

Abstract
Background
Combining information from different studies is an important and useful practice in bioinformatics, including genome-wide association study, rare variant data analysis and other set-based analyses. Many statistical methods have been proposed to combine p-values from independent studies. However, it is known that there is no uniformly most powerful test under all conditions; therefore, finding a powerful test in specific situation is important and desirable.
Results
In this paper, we propose a new statistical approach to combining p-values based on gamma distribution, which uses the inverse of the p-value as the shape parameter in the gamma distribution.
Conclusions
Simulation study and real data application demonstrate that the proposed method has good performance under some situations.
Keywords: Fisher test, Lancaster method, rare variant association test, z-test
Background
To combine information from individual studies, many statistical approaches have been proposed. For example, meta-analysis with fixed or random effects has been intensively used to combine information from separate relevant genome-wide association studies (GWASs). However, in practice sometimes it may not be able to get all the statistics that we need, such as odds ratio and its 95% confidence interval; instead, only p-value from each study is available. In this case, combining p-values from independent studies should be used. In the literature, many statistical methods have been proposed to combine p-values [1-14]. For example, it has been shown that the Fisher test is more robust than the z-test and is commonly used for genetic data [15-26]. On the other hand, if the effects have the same direction and/or similar sizes, z-test is more powerful than the Fisher test. Some studies have shown that the weighted z-tests with weight equals to the sample size or the inverse of the standard error may perform better than the unweighted z-test under certain situations [27]. However, it has also been shown that there is no uniformly most powerful method [1]. Therefore, it is desirable to find a test which is more powerful than others for given situations. For instance, in GWAS meta-analysis, it is very common that the genetic effects of the same single-nucleotide polymorphism (SNP) from different studies are heterogeneous due to various environmental factors and study populations. Therefore, the fixed effect model cannot be applied and a p-value combining method is preferred.
Lancaster generalized Fisher test by giving certain degrees of freedom to individual studies when combine p-values based on the chi-square distribution. When the degrees of freedom (df) equal to two for each study, Lancaster's test is identical to the Fisher test. Recently, Chen and Nadarajah have studied another special case of Lancaster's test where the df is one for each study [14]. They have shown that their test can also be viewed as a weighted z-test with the "weight" equals to the estimated effect, defined as the estimated mean difference divided by the estimated standard error, which can be calculated by , where pi is the one-sided p-value from the ith study and is the inverse of the cumulative density function (CDF) of the standard normal distribution, N(0,1).
Methods based on the gamma distribution (GDM) are also available in the literature. In fact, Lancaster's methods are special cases of GDMs. GDMs are more flexible and potentially can be more powerful in some situations when appropriate parameters (e.g., the shape parameter in the gamma distribution) are chosen. However, it is usually difficult to set appropriate parameters before we see the data. In this paper, we propose a GDM, which adaptively chooses the shape parameter of the gamma distribution for each individual study. We compare the performance of the proposed test with existing methods through simulation studies. We also use real data application to illustrate the use of the new approach.
Methods
Suppose we have K independent studies and their associated p-values pi (i = 1,2,...,K). Under the null hypothesis that there is no effect for all studies, the p-values from individual studies are uniformly distributed between 0 and 1. The weighted z-tests are formulated as follows:
| (1) |
where wi is the weight for study i. When all wi = 1, the above test is the unweighted z-test, also called the Stouffer test [5]. When wi = ni, where ni is the sample size for study i , it is called the Mosteller-Bush test [8]. Other researchers suggested the use of the square root of the sample size or the inverse of the estimated standard error as weight [27].
Other ways to combine p-values are based on the following property: if are K independent random variables and each has a chi-square distribution with df equal to di, then their sum has a chi-square distribution with df equal to the sum of their df's. Fisher [9] found that if K random variables are independent and identically uniformly distributed between 0 and 1, then each has a chi-square distribution with df = 2 and their sum has a chi-square distribution with 2K df, . Based on this fact, Fisher used test statistic and compared it to to calculate the overall p-value. Lancaster [3] generalized Fisher's test by giving different di df for each study. The test statistic under the null hypothesis has a . More specifically, the test statistic is given by:
| (2) |
where is the inverse of .
