterol and adiponectin). However, IR is now also recognized as a common feature of youth and adults with type 1 diabetes, despite lacking hyperinsulinemia and the aforementioned typical features of the metabolic syndrome (2–4). We previously reported significant IR in normal-weight youths with type 1 diabetes, as well as exercise and cardiac and vascular dysfunction compared with nondiabetic youth (2). Thus, the presence of IR may contribute to the early and excess cardiovascular disease seen in type 1 diabetes (2,5–7).
The mechanism(s) of muscle IR in individuals with type 2 diabetes is thought to include decreased glucose transport secondary to decreased mitochondrial function, decreased blood flow, increased circulating free fatty acid (FFA) levels, increased IMCL, signaling defects, or inflammation (8). Decreased mitochondrial function and alterations in lipid metabolism have also been detected in nondiabetic populations with IR including aging, burn trauma, and HIV medication–related lipodystrophy (9–11). In adults with type 1 diabetes, fasting FFAs are elevated and are not fully suppressed by hyperinsulinemia (6,12). In youth with type 1 diabetes, we demonstrated vascular dysfunction and increased FFAs but no increase in IMCL or markers of systemic inflammation compared with nondiabetic youth (2). In contrast, IMCL and inflammatory markers were reported to be increased in adults with type 1 diabetes (2,13,14). Therefore, IR in type 1 diabetes may share some, but not all, mechanistic features of type 2 diabetes.
There is limited information on the role of mitochondrial function in IR in type 1 diabetes. The majority of previous studies used muscle biopsy in adults and suggested mitochondrial dysfunction. Mitochondrial DNA from fasting adults with type 1 diabetes showed decreased expression of mitochondrial oxidative genes compared with control subjects (15). After insulin withdrawal in adults with type 1 diabetes, mitochondrial DNA expression decreased relative to the normoinsulinemic state (16). Adults with type 1 diabetes had decreased energy flux through ATP synthase during a hyperinsulinemic-euglycemic clamp (17). However, the two published studies using 31P spectroscopy methods are contradictory (18,19). The mitochondrial studies to date include only adults with long-standing diabetes duration, where complications such as microvascular disease or obesity may confound the results. Therefore, studies in youth are important to understand the initial pathophysiology and significance.
Assessment of mitochondrial function in youth with type 1 diabetes has not been published to date. Moreover, studies in adults with type 1 diabetes have not examined the role of serum FFA or glucose, muscle insulin sensitivity, or IMCL in mitochondrial dysfunction. We hypothesized that nonobese youth with type 1 diabetes have mitochondrial dysfunction, which relates to IR and elevated FFA. The purpose of this study was to assess muscle IR and mitochondrial function in youth with type 1 diabetes, along with additional established contributors to IR in other populations: FFA, glucose, IMCL, and inflammation.
Research Design and Methods
Subjects
Thirty-eight participants 12–18 years of age were recruited from pediatric clinics at the Children’s Hospital Colorado and the Barbara Davis Center for Childhood Diabetes for a prospective, cross-sectional study. Participants included nonobese (BMI ≤95th percentile for age) volunteers with and without type 1 diabetes. To help reduce variability, we ensured that all participants were untrained (defined as ≤3 h per week of exercise, verified by standardized 3-day activity recall and by 7-day accelerometer recording [Actigraph, Pensacola, FL]), had achieved Tanner Stage 2 or above in puberty (as assessed by physical exam by a pediatric endocrinologist), and were not prescribed medications known to effect IR, blood pressure, or lipids. Youth with type 1 diabetes had an HbA1c of <12% (108 mmol/mol). This study was approved by the University of Colorado Anschutz Medical Campus Institutional Review Board. Parental informed consent and participant assent were obtained from all participants ≤18 years old and participant consent from those aged 18 years old.
Overall Study Design
Participants underwent a screening visit, an overnight inpatient stay prior to a hyperinsulinemic-euglycemic clamp, and an exercise/imaging study visit, which included MRI of the leg for maximal cross-sectional area, proton MRS (1H-MRS) to measure IMCL content, and in-MRI exercise testing with phosphorus MRS (31P-MRS). 31P-MRS studies during exercise have been shown to correlate well with mitochondrial function assessed from muscle biopsy (20). Volunteers consumed an isocaloric diet (55% carbohydrate, 15% protein, and 30% fat) for 3 days prior to admission, and all subjects were free of acute illness. All testing was performed fasting, with no strenuous exercise for the 3 days prior. Blood glucose was checked in subjects with type 1 diabetes immediately prior to the MRI. Body composition (Hologic, Waltham, MA) was assessed by standard DEXA methods (18).
Measure of Insulin Sensitivity
In participants with type 1 diabetes, subcutaneous insulin was replaced with an overnight intravenous insulin infusion to normalize blood glucose levels (goal of 100 mg/dL). A 3.5 to 4.5-h hyperinsulinemic-euglycemic clamp (80 mU/m2 ⋅ min insulin) was performed fasting in the morning to measure IR, similar to our previously described methods (2). Briefly, serum glucose concentrations were maintained at ∼95 mg/dL based on serial blood samples drawn every 5 min and analyzed at the bedside with a YSI (Yellow Springs Instrument, OH). Glucose infusion rate (GIR) during the last stage of the clamp was expressed as milligrams dextrose infused per lean kilogram, corrected for serum blood glucose. A stable isotope 6,6 D2 glucose tracer was included as previously described in a subset of 9 youth with type 1 diabetes and 11 control youth to allow calculations of endogenous glucose release during hyperinsulinemia and isolate muscle insulin sensitivity (4).
