

Abstract
Background
The incidence of Acute Megakaryoblastic Leukemia (AMKL) is 500-fold higher in children with Down Syndrome (DS) compared with non-DS children, but the relevance of trisomy 21 as a specific background of AMKL in DS is still an open issue. Several Authors have determined gene expression profiles by microarray analysis in DS and/or non-DS AMKL. Due to the rarity of AMKL, these studies were typically limited to a small group of samples.
Methods
We generated integrated quantitative transcriptome maps by systematic meta-analysis from any available gene expression profile dataset related to AMKL in pediatric age. This task has been accomplished using a tool recently described by us for the generation and the analysis of quantitative transcriptome maps, TRAM (Transcriptome Mapper), which allows effective integration of data obtained from different experimenters, experimental platforms and data sources. This allowed us to explore gene expression changes involved in transition from normal megakaryocytes (MK, n=19) to DS (n=43) or non-DS (n=45) AMKL blasts, including the analysis of Transient Myeloproliferative Disorder (TMD, n=20), a pre-leukemia condition.
Results
We propose a biological model of the transcriptome depicting progressive changes from MK to TMD and then to DS AMKL. The data indicate the repression of genes involved in MK differentiation, in particular the cluster on chromosome 4 including PF4 (platelet factor 4) and PPBP (pro-platelet basic protein); the gene for the mitogen-activated protein kinase MAP3K10 and the thrombopoietin receptor gene MPL. Moreover, comparing both DS and non-DS AMKL with MK, we identified three potential clinical markers of progression to AMKL: TMEM241 (transmembrane protein 241) was the most over-expressed single gene, while APOC2 (apolipoprotein C-II) and ZNF587B (zinc finger protein 587B) appear to be the most discriminant markers of progression, specifically to DS AMKL. Finally, the chromosome 21 (chr21) genes resulted to be the most over-expressed in DS and non-DS AMKL, as well as in TMD, pointing out a key role of chr21 genes in differentiating AMKL from MK.
Conclusions
Our study presents an integrated original model of the DS AMLK transcriptome, providing the identification of genes relevant for its pathophysiology which can potentially be new clinical markers.
Electronic supplementary material
The online version of this article (doi:10.1186/s12920-014-0063-z) contains supplementary material, which is available to authorized users.
Keywords: Down Syndrome (Trisomy 21), Acute Megakaryoblastic Leukemia (AMKL), Transient Myeloproliferative Disorder (TMD), Megakaryocyte (MK), Gene expression profile, Integrated transcriptome map
Background
Trisomy for human chromosome 21 (chr21) is the most frequent live-born aneuploidy and is the cause of Down syndrome (DS), whose main symptoms include intellectual disability, cardiovascular defects and craniofacial dysmorphisms [1]. The DS phenotype is thought to be associated with an altered expression of the genes located on chr21 [2-7]. Basic research on DS is now rapidly accelerating, and there is the possibility that the results will be beneficial for individuals with DS [8].
Several studies have shown that individuals with DS have a specific cancer risk pattern, or tumor profile: their risk of developing leukemia and testicular cancer is much higher than age-matched controls, while women with DS almost never develop breast cancer [9,10]. In particular, children with DS show an increased prevalence of acute leukemia, both lymphoid (ALL) and myeloid (AML), with relative risk ranging from 10 to 20 times higher than the normal population [11,12]. In nearly half of the cases, these childhood leukemias are classified as megakaryoblastic leukemia (AMKL), a relatively rare subtype of AML also known as AML M7, according to French–American–British (FAB) classification, whose incidence increases by 500-fold in children with DS by the age of 4 years as compared to the chromosomally normal population (reviewed in [13]). This observation strongly suggests that trisomy 21 directly contributes to the neoplastic transformation of hematopoietic cells, in particular in the megakaryocyte lineage cells. Interestingly, acute leukemia cells harboring megakaryocyte markers and presenting in subjects without DS may show trisomy 21 [14]. We also described a cell line derived from blast cells of a patient with type M2 AML which has trisomy 21 and megakaryocyte features [15]. More recently, mutations of the gene encoding for the transcription factor GATA1 have been shown to cooperate with trisomy 21 in initiating megakaryoblastic proliferation in nearly all DS AMKL cases while they are absent in non-DS AMKL [13,16]. GATA1 mutations in DS cells give rise to a short, truncated form of GATA1 (GATA1s) transcription factor that, in this form, is not able to establish normal interactions with other gene regulators [17].
Transient myeloproliferative disorder (TMD) is a clonal pre-leukemia condition, occurring in 10% of children with DS during the neonatal period, presenting at a median age of 3-7 days with accumulation of immature megakaryoblasts [13]. TMD cases usually resolve spontaneosuly, but DS AMKL may develop within 1-4 years in 20-30% of these children. AMKL may develop in non-DS children, usually at an higher age in comparison to DS subjects (median 8 vs. 1.8 years, respectively) and in absence of a trisomy 21 background. Cytogenetic abormalities described in non-DS AMKL cells include trisomy 8 and 1 and monosomy 7 [13].
An open issue is the relevance of trisomy 21 as a specific background for the higher incidence of AMKL in DS. A few previous studies have used gene expression profiling by microarray analysis in order to identify specific transcriptome alterations in DS and/or non-DS AMKL, as well as in TMD [17-24]. Due to the rarity of AMKL, these works often analyze a small number of cases, using a variety of experimental platforms. Results were consequently affected by a small grade of comparability.
One of the first goals of this work was to perform a systematic meta-analysis using any available gene expression profile dataset related to AMKL in pediatric age in order to produce a differential transcriptome map between DS and non-DS related AMKL. This task has been accomplished using a tool recently described by us for the generation and the analysis of quantitative transcriptome maps, TRAM (Transcriptome Mapper) [25], which allows effective integration of data obtained from different experimenters, experimental platforms and data sources. The comparison of 43 DS AMKL samples with 45 non-DS AMKL samples represents the largest study on the subject, highlighting the relevance of trisomy 21 in the development of AMKL in comparison with AMKL originating from non-trisomic cells. Results show significant over- or under-expression of distinct chromosomal segments and of single key genes in the whole genome, as well as on chr21, adding new knowledge compared with that produced by the single works from which the data were originally obtained. In addition, each considered type of leukemia was compared with the expression profile of TMD cells and normal human megakaryoblast/megakaryocyte cells (MK), allowing the building of a model for the disorder in differentiation process that lead to DS and non-DS AMKL. Comparisons with cord blood-derived MK cells (CB MK) have also been performed, due to the fact that leukemias in infants or young children originate from fetal hematopoietic cells [17,18,26,27] and the progenitor cells (fetal/neonatal MKP) are present in the cord blood (CB) [28,29].
For each cell type investigated, reference expression data for about 17,000-26,000 mapped sequences have been generated and validated through a sample comparison with known data. The biological and clinical significance of these data is discussed.
Methods
Literature search
A systematic biomedical literature search was performed up to January 2013 in order to identify articles related to global gene expression profile experiments in AMKL patients (DS AMKL, non-DS AMKL and TMD conditions). A general search using the commonly used acronym "AMKL" retrieved 157 articles.
The MeSH term "Leukemia, Megakaryoblastic, Acute" was also used for a PubMed search in the expression: "Leukemia, Megakaryoblastic, Acute"[Mesh] AND ("Gene Expression Profiling"[MeSH] OR "Oligonucleotide Array Sequence Analysis"[Mesh] OR "Microarray Analysis"[Mesh] OR microarray* OR "Expression profile" OR SAGE).
Database search
Gene Expression Omnibus (GEO) [30] functional genomics repository was searched for: (AMKL[All Fields] OR (AML[All Fields] AND M7[All Fields])) AND "Homo sapiens"[Organism]. A more general search using the expression "Down Syndrome"[MeSH] AND "Homo sapiens" [Organism] was also used.
ArrayExpress database [31] of functional genomics experiments was searched for the terms: ''AMKL'', "Megakaryoblastic", "AML M7".
In order to obtain gene expression profile datasets for normal human MK cells, in addition to the 9 used in the original description of the TRAM software [25], we searched GEO for the expression ("Megakaryocytes" [Mesh] OR Megakaryoblast*) AND "Homo sapiens" [ORGANISM]. The ArrayExpress database was searched for the expressions "Megakaryocyte", "Megakaryocytic", "Megakaryoblast", "MK".
The searches were performed up to January 2013.
Dataset selection
The inclusion criteria of datasets in the analysis were: availability of the raw or pre-processed data; pediatric age of the subject from whom the sample was obtained; diagnosis of DS or non-DS AMKL or TMD.
Exclusion criteria were: exon arrays (hampering data elaboration by TRAM due to exceedingly high number of data rows) or platforms using probes split into several distinct arrays for each sample (hampering intra-sample normalization); lack of identifiers corresponding to those found in the GEO sample records (GSM) or ArrayExpress sample records; platforms assaying an atypical number of genes (i.e. <5.000 or >60.000); cell line derived data; specific subtype of non-DS AMKL, e.g. t(1;22); trisomy 21 in non-DS AMKL samples.
Normal MK samples were considered for the analysis when fulfilling these criteria: late MK colonies (10-14 days) or MK sorted cells, obtained from peripheral blood (PB), bone marrow (BM) or cord blood (CB). MK cultured for less than 10 days or Colony Forming Unit-Megakaryocytic (CFU-MK) were excluded.
In order to obtain a quantitative transcriptome map, values from each dataset were linearized when provided as logarithms. In some cases we used raw files (e.g. File CEL) to be converted into pre-processed data, using the software "Alt Analyze" [32].
TRAM (transcriptome mapper) analysis
TRAM (Transcriptome Mapper) software [25] allows the import of gene expression data recorded in the NCBI (National Center for Biotechnology Information) GEO and EBI (European Bioinformatics Institute) ArrayExpress databases in tab-delimited text format. It also allows the integration of all data by decoding probe set identifiers to gene symbols via UniGene data parsing [33], normalizing data from multiple platforms using intra-sample and inter-sample normalization (scaled quantile normalization) [34], creating graphical representation of gene expression profile through two ways, "Map" and "Cluster" mode, and determining the statistical significance of results. Moreover, TRAM allows to compare two biological conditions identifying critical genomic regions and genes with significant differential expressions.
We created a directory (folder) for each condition, containing all the sample datasets related to the same source and selected for the study: DS AMKL (pool 'A'); non-DS AMKL (pool 'B'); TMD (pool 'C'); normal MK (pool 'D'); normal CB MK (pool 'E').
We ran the whole set of analyses permitted by TRAM (in both "Map" and "Cluster" mode, although we focused on the "Map" mode) using default parameters as described [25]. We used an updated version of TRAM including enhanced resolution of gene identifiers and updated UniGene and Entrez Gene databases (TRAM 1.1, June 2013), in comparison with the original 2011 version [25]. When the gene location cytoband was not available in the Gene database [35], it was manually derived from UCSC Genome Browser [36]. TRAM is freely available at http://apollo11.isto.unibo.it/software. Briefly, gene expression values were assigned to individual loci via UniGene, intra-sample normalized as percentage of the mean value and inter-sample normalized by scaled quantile. The value for each locus, in each biological condition, is the mean value of all available values for that locus. The genome wide gene expression median value was used in order to determine percentiles of expression for each gene.
Using the "Map" mode graphical representation we searched for over/under-expressed genome segments, which have a window size of 500,000 bp and a shift of 250,000 bp. The expression value for each genomic segment is the mean of the expression values of the loci included in that segment. A segment is defined over/under-expressed if it has an expression value which is significantly different between two conditions analyzed, and contains at least 3 individually over/under-expressed genes, e.g. genes which have expression values within the highest and the lowest 2.5th percentile. Significance of the over/under-expression for single genes was determined by running TRAM in "Map" mode with a segment window of 12,500 bp. This window size corresponds to about a quarter of the mean length of a gene, so the significant over/under-expression of a segment almost always corresponds with that of a single gene. A segment or a gene was considered to be statistically significantly over- or under-expressed for q < 0.05, where q is the p-value obtained by the method of hypergeometric distribution [25] and corrected for multiple comparison. When the segment window contains more than one gene, the significance is maintained if the expression value of the over/under-expressed gene prevails over the others.
For the creation of the maps, TRAM software does not consider probes where the expression values is not available, assuming that an expression level has not been measured. Furthermore, it gives 95% of the minimum positive value present in a sample to those expression values equal to or lower than "0", in order to obtain meaningful numbers when we need to obtain a ratio between values in pool 'A' and pool 'B'. Assuming that in these cases an expression level is too low to be detected under the experimental conditions used, this transformation is useful to highlight differential gene expression. Finally, we considered the most over- or under-expressed genes among the genes associated with at least 5 data points.
At chromosomal level, we calculated (in the TRAM "chr" table) the median expression ratio for all the genes located in the same chromosome.
Other analysis
FuncAssociate analysis [37] was used to obtain Gene Ontology attributes in order to functionally characterize large sets of genes derived from the TRAM analysis.
Results
Literature search
A general search using the acronym ''AMKL'' retrieved 157 articles, 6 of them describe gene expression profiling experiments [17,18,20-23].
No additional pertinent item was retrieved using the expression described in the Methods section and including the MeSH Term "Leukemia, Megakaryoblastic, Acute".
Database search
The Gene Expression Omnibus (GEO) [30] search allowed the retrievement of three additional works describing data possibly useful for meta-analysis [19,24,38]. The lack of inclusion of these works in the literature search was due to failure of using the ''AMKL'' acronym and assigning the MeSH Term "Leukemia, Megakaryoblastic, Acute" during the PubMed indexing process (the more general term "Leukemia, Myeloid, Acute" was used).
The more general search using the expression "Down Syndrome"[MeSH] AND "Homo sapiens"[Organism] allowed the addition of one further work [39]. This work analyzed several types of AML samples and did not explicitly mention AMKL or AML M7 in both PubMed and GEO databases.
No further pertinent works related to AMKL were identified by ArrayExpress database [31] search.
Several datasets for normal MK cells global gene expression profile fulfilling the selection criteria were obtained from the works [40] (GEO, 7 samples) and [41] (ArrayExpress, 1 sample), in addition to the 4 sample series identified in the first report of the TRAM software [25] and obtained from different works [42-45], for a total of 19 datasets related to human normal MK cells.
Dataset building
Of the 10 works related to DS or non-DS AMKL retrieved as above described, 7 were considered for the meta-analysis (Table 1). It was not possible to obtain raw data from the Authors of [20], while the only sample of AML M7 described in [38] was related to "Leukemic Stem Cells" cell type and the two AML M7 reported by Tomasson et al. [39] were obtained from elderly patients. Raw data from [23] were kindly provided by Drs. Jeffrey Taub and Yubin Ge.
Table 1.
