

Key Points
In this first ALL GWAS in AYAs, we determined that inherited GATA3 variants strongly influence ALL susceptibility in this age group.
These findings revealed similarities and differences in the genetic basis of ALL susceptibility between young children and AYAs.
Abstract
Acute lymphoblastic leukemia (ALL) in adolescents and young adults (AYA) is characterized by distinct presenting features and inferior prognosis compared with pediatric ALL. We performed a genome-wide association study (GWAS) to comprehensively identify inherited genetic variants associated with susceptibility to AYA ALL. In the discovery GWAS, we compared genotype frequency at 635 297 single nucleotide polymorphisms (SNPs) in 308 AYA ALL cases and 6,661 non-ALL controls by using a logistic regression model with genetic ancestry as a covariate. SNPs that reached P ≤ 5 × 10−8 in GWAS were tested in an independent cohort of 162 AYA ALL cases and 5,755 non-ALL controls. We identified a single genome-wide significant susceptibility locus in GATA3: rs3824662, odds ratio (OR), 1.77 (P = 2.8 × 10−10) and rs3781093, OR, 1.73 (P = 3.2 × 10−9). These findings were validated in the replication cohort. The risk allele at rs3824662 was most frequent in Philadelphia chromosome (Ph)-like ALL but also conferred susceptibility to non–Ph-like ALL in AYAs. In 1,827 non-selected ALL cases, the risk allele frequency at this SNP was positively correlated with age at diagnosis (P = 6.29 × 10−11). Our results from this first GWAS of AYA ALL susceptibility point to unique biology underlying leukemogenesis and potentially distinct disease etiology by age group.
Introduction
Cancer survival rates have been steadily increasing in the United States across age groups except for in adolescents and young adults (AYA; age 16 to 39 years), partly because of the persisting inferior treatment response in hematologic malignancies.1 Particularly with acute lymphoblastic leukemia (ALL), age as a continuous variable is negatively correlated with prognosis in spite of risk-adapted combination chemotherapy.2 In an analysis of 21 626 ALL cases diagnosed between 1990 and 2005 and treated on Children’s Oncology Group (COG) frontline protocols, survival rates decreased significantly with increasing age at diagnosis regardless of treatment era (eg, 94.1% for age 1 to 10, 84.7% for age 10 to 15, and 75.9% for age 15 to 22 years in the 2000-2005 cohort).3 Although pediatric-based treatment regimens have been tested in AYA populations and have resulted in improved survival, the gap in treatment outcome between age groups persists, and ALL remains one of the leading causes of cancer-related deaths in the AYA population.4,5
The inferior prognosis of AYA is likely to be multifactorial and includes socioeconomic factors, medication adherence, clinical trial enrollment and, importantly, age-related differences in ALL tumor and host biology.6 For example, as age increases, there is a progressive rise in prevalence of ALL genetic subtypes with poor prognosis such as Philadelphia chromosome–positive (Ph+),7 Ph-like,8 or intrachromosomal amplification of chromosome 21,9 whereas subtypes with favorable outcome (high hyperdiploidy10 and ETV6-RUNX111) become less common. These comparisons are informative but are also limited because they are primarily driven by ALL features discovered in children and/or older adults. As a result, differences in tumor biology between AYA and childhood ALL may have been underestimated, and genomic profiling studies focusing on the AYAs are likely to reveal novel molecular features unique to this population.
Inherited genetic variations can strongly influence both the susceptibility to ALL12-15 and treatment outcomes.16-20 For example, genome-wide association studies (GWASs) have identified germline genetic variants at ARID5B, IKZF1, CEBPE, PIP4K2A, and CDKN2A/CDKN2B loci with substantial cumulative effects on ALL disease risk in children. These ALL susceptibility genes are involved in lymphoid cell development, cell cycle control, and tumor suppression, collectively affecting leukemogenesis. Although one’s inherited genetic variants remain unchanged over a lifetime, it is possible that the effects of these susceptibility variants vary by age, thus contributing to the age-related differences in ALL incidence and subtype. In fact, when we examined the risk of ALL conferred by the ARID5B variant, there was a clear trend of diminishing effects with increasing age (ie, allelic odds ratio of 2.01, 1.8, and 1.48 in children younger than age 5, 5 to 10, and older than 10 years, respectively).15 However, germline variants related to ALL risk in the AYA population have not been comprehensively examined.
To better understand the potential unique leukemia etiology in AYAs, we conducted the first GWAS to systemically interrogate germline single nucleotide polymorphisms (SNPs) for their contribution to ALL risk in this age group.