MRI and MRS
Imaging Acquisition
Imaging and spectroscopy were performed on a General Electric 3 Tesla magnet with HDx MRI (General Electric, Milwaukee, WI) running version 15M4 software equipped with the General Electric multinuclear spectroscopy accessory of hardware and research software and a custom 1H/31P leg coil (Clinical MR Solutions, Brookfield, WI) (21). The coil was a concentric probe with an inner coil 9 cm in diameter (for 31P) and a 13-cm outer 1H coil for scout imaging and shimming. Cross-sectional area of the calf was measured and based on a sampling depth of 5.5 cm, and the percent of soleus and gastrocnemious muscle sampled was calculated (21).
Spectroscopy
Rates of mitochondrial phosphorylation were assessed by 31P-MRS performed at 51.70 MHz with the 1H/31P coil. The machine was autoshimmed with 1H, and then a 31P scan was performed for resting baseline measurements (long repetition time of 15,000 ms, flip angle of 135 degrees, and 32 scans) to measure a fully relaxed spectrum. The 31P exercise scan was then performed under partially saturated conditions (repetition time 1,000 ms, flip angle 135, 2,048 points). IMCL and extramyocellular lipid content were measured via 1H-MRS as previously described (2).
31P-MRS Exercise Protocol
Strength testing to determine maximal volitional contraction (MVC) was done on a custom-built magnetic resonance–compatible plantar flexion device with force measurement capability as previously described (21–23). The force transducer box was connected to an external readout stage (Omega Engineering, Stamford, CT) and then to a computer for recording of force throughout exercise with data acquisition software (Labview; National Instruments, Austin, TX).
The 31P-MRS exercise protocol consisted of measurements during rest for 90 s, isometric plantar flexion exercise for 90 s at 70% MVC, and recovery for 5 min postexercise. We selected a 90-s isometric exercise bout, as this perturbation has been extensively modeled and used for assessing both aerobic and anaerobic processes (24). Force was monitored continuously throughout the exercise, with verbal feedback to keep the force measurements within the target goal. The average force applied was recorded in kilograms. All participants were able to complete the exercise for 90 s at or near target force. In approximately half of the subjects, an additional exercise test was also performed at 45% MVC to test the effect of submaximal exercise, with at least 15 min of rest between exercise bouts.
Spectroscopy Analysis
Peak positions and areas of interest (phosphocreatine [PCr], inorganic free phosphate [Pi], β-ATP [three peaks], α-ATP [two peaks], and γ-ATP [two peaks], and phosphomonoester) were determined by time domain fitting using jMRUi software (25,26) using AMARES (A Method of Accurate, Robust and Efficient Spectral fitting), a nonlinear least square fitting algorithm using previously built prior knowledge files (27). A representative set of exercise spectra is shown in Fig. 1 for a 70% MVC exercise bout. All exercise spectra were corrected for saturation using the fully relaxed spectra. The jMRUi data were used to calculate metabolic variables as previously described (28). Calculations included the rates of oxidative phosphorylation (OxPhos) after exercise, creatine kinase (CK) reaction, initial PCr synthesis (VPCr), and anaerobic glycolysis (AnGly). ADP, PCr, and Pi time constants were calculated via regression analyses with Sigmaplot (Systat Software, Inc., San Jose, CA). VPCr, the initial rate of PCr resynthesis in the first 10 s after exercise, = (1/PCr time constant) × ([PCr rest] − [PCr end exercise]) in mmol/L/s. Qmax, the apparent mitochondrial capacity, i.e., maximal oxidative ATP production rate, = VPCr[1+(30/[ADP end exercise])] in mmol/L/s. AnGly in millimoles per liter per second is calculated from data collected in the last 10 s of exercise as follows:
Figure 1.

Representative plot of 70% exercise data for a volunteer with type 1 diabetes. Raw 31P-MRS data are shown. Each line represents 2 s of data. Time is on the Z axis, and the graph represents ∼5 min of data collection. The nadir of PCr peak height and maximum of Pi peak height is the end of exercise.
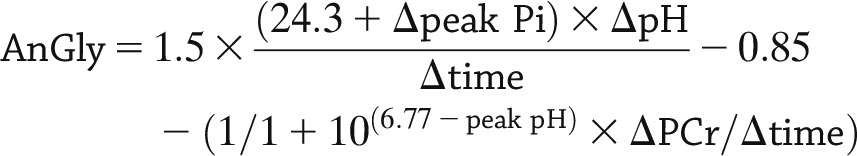 |
OxPhos in millimoles per liter per second is calculated from the slope of the line of ∆PCr/ ∆time during the first 10 s after cessation of exercise. CK represents the ATP production from CK reactions, which are not mitochondrial dependent. CK is calculated as the slope of the line of ∆PCR/∆time during the last 10 s of exercise in millimoles per liter per second. Qmax is based on VPc, end exercise (ADP), and an assumption of ADP control in mitochondria. It represents the apparent mitochondrial maximal oxidative ATP production rate relative to mitochondrial density. Qmax was only calculated from the 70% exercise bout data, which is near maximal.