Main features of the samples used in TRAM analyses
| Study ID | Sample ID | Sample type | Platform | Microarray | Spots | References |
|---|---|---|---|---|---|---|
| Pool 'A' (n=43) - DS AMKL (25,955 mapped loci following analysis by TRAM) | ||||||
| A1…A3 (n=3) | GSM491372…4 | BM Sorted leukemic blasts | GPL570 | Affymetrix U133 Plus 2.0 | 54,675 | [18] |
| A4…A25 (n=22) | GSM94245, GSM94272…92 | BM or PB | GPL96 | Affymetrix U133A | 22,283 | [22] |
| A26…A31 (n=6) | GSM417985…90 | BM or PB Sorted leukemic blasts | GPL570 | Affymetrix U133 Plus 2.0 | 54,675 | [17] |
| A32…A38 (n=7) | E-MEXP-72* | BMMC or PBMC | A-AFFY-33.adf.txt* | Affymetrix U133A | 22,283 | [21] |
| A39…A43 (n=5) | / | BMMC or PBMC | GPL96 | Affymetrix U133A | 22,283 | [23] |
| Pool 'B' (n=45) - non-DS AMKL (26,045 mapped loci following analysis by TRAM) | ||||||
| B1-B2 (n=2) | GSM491370-1 | BM Sorted leukemic blasts | GPL570 | Affymetrix U133 Plus 2.0 | 54,675 | [18] |
| B3…B23 (n=21) | GSM94221-4-5, GSM94227…32, GSM94234-5-7-8, GSM94240-2-3-8, GSM94256-9, GSM94261-2 | BM or PB | GPL96 | Affymetrix U133A | 22,283 | [22] |
| B24…B28 (n=5) | GSM39832-5, GSM39842-4, GSM39863 | PBMC or BMMC | GPL8300 | Affymetrix U95 Version 2 | 12,625 | [19] |
| B29…B35 (n=7) | GSM361502…4, GSM361506…8, GSM361510 | BM or PB | GPL96 | Affymetrix U133A | 22,283 | [24] |
| B36…B40 (n=5) | GSM417991…5 | BM or PB Sorted leukemic blasts | GPL570 | Affymetrix U133 Plus 2.0 | 54,675 | [18] |
| B41…B45 (n=5) | / | BMMC or PBMC | GPL96 | Affymetrix U133A | 22,283 | [23] |
| Pool 'C' (n=20) - TMD (25,955 mapped loci following analysis by TRAM) | ||||||
| C1…C3 (n=3) | GSM491375…7 | BM Sorted leukemic blasts | GPL570 | Affymetrix U133 Plus 2.0 | 54,675 | [18] |
| C4…C11 (n=8) | GSM94293…9, GSM94300 | BM or PB | GPL96 | Affymetrix U133A | 22,283 | [22] |
| C12…C20 (n=9) | E-MEXP-72* | BMMC or PBMC | A-AFFY-33.adf.txt* | Affymetrix U133A | 22,283 | [21] |
| Pool 'D' (n=19) - MK (26,372 mapped loci following analysis by TRAM) | ||||||
| D1-D2 (n=2) | GSM321577-8 | MK (BM) (subj = pool) | GPL96 | Affymetrix U133A | 22,283 | [45] |
| D3…D6 (n=4) | GSM112277-8, GSM112291-2 | MK (PB) (subj = 1, rep. 1) | GPL887 | Agilent 1A | 22,575 | [44] |
| D7-D8 (n=2) | GSM15648, GSM8649 | MK (BM) (subj = 6) | GPL96 | Affymetrix U133A | 22,283 | [42] |
| D9…D11 (n=3) | GSM88014-22-34 | MK (PB) (subj = 1) | GPL887 | Agilent 1A | 22,575 | [43] |
| D12…D18 (n=7) | GSM609746…52 | CB | GPL4685 | Affymetrix HT-HG_U133A | 22,944 | [40] |
| D19 (n=1) | E-MEXP-2146* | CB | A-AFFY-44.adf.txt | Affymetrix U133 Plus 2.0 | 54,675 | [41] |
| Pool 'E' = D12…D19 (n=8) - CB MK (25,577 mapped loci following analysis by TRAM) | ||||||
Samples selected for the meta-analysis of gene expression profiles in DS AMKL (pool 'A'), non-DS AMKL (pool 'B'), TMD (pool 'C'), and megakaryocytic cells (pool 'D' and 'E'). All Sample IDs and Platforms IDs are related to GEO database, other than codes marked with * (ArrayExpress database). Sample type: BM, bone marrow; PB, peripheral blood; BMMC, bone marrow mononuclear cells; PBMC, peripheral blood mononuclear cells; MK, megakaryocytic/megakaryoblast cells, obtained by in vitro differentiation of CD34+ cells; CD34+, undifferentiated CD34+ cells; BM, CB or PB: CD34+ cells derived from bone marrow, cord blood or peripheral blood, respectively. subj = number of subjects from which the sample was derived (in some cases, where subj = pool, the exact number of subjects included in a pool was not available). rep. = biological replicate. Microarray: U133A: Affymetrix Human Genome U133A Array; 1A: Agilent-012097 Human 1A Microarray (V2) G4110B; 22k A: Agilent Human oligo 22k A; HG-Focus: Affymetrix Human HG-Focus Target Array.
Details about Sample identifiers and main sample features are listed in Additional file 1 (available at: http://apollo11.isto.unibo.it/suppl).
At the end, DS AMKL sample pool 'A' included 43 datasets, while non-DS AMKL sample pool 'B' was composed of 45 datasets. A TMD dataset pool 'C' was constructed starting from 20 samples described in some of the DS AMKL related articles [18,21,22]. Age and sex data were available for 29 out of 43 DS AMKL patients (mean age: 20 months; 11 males and 18 females), for 26 out of 45 non-DS AMKL patients (mean age: 19 months; 19 males and 7 females) and for 9 out of 20 TMD patients (mean age: 8 days; 7 males and 2 females). GATA1 mutations giving rise to GATA1s were present in all DS AMKL and TMD samples, and not in non-DS AMKL samples, considering all samples for which this information was provided. Sample identifiers and main sample features are listed in Table 1 and Additional file 1 (available at: http://apollo11.isto.unibo.it/suppl).
Two pools were constructed from the normal MK related dataset selected: pool 'D' included all available MK samples, while pool 'E' was a subset including only CB-derived MK cells (Table 1 and Additional file 1).
Transcriptome differential maps
Datasets were loaded into TRAM and analyzed obtaining 8 transcriptome maps: DS AMKL (pool 'A') vs. non-DS AMKL (pool 'B'); DS AMKL (pool 'A') vs. normal MK (pool 'D'); non-DS AMKL (pool 'B') vs. normal MK (pool 'D'); DS AMKL (pool 'A') vs. normal CB MK (pool 'E') cells; non-DS AMKL (pool 'B') vs. normal CB MK (pool 'E'); DS AMKL (pool 'A') vs. TMD (pool 'C'); TMD (pool 'C') vs. normal MK (pool 'D'); TMD (pool 'C') vs. normal CB MK (pool 'E').
For each comparison between two cell types by TRAM, we describe below or in the corresponding Figures or Tables the total of data points analyzed for each cell type, i.e. gene expression values for all human mapped loci following intra- and inter-sample normalization [25]; the number of loci for which the comparison between the two conditions was possible due to the presence of values for those loci in both sample pools considered; the number and the gene content of each genomic segment containing at least three over- or under-expressed genes and found to be statistically significantly over- or under-expressed in the comparison between the two tissues. Each genomic segment was identified among the 12,373 segments generated using the default window of 500,000 bp with a sliding window of 250,000 bp and following removal of overlapping segments with similar gene content. When the results were reported for the over/under-expressed single genes, we considered only the genes associated at at least 5 data points.
The description of the gene name corresponding to all gene symbols cited here in the text, Figures or Tables is given in the Additional file 2. We performed a PubMed search for the most relevant over- or under-expressed genes using gene symbol or gene description along with MeSH terms related to MK or MK progenitor cells, thrombopoiesis, AMKL, platelets.
Detailed results for each map are provided below, and are also available at: http://apollo11.isto.unibo.it/suppl.
The absolute (not differential) expression values and maps for each cell type (not compared to another cell type) are also available in the complete sets of results at http://apollo11.isto.unibo.it/suppl, but are not discussed here because they include typical housekeeping genes whose over-expression is no longer evident when compensated by the corresponding housekeeping genes in the compared cell type.
Transcriptome map comparison of DS AMKL vs. non-DS AMKL
We first analyzed regional differential expression of pool 'A' (43 DS AMKL samples) versus pool 'B' (45 non-DS AMKL samples) (Table 1). A total of 1,061,761 data points from the pool 'A' and 1,084,700 data points from the pool 'B' were included in the analysis. An 'A'/'B' ratio value was determinable for 25,954 loci having values both in 'A' and 'B' pools (Additional file 3). The main results are shown in Figure 1. Results obtained by the analysis included 3 significantly non-overlapped over-expressed segments (Table 2a). The highest expression ratio between DS AMKL and non-DS AMKL (3.27) was observed in a segment on chromosome 15 (15q21.2), including the known gene HDC (encoding for histidine decarboxylase, which converts L-histidine to histamine). The second segment with the highest expression was located on chromosome 4 (4q31.1) and contained over-expressed genes such as GYPE, GYPB and GYPA, encoding for glycophorin E, B and A (MNS blood group) respectively. The third over-expressed segment spans the cluster of apolipoprotein encoding genes on chromosome 19 (19q13.2).
Figure 1.
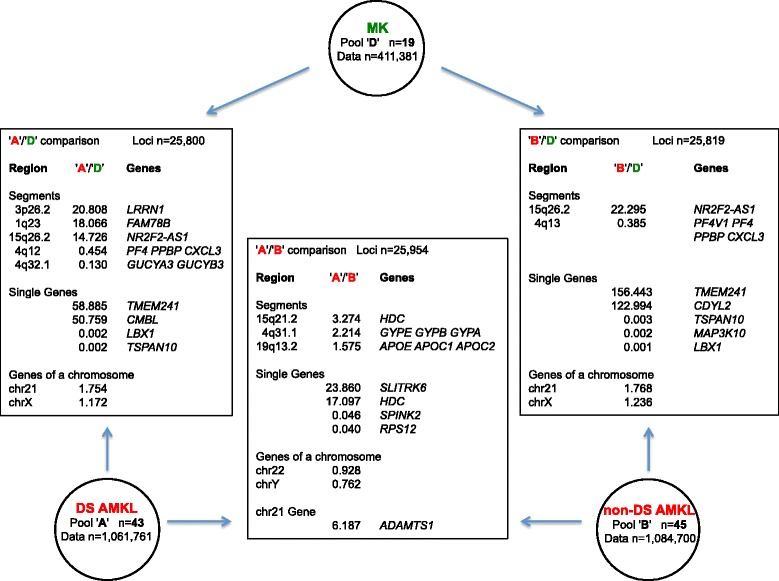
Main results of DS AMKL vs. MK, DS AMKL vs. non-DS AMK, non-DS AMKL vs. MK comparisons. For each comparison the number of loci analyzed, the most over- or under-expressed segments and single genes, and the highest and lowest median expression ratios for all the genes located in the same chromosome are indicated.
Table 2.
Genomic segments significantly over- or under-expressed
| a) DS AMKL vs. non-DS AMKL | |||||
| Chr and location a | Segment start b | Segment end b | 'A'/'B' ratio | q-value | Genes in the segment c |
| Over-expressed segments | |||||
| chr15 15q21.2 | 50,250,001 | 50,750,000 | 3.27 | 0.00233 | ATP8B4- SLC27A2+ HDC+ GABPB1+ FLJ10038+ GABPB1-AS1+ Hs.656448+ Hs.660869+ d USP8+ |
| chr4 4q31.1 | 144,750,001 | 145,250,000 | 2.21 | 0.00024 | GYPE+ Hs.658686- GYPB + GYPA + |
| chr19 19q13.2 | 45,000,001 | 45,500,000 | 1.58 | 0.00461 | ZNF180- PVR+ CEACAM19- BCL3+ CBLC+ BCAM+ PVRL2- Hs.666142- TOMM40+ APOE+ APOC1 + APOC4+ APOC2 + CLPTM1+ |
| b) DS AMKL vs. MK | |||||
| Chr and location | Segment start | Segment end | 'A'/'D' ratio | q-value | Genes in the segment |
| Over-expressed segments | |||||
| chr3 p26.2 | 3,500,001 | 4,000,000 | 20.81 | 0.00006 | Hs.241414+ Hs.587205+ LRRN1 + Hs.128128+ |
| chr1 1q23 | 166,000,001 | 166,500,000 | 18.07 | 0.00004 | Hs.22930+ FAM78B+ Hs.662048+ |
| chr15 15q26.2 | 96,750,001 | 97,250,000 | 14.73 | 0.00001 | Hs.677040+ Hs.661950+ NR2F2-AS1 + NR2F2- Hs.592015+ |
| Under-expressed segments | |||||
| chr4 4q12-q21 | 74,750,001 | 75,250,000 | 0.45 | 0.00079 | PF4 - PPBP - CXCL5- CXCL3 - CXCL2- MTHFD2L- Hs.662627- EREG- |
| chr4 4q32.1 | 156,250,001 | 156,750,000 | 0.13 | 0.00012 | MAP9- GUCY1A3 - Hs.612374- GUCY1B3 - |
| c) Non-DS AMKL vs. MK | |||||
| Chr and location | Segment start | Segment end | 'B'/'D' ratio | q-value | Genes in the segment |
| Over-expressed segments | |||||
| chr15 15q26.2 | 96,750,001 | 97,250,000 | 22.30 | <0.00001 | Hs.677040+ Hs.661950+ NR2F2-AS1 + NR2F2- Hs.592015+ |
| Under-expressed segments | |||||
| chr4 4q13-q21 | 74,500,001 | 75,000,000 | 0.39 | 0.00009 | IL8- CXCL6- PF4V1 - CXCL1- PF4 - PPBP - CXCL5- CXCL3 - CXCL2- |
Data refer to the following comparisons: a) DS AMKL (pool 'A') vs. non-DS AMKL (pool 'B'); b) DS AMKL (pool 'A') vs. MK (pool 'D'); c) non-DS AMKL (pool 'B') vs. MK (pool 'D'). Analysis was performed using default parameters (see Methods section). Segments are sorted by decreasing 'A'/'B' ratio in a), 'A'/'D' ratio in b), 'B'/'D' ratio in c). In the "Map" mode, TRAM displays UniGene EST clusters (with the prefix "Hs." in the case of Homo sapiens) only if they have an expression value. Some segments are not shown for simplicity because they are over-lapping with those highlighted in one of the listed regions. The complete results for these models are available as on line additional material.
aChr: chromosome. The segment location cytoband was derived from that of the first mapped gene within the segment.
bSegment Start/End: chromosomal coordinates for each segment.
cBold and '+': over-expressed gene; bold and '-': under-expressed gene; '+' or '-': gene expression value higher or lower than the median value, respectively.
dThis UniGene cluster contains EST with at least one Alu sequence, according to Repeat Masker [46]. For this reason, it can not be excluded that its over-expression is related to unspecific hybridization by Alu-containg probes.