Methods
Study design and patients
In the discovery GWAS, the ALL cases consisted of 209 adolescents (median age, 17.4 years; range, 16 to 21 years) and 99 young adults (median age, 24.3 years; range, 21 to 39 years) with newly diagnosed B-cell ALL who were treated on the Children’s Oncology Group (COG; N = 202),21 the Alliance-Cancer and Leukemia Group B (N = 56),22 Eastern Cooperative Oncology Group E2993 (N = 29),23 MD Anderson Cancer Center (N = 11) 24-27, and St. Jude Children’s Research Hospital (N = 10) trials.28 The subjects were chosen on the basis of the availability of germline DNA, which was extracted from peripheral blood samples during clinical remission (<5% blasts cells in bone marrow). A total of 6,661 unrelated subjects from the Multi-Ethnic Study of Atherosclerosis (MESA) cohort (dbGaP phs000209.v9) were considered as non-ALL control subjects because the prevalence of adult survivors of childhood ALL is extremely low.14
For the replication analyses, 162 children with ALL age 16 to 21 years from the COG P9900 protocols (COG P9905 [NCT00005596], COG P9904 [NCT0000558529], COG P9906 [NCT0000560330], and COG AALL0232 [NCT0007572521]) were included. A set of 5755 unrelated non-ALL controls were included in the replication analysis: 1228 African Americans (AAs) from the AIDS Linked to Intravenous Experience (ALIVE) cohort,31 880 Hispanic Americans from the Genetics of Asthma in Latino Americans (GALA) study,32 and 3647 European Americans (EAs) from the Genetic Association Informative Network (GAIN) schizophrenia cohort (dbGAP phs000021.v3.p2)33 and GAIN bipolar cohort (phs000017.v3.p134; Figure 1).
Figure 1.

GWAS study design. ALL susceptibility variants were identified by comparing SNP genotype frequency in AYA ALL cases compared with non-ALL controls in the discovery GWAS, followed by replication. CALGB: The Alliance-Cancer and Leukemia Group B; COG: Children’s Oncology Group; ECOG: Eastern Cooperative Oncology Group; MDACC: MD Anderson Cancer Center; SJ: St. Jude Children’s Research Hospital.
The clinical trials were approved by local institutional review boards, and informed consent for trial enrollment and banking of specimens for future research was obtained from parents, guardians, or patients, as appropriate. This study was approved by the St. Jude Children’s Research Hospital institutional review board.
Genotyping and quality control
Genome-wide SNP genotyping was performed by using the Affymetrix Human SNP Array 6.0 for ALL cases in the discovery GWAS, those in the COG P9905, P9904, and AALL0232 cohorts, and for all non-ALL controls (dbGaP MESA, ALIVE, GAIN, and GALA). Genotype calls (coded as 0, 1, and 2 for AA, AB, and BB genotypes) were determined by the Birdseed v2 (Affymetrix SNP 6.0) algorithm.35 Samples for which genotypes were ascertained for less than 95% of SNPs on the array were deemed to have failed and were excluded from the analyses. For the ALL cases in the COG P9906 trial, genome-wide SNP genotyping was performed by using Affymetrix Human SNP Array 500K, and GATA3 SNPs were genotyped by polymerase chain reaction and Sanger sequencing, as described previously.36 We did not observe evidence of potential genotyping errors in the germline DNA because of tumor cell contamination (data not shown).
Prior to GWAS, SNPs were subjected to a series of quality control steps (supplemental Figure 1, available on the Blood Web site). First, we filtered SNPs on the basis of minor allele frequency (MAF) and SNP call rate: for SNPs with an MAF of 1% to 3%, we excluded those with a call rate <99%; for SNPs with an MAF of 3% to 5%, we excluded those with a call rate <98%; for SNPs with an MAF of >5%, we excluded those with a call rate <95%. An additional filtering step was applied in the GWAS involving non-ALL controls: we removed SNPs for which genotype frequencies differed significantly among control groups (ie, dbGaP MESA vs HapMap unrelated CEU or dbGaP MESA vs the GAIN bipolar cohort [dbGaP phs000017.v3]33; P < 10−6 by χ2 test), and the comparison was restricted to EAs. Finally, those SNPs deviating from Hardy-Weinberg equilibrium (P < .01 in EA cases or controls) were also excluded from the analysis. After quality control filters were applied, 635 297 SNPs were included in the GWAS.
Genetic ancestry and population structure
Genetic ancestry was determined by using STRUCTURE (version 2.2.3),37 based on genotypes at 30 000 SNPs randomly selected from the Affymetrix SNP arrays. HapMap samples from descendants of Northern Europeans (CEU; N = 60), West Africans (YRI; N = 60), East Asians (CHB/JPT; N = 90), and Native Americans (NAs; N = 105)38 references were used to represent European, African, Asian, and Native American ancestries, respectively. We assumed that these four ancestries summed to 100% in each genotyped individual. EAs, AAs, NAs, and Asians were defined as having >95% European genetic ancestry, >70% African ancestry, >90% NA ancestry, and >90% Asian ancestry, respectively. Hispanics were individuals for whom NA ancestry was >10% and greater than African ancestry (including genetically defined NAs). The rest of the subjects were grouped as “Others.”