Rather than the chi-square distribution, a more generalized distribution, gamma distribution, can be used. The test statistic based on the gamma distribution can be written as:
| (3) |
where is the inverse gamma distribution with shape parameter αi and scale parameter β. Due to the property of the gamma distribution, for constant shape parameter αi, T will have a gamma distribution with shape parameter equals to and scale parameter equals to β. When all αi = 1, T has an exponential distribution under the null hypothesis. When all αi = v/2 and β = 2, the null distribution of T is a chi-square distribution with . When v = 1, it is the Chen-Nadarajah test [14]; when v = 2, it is the Fisher test.
In this paper, we use β = 1 as the scale parameter which has no effect on power of the test T. For the shape parameter, we will use for the ith study. So the proposed test statistic is:
| (4) |
Notice that since the gamma distribution with shape parameter αi and scale parameter 1 has expected value αi, a small p-value of pi results in a large expected value. Therefore, the proposed test gives larger "weights" to smaller p-values. In addition, since pi is a random variable, the proposed test doesn't follow a gamma distribution any more. However, the p-value can be easily estimated by resampling method. Under the null hypothesis, pi is uniformly distributed between 0 and 1. For the given number of studies, K, we can generate K numbers from uniform distribution U(0,1) and then calculate the statistic t defined in (4). We repeat this step N times (say, N = 108), then the null distribution of T can be approximated by those numbers and the p-value can be estimated by the proportion of the N values which are greater than the observed statistic.
Results
Simulation study
To assess the performance of the proposed test, we conduct a simulation study by comparing it with some existing methods, including the z-test (denoted by Z), weighted z-tests with weights equal to the sample size (Z_n) or the estimated standard error (Z_se), the Chen-Nadarajah (CN) method, the Fisher test (Fisher). In the simulation study, we assume there are K independent studies, where K = 2, 10, or 100. For each study, we simulate data from two normal distributions: , and with sample sizes n1 = n2 = n, respectively. Of the K studies, there are different numbers of studies that have none-zero effects (i.e., ), which may have different values among studies but their sum is a constant. We consider several conditions for allocating effect sizes among the K studies. We first consider sample sizes and variances are fixed. We then assume the sample size, or the variance, or both the sample size and the variance are randomly sampled from given distributions. For random sample size, we assume it follows a Poisson distribution, Poi(λ); for random variances, we assume the standard deviation follows a gamma distribution with shape parameter α and scale parameter β, gamma(α, β). A p-value from a two-sample t-test to compare two group means for each study is obtained and is used to combine those K studies. When K is small (i.e., 2, and 10), we consider situations where there are 1 to K studies having none-zero effects. For K = 100, we consider i (i = 1, 2,..., 10) studies having the same effect size while the remaining 100-i studies having zero effect. We choose significance level 0.05 in the simulation study and use 105 replicates to estimate the type I error rate and the power.
Simulation results
All the methods can control type I error rate (data not shown). Figure 1 plots the power values for each method when there are only two studies (K = 2) with the sum of the two effect sizes equals to 1. Six conditions are considered: the ratios of the effects sizes between study1 and study 2 are 0, 0.01, 0.05, 0.1, 0.5, and 1, respectively. Therefore, the heterogeneity of effects between the two studies decreases from condition 1 to condition 6. Figure 1 (a) shows that the gamma-distribution based methods (Fisher, CN, and New) perform similarly, and all outperform the Z-based methods (Z, Z_n, Z_se) when the effects are not homogenous between the two studies (conditions 1 to 5). When the two studies have the same effect size (condition 6), the power values from all methods are close to each other. We have similar observations: when the sample size is randomly sampled from Poisson distribution Poi(20) (Figure 1 (b)), or when the standard deviation is randomly sampled from gamma distribution, gamma(10, 0.1) (Figure 1 (c)), or when both sample sizes and standard deviations are random samples as in Figure 1 (b) and 1(c) (Figure 1 (d)).