1H-MRS data were analyzed as previously described (2). IMCL concentrations, obtained by reference to the unsuppressed water peak, are reported in institutional concentration units.
Statistical Analysis
The distribution of all variables was examined, and results are presented as mean ± SD, median (minimum, maximum), or proportions, as appropriate. Group comparisons were made using χ2 or Fisher exact test for proportions and the t test or Kruskal-Wallis test for continuous variables. The associations between blood glucose at time of MRI, HbA1c, BMI, BMI Z score, GIR, IMCL, and FFA concentrations during hyperinsulinemia and the three primary outcomes from the 70% exercise were examined (ADP time constant, Qmax, and OxPhos) using multiple regression. The Spearman correlation between each of the outcomes and the covariates was estimated, and only those covariates with a correlation significantly different from zero were included in the regression. P values <0.05 were considered significant. All statistical analyses were performed with SAS Software, version 9.3 (SAS, Cary, NC).
Results
Of the 38 adolescents enrolled, 21 had type 1 diabetes and 17 were nondiabetic control subjects. Participant demographics are shown in Table 1. Groups had similar sex and age distributions. Most participants were in mid- to late puberty. Participants with type 1 diabetes had a mean HbA1c of 8.2% (66 mmol/mol). Of note, markers of inflammation were similar between the groups, with no difference in hs-CRP, interleukin-6, myloperoxidase, aspartate aminotransferase (AST), alanine aminotransferase (ALT), or platelets (Table 1). Fasting LDL cholesterol, HDL cholesterol, and triglycerides were also similar between groups. Fasting FFA concentrations were significantly lower in type 1 diabetes, likely due to the suppressive effects of the overnight insulin infusion used to control glycemia. In addition, in youth with type 1 diabetes, serum FFA concentrations inappropriately failed to further suppress in response to hyperinsulinemia (consistent with adipose IR), and the GIR was significantly lower (10.10 mg/kglean/min [3.01, 20.78] vs. 18.92 mg/kglean/min [9.39, 28.09]; P < 0.01) (consistent with whole-body IR). Rd adjusted for tracer-determined endogenous glucose release in a subset of subjects was also different between groups (12.3 ± 4.4 mg/kglean/min in type 1 diabetic, 19.9 ± 3.9 in control subjects; P < 0.001), consistent with muscle IR. These differences in insulin sensitivity occurred despite similar serum glucose and insulin concentrations during the hyperinsulinemic-euglycemic clamp.
Table 1.
Demographics and metabolic measurements
| Type 1 diabetes (n = 21) | Control (n = 17) | |
|---|---|---|
| Sex (male) | 10 (47.6) | 5 (29.4) |
| Age (years) | 15.0 (13.0, 18.0) | 14.0 (12.0, 18.0) |
| Race** | ||
| White | 18 (85.7) | 9 (52.9) |
| Hispanic | 3 (14.3) | 4 (23.5) |
| Black | 0 | 2 (11.8) |
| Other (more than one) | 0 | 2 (11.8) |
| Tanner stage* | ||
| Missing | 1 (4.8) | 0 |
| 2 | 0 | 3 (17.7) |
| 3 | 1 (4.8) | 0 |
| 4 | 4 (19.1) | 7 (41.2) |
| 5 | 15 (71.4) | 7 (41.2) |
| BMI (kg/m2)** | 25.0 ± 4.2 | 21.4 ± 3.9 |
| Waist-to-hip ratio | 0.9 ± 0.1 | 0.8 ± 0.1 |
| Percent total body muscle mass | 65.9 ± 9.8 | 68.3 ± 8.8 |
| HbA1c (%)*** | 8.2 (6.7, 12.0) | 5.3 (4.7, 5.8) |
| HbA1c (mmol/mol)*** | 66 (50, 108) | 34 (28, 40) |
| Inflammatory markers | ||
| AST (units/L) | 28.5 (19.0, 45.0) | 29.0 (18.0, 62.0) |
| ALT (units/L) | 20.0 (3.0, 38.0) | 19.5 (12.0, 64.0) |
| hs-CRP (mg/L) | 0.49 (0.12, 7.71) | 0.20 (0.03, 20.60) |
| Interleukin-6 (pg/mL) | 1.25 (1.25, 12.50) | 1.25 (1.25, 11.90) |
| Myloperoxidase (mg/L) | 445.0 ± 193.7 | 433.7 ± 173.2 |
| White blood cells (1 kcell/μg) | 6.2 (3.9, 14.3) | 5.8 (3.4, 7.8) |
| Platelets (1 kcell/μg) | 240.7 ± 57.6 | 251.1 ± 51.2 |
| Lipids | ||
| Total cholesterol (mg/dL)** | 140.2 ± 24.9 | 163.2 ± 26.6 |
| Triglycerides (mg/dL) | 74.0 (42.0, 112.0) | 83.0 (44.0, 196.0) |
| HDL (mg/dL) | 47.1 ± 9.2 | 46.8 ± 10.9 |
| LDL (mg/dL)* | 78.5 ± 20.7 | 98.9 ± 26.1 |
| FFA baseline (mmol/L)* | 355.8 ± 226.3 | 563.9 ± 212.1 |
| Clamp measurements | ||
| FFA end clamp (mmol/L)* | 33.0 (7.5, 125.0) | 20.5 (15.0, 35.0) |
| Insulin end clamp (mmol/L) | 181.0 (108.0, 812.0) | 213.3 (158.0, 252.0) |
| Glucose end clamp (mg/dL) | 94.7 ± 4.7 | 99.1 ± 9.4 |
| GIR (mg/kglean/min)*** | 10.10 (3.01, 20.78) | 18.92 (9.39, 28.09) |
| Rd (mg/kglean/min)*** | 12.3 ± 4.4 | 19.9 ± 3.9 |
Data are n (%), mean ± SD, or median (minimum, maximum). P values are from χ2 or Fisher exact test, t test, or Kruskal-Wallis test, as appropriate.