At single gene level, a fold increase higher than 5 was observed in all of the first 20 genes with the greatest expression ratios of DS AMKL vs. non-DS AMKL samples (Table 3a and Additional file 3). In particular, a 24-fold increase was observed for SLITRK6 gene, encoding for a membrane protein to date described as similar to receptor for BDNF (brain-derived neurotrophic factor) and predominantly expressed in neural tissues. Among the genes with the lower 'A'/'B' expression ratios a 169.5-fold decrease was observed for a UniGene EST cluster, Hs.355689.
Table 3.
List of the five most over- or under-expressed genes (all significantly, with q < 0.05)
| a) DS AMKL vs. non-DS AMKL | ||||||||
| Gene | Value 'A' | Value 'B' | 'A'/'B' ratio | Location | Data points | SD | SD | |
| 'A' | 'B' | 'A' | 'B' | |||||
| Over-expressed genes | ||||||||
| SLITRK6 | 465.5 | 19.5 | 23.860 | 13q31.1 | 27 | 21 | 113.0 | 79.6 |
| HDC | 1,119.4 | 65.5 | 17.097 | 15q21-q22 | 36 | 45 | 98.8 | 249.4 |
| ZNF587B | 615.0 | 39.3 | 15.643 | 19q13.43a | 43 | 40 | 427.6 | 32.2 |
| SOSTDC1 | 111.7 | 8.6 | 12.957 | 7p21.1 | 43 | 45 | 176.5 | 108.4 |
| LOC100287628* | 1,381.3 | 107.7 | 12.820 | 16p13.2 | 9 | 7 | 80.2 | 56.8 |
| Under-expressed genes | ||||||||
| Hs.587427* | 19.8 | 407.8 | 0.049 | 7p15.2 | 18 | 14 | 28.9 | 353.3 |
| SPINK2 | 13.1 | 285.1 | 0.046 | 4q12 | 36 | 45 | 81.8 | 115.0 |
| Hs.602709* | 15.8 | 370.2 | 0.043 | 11q13.2a | 9 | 7 | 33.1 | 256.9 |
| RPS12 | 20.7 | 519.1 | 0.040 | 6q23.2 | 43 | 50 | 113.7 | 213.0 |
| Hs.355689* | 3.7 | 624.4 | 0.006 | 18q11.2a | 9 | 7 | 47.3 | 168.4 |
| b) DS AMKL vs. MK | ||||||||
| Gene | Value 'A' | Value 'D' | 'A'/'D' ratio | Location | Data points | SD | SD | |
| 'A' | 'D' | 'A' | 'D' | |||||
| Over-expressed genes | ||||||||
| TMEM241 | 1,036.6 | 17.6 | 58.885 | 18q11.2 | 18 | 8 | 57.7 | 115.8 |
| CMBL | 592.4 | 11.7 | 50.759 | 5p15.2 | 27 | 8 | 142.0 | 30.1 |
| IFI27 | 467.8 | 11.6 | 40.474 | 14q32 | 36 | 82 | 90.0 | 27.6 |
| CSRNP1 | 640.6 | 16.8 | 38.023 | 3p22 | 9 | 8 | 41.3 | 138.3 |
| PTGS2 | 2,205.0 | 59.2 | 37.276 | 1q25.2 q25.3 | 63 | 20 | 156.4 | 131.6 |
| APOC2* b | 431.7 | 11.7 | 37.044 | 19q13.2 | 54 | 28 | 81.4 | 41.6 |
| SLITRK6 b | 465.5 | 13.3 | 35.057 | 13q31.1 | 27 | 10 | 113.0 | 95.6 |
| Under-expressed genes | ||||||||
| MAP3K10 | 5.4 | 2,352.3 | 0.002 | 19q13.2 | 36 | 19 | 47.4 | 135.1 |
| DRD4 | 5.0 | 2,327.6 | 0.002 | 11p15.5 | 36 | 19 | 51.3 | 135.2 |
| DLGAP3 | 4.8 | 2,297.9 | 0.002 | 1p35.3-p34.1 | 9 | 7 | 40.8 | 37.6 |
| LBX1 | 2.7 | 1,358.5 | 0.002 | 10q24 | 36 | 19 | 46.4 | 152.6 |
| TSPAN10* | 7.9 | 4,674.7 | 0.002 | 17q25.3 | 9 | 8 | 55.0 | 46.0 |
| c) Non-DS AMKL vs. MK | ||||||||
| Gene | Value 'B' | Value 'D' | 'B'/'D' ratio | Location | Data points | SD | SD | |
| 'B' | 'D' | 'B' | 'D' | |||||
| Over-expressed genes | ||||||||
| TMEM241 | 2,754.1 | 17.6 | 156.443 | 18q11.2 | 14 | 8 | 89.9 | 115.8 |
| CDYL2 | 1,613.5 | 13.1 | 122.994 | 16q23.2 | 7 | 8 | 128.9 | 69.8 |
| MPV17L | 1,857.8 | 24.4 | 76.280 | 16p13.11 | 7 | 8 | 119.5 | 65.0 |
| ATP6V0D2 | 836.7 | 11.9 | 70.488 | 8q21.3a | 21 | 10 | 169.4 | 82.9 |
| CMBL | 757.9 | 11.7 | 64.938 | 5p15.2 | 21 | 8 | 176.2 | 30.1 |
| Under-expressed genes | ||||||||
| ILDR1 | 13.7 | 4,333.8 | 0.003 | 3q13.33 | 14 | 9 | 98.4 | 63.3 |
| HIST3H3 | 3.9 | 1,313.0 | 0.003 | 1q42 | 40 | 19 | 78.0 | 137.9 |
| TSPAN10* | 12.1 | 4,674.7 | 0.003 | 17q25.3 | 7 | 8 | 88.1 | 46.0 |
| MAP3K10 | 3.9 | 2,352.3 | 0.002 | 19q13.2 | 45 | 19 | 40.1 | 135.1 |
| LBX1 | 2.0 | 1,358.5 | 0.001 | 10q24 | 45 | 19 | 85.7 | 152.6 |
Data refer to the following comparisons: a) DS AMKL (pool 'A') vs. non-DS AMKL (pool 'B'); b) DS AMKL (pool 'A') vs. MK (pool 'D'); c) non-DS AMKL (pool 'B') vs. MK (pool 'D'). Value: mean gene expression value normalized across all the pool samples; data points: number of spots associated to an expression value for the locus; SD: standard deviation for the expression value expressed as percentage of the mean. Full results available as additional material (see text). *The segment window contains more than one gene, but the significance is assumed to be maintained because the expression value of this over- or under-expressed gene prevails over the others.
aCytoband not available in Gene was derived from the UCSC Genome Browser [36].
bThis gene, exceeding the limit of five genes for each list, has been shown for its relevance in the Discussion, being recurrent in other comparisons.
At chromosomal level, we calculated (in the TRAM "Chr" table) the median 'A'/'B' expression ratio for all the genes located in the same chromosome. The highest ratios were near to 1 (0.93 for chr22, 0.92 for chrX and chr21, 0.91 for chr19); other values were in the range from 0.90 (chr17 and chr12) to 0.76 (chrY).
We performed two additional transcriptome maps to investigate specifically sex-biased gene expression patterns (data not shown, results may be regenerated by the user by excluding/including or reimporting samples on the basis of data provided in Additional file 1): in particular, we compared male (pool 'A.1', n=11) vs. female DS AMKL cells (pool 'A.2', n=18). These datasets are derived from the samples for which the knowledge about the sex of the sample donor was available. The results showed a significant statistical correlation of data between male and female gene expression data (r=0.99, p-value<0.0001), showing a large overlap of results between the two transcriptome maps, with the exception of single genes with a well known sex-biased expression pattern. For example, XIST, which is specifically activated in female cells to start the X-inactivation process, turns out to be the most differentially expressed gene between female (value=402.60) and male (value=12.20) DS AMKL cells (ratio=33).
Transcriptome map comparison of DS AMKL or non-DS AMKL vs. normal MK
Regional differential expression of pool 'A' (43 DS AMKL samples) or pool 'B' (45 non-DS AMKL samples) versus pool 'D' (19 normal MK cell samples, 411,381 data points) (Table 1) was investigated. An 'A'/'D' ratio value was determinable for 25,800 loci (Additional file 4). The main results are shown in Figure 1.
For what DS AMKL samples are concerned, results included 5 significantly differentially expressed segments in DS AMKL cells, 3 over- and 2 under-expressed (Table 2b). The highest expression ratio (20.81) between DS AMKL cells and normal MK was observed in the segment at coordinates 3,500,001-4,000,000 on chromosome 3, including the known gene LRRN1, encoding for a type I transmembrane protein. The second segment with highest expression ratio (18.07) was located on chromosome 1 (1q23) and contained FAM78B (family with sequence similarity 78, member B). The third segment was on chromosome 15 and included NR2F2-AS1, a non-coding RNA. The first significantly under-expressed segment (4q32.1) includes genes encoding for subunits of soluble guanylate cyclase (GUCY1A3 and GUCY1B3), while the second spans the cluster of MK specific genes located on chromosome 4 (4q12-q21).
At single gene level, a fold increase higher than 18 was observed in all of the first 20 genes with the greatest expression ratios of DS AMKL vs. MK samples (Table 3b and Additional file 4). In particular, a 59-fold increase was observed for TMEM241, encoding a transmembrane protein of unknown function. Among the genes with the lowest 'A'/'D' expression ratio a 589.7-fold decrease was observed for TSPAN10, encoding for tetraspanin 10.
At chromosomal level, the highest ratio was observed for chr21 (1.75), the lowest (chr17), other values were in the range from 1.68 (chrY) to a value of 1.23 for chr17.
Regarding the non-DS AMKL samples, results obtained by default analysis and derived from 'B'/'D' expression ratio for 25,819 loci included one significantly over- and one significantly under-expressed segment (Table 2c). The highest expression ratio (22.30) between non-DS AMKL and normal MK was observed in the same segment on chr15, significantly over-expressed also in the DS AMKL transcriptome map. This segment was the only significantly over-expressed one in this comparison. Similarly, the only significantly under-expressed segment includes the cluster of MK specific genes on chromosome 4 also found to be under-expressed in DS AMKL samples (Table 2b).
At single gene level, a fold increase higher than 31 was observed in all of the first 20 genes with the greatest expression ratios of non-DS AMKL vs. MK samples (Table 3c and Additional file 5). In particular, a 156-fold increase was observed for TMEM241. Overall, there was a remarkable overlap between the most over- (TMEM241, CMBL, ZNF445, SPRR4) and under-expressed (PF4V1, FLJ22184, FSIP2, PPP1R3B, HIST3H3, PIF1, SPSB4, ILDR1, MAP3K10, DRD4, LBX1, TSPAN10) genes in DS and in non-DS AMKL samples (Additional files 4 and 5).
At chromosomal level, looking at the median 'B'/'D' expression ratio for the genes located in the same chromosome, the highest ratio was observed for chr21 (1.77) and chrY (1.73), followed by chr13 (1.71) and chr10 (1.59); other values were in the range from 1.58 (chr20), to a value of 1.24 (chrX).
Transcriptome map comparison of DS AMKL or non DS AMKL vs. normal CB MK
Regional differential expression of pool 'A' (43 DS AMKL samples) or pool 'B' (45 non-DS AMKL samples) versus pool 'E' (8 normal cord blood (CB)-derived MK cell samples, 191,798 data points) (Table 1) was investigated. The main results are shown in Figure 2.
Figure 2.
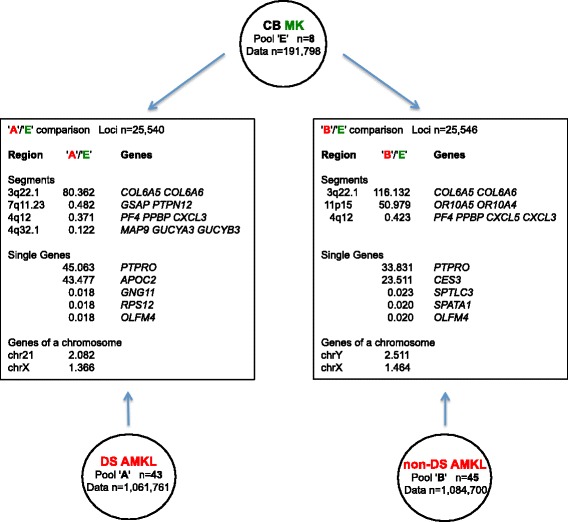
Main results of DS AMKL vs. CB MK and non-DS AMKL vs. CB MK comparisons. For each comparison the number of loci analyzed, the most over- or under-expressed segments and single genes, and the highest and lowest median expression ratios for all the genes located in the same chromosome are indicated.
For what DS AMKL samples are concerned, results derived from 'A'/'E' expression ratio for 25,540 loci (Additional file 6) included 1 significantly over- and 3 under-expressed segments in DS AMKL cells (Table 4a). A remarkable expression ratio (80.36) between DS AMKL cells and normal CB MK was observed for a segment on chromosome 3 (3q22.1), including collagen-encoding COL6A5 and COL6A6 known loci. The three significantly under-expressed segments included the region on chromosome 4 (4q12-q21) with PF4, PPBP and CXCL3 loci implied in MK differentiation.
Table 4.
Genomic segments significantly over- or under-expressed
| a) DS AMKL vs. normal CB MK | |||||
| Chr and location a | Segment start b | Segment end b | 'A'/'E' ratio | q-value | Genes in the segment c |
| Over-expressed segments | |||||
| chr3 3q22.1 | 130,000,001 | 130,500,000 | 80.36 | 0.00015 | COL6A5 + COL6A6 + Hs.596709+ Hs.596805+ PIK3R4- |
| Under-expressed segments | |||||
| chr7 7q11.23 | 77,000,001 | 77,500,000 | 0.48 | 0.00117 | GSAP - PTPN12 - LOC101059910- RSBN1L-AS1- RSBN1L- Hs.594486- Hs.720279- TMEM60- PHTF2- |
| chr4 4q12-q21 | 74,750,001 | 75,250,000 | 0.37 | 0.00119 | PF4 - PPBP - CXCL5- CXCL3 - CXCL2- MTHFD2L- Hs.662627- EREG- |
| chr4 4q32.1 | 156,250,001 | 156,750,000 | 0.12 | 0.00000 | MAP9 - GUCY1A3 - Hs.612374 - GUCY1B3 - |
| b) Non-DS AMKL vs. normal CB MK | |||||
| Chr and location | Segment start | Segment end | 'B'/'E' ratio | q-value | Genes in the segment |
| Over-expressed segments | |||||
| chr3 3q22.1 | 130,000,001 | 130,500,000 | 116.13 | 0.00030 | COL6A5 + COL6A6 + Hs.596709+ Hs.596805+ PIK3R4- |
| chr11 11p15 | 6,750,001 | 7,250,000 | 50.98 | 0.00117 | OR6A2+ OR10A5 + OR10A4 + ZNF215- ZNF214- Hs.445849+ NLRP14+ RBMXL2- LOC100506238+ |
| Under-expressed segments | |||||
| chr4 4q12-q21 | 74,750,001 | 75,250,000 | 0.42 | 0.00003 | PF4 - PPBP - CXCL5 - CXCL3 - CXCL2- MTHFD2L- Hs.662627- EREG- |
Data refer to the following comparisons: a) DS AMKL (pool 'A') vs. normal CB MK (pool 'E'); b) non-DS AMKL (pool 'B') vs. normal CB MK (pool 'E'). Analysis was performed using default parameters (see Methods section). Segments are sorted by increasing 'A'/'E' ratio in a), 'B'/'E' ratio in b). In the "Map" mode, TRAM displays UniGene EST clusters (with the prefix "Hs." in the case of H. sapiens) only if they have an expression value. Some segments are not shown for simplicity because they are over-lapping with those highlighted in one of the listed regions. The complete results for this model are available as on line additional material.
aChr: chromosome. The segment location cytoband was derived from that of the first mapped gene within the segment.
bSegment Start/End: chromosomal coordinates for each segment.
cBold and '+': over-expressed gene; bold and '-': under-expressed gene; '+' or '-': gene expression value higher or lower than the median value, respectively.