Figure 1.
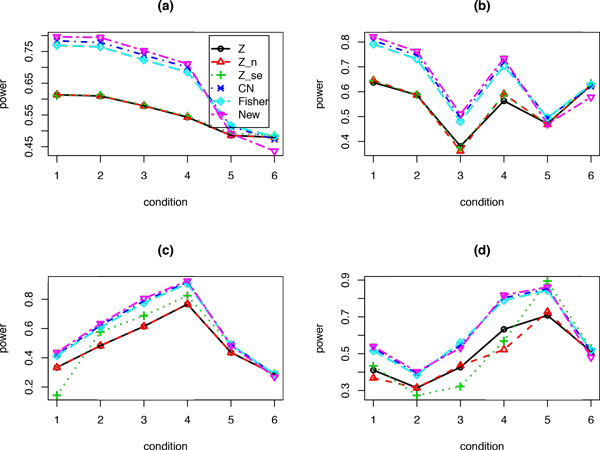
Power values for each method under six different conditions: the total effect size is 1 and the ratios of the K = 2 effect sizes between study 1 and study 2 are 0, 0.01, 0.05, 0.1, 0.5, and 1. (a) Sample sizes n = 20, and standard deviation σ = 1 for each study; (b) Same as (a), but the sample size n is a random sample from Poi(20); (c) Same as (a), but the standard deviation σ is a random sample from gamma(10, 0.1); (d) Same as (b), but the standard deviations σ is a random sample from gamma(10, 0.1).
Figure 2 plots the estimated power values for each method when there are i (i = 1,2,..., 10) studies having none-zero effects and the sum of those effect sizes is 2. For those i studies with none-zero effects, we assume the mean of the second group equals to 2/i. Figure 2 (a) shows that when only a few studies have none-zero effects (e.g., i = 1, 2, 3) the gamma-distribution based methods, including the proposed test perform better than those Z-based methods. However, when the effects among those studies become more homogenous, the proposed test has slightly lower power values. The sample size n = 20 and standard deviation σ = 1 are assumed for each study in Figure 2 (a). We have similar observations when sample size n is a random sample from Poi(20) (Figure 2 (b)), or when standard deviation σ is randomly sampled from a gamma(10, 0.1) (Figure 2 (c)), or when both sample size and standard deviation are random samples as in Figure 2 (b) and 2(c) (Figure 2 (d)).
Figure 2.
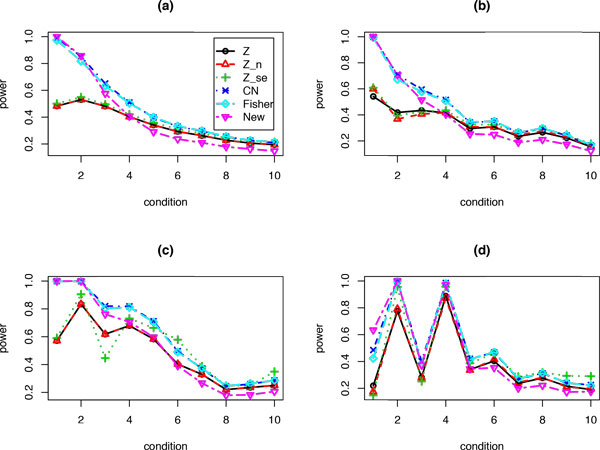
Power values for each method under 10 different conditions: for situation i, there are i out of K = 10 studies each has the same effect size 2/i and the remaining 10-i studies have zero effect. (a) Sample size n = 20, and standard deviation σ = 1 for each study; (b) Same as (a), but the sample size n is a random sample from Poi(20); (c) Same as (a), but the standard deviation σ is a random sample from gamma(10, 0.1); (d) Same as (b), but standard deviation σ is a random sample from gamma(10, 0.1).