We also performed principal component analysis of ALL cases and controls in the discovery GWAS cohort, including all SNPs that passed the quality control and observed comparable population structure between cases and controls (supplemental Figure 2). In addition, we exhaustively examined potential relatedness within ALL cases and within controls included in the discovery GWAS by computing pairwise identity by descent probabilities. No evidence of first or second-degree relationships was identified.
ALL somatic genomic lesions
In the discovery cohort, the ALL genetic subtypes included high hyperdiploid (>50 chromosomes), ETV6-RUNX1, TCF3-PBX1, MLL-rearranged, BCR-ABL1 (Ph+), and Ph-like (with or without CRLF2 rearrangements). Ph-like and ERG-deregulated ALL were defined by Predictive Analysis of Microarrays.21,39 In the COG P9900 series, ALL subtypes included ETV6-RUNX1, TCF3-PBX1, hyperdiploid, and MLL-rearranged, with the remainder of cases considered as B-other. GATA3 expression was quantified in 237 ALL blasts in 237 AYA cases, using Affymetrix U133A array.8
Statistical analysis
In the discovery GWAS, the association test between genotypes at each of the 635 297 SNPs and ALL susceptibility was tested by comparing genotype frequency between AYA ALL cases and non-ALL controls using a logistic regression test under an additive model, including European, African, and NA ancestry (as continuous variables) as covariates using PLINK (v1.07).40 Population stratification was assessed by the construction of a quantile-quantile plot (supplemental Figure 3), and there was only a minimal inflation at the upper tail of the distribution (λ = 1.02). SNPs that reached the association P ≤ 5 × 10−8 in the discovery GWAS were evaluated in the independent replication series (1-tailed test). In both discovery and replication groups, we also tested GATA3 SNPs separately in EAs, AAs, and Hispanic Americans.
R (version 2.15.1) statistical software was used for the rest of the analyses unless indicated otherwise. Statistical tests were chosen as appropriate and according to the phenotype distribution (eg, normally or binomially distributed for continuous or categorical variables, respectively). Associations of SNP genotype with somatic lesions and age were estimated by logistic regression and linear regression test, respectively, after adjusting for genetic ancestry. Associations of GATA3 SNP genotype with GATA3 gene expression was assessed by linear regression model, adjusting for genetic ancestry.
Results
AYA ALL GWAS
In the discovery GWAS, we compared genotype frequency at 635 297 SNPs between 308 AYA ALL cases and 6,661 non-ALL controls (Figure 1). After adjusting for genetic ancestry, only two SNPs at 10p14 within the GATA3 gene reached genome-wide significance: rs3824662 (odds ratio [OR], 1.77; 95% confidence interval [CI], 1.48 to 2.12; P = 2.84 × 10−10) and rs3781093 (OR, 1.73; 95% CI, 1.44 to 2.08; P = 3.20 × 10−9; Table 1 and Figure 2). These two SNPs were in strong linkage disequilibrium (r2 = 0.94; D′ = 1 in HapMap CEU; supplemental Figure 4), representing a single susceptibility locus. The A allele at rs3824662 was significantly overrepresented in ALL cases compared with controls (35% vs 20%) and was consistent across race/ethnicity (ie, EAs, 30% vs 17% [P = 1.09 × 10−5]; Hispanics, 50% vs 33% [P = .0008]; and AAs, 20% vs 10% [P = .07]; Figure 3A). rs3781093 was significantly associated with ALL risk in EAs and Hispanics, but not in individuals of African descent in whom it was no longer in linkage disequilibrium (r2 = 0.006; D′ = 0.16) with rs3824662 (supplemental Figure 5A).
Table 1.
Association of GATA3 SNPs with AYA ALL susceptibility in the discovery GWAS and replication cohort
| Chr | Position* | SNP | Alleles† | Cohort | RAF§ in AYA cases (%) | Genotype count in cases‡ | RAF§ in non-ALL controls (%) | Genotype count in controls‡ | AYA ALL vs non-ALL | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RR | RW | WW | RR | RW | WW | P|| | OR|| | 95% CI | |||||||
| 10 | 8144214 | rs3824662 | A/C | Discovery | 35 | 36 | 145 | 125 | 20 | 341 | 2078 | 4242 | 2.84 × 10-10 | 1.77 | 1.48-2.12 |
| Replication | 39 | 24 | 78 | 59 | 18 | 226 | 1651 | 3876 | 1.52 × 10-10 | 2.21 | 1.72-2.83 | ||||
| 10 | 8141933 | rs3781093 | C/T | Discovery | 33 | 34 | 137 | 136 | 22 | 380 | 2254 | 4025 | 3.20 × 10-9 | 1.73 | 1.44-2.08 |
| Replication | 35 | 18 | 75 | 64 | 18 | 236 | 1702 | 3809 | 1.00 × 10-7 | 1.96 | 1.52-2.54 | ||||
Association of SNP genotype and ALL was evaluated by comparing allele frequency between ALL and non-ALL, after adjusting for genetic ancestry.