At single gene level, a fold increase higher than 15.7 was observed in all of the first 20 genes with the greatest expression ratios of DS AMKL cells vs. CB MK (Table 5a and Additional file 6). In particular, a 45-fold increase was observed for the tyrosine phosphatase receptor gene (PTPRO), known to be involved in megakaryocytopoiesis.
Table 5.
List of the five most over- or under-expressed genes (all significantly, with q <0.05)
| a) DS AMKL vs. normal CB MK | ||||||||
| Gene | Value 'A' | Value 'E' | Ratio 'A'/'E' | Location | Data points | SD | SD | |
| 'A' | 'E' | 'A' | 'E' | |||||
| Over-expressed genes | ||||||||
| PTPRO | 679.7 | 15.1 | 45.063 | 12p13-p12 | 81 | 9 | 133.0 | 60.8 |
| APOC2 a | 431.7 | 9.9 | 43.477 | 19q13.2 | 54 | 10 | 81.4 | 43.4 |
| IFI27 | 467.8 | 11.5 | 40.742 | 14q32 | 36 | 8 | 90.0 | 57.1 |
| HDC | 1,119.4 | 34.2 | 32.716 | 15q21-q22 | 36 | 8 | 98.8 | 168.1 |
| VIPR2 ab | 346.2 | 13.5 | 25.582 | 7q36.3 | 115 | 23 | 334.8 | 49.3 |
| Under-expressed genes | ||||||||
| CDKL1 | 4.6 | 174.3 | 0.027 | 14q21.3 | 45 | 9 | 65.3 | 99.0 |
| ASAP2 | 19.1 | 734.1 | 0.026 | 2p24 | 36 | 8 | 95.4 | 67.3 |
| GNG11 | 18.9 | 1,042.3 | 0.018 | 7q21 | 36 | 8 | 119.7 | 81.2 |
| RPS12 | 20.7 | 1,157.2 | 0.018 | 6q23.2 | 43 | 15 | 113.7 | 112.4 |
| OLFM4 | 8.9 | 499.3 | 0.018 | 13q14.3 | 36 | 7 | 83.9 | 121.5 |
| b) Non-DS AMKL vs. normal CB MK | ||||||||
| Gene | Value 'B' | Value 'E' | Ratio 'B'/'E' | Location | Data points | SD | SD | |
| 'B' | 'E' | 'B' | 'E' | |||||
| Over-expressed genes | ||||||||
| PTPRO b | 510.3 | 15.1 | 33.831 | 12p13-p12 | 97 | 9 | 137.8 | 60.8 |
| CES3 b | 287.4 | 12.2 | 23.511 | 16q22.1 | 47 | 9 | 335.8 | 73.8 |
| SCD5 ab | 544.7 | 27.2 | 19.992 | 4q21.22 | 61 | 18 | 290.3 | 73.9 |
| TOP3B ab | 150.2 | 7.5 | 19.922 | 22q11.22 | 60 | 14 | 431.8 | 19.7 |
| MT1E ab | 237.1 | 11.9 | 19.899 | 16q13 | 45 | 8 | 92.5 | 27.3 |
| Under-expressed genes | ||||||||
| Hs.23729a | 2.3 | 85.1 | 0.027 | 1p13.2c | 40 | 8 | 121.0 | 89.5 |
| PDE6C | 2.2 | 88.2 | 0.025 | 10q24 | 45 | 8 | 112.9 | 105.9 |
| SPTLC3 | 4.8 | 203.7 | 0.023 | 20p12.1 | 47 | 9 | 86.5 | 110.7 |
| SPATA1 | 7.0 | 356.2 | 0.020 | 1p22.3 | 40 | 7 | 127.4 | 77.9 |
| OLFM4 | 9.7 | 499.3 | 0.020 | 13q14.3 | 45 | 7 | 105.1 | 121.5 |
Data refer to the following comparisons: a) DS AMKL (pool 'A') vs. normal CB MK (pool 'E'); b) non-DS AMKL (pool 'B') vs. normal CB MK (pool 'E'). Value: mean gene expression value normalized across all the pool samples; data points: number of spots associated to an expression value for the locus; 'SD': standard deviation for the expression value expressed as percentage of the mean. Full results available as additional material (see text).
aThe segment window contains more than one gene, but the significance is assumed to be maintained because the expression value of this over- or under-expressed gene prevails over the others.
bAccording to the criteria detailed in Methods section, this gene is one of the five most over-expressed ones, but the value is not statistically significant due to the presence of a lot of loci associated with less than 5 data points in the CB MK integrated dataset.
cCytoband not available in Gene was derived from the UCSC Genome Browser [36].
At chromosomal level, the highest ratio was observed for chr21 (2.08), followed by chrY (1.89); other values were in the range from 1.84 (chr22) to 1.37 (chrX).
Regarding the non-DS AMKL samples, results derived from 'B'/'E' expression ratio (for 25,546 loci) included 2 significantly over- and 1 under-expressed segments in non-DS AMKL (Table 4b). A remarkable expression ratio between non-DS AMKL and normal CB MK (116.13) was observed in the same segment on chromosome 3 (3q22.1), including collagen-encoding COL6A5 and COL6A6 known loci that was observed in DS AMKL samples. The second segment was specific of non-DS AMKL samples and included the two olfactive receptor genes OR10A5 and OR10A4. The only significantly under-expressed segment included the region on chromosome 4 (4q12-q21) highly enriched in MK-specific loci (PF4, PPBP, CXCL5 and CXCL3) as in the case of DS AMKL samples, and was extended to PF4V1 locus.
At single gene level, a fold increase higher than 14.7 was observed in all of the first 20 genes with the greatest expression ratios of non-DS AMKL vs. CB MK samples (Table 5b and Additional file 7). In particular, a 33.8-fold increase was observed for PTPRO, encoding a tyrosine phosphatase receptor. Overall, there was some overlapping between the most over- and under-expressed genes in DS and in non-DS AMKL samples (Additional file 6 and Additional file 7).
At chromosomal level, regarding the median 'B'/'E' expression ratio for the genes located in the same chromosome, the highest ratio was observed for chrY (2.51), followed by chr21 (2.19); other values were in the range from 2.03 (chr20) to 1.46 (chrX).
Transcriptome map comparison of DS AMKL vs. TMD
Regional differential expression of pool 'A' (43 DS AMKL samples) versus pool 'C' (20 TMD samples, 398,162 data points) (Table 1) was investigated. The main results are shown in Figure 3.
Figure 3.
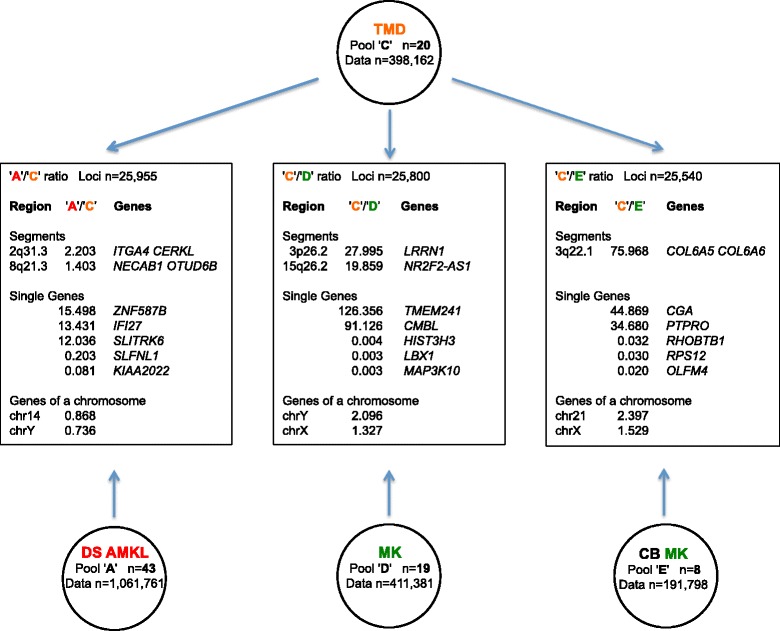
Main results of DS AMKL vs. TMD, TMD vs. MK, TMD vs. CB MK comparisons. For each comparison the number of loci analyzed, the most over- or under-expressed segments and single genes, and the highest and lowest median expression ratios for all the genes located in the same chromosome are indicated.
Results obtained by default analysis and derived from 'A'/'C' expression ratio for 25,955 loci (Additional file 8) included 2 significantly over-expressed segments in DS AMKL (Table 6a). The highest expression ratio (2.20) between DS AMKL and TMD was observed in a segment on chromosome 2 (2q31.3), including the known gene ITGA4, encoding an alpha 4 chain of integrin protein and CERKL, a gene responsible for retinitis pigmentosa and involved in the protection of cells from apoptosis induced by oxidative stress [47]. The second segment with the highest expression ratio (1.40) was located on chromosome 8 (8q21.3) and contained the NECAB1 and OTUD6B genes, encoding for neuronal Ca(2+)-binding protein and the deubiquitinating enzyme, respectively.
Table 6.
Genomic segments significantly over- or under-expressed
| a) DS AMKL vs. TMD | |||||
| Chr a and location | Segment start b | Segment end b | 'A'/'C' ratio | q-value | Genes in the segment c |
| Over-expressed segments | |||||
| chr2 2q31.3 | 182,000,001 | 182,500,000 | 2.20 | 0.00045 | ITGA4 + Hs.660611- Hs.658786+ CERKL+ Hs.72981+ |
| chr8 8q21.3 | 91,750,001 | 92,250,000 | 1.40 | 0.00051 | NECAB1 + Hs.743640+ LOC100127983+ TMEM55A+ LOC100506365- OTUD6B+ LRRC69+ |
| b) TMD vs. normal MK | |||||
| Chr and location | Segment start | Segment end | 'C'/'D' ratio | q-value | Genes in the segment |
| Over-expressed segments | |||||
| chr3 3p26.2 | 3,500,001 | 4,000,000 | 28.00 | 0.00006 | Hs.241414+ d Hs.587205+ LRRN1 + Hs.128128+ |
| chr15 15q26.2 | 96,750,001 | 97,250,000 | 19.86 | 0.00000 | Hs.677040+ d Hs.661950+ d NR2F2-AS1 + NR2F2- Hs.592015+ |
| c) TMD vs. normal CB MK | |||||
| Chr and location | Segment start | Segment end | 'C'/'E' ratio | q-value | Genes in the segment |
| chr3 3q22.1 |
130,000,001 | 130,500,000 | 75.97 | 0.00015 | COL6A5 + COL6A6 + Hs.596709+ Hs.596805+ PIK3R4- |
Data refer to the following comparisons: a) DS AMKL (pool 'A') vs. TMD (pool 'C'); b) TMD (pool 'C') vs. normal MK (pool 'D'); c) TMD (pool 'C') vs. normal CB MK (pool 'E'). Analysis was performed using default parameters (see Methods section). Segments are sorted by increasing 'A'/'C' ratio in a), 'C'/'D' ratio in b) and 'C'/'E' ratio in c). In the "Map" mode, TRAM displays UniGene EST clusters (with the prefix "Hs." in the case of H. sapiens) only if they have an expression value. Some segments are not shown for simplicity because they are over-lapping with those highlighted in one of the listed regions. The complete results for this model are available as additional material on line.
aChr: chromosome. The segment location cytoband was derived from that of the first mapped gene within the segment.
bSegment Start/End: chromosomal coordinates for each segment.
cBold and '+': over-expressed gene; bold and '-': under-expressed gene; '+' or '-': gene expression value higher or lower than the median value, respectively.
dThis UniGene cluster contains EST with at least one Alu sequence, according to Repeat Masker [46]. For this reason, it can not be excluded that its over-expression is related to non-specific hybridization by Alu-containg probes.
At single gene level, a fold increase ranged from 15.5 to 3.5 for the first 20 genes with the greatest expression ratios of DS AMKL vs. TMD samples (Table 7a and Additional file 8). The highest fold increases were observed for the ZNF587B (15.5) and IFI27 (13.4) genes, encoding for a zinc finger protein and the interferon alpha-inducible protein 27, respectively. The lowest 'A'/'C' expression ratios were observed for KIAA2022 (10-fold decrease) and SLFNL1 (5-fold decrease) genes.
Table 7.