*0.01 ≤ P ≤ 0.05;
**0.0001 ≤ P < 0.01;
***P < 0.0001. kcell, representing 1,000 cells.
The leg muscle cross-sectional area was slightly larger in the group with diabetes (3,681 ± 464 vs. 2,948 ± 776 mm2). However, the force generated per area was similar between groups (Table 2). The soleus represented the majority of muscle sampled in both groups (76 ± 7 type 1 diabetic vs. 81 ± 6% control subjects). Group means of raw data from 31P-MRS, including PCr, free phosphate, ADP concentrations, and pH, are shown in Fig. 2. Youth with type 1 diabetes had similar perturbations in PCr (Fig. 2B) and pH (Fig. 2D), but the peak ADP concentration was approximately half that of control subjects (Fig. 2C), and thus free phosphate concentrations were also lower in subjects with type 1 diabetes (Fig. 2A). 31P-MRS calculated measures for both 45% and 70% exercise are shown in Table 2. Youth with type 1 diabetes generated a similar isometric force relative to the size of their calf muscle for both levels of exertion, indicating equal relative workloads (for 70% exercise: 27.67 ± 5.44 vs. 24.69 ± 5.74 kg/cm2), and the mean force over the entire 70% exercise bout was identical, as shown in Fig. 3. However, during 70% exercise, youth with type 1 diabetes had a significantly longer ADP time constant (23.38 ± 5.26 vs. 18.80 ± 3.90 s; P < 0.01), indicating slower mitochondrial rates of conversion of ADP to ATP and a lower rate of oxidative phosphorylation (0.09 mmol/L/s [0.01, 0.43] vs. 0.21 mmol/L/s [0.10, 0.37]; P < 0.01). The Qmax was also lower in youth with type 1 diabetes (0.43 mmol/L/s [0.09, 1.10] vs. 0.54 mmol/L/s [0.41, 2.50]; P < 0.05). The rate of AnGly was higher in youth with type 1 diabetes (0.37 mmol/L/s [0.09, 0.69] vs. 0.22 mmol/L/s [0.06, 0.51]; P < 0.05). Rates of the CK reaction were similar between groups. Overall, results during 45% exercise were similar to those seen with 70% exercise (Fig. 4). IMCL was similar between groups, with 424.18 (150.05, 1,260.00) in type 1 diabetic and 464.80 (197.32, 1,490.00) in control subjects. Mean blood glucose at the time of MRS in the youth with type 1 diabetes was 134 mg/dL (98, 394).
Table 2.
Markers of muscle metabolism during exercise
| Type 1 diabetes | Control | |
|---|---|---|
| 45% exercise | 11 | 16 |
| Force/area (kg/cm2) | 18.60 ± 3.21 | 18.05 ± 4.23 |
| AnGly (mmol/L/s) | 0.33 (0.04, 0.64) | 0.28 (0.10, 0.67) |
| CK ATP production (mmol/L/s) | 0.07 ± 0.06 | 0.07 ± 0.06 |
| OxPhos (mmol/L/s)* | 0.05 (0.00, 0.32) | 0.14 (0.03, 0.28) |
| Total ATP production (mmol/L) | 0.47 ± 0.19 | 0.52 ± 0.17 |
| ADP time constant (s) | 21.71 ± 5.34 | 17.61 ± 5.64 |
| PCr time constant (s) | 27.51 (15.53, 41.02) | 22.50 (13.52, 39.15) |
| VPCr (mmol/L//s)** | 0.11 ± 0.05 | 0.24 ± 0.13 |
| 70% exercise | 21 | 17 |
| Force/area (kg/cm2) | 27.67 ± 5.44 | 24.69 ± 5.73 |
| AnGly (mmol/L/s)* | 0.37 (0.09, 0.69) | 0.22 (0.06, 0.51) |
| CK ATP production (mmol/L/s) | 0.08 ± 0.07 | 0.08 ± 0.05 |
| OxPhos (mmol/L/s)** | 0.09 (0.01, 0.43) | 0.21 (0.10, 0.37) |
| Total ATP production (mmol/L) | 0.56 ± 0.24 | 0.56 ± 0.09 |
| ADP time constant (s)** | 23.38 ± 5.26 | 18.80 ± 3.90 |
| PCr time constant (s) | 29.18 (17.45, 51.26) | 30.15 (8.14, 39.00) |
| VPCr (mmol/s)** | 0.23 ± 0.16 | 0.42 ± 0.27 |
| Qmax (mmol/s)* | 0.43 (0.09, 1.10) | 0.54 (0.41, 2.50) |
Data are means ± SD, n, or median (minimum, maximum). P values are from t test or Kruskal-Wallis test, as appropriate.