Chr, chromosome.
Chromosomal locations are based on hg18.
Bold indicates risk allele for ALL.
Genotype is denoted by RR (homozygous for the risk allele), RW (heterozygous), or WW (homozygous for the wild-type allele).
RAF, risk allele frequency (allele A at rs38246623 and allele C at rs3781093).
P values were estimated by the logistic regression test and OR represents the increase in risk of developing ALL for each copy of the risk allele compared with participants who don’t carry the risk allele.
Figure 2.

Genome-wide association of SNP genotype with ALL susceptibility in AYAs. The association between genotype and ALL susceptibility was evaluated by using a logistic regression model for 635 297 SNPs in 308 AYA ALL cases and 6661 non-ALL controls. P values (y-axis) were plotted against respective chromosomal position of each SNP (x-axis). Points above the blue horizontal line indicate SNPs achieving the genome-wide significant threshold (P < 5 × 10−8). Gene symbol was indicated for the GATA3 locus at 10p14.
Figure 3.

Association of GATA3 SNP rs3824662 with ALL in AYAs by race/ethnicity. In the discovery group (A) the A allele at rs3824662 was overrepresented in AYA ALL cases relative to non-ALL controls. This association was true within the European Americans (>95% European genetic ancestry), African Americans (>70% African ancestry), or Hispanic Americans (>10% Native American genetic ancestry, and Native American ancestry > African genetic ancestry). (B) Similar association was confirmed in the replication group (1-tailed test). Genetic ancestry was determined by using STRUCTURE (version 2.2.3) with HapMap CEU, YRI, CHB/JPT, and indigenous Native Americans as reference populations.
To validate the association signals at these GATA3 SNPs, we tested an independent set of 162 AYA ALL cases enrolled in COG P9900 and ALL0232 protocols and an additional 5,755 non-ALL controls. In the replication analysis, risk alleles at both GATA3 SNPs were consistently overrepresented in AYA ALL cases compared with non-ALL controls: rs3824662 (OR, 2.21; 95% CI, 1.72 to 2.83; P = 1.52 × 10−10) and rs3781093 (OR, 1.96; 95% CI, 1.52 to 2.54; P = 1.0 × 10−7; Table 1, Figure 3B, and supplemental Figure 5B). rs3824662 was validated across race/ethnicity in the replication group (ie, EAs, 35% vs 18% [P = 2.0 × 10−7]; Hispanics, 55% vs 39% [P = .005]; and AAs, 13% vs 9% [P = .035]; Figure 3B). In contrast, rs3781093 was significant in EAs and Hispanics but not in AAs (supplemental Figure 5B).
We also examined the association signals in AYAs for susceptibility loci previously identified in pediatric populations (supplemental Table 1). ARID5B, IKZF1, and PIP4K2A variants were nominally significant in AYAs in the discovery GWAS and/or in the replication analyses. In contrast, CEBPE and CDKN2A/CDKN2B were not associated with ALL risk in AYAs in either discovery or replication cohorts. These results imply both similarities and differences in genetic predisposition to ALL between children and AYAs.15
GATA3 SNP rs3824662 and ALL subtypes in AYAs
We further analyzed the association of the GATA3 SNP rs3824662 with somatic ALL genomic abnormalities. Among the AYA ALL cases in the discovery cohort, the risk allele at rs3824662 was underrepresented among hyperdiploid ALL cases (22% vs 37%; P = .03; Figure 4 and supplemental Table 2), with a similar trend for TCF3-PBX1 and ETV6-RUNX1 ALL albeit not statistically significant. In contrast, the ALL risk allele frequency of rs3824662 was higher in AYA ALL cases with the Ph-like gene expression profile than in those without this signature (48% vs 32%; P = .02; Figure 4 and supplemental Table 2). This was consistent with our prior reports of GATA3 as a susceptibility gene for Ph-like ALL,36 although there was no overlap in cases included in the current AYA ALL GWAS and those in our previous Ph-like ALL GWAS.36 Within Ph-like ALL, there was a trend with A allele further enriched in cases involving CRLF2 rearrangements (P = .06; Figure 4).
Figure 4.

GATA3 SNP genotype and ALL genetic subtypes in AYAs. The allele frequency of rs3824662 varied substantially by ALL somatic genomic abnormalities, with the ALL risk allele underrepresented in hyperdiploid cases and more common in the Ph-like subtype. Numbers are based on the ALL cases included in the discovery GWAS (N = 308). The frequency of A allele at rs3824662 was 20% among unrelated non-ALL controls (MESA).