List of the five genes most over- or under-expressed (all significantly, with q <0.05)
| a) DS AMKL vs. TMD | ||||||||
| Gene | Value 'A' | Value 'C' | Ratio 'A'/'C' | Location | Data points | SD | SD | |
| 'A' | 'C' | 'A' | 'C' | |||||
| Over-expressed genes | ||||||||
| ZNF587B | 615.0 | 39.7 | 15.498 | 19q13.43c | 43 | 20 | 427.6 | 20.9 |
| IFI27 | 467.8 | 34.8 | 13.431 | 14q32 | 36 | 11 | 90.0 | 91.4 |
| SLITRK6 | 465.5 | 38.7 | 12.036 | 13q31.1 | 27 | 9 | 113.0 | 84.5 |
| BST2 a | 117.6 | 14.8 | 7.967 | 19p13.1 | 36 | 11 | 260.0 | 98.8 |
| ZNF521 | 143.0 | 19.6 | 7.309 | 18q11.2 | 18 | 6 | 113.5 | 58.6 |
| Under-expressed genes | ||||||||
| ALS2CR12 | 4.2 | 15.8 | 0.268 | 2q33.1 | 18 | 6 | 80.3 | 103.2 |
| GBP5 | 11.6 | 43.8 | 0.266 | 1p22.2 | 18 | 6 | 75.7 | 110.6 |
| GHRH | 14.7 | 67.1 | 0.219 | 20q11.2 | 43 | 20 | 97.7 | 257.0 |
| SLFNL1 a | 18.9 | 92.9 | 0.203 | 1p34.2 | 18 | 6 | 90.3 | 122.1 |
| KIAA2022 | 24.3 | 297.9 | 0.081 | Xq13.3 | 27 | 9 | 52.2 | 260.9 |
| b) TMD vs. normal MK | ||||||||
| Gene | Value 'C' | Value 'D' | Ratio 'C'/'D' | Location | Data points | SD | SD | |
| 'C' | 'D' | 'C' | 'D' | |||||
| Over-expressed genes | ||||||||
| TMEM241 | 2,224.4 | 17.6 | 126.356 | 18q11.2 | 6 | 8 | 57.1 | 115.8 |
| CMBL | 1,063.5 | 11.7 | 91.126 | 5p15.2 | 9 | 8 | 168.8 | 30.1 |
| PTGS2 | 2,256.4 | 59.2 | 38.145 | 1q25.2-q25.3 | 20 | 20 | 147.6 | 131.6 |
| CGA | 360.6 | 10.0 | 36.238 | 6q12-q21 | 14 | 19 | 159.7 | 27.3 |
| TMEFF2 | 755.1 | 22.5 | 33.605 | 2q32.3 | 9 | 9 | 190.3 | 44.3 |
| Under-expressed genes | ||||||||
| ILDR1 | 18.3 | 4,333.8 | 0.004 | 3q13.33 | 6 | 9 | 58.9 | 63.3 |
| DRD4 | 9.3 | 2,327.6 | 0.004 | 11p15.5 | 11 | 19 | 52.3 | 135.2 |
| HIST3H3 | 5.2 | 1,313.0 | 0.004 | 1q42 | 11 | 19 | 36.2 | 137.9 |
| LBX1 | 4.3 | 1,358.5 | 0.003 | 10q24 | 11 | 19 | 26.6 | 152.6 |
| MAP3K10 | 7.4 | 2,352.3 | 0.003 | 19q13.2 | 11 | 19 | 28.8 | 135.1 |
| c) TMD vs. normal CB MK | ||||||||
| Gene | Value 'C' | Value 'E' | Ratio 'C'/'E' | Location | Data points | SD | SD | |
| 'C' | 'E' | 'C' | 'E' | |||||
| Over-expressed genes | ||||||||
| CGA | 360.6 | 8.0 | 44.869 | 6q12-q21 | 14 | 8 | 159.7 | 39.9 |
| PTPRO b | 523.1 | 15.1 | 34.680 | 12p13-p12 | 25 | 9 | 116.8 | 60.8 |
| CLCA1 b | 319.2 | 9.9 | 32.380 | 1p22.3 | 20 | 8 | 116.7 | 17.5 |
| APOC2 ab | 301.6 | 9.9 | 30.372 | 19q13.2 | 17 | 10 | 66.6 | 43.4 |
| HDC ab | 918.0 | 34.2 | 26.830 | 15q21-q22 | 11 | 8 | 59.9 | 168.1 |
| Under-expressed genes | ||||||||
| CDKL1 | 5.9 | 174.3 | 0.034 | 14q21.3 | 14 | 9 | 82.0 | 99.0 |
| GNG11 | 34.3 | 1,042.3 | 0.033 | 7q21 | 11 | 8 | 80.1 | 81.2 |
| RHOBTB1 | 7.7 | 241.3 | 0.032 | 10q21.2 | 20 | 8 | 49.7 | 97.4 |
| RPS12 | 34.6 | 1,157.2 | 0.030 | 6q23.2 | 20 | 15 | 77.4 | 112.4 |
| OLFM4 | 10.0 | 499.3 | 0.020 | 13q14.3 | 11 | 7 | 56.0 | 121.5 |
Data refer to the following comparisons: a) DS AMKL (pool 'A') vs. TMD (pool 'C'); b) TMD (pool 'C') vs. normal MK (pool 'D'); c) TMD (pool 'C') vs. normal CB MK (pool 'E'). Value: mean gene expression value normalized across all the pool samples; data points: number of spots associated to an expression value for the locus; 'SD': standard deviation for the expression value expressed as percentage of the mean. Full results available as additional material (see text).
aThe segment window contains more than one gene, but the significance is assumed to be maintained because the expression value of this over- or under-expressed gene prevails over the others.
bAccording to the criteria detailed in Methods section, this gene is one of the five most over-expressed ones, but the value is not statistically significant due to the presence of a lot of loci associated with less than 5 data points in the CB MK integrated dataset.
cCytoband not available in Gene was derived from the UCSC Genome Browser [36].
At chromosomal level, the highest ratio was observed for chr14 (0.87, followed by 0.86 for chr5, chr21, chr8, chr12 and chr16), the lowest for chrY (0.74).
Transcriptome map comparison of TMD vs. normal MK or CB MK cells
Regional differential expression of pool 'C' (20 TMD samples) versus pool 'D' (19 MK samples) or 'E' (8 CB MK samples) (Table 1) was investigated. The main results are shown in Figure 3.
For what MK samples are concerned, results obtained by default analysis and derived from 'C'/'D' expression ratios for 25,800 loci (Additional file 9) included 2 significantly over-expressed segments in TMD cells (Table 6b).
The highest expression ratio (28.0) between TMD and normal MK was observed in a segment on chromosome 3 including the known gene LRRN1, already observed as over-expressed in comparison of DS AMKL vs. normal MK (Table 2b). The second segment with the highest expression ratio (19.9) was located on chromosome 15, and contained the locus NR2F2-AS1, encoding for an antisense mRNA, already observed as over-expressed in comparison of DS AMKL and non-DS AMKL vs. normal MK (Table 2b and 2c).
At single gene level, the fold increase was higher than 16.5 for the first 20 genes with the greatest expression ratios (Table 7b and Additional file 9), with the highest fold increases for TMEM241 (126.4) and the cysteine hydrolase gene (CMBL) (91.1). The lowest 'C'/'D' expression ratios were observed for a member of the serine/threonine kinase family (MAP3K10) and the homeobox gene (LBX1) (both with 333-fold decrease).
At chromosomal level, the highest ratio was observed for chrY (2.1) and chr21 (1.93), followed by chr13 (1.71), the lowest for chrX (1.33).
As far as the comparison of TMD with CB MK samples is concerned, results derived from the 'C'/'E' expression ratio for 25,540 loci (Additional file 10) included only 1 significantly over-expressed segment in TMD cells (Table 6c). The segment with a significant high expression ratio (76.0) between TMD and normal CB MK cells was on chromosome 3 (3q22.1), including the known genes COL6A5 and COL6A6 and already observed as over-expressed in DS as well in non-DS AMKL samples in comparison with CB MK samples.
At single gene level, a fold increase ranged from 44.9 to 15.1 for the first 20 genes with the greatest expression ratios (Table 7c and Additional file 10). The highest fold increases were observed for CGA (44.9), encoding for the alpha chain of the glycoprotein hormones and PTPRO (34.7), as already observed in non-DS AMKL vs. CB MK comparison. The lowest 'C'/'E' expression ratio was observed for OLFM4 (50-fold decrease), encoding olfactomedin 4, an antiapoptotic factor that promotes tumor growth.
At chromosomal level, the highest ratio was observed for chr21 (2.40), followed by chrY (2.37) and chr20 (2.13), the lowest for chrX (1.53).
Comparison with previously published data
As a result of the analysis above described, a reference integrated map for the expression of about 26,000 mapped sequences (~75% known genes and ~25% expression sequence tags - ESTs) was de facto generated for five cell types (DS AMKL cells, non-DS AMKL cells, TMD cells, MK and CB MK). This gave us the opportunity to compare our data with the expression values of specific known genes from previously published works about the considered cell types.
Following analysis of the main literature about AMKL, we selected 38 genes of interest and have tabulated their expression values desumed from our 8 differential maps, comparing these values to the ones previously described in different experimental settings (Table 8).
Table 8.
Comparison with previously published data
| Gene | Location | References | 'A' /' B' | 'A' /'D' | 'B' /'D' | 'A '/' E' | 'B' /' E' | 'A' /' C' | 'C' / 'D' | 'C '/' E' |
|---|---|---|---|---|---|---|---|---|---|---|
| MK markers | ||||||||||
| BST2 | 19p13.11 | [23] | 0.53 | 5.36 | 10.07 | 7.21 | 13.56 | 7.97 | 0.67 | 0.91 |
| GATA1 | Xp11.23 | [13,22,48] | 3.39 | 1.45 | 0.43 | 1.97 | 0.58 | 1.19 | 1.22 | 1.66 |
| GP1BA | 17p13.2 | [43] | 0.46 | 0.07 | 0.14 | 0.25 | 0.53 | 0.81 | 0.08 | 0.30 |
| HBG1 | 11p15.5 | [22] | 2.36 | 1.40 | 0.59 | 1.07 | 0.45 | 0.78 | 1.79 | 1.37 |
| HBG2 | 11p15.5 | [22] | 2.47 | 1.31 | 0.53 | 1.06 | 0.43 | 0.71 | 1.84 | 1.48 |
| ITGA2B | 17q21.32 | [23] | 0.82 | 0.63 | 0.77 | 1.52 | 1.84 | 1.05 | 0.60 | 1.44 |
| ITGB3 | 17q21.32 | [23] | 0.75 | 0.08 | 0.10 | 0.10 | 0.13 | 0.65 | 0.12 | 0.15 |
| PF4 | 4q12-q21 | [25] | 0.90 | 0.06 | 0.06 | 0.11 | 0.12 | 0.44 | 0.13 | 0.25 |
| PPBP | 4q12-q13 | [25] | 1.45 | 0.10 | 0.07 | 0.13 | 0.09 | 0.28 | 0.34 | 0.44 |
| TAL1 | 1p32 | [23] | 0.85 | 0.19 | 0.22 | 0.16 | 0.19 | 1.05 | 0.18 | 0.16 |
| Chr21 genes | ||||||||||
| ADAMTS1 | 21q21.2 | [49] | 6.19 | 7.02 | 1.14 | 7.14 | 1.15 | 1.82 | 3.85 | 3.91 |
| BACH1 | 21q22.11 | [22] | 1.86 | 1.44 | 0.77 | 1.04 | 0.56 | 1.55 | 0.93 | 0.67 |
| DYRK1A | 21q22.13 | [50] | 1.61 | 0.87 | 0.54 | 0.63 | 0.39 | 1.14 | 0.77 | 0.55 |
| ERG | 21q22.3 | [18,22] | 0.68 | 3.47 | 5.12 | 3.10 | 4.57 | 1.42 | 2.44 | 2.18 |
| ETS2 | 21q22.2 | [18,22] | 0.93 | 1.11 | 1.20 | 1.39 | 1.49 | 0.73 | 1.52 | 1.90 |
| GABPA | 21q21.3 | [18,22] | 1.39 | 2.45 | 1.76 | 1.52 | 1.10 | 1.55 | 1.58 | 0.98 |
| RUNX1 | 21q22.3 | [18,22] | 0.71 | 1.08 | 1.51 | 0.84 | 1.18 | 1.06 | 1.01 | 0.80 |
| SOD1 | 21q22.11 | [51] | 1.88 | 1.52 | 0.81 | 2.78 | 1.48 | 1.52 | 1.00 | 1.84 |
| SON | 21q22.11 | [22] | 1.28 | 1.39 | 1.08 | 1.03 | 0.80 | 1.44 | 0.96 | 0.71 |
| Other genes | ||||||||||
| APOC1 | 19q13.2 | [20,22,23] | 4.37 | 10.04 | 2.30 | 16.26 | 3.72 | 1.79 | 5.63 | 9.11 |
| APOC2 | 19q13.2 | [22] | 4.36 | 37.04 | 8.49 | 43.48 | 9.96 | 1.43 | 25.88 | 30.37 |
| APOE | 19q13.2 | [22,23] | 2.18 | 3.57 | 1.64 | 3.24 | 1.48 | 1.56 | 2.30 | 2.08 |
| CDA | 1p36.2-p35 | [23] | 0.39 | 0.40 | 1.03 | 0.88 | 2.24 | 0.88 | 0.46 | 1.00 |
| DICER1 | 14q32.13 | [18] | 0.80 | 0.41 | 0.51 | 0.27 | 0.34 | 0.99 | 0.41 | 0.27 |
| FCER1A | 1q23 | [22,23] | 6.32 | 4.28 | 0.68 | 8.86 | 1.40 | 1.32 | 3.25 | 6.72 |
| GYPA | 4q31.21 | [23] | 3.40 | 1.56 | 0.46 | 0.89 | 0.26 | 1.19 | 1.31 | 0.75 |
| GYPB | 4q31.21 | [23] | 2.81 | 1.28 | 0.46 | 0.89 | 0.31 | 1.27 | 1.01 | 0.70 |
| GYPE | 4q31.1 | [23] | 2.07 | 1.63 | 0.79 | 1.71 | 0.82 | 1.72 | 0.95 | 0.99 |
| HDC | 15q21-q22 | [22,23] | 17.10 | 9.95 | 0.58 | 32.72 | 1.91 | 1.22 | 8.16 | 26.83 |
| IGF1R | 15q26.3 | [17] | 0.76 | 1.18 | 1.55 | 0.94 | 1.24 | 1.21 | 0.98 | 0.78 |
| ITGAL | 16p11.2 | [52] | 0.79 | 0.21 | 0.26 | 0.09 | 0.11 | 1.02 | 0.20 | 0.09 |
| KIT | 4q11-q12 | [22] | 1.66 | 3.34 | 2.01 | 3.08 | 1.85 | 1.22 | 2.74 | 2.53 |
| MPL | 1p34 | [53] | 1.11 | 0.31 | 0.28 | 0.22 | 0.20 | 1.23 | 0.25 | 0.18 |
| MTOR | 1p36.22 | [17] | 0.87 | 1.57 | 1.82 | 1.94 | 2.24 | 0.89 | 1.76 | 2.17 |
| MYCN | 2p24.3 | [21] | 0.35 | 1.00 | 2.87 | 0.77 | 2.19 | 0.34 | 2.99 | 2.28 |
| PRAME | 22q11.22 | [21] | 1.60 | 11.59 | 7.24 | 15.61 | 9.76 | 4.18 | 2.77 | 3.73 |
| SMOX | 20p13 | [23] | 1.37 | 0.76 | 0.55 | 1.02 | 0.75 | 0.79 | 0.96 | 1.30 |
| ST18 | 8q11.23 | [18] | 0.90 | 0.74 | 0.82 | 0.74 | 0.82 | 0.78 | 0.96 | 0.95 |
Expression values of genes known for their role in MK differentiation and DS or non-DS AMKL development in each of the eight comparisons: DS AMKL (pool 'A') vs. non-DS AMKL (pool 'B'); DS AMKL (pool 'A') vs. normal MK (pool 'D'); non-DS AMKL (pool 'B') vs. normal MK (pool 'D'); DS AMKL (pool 'A') vs. normal CB MK (pool 'E') cells; non-DS AMKL (pool 'B') vs. normal CB MK (pool 'E'); DS AMKL (pool 'A') vs. TMD (pool 'C'); TMD (pool 'C') vs. normal MK (pool 'D'); TMD (pool 'C') vs. normal CB MK (pool 'E'). Data extracted from the full tables showing expression values and their ratio for about 17-26,000 loci for each comparison. In bold: expression ratio values are consistent with data available in the literature (see references indicated). The other values are reported for completeness. Descriptions of the gene symbols are given in Additional file 2.