0.01 ≤ P ≤ 0.0.5;
0.0001 ≤ P < 0.01;
P < 0.0001.
Figure 2.

Average muscle metabolite concentrations before, during, and after 45% exercise. A: Inorganic phosphate. B: PCr concentrations. C: ADP concentrations. D: Intracellular pH.
Figure 3.

Average muscle metabolite concentrations before, during, and after 70% exercise. A: Inorganic phosphate. B: PCr concentrations. C: ADP concentrations. D: Intracellular pH.
Figure 4.

Plantar flexion force before, during, and after 70% exercise. A: Average force during the 45% bout. B: Average force exerted during the 70% bout. The forces are not different between subject groups.
Results from regression analyses for the primary mitochondrial end points (ADP time constant, OxPhos, and Qmax from 70% exercise) are shown in Table 3. Variables considered in the initial Spearman correlation analysis for each regression were HbA1c, GIR, blood glucose at time of MRI, and FFA concentration after hyperinsulinemia (end FFA). HbA1c, GIR, and end FFA concentration were univariately associated with ADP time constant, but only GIR was independently associated, with a lower GIR associated with a higher ADP time constant. HbA1c and GIR were univariately associated with oxidative phosphorylation, but only GIR was independently associated with oxidative phosphorylation, with a lower GIR associated with lower oxidative phosphorylation. Finally, HbA1c, GIR, and IMCL were univariately correlated with Qmax, but only IMCL was independently associated with Qmax. Of note, a higher IMCL was associated with a higher or more efficient Qmax. Spearman analysis was performed with Rd corrected for hepatic glucose output to isolate muscle IR in a subgroup with similar results to the unadjusted GIR, but regression analysis could not be performed owing to the smaller sample size.
Table 3.
Parameter estimates and P values for regression with mitochondrial outcomes from 70% MVC exercise
| Outcome | Covariate | Parameter estimate | SE of parameter estimate | P |
|---|---|---|---|---|
| ADP TC | Intercept | 25.23 | 4.96 | <0.0001 |
| ADP TC | HbA1c | 0.18 | 0.46 | 0.7025 |
| ADP TC | GIR | −0.36 | 0.15 | 0.0261 |
| ADP TC | End FFA concentration | −0.01 | 0.04 | 0.8735 |
| OxPhos | Intercept | 0.26 | 0.29 | 0.3929 |
| OxPhos | HbA1c | 0.001 | 0.028 | 0.9471 |
| OxPhos | GIR | 0.02 | 0.01 | 0.0382 |
| Qmax | Intercept | 0.44 | 0.23 | 0.0802 |
| Qmax | HbA1c | −0.02 | 0.02 | 0.3431 |
| Qmax | GIR | 0.01 | 0.01 | 0.1933 |
| Qmax | IMCL | 0.0002 | 0.0001 | 0.0159 |
Items in bold indicate statistical significance.
Discussion
The existence of significant IR in individuals with type 1 diabetes is increasingly recognized; yet, the relationship between mitochondrial function and IR has not been explored in youth with type 1 diabetes. We found that nonobese youth with type 1 diabetes have muscle mitochondrial dysfunction compared with nondiabetic youth of similar age, pubertal stage, BMI, and level of habitual physical activity, which correlates inversely with insulin sensitivity. Specifically, youth with type 1 diabetes had a slower rate of conversion of ADP back to ATP after cessation of near-maximal isometric exercise, despite relatively lower ADP production during exercise. Thus, youth with type 1 diabetes seem to use AnGly as a fuel source rather than OxPhos. These findings are consistent after both moderate and submaximal exercise loads, showing mitochondrial dysfunction even at levels of exercise performed in everyday life. Of note, these changes in mitochondrial function were found in the absence of other features of the metabolic syndrome such as dyslipidemia or increased IMCL and did not relate to markers of glycemic control or BMI. Thus, youth with type 1 diabetes have a unique phenotype of IR and mitochondrial dysfunction but lack many other features typical of IR.
This is the first report of mitochondrial dysfunction in youth with type 1 diabetes, and mitochondrial studies in adults with type 1 diabetes using either nutritional or exercise perturbations are conflicting to date. For example, Kacerovsky et al. (17) measured ATP flux with 31P-MRS fasted and during a hyperinsulinemic-euglycemic clamp. They found that insulin-stimulated glucose flux through ATP synthase during the clamp was reduced by 25% in type 1 diabetes, and glucose 6 phosphate concentrations were reduced by 42% (17). Since the glucose 6 phosphate was reduced more than the ATP synthase, one interpretation is that delivery of glucose to the cell is impaired, rather than the mitochondria itself being dysfunctional. The authors found a tight relationship between HbA1c, Rd, and ATP flux and thus concluded that despite tight glycemic control (mean HbA1c of 6.89 ± 0.4% [52 ± 1 mmol/mol] in the type 1 diabetic participants), glucotoxicity was the cause of the findings. Alternatively, Antonetti et al. (15) found that mitochondrial gene expression, specifically, cytochorome oxidase I, cytochodrome oxidase III, NADH dehydrogenase IV, and 12s mRNA gene expression, was actually increased in muscle biopsies from patients with type 1 diabetes and vascular disease, suggesting increased mitochondrial function, perhaps in response to hypoxemia. In men with type 1 diabetes, postexercise PCr recovery assessed with 31P-MRS was slower after 120 s of 75% maximal plantar flexion; however, the participants with diabetes worked harder than the control subjects (19). Item et al. (18) found that mitochondrial capacity as assessed with 31P-MRS during 85% plantar flexion for 30 s was not impaired in women with type 1 diabetes relative to control subjects and related to HbA1c. However, this short exercise paradigm may not be of an adequate duration to test OxPhos, as initial force generation with muscle occurs via the PCr reaction (29). In summary, three of five studies in adults indicated that mitochondrial function, stimulated by either insulin or exercise, may be impaired in type 1 diabetes, and our data add to this body of evidence that the defect occurs early in diabetes development. What remains unclear, however, is whether the mitochondria themselves are impaired or whether nutrient delivery (i.e., reduced muscle blood flow secondary to lack of insulin action) or availability is the cause of the resulting poor mitochondrial function. Finally, it is unclear whether abnormalities in mitochondrial function seen in adults will be the same as those seen in youth.