Importantly, even after excluding Ph-like ALL cases, the risk allele at rs3824664 was still more common in AYA ALL cases compared with non-ALL controls (rs3824662: OR, 1.56 [95% CI, 1.25 to 1.96; P = 8.13 × 10−5]; rs3781093: OR, 1.53 [95% CI, 1.21 to 1.92; P = .0002]; supplemental Figure 6). This suggested that the influence of the GATA3 variant on ALL susceptibility in AYAs extends beyond the predisposition to Ph-like subtype.
GATA3 SNP rs3824662 and age at ALL diagnosis
Finally, we examined the distribution of the GATA3 SNP genotype by age at diagnosis in a cohort of largely unselected patients enrolled on the COG P9900 protocols (N = 1,827, age 0.1 to 21 years). When we divided patients into four consecutive age groups (<5, 5 to 10, 10 to 15, and >15 years), we observed a clear progressive increase in the risk allele frequency at rs3824662 (P = 6.29 × 10−11; Figure 5) with increasing allelic ORs (ie, relative risk of ALL conferred by each copy of the A allele at rs3824662; Figure 5, inset plot): 0.96 (95% CI, 0.85 to 1.09), 1.26 (95% CI, 1.08 to 1.48), 1.48 (95% CI, 1.19 to 1.84), and 2.40 (95% CI, 1.81 to 3.19). Similar correlation between GATA3 genotype frequency and age was evident irrespective of genetic ancestry, but the GATA3 risk allele was markedly more common in Hispanics (ie, individuals with high NA genetic ancestry; Figure 5). To examine whether the association with age is confounded by ALL genetic subtype, we compared rs3824662 allele frequency by age in the COG P9900 protocols after stratifying ALL cases into TCF3-PBX1, ETV6-RUNX1, high hyperdiploid, MLL-rearranged, and B-other. There was a trend for the risk allele at this SNP to be more frequent in patients older than age 16 years relative to those younger than age 16 years in 5 subtypes examined, although with a limited sample size (supplemental Figure 7). This suggests that GATA3 germline variants confer a general ALL disease risk in AYAs. In contrast, the frequency of ALL risk variant in ARID5B (rs10821936) decreased progressively with increasing age at diagnosis in the COG P9900 cohort (P = .006), whereas PIP4K2A, CDKN2A/2B, IKZF1, and CEBPE variants were not related to age (P > .05; data not shown).
Figure 5.
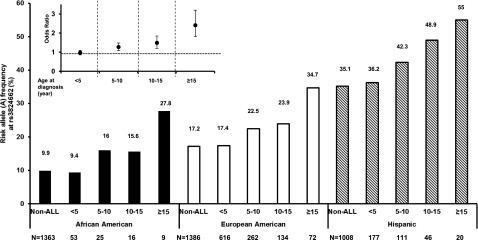
GATA3 SNP rs3824662 and age at ALL diagnosis. In a largely unselected cohort of ALL cases enrolled on the Children’s Oncology Group (COG) P9900 trials (N = 1,827), the frequency of ALL risk allele at rs3824662 was positively correlated with patient age at diagnosis consistently across race/ethnicity. Inset figure: the relative risk of ALL (odds ratio) conferred by each copy of the A allele at rs3824662 increased progressively with age, as estimated by logistic regression after adjusting for genetic ancestry. Horizontal dotted line is odds ratio of 1. Unrelated participants from the Multi-ethnic Study of Atherosclerosis (MESA) were considered as non-ALL controls.
Discussion
Because ALL is the most common cancer in children, previous susceptibility GWAS studies understandably focused on pediatric populations. We hypothesized that ALL in AYAs has distinct tumor biology and genetic etiology, which potentially contribute to the disparities in treatment outcomes by age. To this end, we performed the first GWAS of ALL susceptibility specifically in the AYA population and identified a single genome-wide significant risk locus within the GATA3 gene on 10p14.