The wide agreement of expression ratio values for specific genes between our data, generated by systematic meta-analysis of hundred of thousands of gene expression values from any gene expression profile available, and the data obtained by different marker-specific methods in published quantitative studies, is relevant for the validation of our maps that may so be used for exploring any other expression ratio in the considered biological conditions.
Discussion
We have presented here a comprehensive analysis of transcriptome in human DS AMKL cells. Integration of data from different sources, including data obtained from different Authors using a variety of platforms, was made possible by a recent approach described by us for creation and analysis of transcriptome maps [25]. While most approaches are aimed to separate gene expression profiles related to the same biological source in subclasses, the TRAM tool provides means to integrate and summarize a pool of samples of the same biological origin leading to a global picture of gene expression for that condition. Moreover, TRAM identifies critical genomic regions and genes with significant differential expressions between two biological conditions.
Several Authors have determined gene expression profiles for DS or non-DS AMKL samples or have explicitly compared these two leukemic conditions. However, due to the rarity of the M7 subtype of leukemia and the need to limit the analysis to pediatric age because DS AMKL occurs almost exclusively in children, these studies were typically limited to small group of samples. In addition, most platforms used in the microarray studies are affected by omissions or errors in mapping a certain percentage of probes to specific loci in the genome. In our analysis, the use of a new version of the TRAM software (TRAM 1.1) allowed us to map thousands of previously uncharacterized microarray probes and to avoid the errors in probe assignment to human loci often present in the data supplied by the manufacturer along with the platforms.
Our data are derived from systematic integration of data from multiple sources at locus level (up-to-date rigorous assignment of each microarray probe to a specific human locus/transcript/EST cluster), map level (up-to-date fine mapping of each transcript on the genome map) and expression value level (assignment of a reference value to each locus in each cell type following an intra- and inter-sample normalization pipeline exploiting both parametric and non-parametric calculations). The combination of many gene expression profile datasets from different sources poses the problem of the batch effect, i.e. the systematic differences between batches (groups) of samples in microarray experiments due to purely technical reasons. However, the intrinsic resistance of the TRAM approach to the batch effect has been discussed previously [25], and it is indirectly confirmed by the clear biological meaning of the differential expression highlighted by the tool when comparison with previous direct key experimental knowledge is possible in several different types of tissues and organs [25,34,54].
A systematic comparison of AMKL originated from trisomy 21 cells versus non-trisomy 21 cells should highlight specific mechanisms [55] related to the presence of an extra copy of chr21 in DS children developing AMKL. Moreover, we presented a comparison with normal MK cells that has never been performed in other analyses about AMKL. Our global quantitative models of the transcriptome in the AMKL cells could also be useful to test hypotheses for correlations between any parameter associated to the condition (e.g., specific mutations or phenotype aspects) and specific changes in gene expression.
Our results, obtained in an integrated and open setting without any a priori assumption, show several previously unidentified aspects regarding specificity of AMKL originated by trisomy 21 cells.
First, there are only a few genomic regions significantly over- or under-expressed when comparing DS versus non-DS AMKL samples. This finding suggests that transcriptome maps of these two conditions are similar while on the other hand allows to focus to a small set of regions that appears to be critical in order to differentiate these disease conditions. Relevant differences regarding genes were reported in Table 2a (genomic segments) and Table 3a (single genes), with potential implications for the identification of diagnostic or therapeutic targets. There are three main regions over-expressed in DS AMKL vs. non-DS AMKL (Table 2a). The first one (15q21.2) contains HDC gene, whose mRNA is translated in the enzyme converting L-histidine to histamine produced by only a few cell types [56]; HDC mRNA increase has been shown to be associated to basophilic rather than to MK differentiation of pluripotent hematopoietic cells [57]. These observations led to the discovery of a skewing toward a potential basophilic differentiation for DS AMKL not highlighted in the original works from which the data were derived. Supporting this hypothesis, FCER1A mRNA, encoding the alpha subunit of the high-affinity IgE receptor, the initiator of the allergic response and strongly typical of basophilic differentiation, is 6.3 times over-expressed (the 8th most over-expressed known gene) in DS vs. non-DS AMKL, reinforcing the notion that in DS AMKL but not in non-DS AMKL the leukemic dedifferentiation involved the possibility of redirection toward basophilic differentiation. Remarkably, in [58] is demonstrated by an electron microscopy analysis that AMKL blast cells from children with DS may contain basophil-like granules which were almost totally absent in blasts from children with non-DS AMKL or adults with AMKL, so that our data allow to the visualization of the molecular correlation at the level of the whole transcriptome of a morphological feature observed more than 20 years ago. Two other genomic regions are over-expressed in DS vs. non-DS AMKL blasts. The first is the region of glycophorins genes (GYPE, GYPA, GYPB) on chr4, erythroid surface markers [59]: this reinforces the concept of a disturbance of multilineage myeloid hematopoiesis in DS AMKL and has been observed by flow cytometry in the non-neoplastic hematopoiesis itself in trisomy 21 [60]. The other is the region of apolipoproteins genes (APOC1, APOC2, APOE) on chr19 that has been described as a signature of progression from TMD to DS AMKL in a gene expression profile ([20], it was not possible to include this in our analysis), further underlining its specificity for DS vs. non-DS MK blast cells.
When grouping expression values by chromosome, the chromosome with the greatest global RNA output was chr21 in both TMD and DS AMKL vs. normal CB MK; we observed the same result in both DS and non-DS AMKL vs. normal MK comparisons. These data suggest that over-expression of chr21 genes is a key factor in AMKL development. In particular, ADAMTS1, encoding a protease known to inhibit angiogenesis [61] is the most over-expressed chr21 gene in DS vs. non-DS AMKL comparison. It is interesting to notice that this gene has been correlated to pediatric leukemias (ALL and DS AML) [49,62] but its exact role in leukemogenesis is still to be discovered. Our quantitative approach summarizing all values for each locus may clearly highlight mean expression ratio near to 1.5:1 for several chr21 genes when comparing DS AMKL vs. non-DS AMKL. This observation is consistent with the presence of an additional copy of the considered genes in trisomic cells. For example, GABPA expression presents a ratio close to 1.5:1 expected in trisomy of chr21, as well as other chr21 genes (DYRK1A, SON, BACH1) (Table 8). Even relatively small but significant differences (around 1.5-fold) in expression of numerous genes likely produce an aggregate effect, as observed in [63], where the same genes seem to be candidates to explain the impact of trisomy 21 in hematopoiesis abnormalities. For this reason it could be interesting to start from significant and robust meta-analysis data to plan functional approaches in the future. Moreover, our data are consistent with the previous observation that RUNX1, ERG and ETS2 oncogenes, although located on chr21, are not over-expressed in DS vs. non-DS AMKL [18,22]. It has also been recently demonstrated that they are not located on a chr21 duplicated minimal region in two cases of AML of M0 subtype (FAB classification) [64]. As regard oncogenes, if chr21 oncogenes cited above appear not to be over-expressed in DS AMKL, TRIB1 (chr8) may be a novel important oncogene for DS AMKL and its mutation is an earlier genetic event in leukemogenesis [65]. In particular, it has been shown that a mutation of TRIB1 [65], a myeloid oncogene whose protein product is able to enhance ERK phosphorylation and to promote degradation of C/EBP family transcription factors, is a gain-of-function mutation remaining in leukocytes of the remission stage in which GATA1 mutation disappeared. Our results show a mean value of 1.40 for the human TRIB1 expression ratio between DS and non-DS AMKL samples, with a higher over-expression observed when comparing leukemic samples to normal MK (ratio 3.15 for DS AMKL and 2.26 for non-DS AMKL).
Although the expression of chr21 genes as a whole, or of some individual chr21 genes, may be coherent with the 3:2 gene expression ratio model in the comparison between trisomic and euploid cells, we note that discrepancies from this ideal model seen in our data for some of the comparisons we have made (Figures 1, 2 and 3) may be ascribed to the complexity of gene regulation in the aneuploid state [66,67], to individual variability [68] as well as to the general dysregulation typical of the neoplastic state for which specific cell types we analyze is concerned.
Additional biologically relevant findings came from comparison of each type of megakaryoblastic leukemic condition with normal MK cells. Due to the role of the microenvironment in the hemopoiesis, including hemopoiesis in DS [69], it is expected that DS MK would present growth alterations due to trisomy 21 in both hemopoietic and microenvironment cells. From this point of view, DS MK cells would be the ideal control for the progression of a trisomic cell toward TMD and AMKL, however no gene expression profile dataset was available for this cell type. We propose here a biological model of the transcriptome depicting progressive changes from normal MK to TMD and then to DS AMKL, able to underline both shared and unique transcriptome map patterns for DS and non-DS variants of AMKL (Figure 4).
Figure 4.
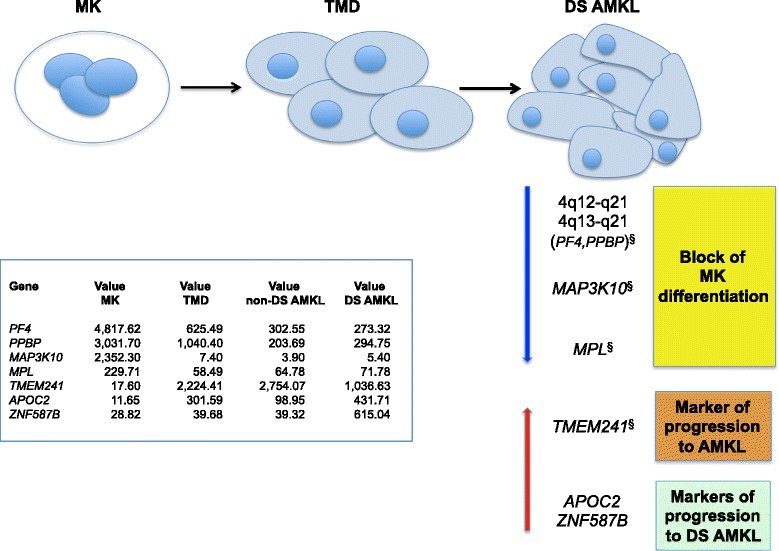
Biological model of the transcriptome depicting progressive changes from MK to TMD then to DS AMKL. Downward pointing arrow indicates the repression of genes involved in MK differentiation; upward pointing arrow indicates the over-expression of potential molecular markers of progression to AMKL. Value: mean gene expression value normalized across all the pool samples. §Observed both in DS and non-DS AMKL.
Noteworthy, the genomic segment on chr4 known to contain a cluster of genes highly specific for MK differentiation [25,70], was the highest significantly under-expressed segment in both DS and non-DS AMKL in comparison with normal MK. In particular, the more strongly under-expressed region 4q12-q21 contains a cluster of genes, including PF4 (encoding for the platelet factor 4, a main component of platelet alpha granules) [71-73], and PPBP (encoding for beta-thromboglobulin) [70], that are the most up-regulated transcripts in the megakaryocytic differentiation from CD34+ hematopoietic progenitors [25]. This finding highlights a common final outcome of the block of MK cells differentiation in both DS and non-DS AMKL. It should be underlined that this result came from systematic ab initio analysis of more than 12,000 segments on the human genome including about 26,000 mapped loci, thus highlighting that this region critical for the MK differentiation is actually the more repressed in absolute when comparing transcriptome maps of AMKL (DS or non-DS) and normal MK cells.
In addition, the most under-expressed gene in TMD blasts when compared to normal MK cells is MAP3K10, encoding an activity of mitogen-activated protein kinase kinase kinase (MAPKKK). It is known that the mitogen-activated protein kinase (MAPK) pathway is involved in and is sufficient for megakaryocytic differentiation [74,75]. MAPK activity is present in several tens of human proteins, and we have identified the member MAP3K10 as the critically repressed gene in the block of MK differentiation in the development of leukemia with MK features in that it appears down-regulated 300-fold in TMD cells and 500-fold in both DS and non-DS AMKL compared with normal MK samples (Tables 3 and 7). Finally, transcript for MPL, the receptor of thrombopoietin which is the primary regulator of normal thrombopoiesis (the formation of platelets) [53,73,76], is decreased by ~70% in either TMD, DS and non-DS AMKL cells vs. normal MK cells.
An exceedingly high over-expression of the gene located on chromosome 18 and encoding for the uncharacterized membrane protein TMEM241 has been found in both DS (59-fold) and non-DS (156-fold) AMKL cells vs. normal MK. Although this probe was not present in all considered experimental platforms, its extreme differential expression makes it a candidate for further studies as a marker of progression from normal MK to AMKL blasts, also due to its 126-fold over-expression in TMD vs. MK cells.
Moreover, we identified several signatures of progression specifically to DS AMKL. Remarkably, segments and genes up- or down-regulated in TMD in comparison with normal MK cells were highly similar to those specifically found in DS AMKL, underlining striking similarities between TMD and DS AMKL at the level of the whole transcriptome (already noted in [21], in their smaller set). On the other hand, a direct comparison between TMD and DS AMKL shows specific potential markers of progression to DS AMKL. As cited above, apolipoproteins genes (APOC1, APOC2, APOE) have been described as a signature of progression from TMD to DS AMKL [20] and it is interesting to notice that APOC2 is among the 20 most expressed genes in the comparison between TMD and MK (25.88-fold increase), showing a progressive increase of expression from normal MK to TMD and then to DS AMKL. In our analysis, ZNF587B appears to be the most discriminant marker between TMD and DS AMKL. Again, this observation offers the opportunity to integrate and discuss single genes and pathways previously described as abnormally expressed in DS or non-DS AMKL (Table 8). For example, the PRAME gene, encoding for a tumor antigen [21] was identified as a specific marker for DS AMKL blasts (n=7), with no expression in TMD (n=9). While our meta-analysis on PRAME expression data points (36 for DS AMKL and 11 for TMD) confirmed a clear over-expression of PRAME in DS AMKL (4.2-fold increase, Table 8), it was not the most discriminant marker, that was exactly ZNF587B, while PRAME was the 33rd out of 25,955 transcripts ordered by decreasing DS AMKL vs. TMD expression ratio (Additional file 8).