Figure 3 plots the power values for each method when there are 100 independent studies but only i(i = 1, 2,..., 10) studies have none-zero effect sizes. For those i studies with none-zero effects, we assume the mean for the second group equals to 2/i. In Figure 3 (a) we set sample size n = 20 and σ = 1 for each study. When there are only one or two studies having none-zero effects, the proposed test has much higher power values than the Fisher test and the CN method, which in turn are more powerful than the Z, Z_n, and Z_se tests. When the number of significant studies increases, all of the methods have close power values. The same pattern can be observed when n is random samples from Poi(20) (Figure 3 (b)), or when σ is randomly sampled from a gamma(10, 0.1) (Figure 3 (c)), or when both sample size and standard deviation are random samples as in Figure 3 (b) and 3(c) (Figure 3 (d)).
Figure 3.
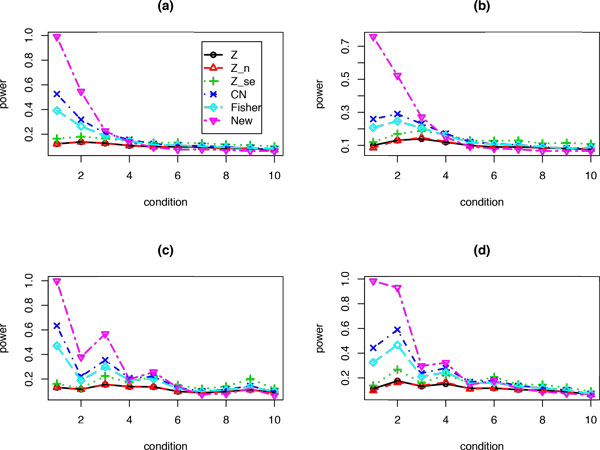
Power values for each method under 10 different conditions: for situation i, there are i out of K = 100 studies each has the same effect size 2/i and the remaining 100-i studies have zero effect. (a) Sample size n = 20, and standard deviation σ = 1 for each study; (b) Same as (a), but the sample size n is a random sample from Poi(20); (c) Same as (a), but the standard deviation σ is a random sample from gamma(10, 0.1); (d) Same as (b), but the standard deviation σ is a random sample from gamma(10, 0.1).
Real data application
We apply the proposed approach to a meta-analysis of Genome-wide association study (GWAS). The data include 5 independent case-control studies where we wanted to test whether there is an association between the SNP rs17110747-A and major depression [28]. Table 1 is the count data from these studies. The 5 p-values are 0.94, 0.0015, 0.97, 0.79, and 0.81. The overall p-values from the Z, Z_n, Z_se, Fisher, CN, and the proposed test are 0.84, 0.53, 0.29, 0.17, 0.062, and 0.0081, respectively. Only the proposed test has p-value less than 0.05.
Table 1.
Count data from the five independent studies investigating the association between SNP rs17110747-A and major depression.
| study | case | control | ||
|---|---|---|---|---|
| event | total | event | total | |
| 1 | 11 | 270 | 25 | 630 |
| 2 | 244 | 1016 | 282 | 926 |
| 3 | 49 | 234 | 35 | 166 |
| 4 | 79 | 600 | 76 | 600 |
| 5 | 71 | 290 | 86 | 340 |
Discussion and conclusions
Combining information from individual studies is an important and useful tool, especially for set-based approaches. For example, in studying the effect of rare variants on diseases, a set of rare variants are tested simultaneously, and their p-values are combined to test for the association between the rare variants and the disease [29]. However, most of the rare variants may have no or little effects while a few of them may have large effects. In this case, the proposed test will be more powerful than other methods if combining p-value methods are used. However, it should be pointed out that, the rare variants from a set (e.g., gene) maybe correlated, and the proposed test needs to be modified accordingly. A permutation-based test can be applied to estimate the p-value. We first calculate the statistic based on the proposed test (4). Then we permute the disease status (case or control); for each permutation, we use the proposed test to calculate a statistic. After a large number of permutations, the p-value will be estimated as the proportion of the statistics from the permutations excessing the observed statistic from the original data. To assess the performance of the proposed test in rare variant association studies, real data are needed. This will be a topic of our future research.