The susceptibility to ALL varies substantially by age. ALL risk first peaks between 2 and 5 years after birth, followed by gradual decrease into adulthood, but rises again in older individuals (older than age 70 years), suggesting that differential combinations of environmental and genetic factors contribute to leukemogenesis at different ages. For example, it has been hypothesized that infection (and supposedly acquired immunity) may ameliorate susceptibility to ALL in young children,41,42 which may not be important in ALL that occurs later in life. Similarly, the in utero occurrence of genomic lesions is characteristic in many (if not most) pediatric ALL cases,43,44 whereas such early origin of presumed initiating events may not be evident in AYAs ALL. Age-dependent differences in lymphocyte development and function are well documented in human and mouse systems,45 and rapid growth of hematopoietic cells may render them particularly susceptible to oncogenic assaults.46 Thus, it can be postulated that specific ALL susceptibility genes are required during a particular stage of hematopoietic development and preferentially influence ALL risk within a certain age range. For example, loss of Arid5b in mice resulted in reduction of lymphoid cells in bone marrow within 3 weeks after birth, but the effect became blunted by 6 weeks.47 In fact, germline ARID5B variants also exhibited increasing influence on ALL predisposition in children as age decreases.15
GATA3 encodes for a transcription factor critical for lymphoid cell lineage commitment and early T-cell differentiation,48 and loss-of-function somatic mutations have been discovered in early T-cell precursor ALL.49 Germline polymorphisms in GATA3, however, appear more important for B-cell malignancies.50 We recently reported that rs3824662 was significantly associated with susceptibility to Ph-like ALL in children and risk of relapse.36 A contemporaneous study by Migliorini et al17 reported the same ALL susceptibility variant in GATA3 in children of European descent and associated it with relapse. Particularly of note, GATA3 risk variants also appeared enriched in older children, even within their predominantly pediatric cohort. The association of rs3824662 with ALL relapse17,36 is in line with the negative prognosis by age and higher frequency of the GATA3 variant in AYAs with ALL. Nevertheless, it is unclear whether poor prognosis conferred by a GATA3 variant was driven by its association with a high-risk subtype (ie, Ph-like ALL), novel somatic genomic aberrations specific to AYA, and/or host biology related to antileukemic drug response. Interestingly, in AYA cases included in the discovery GWAS, the number of the risk allele at rs3824662 was significantly associated with GATA3 expression in ALL blasts (P = .02; supplemental Figure 8), consistent with our previous report in pediatric ALL cases of this variant functioning as a cis-acting regulatory element of GATA3 transcription.36
The overrepresentation of the GATA3 variant in AYAs is consistent with its association with Ph-like ALL36 for which the frequency increases with age.8 However, the risk variant at rs3824662 remained significantly associated with susceptibility to AYA ALL cases without Ph-like expression pattern, suggesting the link to Ph-like ALL contributed only partly to the genome-wide significant association signal at rs3824662. In fact, the GATA3 risk allele tended to be more common in ALL patients age 16 years or older than in those age younger than 16 years consistently across different genetic subtypes, plausibly conferring a general ALL risk in AYAs. It remains unknown how the GATA3 variants influence the risk of developing ALL in older adults, including the elderly (>60 years). Future studies including this age group may provide insights on molecular etiology of ALL across the age spectrum. It is also noteworthy that MLL-rearranged cases had the second highest GATA3 risk variant frequency (Figure 4), although the number of patients was relatively small and the difference did not reach statistical significance. Future studies are warranted to comprehensively characterize potential interactions of germline GATA3 variants with somatic genomic lesions in ALL.
In conclusion, our GWAS identified inherited GATA3 genetic variants that strongly influence ALL susceptibility in adolescents and young adults, shedding new light on potential age-related differences in ALL biology and treatment outcome.
Acknowledgments
The authors thank the patients and parents who participated in the clinical trials included in this study and M. Shriver (Pennsylvania State University) for sharing SNP genotype data for the Native American references. Genome-wide genotyping of COG P9904/P9905 samples was performed by the Center for Molecular Medicine with the generous financial support from the Jeffrey Pride Foundation and the National Childhood Cancer Foundation.
This work was supported by the National Institutes of Health, National Cancer Institute grants CA145707, CA156449, CA21765, CA36401, CA98543, CA114766, CA98413, CA140729, CA176063, and HHSN261200800001E, the National Institute of General Medical Sciences grant GM92666, in part by the intramural Program of the National Cancer Institute, by a Stand Up to Cancer Innovative Research Grant, and by the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital, by a St. Jude Children’s Research Hospital Academic Programs Special Fellowship and a Spanish Ministry of Education Fellowship Grant (V.P.-A.), by the American Society of Hematology Scholar Award and by the Order of St. Francis Foundation (J.J.Y.), and by a Leukemia and Lymphoma Society Fellow Award and Alex’s Lemonade Stand Foundation Young Investigator Award (K.G.R.). S.P.H. is the Ergen Family Chair in Pediatric Cancer, C.G.M. is a Pew Scholar in the Biomedical Sciences and a St. Baldrick’s Scholar, and H.Z. is a St. Baldrick’s International Scholar.