Finally, since leukemias in infants or young children originate from fetal hematopoietic cells [17,18,26,27] and the progenitors (fetal/neonatal MKP) are present in the cord blood [28,29], comparisons with CB MK cells have been also performed. Data from DS and non-DS AMKL vs. CB MK comparisons confirmed the repression of the clusters of genes expressed in MK. The over-expression of a region with collagen genes emerged both in DS and non-DS AMKL as well as that of the single gene PTPRO (Table 5a and 5b), encoding for a tyrosine phosphatase receptor known to be involved in megakaryocytopoiesis and whose mRNA targeting by antisense oligonucleotides results in inhibited MK progenitor proliferation [77]. On the other hand, the difference between DS and non-DS AMKL vs. CB MK is shown by the over-expression of the two olfactive receptor genes OR10A5 and OR10A4 (Table 4) only in non-DS derived cells.
The analysis of enrichment in specific gene functions using the tool FuncAssociate, for the 100 most over- or under-expressed genes in the comparison of DS vs. non-DS AMKL and of both of them vs. MK cells gave no significant results, other than a significant enrichment in genes involved in sequence-specific DNA-binding in non-DS AMKL vs. MK cells (data not shown), highlighting the relevance of remodeling the transcription factor network in leukemia.
Conclusions
Our results provide a systematic meta-analysis using any available gene expression profile dataset related to AMKL in pediatric age. These allow to identify more general trends and to produce a highly coherent view of the transcriptome depicting progressive changes from MK to TMD and then to DS AMKL. We believe that the originality of our results is due to several concurrent original features of the TRAM 1.1 platform. Advantages and relevant differences are: integration of the largest possible number of samples; integrated analysis of the largest possible number of genes (the integration of different platforms led us to assess expression ratio for about 26,000 loci, quantitating almost 4,000 genes in addition to the widely used platform U133A when used alone); absence of a priori filtering (in several works, this led to actual analysis of less than 50% of the genes present on the experimental platform); characterization at regional/map level in the study of gene expression (usually absent in the works from which data were obtained), relevant with regard to the study of an aneuploidy such as trisomy 21.
These results provide a new integrated model of the whole human transcriptome in DS and non DS AMKL, TMD and normal human MK cells, providing hints about pathophysiology of AMKL and also being useful to highlight possible clinical markers.
Acknowledgments
We would like to give special thanks to the Fondazione Umano Progresso, Milano, Italy, for supporting the research on trisomy 21 conducted at the DIMES Department. MC fellowship is funded by a donation from the company Illumia, Bologna, Italy, that we greatly thank for their interest in our research. We are grateful to Rotary Club, Cesena, Italy (President: Ing. Giuliano Arbizzani) for the generous donation of the thermal cycler “GenePro” (Bioer). We thank all the other people that very kindly contributed by individual donations to support part of the work that we are conducting on the subject. In particular, we are profoundly grateful to Matteo and Elisa Mele; to the Costa, Dal Monte, Ghignone, and Morini families as well as to the architects of the Jérôme Lejeune exhibtion at the Rimini Meeting, 2012; to Rina Bini; to the “Gruppo Arzdore” and the “Associazione Turistica Pro Loco di Dozza” (Dozza, Bologna, Italy) for their generous support to our trisomy 21 research. We are grateful to Kirsten Welter for her expert revision of the manuscript.
We are grateful to Drs. Jeffrey Taub and Yubin Ge for kindly providing us the raw data of their study [23] in order to use them in the meta-analysis.
Abbreviations
- AMKL
Acute Megakaryoblastic Leukemia
- DS
Down Syndrome
- MK
Megakaryocytes
- TMD
Transient Myeloproliferative Disorder
- CB
Cord Blood
Additional files
Table listing samples selected for the meta-analysis of gene expression profiles in DS AMKL (pool 'A'), non-DS AMKL (pool 'B'), TMD (pool 'C'), and MK (pool 'D' and 'E') cells. All known data about patients whose samples were included in the meta-analysis are described.
Table listing the genes cited in the paper. We indicate gene symbol, gene name and location based on Gene database [35] or UCSC Genome Browser [36].
Table listing the 25,954 mapped loci of Transcriptome Map Comparison of DS vs. non-DS AMKL Cells (Pool 'A' vs Pool 'B') sorted in descending order of 'A'/'B' expression value. N/A: not available.
Table listing the 25,800 mapped loci of Transcriptome Map Comparison of DS AMKL vs. MK Cells (Pool 'A' vs Pool 'D') sorted in descending order of 'A'/'D' expression value. N/A: not available.
Table listing the 25,819 mapped loci of Transcriptome Map Comparison of non-DS AMKL vs. MK Cells (Pool 'B' vs Pool 'D') sorted in descending order of 'B'/'D' expression value. N/A: not available.
Table listing the 25,540 mapped loci of Transcriptome Map Comparison of DS AMKL vs. CB MK Cells (Pool 'A' vs Pool 'E') sorted in descending order of 'A'/'E' expression value. N/A: not available.
Table listing the 25,546 mapped loci of Transcriptome Map Comparison of non-DS AMKL vs. CB MK Cells (Pool 'B' vs Pool 'E') sorted in descending order of 'B'/'E' expression value. N/A: not available.
Table listing the 25,955 mapped loci of Transcriptome Map Comparison of DS AMKL vs. TMD Cells (Pool 'A' vs Pool 'C') sorted in descending order of 'A'/'C' expression value. N/A: not available.
Table listing the 25,800 mapped loci of Transcriptome Map Comparison of TMD vs. MK Cells (Pool 'C' vs Pool 'D') sorted in descending order of 'C'/'D' expression value. N/A: not available.
Table listing the 25,540 mapped loci of Transcriptome Map Comparison of TMD vs. CB MK Cells (Pool 'C' vs Pool 'E') sorted in descending order of 'C'/'E' expression value. N/A: not available.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MCP collected the data for the meta-analysis and performed the TRAM analysis. AP, MC, ACB and LV contributed to the data interpretation. LV discussed the biology of MK and blast cells. PS conceived the study and participated in its design and coordination. MCP and PS wrote the manuscript. All authors contributed to the Discussion and read and approved the final manuscript.
Contributor Information
Maria Chiara Pelleri, Email: mariachiara.pelleri2@unibo.it.
Allison Piovesan, Email: allison.piovesan2@unibo.it.
Maria Caracausi, Email: maria.caracausi2@unibo.it.
Anna Concetta Berardi, Email: annacberardi@yahoo.it.
Lorenza Vitale, Email: lorenza.vitale@unibo.it.
Pierluigi Strippoli, Email: pierluigi.strippoli@unibo.it.
References
- 1.Gardiner KJ. Molecular basis of pharmacotherapies for cognition in Down syndrome. Trends Pharmacol Sci. 2010;31:66–73. doi: 10.1016/j.tips.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sinet PM, Allard D, Lejeune J, Jerome H: Letter: Gene dosage effect in trisomy 21.Lancet 1975, 1:276. [DOI] [PubMed]
- 3.Gardiner K, Costa AC. The proteins of human chromosome 21. Am J Med Genet C Semin Med Genet. 2006;142C:196–205. doi: 10.1002/ajmg.c.30098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roper RJ, Reeves RH: Understanding the basis for Down syndrome phenotypes.PLoS Genet 2006, 2:e50. [DOI] [PMC free article] [PubMed]
- 5.Pritchard M, Reeves RH, Dierssen M, Patterson D, Gardiner KJ. Down syndrome and the genes of human chromosome 21: current knowledge and future potentials. Report on the Expert workshop on the biology of chromosome 21 genes: towards gene-phenotype correlations in Down syndrome. Cytogenet Genome Res. 2008;121:67–77. doi: 10.1159/000124384. [DOI] [PubMed] [Google Scholar]
- 6.Korenberg JR. Down syndrome: the crucible for treating genomic imbalance. Genet Med. 2009;11:617–619. doi: 10.1097/GIM.0b013e3181b765e7. [DOI] [PubMed] [Google Scholar]
- 7.Patterson D. Molecular genetic analysis of Down syndrome. Hum Genet. 2009;126:195–214. doi: 10.1007/s00439-009-0696-8. [DOI] [PubMed] [Google Scholar]
- 8.Antonarakis SE, Epstein CJ. The challenge of Down syndrome. Trends Mol Med. 2006;12:473–479. doi: 10.1016/j.molmed.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 2000;355:165–169. doi: 10.1016/S0140-6736(99)05264-2. [DOI] [PubMed] [Google Scholar]
- 10.Patja K, Pukkala E, Sund R, Iivanainen M, Kaski M. Cancer incidence of persons with Down syndrome in Finland: a population-based study. Int J Cancer. 2006;118:1769–1772. doi: 10.1002/ijc.21518. [DOI] [PubMed] [Google Scholar]
- 11.Hitzler JK, Zipursky A. Origins of leukaemia in children with Down syndrome. Nat Rev Cancer. 2005;5:11–20. doi: 10.1038/nrc1525. [DOI] [PubMed] [Google Scholar]
- 12.Massey GV. Transient leukemia in newborns with Down syndrome. Pediatr Blood Cancer. 2005;44:29–32. doi: 10.1002/pbc.20141. [DOI] [PubMed] [Google Scholar]
- 13.Khan I, Malinge S, Crispino J. Myeloid leukemia in Down syndrome. Crit Rev Oncog. 2011;16:25–36. doi: 10.1615/CritRevOncog.v16.i1-2.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paolini R, Bonaldi L, Bianchini E, Ramazzina E, Cella G. Spontaneous evolution of essential thrombocythaemia into acute megakaryoblastic leukaemia with trisomy 8, trisomy 21 and cutaneous involvement. Eur J Haematol. 2003;71:466–469. doi: 10.1046/j.0902-4441.2003.00139.x. [DOI] [PubMed] [Google Scholar]
- 15.Bonsi L, Grossi A, Strippoli P, Tumietto F, Tonelli R, Vannucchi AM, Ronchi A, Ottolenghi S, Visconti G, Avanzi GC, Pegoraro L, Bagnara GP. An erythroid and megakaryocytic common precursor cell line (B1647) expressing both c-mpl and erythropoietin receptor (Epo-R) proliferates and modifies globin chain synthesis in response to megakaryocyte growth and development factor (MGDF) but not to erythropoietin (Epo) Br J Haematol. 1997;98:549–559. doi: 10.1046/j.1365-2141.1997.2793096.x. [DOI] [PubMed] [Google Scholar]
- 16.Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, Crispino JD. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002;32:148–152. doi: 10.1038/ng955. [DOI] [PubMed] [Google Scholar]
- 17.Klusmann JH, Godinho FJ, Heitmann K, Maroz A, Koch ML, Reinhardt D, Orkin SH, Li Z. Developmental stage-specific interplay of GATA1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis. Genes Dev. 2010;24:1659–1672. doi: 10.1101/gad.1903410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klusmann JH, Li Z, Böhmer K, Maroz A, Koch ML, Emmrich S, Godinho FJ, Orkin SH, Reinhardt D. miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukemia. Genes Dev. 2010;24:478–490. doi: 10.1101/gad.1856210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yagi T, Morimoto A, Eguchi M, Hibi S, Sako M, Ishii E, Mizutani S, Imashuku S, Ohki M, Ichikawa H. Identification of a gene expression signature associated with pediatric AML prognosis. Blood. 2003;102:1849–1856. doi: 10.1182/blood-2003-02-0578. [DOI] [PubMed] [Google Scholar]
- 20.Lightfoot J, Hitzler JK, Zipursky A, Albert M, Macgregor PF. Distinct gene signatures of transient and acute megakaryoblastic leukemia in Down syndrome. Leukemia. 2004;18:1617–1623. doi: 10.1038/sj.leu.2403466. [DOI] [PubMed] [Google Scholar]
- 21.McElwaine S, Mulligan C, Groet J, Spinelli M, Rinaldi A, Denyer G, Mensah A, Cavani S, Baldo C, Dagna-Bricarelli F, Hann I, Basso G, Cotter FE, Nizetic D. Microarray transcript profiling distinguishes the transient from the acute type of megakaryoblastic leukaemia (M7) in Down’s syndrome, revealing PRAME as a specific discriminating marker. Br J Haematol. 2004;125:729–742. doi: 10.1111/j.1365-2141.2004.04982.x. [DOI] [PubMed] [Google Scholar]
- 22.Bourquin JP, Subramanian A, Langebrake C, Reinhardt D, Bernard O, Ballerini P, Baruchel A, Cavé H, Dastugue N, Hasle H, Kaspers GL, Lessard M, Michaux L, Vyas P, van Wering E, Zwaan CM, Golub TR, Orkin SH. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci U S A. 2006;103:3339–3344. doi: 10.1073/pnas.0511150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge Y, Dombkowski AA, LaFiura KM, Tatman D, Yedidi RS, Stout ML, Buck SA, Massey G, Becton DL, Weinstein HJ, Ravindranath Y, Matherly LH, Taub JW. Differential gene expression, GATA1 target genes, and the chemotherapy sensitivity of Down syndrome megakaryocytic leukemia. Blood. 2006;107:1570–1581. doi: 10.1182/blood-2005-06-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radtke I, Mullighan CG, Ishii M, Su X, Cheng J, Ma J, Ganti R, Cai Z, Goorha S, Pounds SB, Cao X, Obert C, Armstrong J, Zhang J, Song G, Ribeiro RC, Rubnitz JE, Raimondi SC, Shurtleff SA, Downing JR. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc Natl Acad Sci U S A. 2009;106:12944–12949. doi: 10.1073/pnas.0903142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lenzi L, Facchin F, Piva F, Giulietti M, Pelleri MC, Frabetti F, Vitale L, Casadei R, Canaider S, Bortoluzzi S, Coppe A, Danieli GA, Principato G, Ferrari S, Strippoli P: TRAM (Transcriptome Mapper): database-driven creation and analysis of transcriptome maps from multiple sources.BMC Genomics 2011, 12:121. [DOI] [PMC free article] [PubMed]
- 26.Chou ST, Opalinska JB, Yao Y, Fernandes MA, Kalota A, Brooks JS, Choi JK, Gewirtz AM, Danet-Desnoyers GA, Nemiroff RL, Weiss MJ. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood. 2008;112:4503–4506. doi: 10.1182/blood-2008-05-157859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tunstall-Pedoe O, Roy A, Karadimitris A, de la Fuente J, Fisk NM, Bennett P, Norton A, Vyas P, Roberts I. Abnormalities in the myeloid progenitor compartment in Down syndrome fetal liver precede acquisition of GATA1 mutations. Blood. 2008;112:4507–4511. doi: 10.1182/blood-2008-04-152967. [DOI] [PubMed] [Google Scholar]
- 28.Olson TA, Levine RF, Mazur EM, Wright DG, Salvado AJ. Megakaryocytes and megakaryocyte progenitors in human cord blood. Am J Pediatr Hematol Oncol. 1992;14:241–247. doi: 10.1097/00043426-199208000-00011. [DOI] [PubMed] [Google Scholar]
- 29.Liu ZJ, Sola-Visner M. Neonatal and adult megakaryopoiesis. Curr Opin Hematol. 2011;18:330–337. doi: 10.1097/MOH.0b013e3283497ed5. [DOI] [PubMed] [Google Scholar]
- 30.Barrett T, Edgar R. Gene expression omnibus: microarray data storage, submission, retrieval, and analysis. Methods Enzymol. 2006;411:352–369. doi: 10.1016/S0076-6879(06)11019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarkans U, Parkinson H, Lara GG, Oezcimen A, Sharma A, Abeygunawardena N, Contrino S, Holloway E, Rocca-Serra P, Mukherjee G, Shojatalab M, Kapushesky M, Sansone SA, Farne A, Rayner T, Brazma A. The ArrayExpress gene expression database: a software engineering and implementation perspective. Bioinformatics. 2005;21:1495–1501. doi: 10.1093/bioinformatics/bti157. [DOI] [PubMed] [Google Scholar]
- 32.Emig D, Salomonis N, Baumbach J, Lengauer T, Conklin BR, Albrecht M. AltAnalyze and DomainGraph: analyzing and visualizing exon expression data. Nucleic Acids Res. 2010;38:W755–W762. doi: 10.1093/nar/gkq405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lenzi L, Frabetti F, Facchin F, Casadei R, Vitale L, Canaider S, Carinci P, Zannotti M, Strippoli P. UniGene Tabulator: a full parser for the UniGene format. Bioinformatics. 2006;22:2570–2571. doi: 10.1093/bioinformatics/btl425. [DOI] [PubMed] [Google Scholar]
- 34.Piovesan A, Vitale L, Pelleri MC, Strippoli P. Universal tight correlation of codon bias and pool of RNA codons (codonome): The genome is optimized to allow any distribution of gene expression values in the transcriptome from bacteria to humans. Genomics. 2013;101:282–289. doi: 10.1016/j.ygeno.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Gene database (http://www.ncbi.nlm.nih.gov/gene).