As mentioned earlier, no method is uniformly most powerful when combing p-values. However, based on our simulation studies, the proposed test is more powerful when the effects among the studies are more heterogeneous. When the effects are homogeneous, perhaps the Z-based tests are more powerful. Without the information about the effect sizes, robust methods, such as the CN and Fisher tests are recommended.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ZC and MQY conceived and guided the project. ZC designed the project and conducted the study. WY, QL, JL, and JYY participated in the study through computational implementation, data analysis and discussions. ZC wrote the manuscript, and ZC and MQY finalized the manuscript, which was read and approved by all authors.
Contributor Information
Zhongxue Chen, Email: zc3@indiana.edu.
William Yang, Email: willyang@indiana.edu.
Qingzhong Liu, Email: liu@shsu.edu.
Jack Y Yang, Email: jyang@hadron.mgh.harvard.edu.
Jing Li, Email: jl204@indiana.edu.
Mary Qu Yang, Email: mqyang@ualr.edu.
Acknowledgements
The research was supported by Indiana University Institutional Research Awards and National Institutes of Health (NIH). ZC would like to thank the institutional faculty research grant awards from the Indiana University Bloomington School of Public Health. MQY would like to thank the support from NIH/NIGMS 5P20GM10342913 and ASTA Award # 15-B-23.
Declaration
The funding for publication of the article has come from Indiana University Bloomington institutional research grant awards to ZC.
This article has been published as part of BMC Bioinformatics Volume 15 Supplement 17, 2014: Selected articles from the 2014 International Conference on Bioinformatics and Computational Biology. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcbioinformatics/supplements/15/S17.
References
- Birnbaum A. Combining independent tests of significance. J Amer Statist Assoc. 1954;49(267):559–574. [Google Scholar]
- Good IJ. On the weighted combination of significance tests. J R Stat Soc Ser B Stat Methodol. 1955;17(2):264–265. [Google Scholar]
- Lancaster H. The combination of probabilities: an application of orthonormal functions. Austral J Statist. 1961;3:20–33. doi: 10.1111/j.1467-842X.1961.tb00058.x. [DOI] [Google Scholar]
- Pearson ES. The probability integral transformation for testing goodness of fit and combining independent tests of significance. Biometrika. 1938;30(1/2):134–148. doi: 10.2307/2332229. [DOI] [Google Scholar]
- Stouffer SA, Suchman EA, DeVinney LC, Star SA, Williams RMJ. In. The American Soldier, Vol 1: Adjustment during Army Life. Princeton: Princeton University Press; 1949. [Google Scholar]
- Tippett L. The Methods of Statistics. London: Williams and Norgate Ltd; 1931. [Google Scholar]
- Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher's approach. J Evol Biol. 2005;18(5):1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- Mosteller F, Bush RR. In: Handbook of Social Psychology. Lindzey G. Cambridge, editor. Mass: Addison-Wesley; 1954. Selected quantitative techniques; pp. 289–334. [Google Scholar]
- Fisher RA. Statistical Methods for Research Workers. Edinburgh: Oliver and Boyd; 1932. [Google Scholar]
- Liptak T. On the combination of independent tests. Magyar Tud Akad Mat Kutato Int Kozl. 1958;3:171–197. [Google Scholar]
- Chen Z, Nadarajah S. Comments on 'Choosing an optimal method to combine p - values' by Sungho Won, Nathan Morris, Qing Lu and Robert C. Elston, Statistics in Medicine 2009; 28: 1537-1553. Statistics in Medicine. 2011;30(24):2959–2961. doi: 10.1002/sim.4222. [DOI] [PubMed] [Google Scholar]
- Chen Z. Is the weighted z - test the best method for combining probabilities from independent tests? Journal of Evolutionary Biology. 2011;24(4):926–930. doi: 10.1111/j.1420-9101.2010.02226.x. [DOI] [PubMed] [Google Scholar]