The study sponsors were not directly involved in the design of the study, the collection, analysis, and interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: V.P.-A., K.G.R., H.X., C.G.M., and J.J.Y. conceived of and designed the study; R.C.H., D.P.-T., I-M.C., W.L.C., N.A.H., A.J.C., E.A.R., J.M.G.-F., G.M., C.D.B., K.M., J.K., W.S., S.M.K., M.K., E.P., J.M.R., S.M.L., M.S.T., M.D., E.G.B., D.G.T., F.Y., Y.W., C.-H.P., S.J., M.V.R., W.E.E., D.S.G., M.L.L., S.P.H., and C.L.W. provided study materials or patients; V.P.-A., K.G.R., H.X., M.D., I-M.C., C.L.W., R.C.H., M.V.R., and W.E.E. collected and assembled data; V.P.-A., H.X., C.S., W.Y., H.Z., M.D., R.C.H., I-M.C., and J.J.Y. analyzed and interpreted data; V.P.-A., H.X., C.G.M., and J.J.Y. wrote the manuscript; and all authors gave final approval for the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jun J. Yang, Department of Pharmaceutical Sciences, MS 313, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, Memphis, TN 38105-3678; e-mail: jun.yang@stjude.org; and Charles G. Mullighan, Department of Pathology, MS 342, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, Memphis, TN 38105-3678; e-mail:charles.mullighan@stjude.org.
References
- 1.Lukenbill J, Advani AS. The treatment of adolescents and young adults with acute lymphoblastic leukemia. Curr Hematol Malig Rep. 2013;8(2):91–97. doi: 10.1007/s11899-013-0159-0. [DOI] [PubMed] [Google Scholar]
- 2.Place AE, Frederick NN, Sallan SE. Therapeutic approaches to haematological malignancies in adolescents and young adults. Br J Haematol. 2014;164(1):3–14. doi: 10.1111/bjh.12556. [DOI] [PubMed] [Google Scholar]
- 3.Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol. 2012;30(14):1663–1669. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ram R, Wolach O, Vidal L, Gafter-Gvili A, Shpilberg O, Raanani P. Adolescents and young adults with acute lymphoblastic leukemia have a better outcome when treated with pediatric-inspired regimens: systematic review and meta-analysis. Am J Hematol. 2012;87(5):472–478. doi: 10.1002/ajh.23149. [DOI] [PubMed] [Google Scholar]
- 5.Schafer ES, Hunger SP. Optimal therapy for acute lymphoblastic leukemia in adolescents and young adults. Nat Rev Clin Oncol. 2011;8(7):417–424. doi: 10.1038/nrclinonc.2011.77. [DOI] [PubMed] [Google Scholar]
- 6.Gramatges MM, Rabin KR. The adolescent and young adult with cancer: state of the art— acute leukemias. Curr Oncol Rep. 2013;15(4):317–324. doi: 10.1007/s11912-013-0325-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aricò M, Schrappe M, Hunger SP, et al. Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol. 2010;28(31):4755–4761. doi: 10.1200/JCO.2010.30.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–1015. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harrison CJ. Cytogenetics of paediatric and adolescent acute lymphoblastic leukaemia. Br J Haematol. 2009;144(2):147–156. doi: 10.1111/j.1365-2141.2008.07417.x. [DOI] [PubMed] [Google Scholar]
- 10.Chessells JM, Hall E, Prentice HG, Durrant J, Bailey CC, Richards SM. The impact of age on outcome in lymphoblastic leukaemia; MRC UKALL X and XA compared: a report from the MRC Paediatric and Adult Working Parties. Leukemia. 1998;12(4):463–473. doi: 10.1038/sj.leu.2400959. [DOI] [PubMed] [Google Scholar]
- 11.Rubnitz JE, Wichlan D, Devidas M, et al. Children’s Oncology Group. Prospective analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia: a Children’s Oncology Group study. J Clin Oncol. 2008;26(13):2186–2191. doi: 10.1200/JCO.2007.14.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1006–1010. doi: 10.1038/ng.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sherborne AL, Hosking FJ, Prasad RB, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet. 2010;42(6):492–494. doi: 10.1038/ng.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Treviño LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1001–1005. doi: 10.1038/ng.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu H, Yang W, Perez-Andreu V, et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013;105(10):733–742. doi: 10.1093/jnci/djt042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu H, Cheng C, Devidas M, et al. ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol. 2012;30(7):751–757. doi: 10.1200/JCO.2011.38.0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Migliorini G, Fiege B, Hosking FJ, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood. 2013;122(19):3298–3307. doi: 10.1182/blood-2013-03-491316. [DOI] [PubMed] [Google Scholar]