We found that mitochondrial function was related to IR but not glycemia. Both ADP time constant and OxPhos were related to IR but not to markers of glucose control including HbA1c or blood glucose at the time of exercise testing. Previous studies proposed that IR in type 1 diabetes was primarily related to hyperglycemia (30,31). However, this assumption has been challenged by more recent studies in both animal models and adults with type 1 diabetes in reasonable glycemic control (4,6,32). We previously found that neither HbA1c nor 3-day glucose readings with continuous glucose monitoring relate to IR in adolescents with type 1 diabetes (2). In studies of nondiabetic patients that included assessments of both IR and mitochondrial function, glycemia did not relate to either IR or mitochondrial function (10,15,33,34). In adults with type 2 diabetes, mitochondrial function was impaired relative to control subjects but was only affected by extreme hyperglycemia, and an HbA1c in the range similar to that of our patient population did not relate to mitochondrial function (34). Thus, it may be that when glucose concentrations are managed near goal clinical glucose ranges, IR and mitochondrial function are not strongly influenced by hyperglycemia.
We found that mitochondrial dysfunction was tightly associated with muscle IR as measured with a hyperinsulinemic-euglycemic clamp but not serum FFA or IMCL. Moreover, we previously demonstrated that IMCL is not elevated in pediatric type 1 diabetes, and others have demonstrated that IMCL does not relate to IR in adults with type 1 diabetes (2,35). In this cohort, we similarly found no elevation in IMCL in youth with type 1 diabetes and that IMCL was associated positively with increased Qmax. This positive relationship of IMCL with Qmax is consistent with the athlete’s paradox, where IMCL rises in healthy subjects as individuals are more trained, and it is negatively correlated with insulin sensitivity (36). Finally, we did find that youth with type 1 diabetes had higher serum FFA during hyperinsulinemia; yet, we did not find an association between FFA suppression and mitochondrial function. However, the serum insulin concentrations achieved during the clamp were supraphysiologic at 200 IU/mL. It is possible that in daily life, serum FFA over 24 h are higher in youth with type 1 diabetes than in control subjects due to inadequate insulin concentrations for suppression of lipolysis and that these FFA elevations may decrease muscle blood flow and secondarily nutrient delivery or directly impair mitochondrial function.
The interpretability of exercise data is always dependent on the quality of the exercise paradigm and the muscle sampled. The raw ADP data shown in Fig. 3C show that the youth with type 1 diabetes produced much less ADP that the control volunteers during a 70% exercise perturbation. The simplest explanation for this discrepancy is that the youth with type 1 diabetes did not perform as much physical work as the control group. However, the force data indicate that participants with type 1 diabetes pushed with a force equal to that of control subjects, and when normalized for leg muscle area, the youth with type 1 diabetes pushed an average of 3 kg harder. Further, the pH curves are similar between the groups, indicating that the intracellular perturbation was equivalent, and the intracellular environment did not become acidotic enough to prevent optimal mitochondrial function (32). Finally, in our study a subset of patients performed two exercise bouts with the second at 45% of MVC. The overall results from this subset are consistent with the 70% results, indicating that the difference between groups is not likely reflective of unequal exercise levels or ischemia induced from maximal contraction. Furthermore, these results illustrate that mitochondrial deficiencies are seen in near-maximal as well as submaximal exercise levels in this population, implying that the mitochondrial dysfunction in type 1 diabetic youth could impact exercise performance in youth. Analysis of the imaging data indicates that the majority of muscle sampled was the soleus, a primarily oxidative muscle type. Thus, it appears that youths with type 1 diabetes are generating the force for the exercise via more nonoxidative pathways, such as AnGly.
There are several limitations to a broader applicability of our findings to all youth with type 1 diabetes. Our sample size was relatively small, as these studies are complex and costly. To be certain that our findings were not due to BMI, we performed a subanalysis of only those volunteers with a BMI <85th percentile and found identical results as presented (data not shown). Thus, we do not believe that our findings of a difference in the youth with type 1 diabetes are an artifact of adiposity. Owing to the inclusion criteria, the study cannot be considered generalizable to physically active or obese youth with type 1 diabetes. Moreover, the impact of FFAs in type 1 diabetes on mitochondrial function requires further study, as we only assessed FFA suppression of high-dose insulin in this analysis.