- Loughin TM. A systematic comparison of methods for combining p-values from independent tests. Computational statistics & data analysis. 2004;47(3):467–485. doi: 10.1016/j.csda.2003.11.020. [DOI] [Google Scholar]
- Chen Z, Nadarajah S. On the optimally weighted z-test for combining probabilities from independent studies. Computational Statistics & Data Analysis. 2014;70:387–394. [Google Scholar]
- Chen Z. A new association test based on Chi - square partition for case - control GWA studies. Genetic Epidemiology. 2011;35(7):658–663. doi: 10.1002/gepi.20615. [DOI] [PubMed] [Google Scholar]
- Chen Z. Association tests through combining p-values for case control genome-wide association studies. Statistics and Probability Letters. 2013;83(8):1854–1862. doi: 10.1016/j.spl.2013.04.021. [DOI] [Google Scholar]
- Chen Z, Huang H, Liu J, Ng HKT, Nadarajah S, Huang X, Deng Y. Detecting differentially methylated loci for Illumina Array methylation data based on human ovarian cancer data. BMC Medical Genomics 2013, 2013;6(Suppl 1):S9. doi: 10.1186/1755-8794-6-S1-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Huang H, Ng HKT. Design and Analysis of Multiple Diseases Genome-wide Association Studies without Controls. GENE. 2012;510(1):87–92. doi: 10.1016/j.gene.2012.07.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Huang H, Ng HKT. Testing for Association in Case-Control Genome-wide Association Studies with Shared Controls. Statistical Methods in Medical Research, Published online before print February 1, 2013, doi: 101177/0962280212474061. 2013. [DOI] [PubMed]
- Chen Z, Liu Q, Nadarajah S. A new statistical approach to detecting differentially methylated loci for case control Illumina array methylation data. Bioinformatics. 2012;28(8):1109–1113. doi: 10.1093/bioinformatics/bts093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Ng HKT. A Robust Method for Testing Association in Genome-Wide Association Studies. Human Heredity. 2012;73(1):26–34. doi: 10.1159/000334719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Chen Z, Huang X. Age-adjusted nonparametric detection of differential DNA methylation with case--control designs. BMC Bioinformatics. 2013;14(1):86. doi: 10.1186/1471-2105-14-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Ng HKT, Li J, Liu Q, Huang H. Detecting associated single-nucleotide polymorphisms on the × chromosome in case control genome-wide association studies. Statistical methods in medical research. 2014. Published online before print September 24, 2014, doi: 2010.1177/0962280214551815. [DOI] [PubMed]
- Chen Z, Huang H, Ng HKT. An Improved Robust Association Test for GWAS with Multiple Diseases. Statistics & Probability Letters. 2014;91:153–161. [Google Scholar]
- Chen Z, Huang H, Liu Q. Detecting differentially methylated loci for multiple treatments based on high-throughput methylation data. BMC Bioinformatics. 2014;15:142. doi: 10.1186/1471-2105-15-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. A new association test based on disease allele selection for case-control genome-wide association studies. BMC Genomics. 2014;15:358. doi: 10.1186/1471-2164-15-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaykin D. Optimally weighted Z - test is a powerful method for combining probabilities in meta - analysis. Journal of Evolutionary Biology. 2011;24(8):1836–1841. doi: 10.1111/j.1420-9101.2011.02297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Pan Z, Jiao Z, Li F, Zhao G, Wei Q, Pan F, Evangelou E. TPH2 gene polymorphisms and major depression-a meta-analysis. PloS one. 2012;7(5):e36721. doi: 10.1371/journal.pone.0036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LS, Hsu L, Gamazon ER, Cox NJ, Nicolae DL. An exponential combination procedure for set-based association tests in sequencing studies. The American Journal of Human Genetics. 2012;91(6):977–986. doi: 10.1016/j.ajhg.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]