- 18.Yang JJ, Cheng C, Devidas M, et al. Genome-wide association study identifies germline polymorphisms associated with relapse of childhood acute lymphoblastic leukemia. Blood. 2012;120(20):4197–4204. doi: 10.1182/blood-2012-07-440107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang JJ, Cheng C, Yang W, et al. Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA. 2009;301(4):393–403. doi: 10.1001/jama.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang JJ, Cheng C, Devidas M, et al. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet. 2011;43(3):237–241. doi: 10.1038/ng.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153–166. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stock W, La M, Sanford B, et al. Children’s Cancer Group; Cancer and Leukemia Group B studies. What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children’s Cancer Group and Cancer and Leukemia Group B studies. Blood. 2008;112(5):1646–1654. doi: 10.1182/blood-2008-01-130237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lazarus HM, Richards SM, Chopra R, et al. Medical Research Council (MRC)/National Cancer Research Institute (NCRI) Adult Leukaemia Working Party of the United Kingdom and the Eastern Cooperative Oncology Group. Central nervous system involvement in adult acute lymphoblastic leukemia at diagnosis: results from the international ALL trial MRC UKALL XII/ECOG E2993. Blood. 2006;108(2):465–472. doi: 10.1182/blood-2005-11-4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kantarjian H, Thomas D, O’Brien S, et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer. 2004;101(12):2788–2801. doi: 10.1002/cncr.20668. [DOI] [PubMed] [Google Scholar]
- 25.Ravandi F, O’Brien S, Thomas D, et al. First report of phase 2 study of dasatinib with hyper-CVAD for the frontline treatment of patients with Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia. Blood. 2010;116(12):2070–2077. doi: 10.1182/blood-2009-12-261586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas DA, Faderl S, Cortes J, et al. Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood. 2004;103(12):4396–4407. doi: 10.1182/blood-2003-08-2958. [DOI] [PubMed] [Google Scholar]
- 27.Thomas DA, O’Brien S, Faderl S, et al. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol. 2010;28(24):3880–3889. doi: 10.1200/JCO.2009.26.9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360(26):2730–2741. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borowitz MJ, Devidas M, Hunger SP, et al. Children’s Oncology Group. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood. 2008;111(12):5477–5485. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115(26):5312–5321. doi: 10.1182/blood-2009-09-245944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Troyer JL, Nelson GW, Lautenberger JA, et al. Genome-wide association study implicates PARD3B-based AIDS restriction. J Infect Dis. 2011;203(10):1491–1502. doi: 10.1093/infdis/jir046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burchard EG, Avila PC, Nazario S, et al. Genetics of Asthma in Latino Americans (GALA) Study. Lower bronchodilator responsiveness in Puerto Rican than in Mexican subjects with asthma. Am J Respir Crit Care Med. 2004;169(3):386–392. doi: 10.1164/rccm.200309-1293OC. [DOI] [PubMed] [Google Scholar]
- 33.Shi J, Levinson DF, Duan J, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460(7256):753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purcell SM, Wray NR, Stone JL, et al. International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korn JM, Kuruvilla FG, McCarroll SA, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008;40(10):1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez-Andreu V, Roberts KG, Harvey RC, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013;45(12):1494–1498. doi: 10.1038/ng.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao X, Bigham AW, Mei R, et al. A genomewide admixture mapping panel for Hispanic/Latino populations. Am J Hum Genet. 2007;80(6):1171–1178. doi: 10.1086/518564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA. 2002;99(10):6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381(9881):1943–1955. doi: 10.1016/S0140-6736(12)62187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006;6(3):193–203. doi: 10.1038/nrc1816. [DOI] [PubMed] [Google Scholar]
- 43.Greaves MF. Biological models for leukaemia and lymphoma. IARC Sci Publ. 2004;(157):351–372. [PubMed] [Google Scholar]
- 44.Greaves M. Pre-natal origins of childhood leukemia. Rev Clin Exp Hematol. 2003;7(3):233–245. [PubMed] [Google Scholar]
- 45.Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol. 2004;5(2):133–139. doi: 10.1038/ni1033. [DOI] [PubMed] [Google Scholar]
- 46.Rizo A, Vellenga E, de Haan G, Schuringa JJ. Signaling pathways in self-renewing hematopoietic and leukemic stem cells: do all stem cells need a niche? Hum Mol Genet. 2006;15(Spec No 2):R210–R219. doi: 10.1093/hmg/ddl175. [DOI] [PubMed] [Google Scholar]
- 47.Lahoud MH, Ristevski S, Venter DJ, et al. Gene targeting of Desrt, a novel ARID class DNA-binding protein, causes growth retardation and abnormal development of reproductive organs. Genome Res. 2001;11(8):1327–1334. doi: 10.1101/gr.168801. [DOI] [PubMed] [Google Scholar]
- 48.Yagi R, Zhu J, Paul WE. An updated view on transcription factor GATA3-mediated regulation of Th1 and Th2 cell differentiation. Int Immunol. 2011;23(7):415–420. doi: 10.1093/intimm/dxr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Enciso-Mora V, Broderick P, Ma Y, et al. A genome-wide association study of Hodgkin’s lymphoma identifies new susceptibility loci at 2p16.1 (REL), 8q24.21 and 10p14 (GATA3). Nat Genet. 2010;42(12):1126–1130. doi: 10.1038/ng.696. [DOI] [PMC free article] [PubMed] [Google Scholar]