In summary, we found that youth with type 1 diabetes have decreased mitochondrial OxPhos after exercise, which correlates with IR but not acute or chronic glycemia, IMCL, or FFA. As future therapeutics are developed to directly improve insulin sensitivity, those that involve mitochondrial function should also be considered for individuals with type 1 diabetes.
Article Information
Acknowledgments. The authors thank the participants and their families for participating.
Funding. J.E.B.R. was funded by VA Merit, the Denver Research Institute, and grant 5P01HL014985 from the Center for Women’s Health Research. J.G.R. was funded by American Diabetes Association (ADA) grant 1-12-CT-64 and by the Center for Women’s Health Research. K.J.N. was funded by National Center for Research Resources (NCRR) grant K23-RR-020038-01, National Institutes of Health (NIH)/NCRR Colorado Clinical and Translational Science Institute (CTSI) Co-Pilot grant TL1-RR-025778, NIH/National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant 1R56-DK-088971-01, JDRF grant 5-2008-291, and ADA grant 7-11-CD-08. M.C.-G. was funded by the Thrasher Pediatric Research Foundation Mentored Pilot grant, NIH/NCRR Colorado CTSI Co-Pilot grant TL1-RR-025778, Pediatric Endocrinology Fellowship training grant NIDDK T32-DK-063687, and a Pediatric Endocrine Society Fellowship. This research was also supported by Adult Clinical Translational Research Center NIH grant M01-RR0051, Pediatric Clinical Translational Research Center NIH grant 5MO1-RR-00069, and NIH/NCRR Colorado CTSI grant UL1-RR-025780.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. M.C.-G. researched data and wrote the manuscript. B.R.N., M.S.B., A.D.B., and B.B. researched data and edited the manuscript. B.D. performed data analysis and edited the manuscript. J.G.R. and J.E.B.R. assisted with study design, researched data, contributed to discussion, and edited the manuscript. L.P. performed all statistical analysis and edited the manuscript. K.J.N. researched data, contributed to discussion, and edited the manuscript. M.C.-G. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the 72nd Scientific Sessions of the American Diabetes Association, Philadelphia, PA, 8–12 June 2012.
References
- 1.Dabelea D, Bell RA, D’Agostino RB Jr, et al. ; Writing Group for the SEARCH for Diabetes in Youth Study Group . Incidence of diabetes in youth in the United States. JAMA 2007;297:2716–2724 [DOI] [PubMed] [Google Scholar]
- 2.Nadeau KJ, Regensteiner JG, Bauer TA, et al. Insulin resistance in adolescents with type 1 diabetes and its relationship to cardiovascular function. J Clin Endocrinol Metab 2010;95:513–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orchard TJ, Olson JC, Erbey JR, et al. Insulin resistance-related factors, but not glycemia, predict coronary artery disease in type 1 diabetes: 10-year follow-up data from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes Care 2003;26:1374–1379 [DOI] [PubMed] [Google Scholar]
- 4.Bergman BC, Howard D, Schauer IE, et al. Features of hepatic and skeletal muscle insulin resistance unique to type 1 diabetes. J Clin Endocrinol Metab 2012;97:1663–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maahs DM, Nadeau K, Snell-Bergeon JK, et al. Association of insulin sensitivity to lipids across the lifespan in people with Type 1 diabetes. Diabet Med 2011;28:148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schauer IE, Snell-Bergeon JK, Bergman BC, et al. Insulin resistance, defective insulin-mediated fatty acid suppression, and coronary artery calcification in subjects with and without type 1 diabetes: The CACTI study. Diabetes 2011;60:306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson JC, Erbey JR, Williams KV, et al. Subclinical atherosclerosis and estimated glucose disposal rate as predictors of mortality in type 1 diabetes. Ann Epidemiol 2002;12:331–337 [DOI] [PubMed] [Google Scholar]
- 8.Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med 2006;119(Suppl. 1):S10–S16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 2003;300:1140–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cree MG, Fram RY, Herndon DN, et al. Human mitochondrial oxidative capacity is acutely impaired after burn trauma. Am J Surg 2008;196:234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cree MG, Newcomer BR, Herndon DN, et al. PPAR-alpha agonism improves whole body and muscle mitochondrial fat oxidation, but does not alter intracellular fat concentrations in burn trauma children in a randomized controlled trial. Nutr Metab (Lond) 2007;4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rickels MR, Naji A, Teff KL. Insulin sensitivity, glucose effectiveness, and free fatty acid dynamics after human islet transplantation for type 1 diabetes. J Clin Endocrinol Metab 2006;91:2138–2144 [DOI] [PubMed] [Google Scholar]
- 13.Perseghin G, Lattuada G, Danna M, et al. Insulin resistance, intramyocellular lipid content, and plasma adiponectin in patients with type 1 diabetes. Am J Physiol Endocrinol Metab 2003;285:E1174–E1181 [DOI] [PubMed] [Google Scholar]