- 36.University of California Santa Cruz (UCSC) Genome Browser. (http://genome-euro.ucsc.edu/cgi-bin/hgGateway).
- 37.Berriz GF, King OD, Bryant B, Sander C, Roth FP. Characterizing gene sets with FuncAssociate. Bioinformatics. 2003;19:2502–2504. doi: 10.1093/bioinformatics/btg363. [DOI] [PubMed] [Google Scholar]
- 38.Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, Takenaka K, Teshima T, Tanaka T, Inagaki Y, Akashi K. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7:708–717. doi: 10.1016/j.stem.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 39.Tomasson MH, Xiang Z, Walgren R, Zhao Y, Kasai Y, Miner T, Ries RE, Lubman O, Fremont DH, McLellan MD, Payton JE, Westervelt P, DiPersio JF, Link DC, Walter MJ, Graubert TA, Watson M, Baty J, Heath S, Shannon WD, Nagarajan R, Bloomfield CD, Mardis ER, Wilson RK, Ley TJ. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood. 2008;111:4797–4808. doi: 10.1182/blood-2007-09-113027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nover-shtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, Habib N, Yosef N, Chang CY, Shay T, Frampton GM, Drake AC, Leskov I, Nilsson B, Preffer F, Dombkowski D, Evans JW, Liefeld T, Smutko JS, Chen J, Friedman N, Young RA, Golub TR, Regev A, Ebert BL. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Felli N, Cianetti L, Pelosi E, Carè A, Liu CG, Calin GA, Rossi S, Peschle C, Marziali G, Giuliani A: Hematopoietic differentiation: a coordinated dynamical process towards attractor stable states.BMC Syst Biol 2010, 4:85. [DOI] [PMC free article] [PubMed]
- 42.Tenedini E, Fagioli ME, Vianelli N, Tazzari PL, Ricci F, Tagliafico E, Ricci P, Gugliotta L, Martinelli G, Tura S, Baccarani M, Ferrari S, Catani L. Gene expression profiling of normal and malignant CD34-derived megakaryocytic cells. Blood. 2004;104:3126–3135. doi: 10.1182/blood-2003-07-2597. [DOI] [PubMed] [Google Scholar]
- 43.Fuhrken PG, Chen C, Miller WM, Papoutsakis ET. Comparative, genome-scale transcriptional analysis of CHRF-288-11 and primary human megakaryocytic cell cultures provides novel insights into lineage-specific differentiation. Exp Hematol. 2007;35:476–489. doi: 10.1016/j.exphem.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 44.Giammona LM, Fuhrken PG, Papoutsakis ET, Miller WM. Nicotinamide (vitamin B3) increases the polyploidisation and proplatelet formation of cultured primary human megakaryocytes. Br J Haematol. 2006;135:554–566. doi: 10.1111/j.1365-2141.2006.06341.x. [DOI] [PubMed] [Google Scholar]
- 45.Ferrari F, Bortoluzzi S, Coppe A, Basso D, Bicciato S, Zini R, Gemelli C, Danieli GA, Ferrari S. Genomic expression during human myelopoiesis. BMC Genomics. 2007;8:264–283. doi: 10.1186/1471-2164-8-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Repeat Masker (http://www.repeatmasker.org/).
- 47.Tuson M, Garanto A, Gonzàlez-Duarte R, Marfany G. Overexpression of CERKL, a gene responsible for retinitis pigmentosa in humans, protects cells from apoptosis induced by oxidative stress. Mol Vis. 2009;15:168–180. [PMC free article] [PubMed] [Google Scholar]
- 48.Caldwell JT, Edwards H, Dombkowski AA, Buck SA, Matherly LH, Ge Y, Taub JW: Over-expression of GATA1 confers resistance to chemotherapy in acute megakaryocytic Leukemia.PLoS One 2013, 8:e68601. [DOI] [PMC free article] [PubMed]
- 49.Roela RA, Carraro DM, Brentani HP, Kaiano JH, Simao DF, Guarnieiro R, Lopes LF, Borojevic R, Brentani MM. Gene stage-specific expression in the microenvironment of pediatric myelodysplastic syndromes. Leuk Res. 2007;31:579–589. doi: 10.1016/j.leukres.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 50.Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, Crispino JD. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest. 2012;122:948–962. doi: 10.1172/JCI60455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taub JW, Huang X, Matherly LH, Stout ML, Buck SA, Massey GV, Becton DL, Chang MN, Weinstein HJ, Ravindranath Y. Expression of chromosome 21-localized genes in acute myeloid leukemia: differences between Down syndrome and non-Down syndrome blast cells and relationship to in vitro sensitivity to cytosine arabinoside and daunorubicin. Blood. 1999;94:1393–1400. [PubMed] [Google Scholar]
- 52.Boztug H, Schumich A, Pötschger U, Mühlegger N, Kolenova A, Reinhardt K, Dworzak M. Blast cell deficiency of CD11a as a marker of acute megakaryoblastic leukemia and transient myeloproliferative disease in children with and without Down syndrome. Cytometry B Clin Cytom. 2013;84:370–378. doi: 10.1002/cyto.b.21082. [DOI] [PubMed] [Google Scholar]
- 53.Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest. 2005;115:3339–3347. doi: 10.1172/JCI26674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caracausi M, Vitale L, Pelleri MC, Piovesan A, Bruno S, Strippoli P. A quantitative transcriptome reference map of the normal human brain. Neurogenetics. 2014;15:267–287. doi: 10.1007/s10048-014-0419-8. [DOI] [PubMed] [Google Scholar]
- 55.Strippoli P, Pelleri MC, Caracausi M, Vitale L, Piovesan A, Locatelli C, Mimmi MC, Berardi AC, Ricotta D, Radeghieri A, Barisani D, Basik M, Monaco MC, Ghezzo A, Seri M, Cocchi G: An integrated route to identifying new pathogenesis-based therapeutic approaches for trisomy 21 (Down Syndrome) following the thought of Jérôme Lejeune.Science Postprint 2013, 1:e00010.
- 56.Melgarejo E, Medina MA, Sánchez-Jiménez F, Urdiales JL. Targeting of histamine producing cells by EGCG: a green dart against inflammation? J Physiol Biochem. 2010;66:265–270. doi: 10.1007/s13105-010-0033-7. [DOI] [PubMed] [Google Scholar]
- 57.Dy M, Pacilio M, Arnould A, Machavoine F, Mayeux P, Hermine O, Bodger M, Schneider E. Modulation of histidine decarboxylase activity and cytokine synthesis in human leukemic cell lines: relationship with basophilic and/or megakaryocytic differentiation. Exp Hematol. 1999;27:1295–1305. doi: 10.1016/S0301-472X(99)00070-3. [DOI] [PubMed] [Google Scholar]
- 58.Eguchi M, Ozawa T, Sakakibara H, Sugita K, Iwama Y, Furukawa T. Ultrastructural and ultracytochemical differences between megakaryoblastic leukemia in children and adults. Analysis of 49 patients. Cancer. 1992;70:451–458. doi: 10.1002/1097-0142(19920715)70:2<451::AID-CNCR2820700213>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 59.Nakahata T, Okumura N. Cell surface antigen expression in human erythroid progenitors: erythroid and megakaryocytic markers. Leuk Lymphoma. 1994;13:401–409. doi: 10.3109/10428199409049629. [DOI] [PubMed] [Google Scholar]
- 60.Maclean GA, Menne TF, Guo G, Sanchez DJ, Park IH, Daley GQ, Orkin SH. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc Natl Acad Sci U S A. 2012;109:17567–17572. doi: 10.1073/pnas.1215468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reynolds LE, Watson AR, Baker M, Jones TA, D’Amico G, Robinson SD, Joffre C, Garrido-Urbani S, Rodriguez-Manzaneque JC, Martino-Echarri E, Aurrand-Lions M, Sheer D, Dagna-Bricarelli F, Nizetic D, McCabe CJ, Turnell AS, Kermorgant S, Imhof BA, Adams R, Fisher EM, Tybulewicz VL, Hart IR, Hodivala-Dilke KM. Tumour angiogenesis is reduced in the Tc1 mouse model of Down’s syndrome. Nature. 2010;465:813–817. doi: 10.1038/nature09106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang HH, McGeachie M, Alterovitz G, Ramoni MF: Mapping transcription mechanisms from multimodal genomic data.BMC Bioinformatics 2010, 11(Suppl 9):S2. [DOI] [PMC free article] [PubMed]
- 63.Chou ST, Byrska-Bishop M, Tober JM, Yao Y, Vandorn D, Opalinska JB, Mills JA, Choi JK, Speck NA, Gadue P, Hardison RC, Nemiroff RL, French DL, Weiss MJ. Trisomy 21-associated defects in human primitive hematopoiesis revealed through induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012;109:17573–17578. doi: 10.1073/pnas.1211175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Canzonetta C, Hoischen A, Giarin E, Basso G, Veltman JA, Nacheva E, Nizetic D, Groet J. Amplified segment in the ‘Down syndrome critical region’ on chr21 shared between Down syndrome and euploid AML-M0 excludes RUNX1, ERG and ETS2. Br J Haematol. 2012;157:197–200. doi: 10.1111/j.1365-2141.2011.08985.x. [DOI] [PubMed] [Google Scholar]
- 65.Yokoyama T, Toki T, Aoki Y, Kanezaki R, Park MJ, Kanno Y, Takahara T, Yamazaki Y, Ito E, Hayashi Y, Nakamura T. Identification of TRIB1 R107L gain-of-function mutation in human acute megakaryocytic leukemia. Blood. 2012;119:2608–2611. doi: 10.1182/blood-2010-12-324806. [DOI] [PubMed] [Google Scholar]
- 66.Prandini P, Deutsch S, Lyle R, Gagnebin M, Delucinge Vivier C, Delorenzi M, Gehrig C, Descombes P, Sherman S, Dagna Bricarelli F, Baldo C, Novelli A, Dallapiccola B, Antonarakis SE. Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance. Am J Hum Genet. 2007;81:252–263. doi: 10.1086/519248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet. 2004;5:725–738. doi: 10.1038/nrg1448. [DOI] [PubMed] [Google Scholar]
- 68.Deutsch S, Lyle R, Dermitzakis ET, Attar H, Subrahmanyan L, Gehrig C, Parand L, Gagnebin M, Rougemont J, Jongeneel CV, Antonarakis SE. Gene expression variation and expression quantitative trait mapping of human chromosome 21 genes. Hum Mol Genet. 2005;14:3741–3749. doi: 10.1093/hmg/ddi404. [DOI] [PubMed] [Google Scholar]
- 69.Roberts I, Izraeli S. Haematopoietic development and leukaemia in Down syndrome. Br J Haematol. 2014;167:587–599. doi: 10.1111/bjh.13096. [DOI] [PubMed] [Google Scholar]
- 70.Tunnacliffe A, Majumdar S, Yan B, Poncz M. Genes for beta-thromboglobulin and platelet factor 4 are closely linked and form part of a cluster of related genes on chromosome 4. Blood. 1992;79:2896–2900. [PubMed] [Google Scholar]
- 71.Gear AR, Camerini D. Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, and host defense. Microcirculation. 2003;10:335–350. doi: 10.1080/713773647. [DOI] [PubMed] [Google Scholar]
- 72.Slungaard A. Platelet factor 4: a chemokine enigma. Int J Biochem Cell Biol. 2005;37:1162–1167. doi: 10.1016/j.biocel.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 73.Bluteau D, Lordier L, Di Stefano A, Chang Y, Raslova H, Debili N, Vainchenker W. Regulation of megakaryocyte maturation and platelet formation. J Thromb Haemost. 2009;7(Suppl 1):227–234. doi: 10.1111/j.1538-7836.2009.03398.x. [DOI] [PubMed] [Google Scholar]
- 74.Melemed AS, Ryder JW, Vik TA. Activation of the mitogen-activated protein kinase pathway is involved in and sufficient for megakaryocytic differentiation of CMK cells. Blood. 1997;90:3462–3470. [PubMed] [Google Scholar]
- 75.Herrera R, Hubbell S, Decker S, Petruzzelli L. A role for the MEK/MAPK pathway in PMA-induced cell cycle arrest: modulation of megakaryocytic differentiation of K562 cells. Exp Cell Res. 1998;238:407–414. doi: 10.1006/excr.1997.3847. [DOI] [PubMed] [Google Scholar]
- 76.Tonelli R, Scardovi AL, Pession A, Strippoli P, Bonsi L, Vitale L, Prete A, Locatelli F, Bagnara GP, Paolucci G. Compound heterozygosity for two different amino-acid substitution mutations in the thrombopoietin receptor (c-mpl gene) in congenital amegakaryocytic thrombocytopenia (CAMT) Hum Genet. 2000;107:225–233. doi: 10.1007/s004390000357. [DOI] [PubMed] [Google Scholar]
- 77.Taniguchi Y, London R, Schinkmann K, Jiang S, Avraham H. The receptor protein tyrosine phosphatase, PTP-RO, is upregulated during megakaryocyte differentiation and is associated with the c-Kit receptor. Blood. 1999;94:539–549. [PubMed] [Google Scholar]