- 14.Snell-Bergeon JK, Nadeau K. Cardiovascular disease risk in young people with type 1 diabetes. J Cardiovasc Transl Res 2012;5:446–462 [DOI] [PubMed] [Google Scholar]
- 15.Antonetti DA, Reynet C, Kahn CR. Increased expression of mitochondrial-encoded genes in skeletal muscle of humans with diabetes mellitus. J Clin Invest 1995;95:1383–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karakelides H, Asmann YW, Bigelow ML, et al. Effect of insulin deprivation on muscle mitochondrial ATP production and gene transcript levels in type 1 diabetic subjects. Diabetes 2007;56:2683–2689 [DOI] [PubMed] [Google Scholar]
- 17.Kacerovsky M, Brehm A, Chmelik M, et al. Impaired insulin stimulation of muscular ATP production in patients with type 1 diabetes. J Intern Med 2011;269:189–199 [DOI] [PubMed] [Google Scholar]
- 18.Item F, Heinzer-Schweizer S, Wyss M, et al. Mitochondrial capacity is affected by glycemic status in young untrained women with type 1 diabetes but is not impaired relative to healthy untrained women. Am J Physiol Regul Integr Comp Physiol 2011;301:R60–R66 [DOI] [PubMed] [Google Scholar]
- 19.Crowther GJ, Milstein JM, Jubrias SA, Kushmerick MJ, Gronka RK, Conley KE. Altered energetic properties in skeletal muscle of men with well-controlled insulin-dependent (type 1) diabetes. Am J Physiol Endocrinol Metab 2003;284:E655–E662 [DOI] [PubMed] [Google Scholar]
- 20.Lanza IR, Bhagra S, Nair KS, Port JD. Measurement of human skeletal muscle oxidative capacity by 31P-MR spectroscopy: a cross-validation with in vitro measurements. J Magn Reson Imaging 2011;34:1143–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cree-Green M, Newcomer BR, Brown M, et al. Method for controlled mitochondrial perturbation during phosphorus MRS in children. Med Sci Sports Exerc. 26 February 2014 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larson-Meyer DE, Newcomer BR, Hunter GR, Hetherington HP, Weinsier RL. 31P MRS measurement of mitochondrial function in skeletal muscle: reliability, force-level sensitivity and relation to whole body maximal oxygen uptake. NMR Biomed 2000;13:14–27 [DOI] [PubMed] [Google Scholar]
- 23.Bamman MM, Caruso JF. Resistance exercise countermeasures for space flight: implications of training specificity. J Strength Cond Res 2000;14:45–9 [PubMed]
- 24.Sirikul B, Hunter GR, Larson-Meyer DE, Desmond R, Newcomer BR. Relationship between metabolic function and skeletal muscle fatigue during a 90 s maximal isometric contraction. Appl Physiol Nutr Metab 2007;32:394–399 [DOI] [PubMed]
- 25.van den Boogaart A. MRUI MANUAL V. 96.3. A User's Guide to the Magnetic Resonance User Interface Software Package. Delft, Netherlands, Delft Technical University Press, 1997 [Google Scholar]
- 26.Klose U. In vivo proton spectroscopy in presence of eddy currents. Magn Reson Med 1990;14:26–30 [DOI] [PubMed] [Google Scholar]
- 27.Rico-Sanz J, Thomas EL, Jenkinson G, Mierisová S, Iles R, Bell JD. Diversity in levels of intracellular total creatine and triglycerides in human skeletal muscles observed by 1H-MRS. J Appl Physiol (1985) 1999;87:2068–2072 [DOI] [PubMed] [Google Scholar]
- 28.Newcomer BR, Boska MD. Adenosine triphosphate production rates, metabolic economy calculations, pH, phosphomonoesters, phosphodiesters, and force output during short-duration maximal isometric plantar flexion exercises and repeated maximal isometric plantar flexion exercises. Muscle Nerve 1997;20:336–346 [DOI] [PubMed] [Google Scholar]
- 29.Spriet LL. Anaerobic metabolism in human skeletal muscle during short-term, intense activity. Can J Physiol Pharmacol 1992;70:157–165 [DOI] [PubMed] [Google Scholar]
- 30.Yki-Järvinen H, Helve E, Koivisto VA. Hyperglycemia decreases glucose uptake in type I diabetes. Diabetes 1987;36:892–896 [DOI] [PubMed] [Google Scholar]
- 31.Vuorinen-Markkola H, Koivisto VA, Yki-Jarvinen H. Mechanisms of hyperglycemia-induced insulin resistance in whole body and skeletal muscle of type I diabetic patients. Diabetes 1992;41:571–580 [DOI] [PubMed] [Google Scholar]
- 32.Liu H-Y, Cao SY, Hong T, Han J, Liu Z, Cao W. Insulin is a stronger inducer of insulin resistance than hyperglycemia in mice with type 1 diabetes mellitus (T1DM). J Biol Chem 2009;284:27090–27100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest 2000;106:171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rabøl R, Højberg PM, Almdal T, et al. Effect of hyperglycemia on mitochondrial respiration in type 2 diabetes. J Clin Endocrinol Metab 2009;94:1372–1378 [DOI] [PubMed] [Google Scholar]
- 35.Thankamony A, Tossavainen PH, Sleigh A, et al. Short-term administration of pegvisomant improves hepatic insulin sensitivity and reduces soleus muscle intramyocellular lipid content in young adults with type 1 diabetes. J Clin Endocrinol Metab 2014;99:639–647 [DOI] [PubMed] [Google Scholar]
- 36.Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab 2001;86:5755–5761 [DOI] [PubMed] [Google Scholar]