

Abstract
Background
PR-104 is rapidly hydrolyzed to PR-104A in vivo, which is activated by reduction to the corresponding 5-hydroxylamine (PR-104H) and amine (PR-104M) to produce DNA interstrand cross-links. PR-104 activation can occur via hypoxia-dependent reductases and also independently of hypoxia by aldo-keto reductase (AKR) 1C3.
Procedures
PR-104A was tested against the PPTP in vitro panel (10 nM to 100 μM), and PR-104 in vivo using a weekly × 6 schedule at its maximum tolerated dose (MTD) of 550 mg/kg. Subsequently PR-104 was tested at 270 and 110 mg/kg. Pharmacokinetics for PR-104 and its metabolites were determined, as were levels of AKR1C3 RNA and protein in xenografts.
Results
In vitro, the leukemia models were most sensitive to PR-104A. In vivo, PR-104 induced objective responses at its MTD in 21/34 solid tumor models and maintained complete responses against 7/7 acute lymphoblastic leukemia (ALL) models. At 270 mg/kg and lower dose levels, PR-104 did not induce solid tumor regressions, suggesting a steep dose–response relationship. Pharmacokinetic analysis suggests higher systemic exposures to PR-104A and its metabolites in mice compared to those achievable in patients. Levels of AKR1C3 protein did not correlate with tumor responsiveness.
Conclusions
As monotherapy, PR-104 demonstrated a high level of activity against both solid tumor and ALL models at its MTD, but the activity was almost completely lost at half the MTD dose for solid tumors. Pharmacokinetic data at the PR-104 MTD from human trials suggest that PR-104 metabolites may not reach the plasma exposures in children that were associated with high-level preclinical activity.
Keywords: developmental therapeutics, preclinical testing, PR-104
INTRODUCTION
Severe hypoxia is a characteristic unique to solid tumors [1]. These regions of hypoxia often surround necrotic zones and harbor viable cells that are resistant to both radiation and chemotherapy. Hypoxia also contributes to the invasive and metastatic characteristics of aggressive cancers through promoting genetic instability and facilitating the accumulation of mutations that can ultimately give rise to drug resistance [2,3]. Resistance to chemotherapy is in part due to diffusion limitations to hypoxic areas where blood flow is reduced; many antitumor agents cannot penetrate beyond 50–100 μm from capillaries, thereby failing to reach cells in the hypoxic regions [4]. In hypoxic zones of tumors the lower nutrient and oxygen supply to cells causes them to proliferate more slowly, so hypoxic tumor cells exhibit greater resistance to chemotherapies and radiation which target rapidly dividing cells or require oxygen for efficacy. Resistance is also a consequence of selection of cells under hypoxia that have attenuated function of the TP53 tumor suppressor [5]. Consequently, drug-induced apoptosis is decreased. Further, agents that mimic radiation, for which killing is oxygen-dependent, have reduced activity against cells in hypoxic regions.
While the selective antitumor activity of most cytotoxic agents is based, in part, on the killing of rapidly proliferating tumor cells, selective activation of drugs in the tumor environment has been attempted using several approaches. These tumor-activated prodrugs (TAPs) include tumor-specific delivery techniques, such as activation of prodrugs by exogenous enzymes delivered to tumor cells conjugated to antibodies (ADEPT) or generated within tumor cells from DNA constructs containing the corresponding gene (GDEPT) [6].
The hypoxic tumor environment offers an opportunity to develop tumor-selective agents that are selectively activated only in tumor tissue. Attempts to exploit tumor hypoxia focus on agents that may destabilize hypoxia-inducible factor 1 alpha (HIF1α) and on hypoxia-activated prodrugs (HAPs). Approaches to implementing the latter strategy have been based on the reduction of quinones, N-oxides, and nitroaromatics by endogenous enzymes or radiation. Several HAPs are being developed for clinical use, including TH-302 and PR-104 [7].
PR-104 is a preprodrug currently in clinical trials that was developed to target hypoxic tumor tissues [8]. It is a water-soluble phosphate ester that is converted in vivo to the corresponding 3,5-dinitrobenzamide mustard prodrug, PR-104A. In tumor tissue PR-104A is activated by nitroreduction to the corresponding DNA cross-linking 5-hydroxylamine (PR-104H) and amine (PR-104M) metabolites [9]. While PR-104 is reduced selectively in hypoxic cells by one-electron reductases [10], aerobic activation can also occur and is catalyzed in humans by the oxygen-independent two-electron reductase aldo-keto reductase family 1, member C3 (3-alpha hydroxysteroid dehydrogenase, type II; AKR1C3) [11]. A recent report has also documented aerobic activation in normal mouse tissues, though the responsible reductase(s) has yet to be identified [9].
Phase I clinical trials in adults with advanced solid tumors have used dosing every 3 weeks at dose levels ranging from 135 to 1,400 mg/m2 [12]. The maximum tolerated dose (MTD) was 1,100 mg/m2, with fatigue being the limiting toxicity. Above the MTD dose-limiting toxicities were febrile neutropenia and infection with normal absolute neutrophil count. Although no patients met criteria for objective response, reductions in tumor size were observed in patients treated with doses ≥550 mg/m2. Pharmacokinetic studies indicated rapid conversion of PR-104 to PR-104A that was dose proportional for both chemical species.
Bifunctional alkylating agents such as cyclophosphamide or ifosfamide play a major role in the curative therapy of solid tumors, brain tumors, and hematopoietic malignancies of childhood. However, resistance to these agents, both intrinsic and acquired, occurs in patients that relapse. Given that PR-104 has the potential to provide targeted delivery of bifunctional alkylating agents (PR-104H and PR-104M) to tumors with hypoxia or with AKR1C3 expression, here we have evaluated its activity against the PPTP’s in vitro and in vivo panels.
MATERIALS AND METHODS
In Vitro Testing
In vitro testing was performed using DIMSCAN, a semiautomatic fluorescence-based digital image microscopy system that quantifies viable (using fluorescein diacetate [FDA]) cell numbers in tissue culture multiwell plates [13]. Cells were incubated in the presence of the prodrug PR-104A for 96 hr under aerobic conditions (21% O2) at concentrations from 10 nM to 100 μM and analyzed as previously described [14].
In Vivo Tumor Growth Inhibition Studies
CB17SC-F scid−/− female mice (Taconic Farms, Germantown NY) were used to propagate subcutaneously implanted kidney/rhabdoid tumors, sarcomas (Ewing, osteosarcoma, rhabdomyosarcoma), neuroblastoma, and non-glioblastoma brain tumors, while BALB/c nu/nu mice were used for glioma models, as previously described [14–17]. Human leukemia cells were propagated by intravenous inoculation in female non-obese diabetic (NOD)/scid−/− mice as described previously [18]. Female mice were used irrespective of the patient gender from which the original tumor was derived. All mice were maintained under barrier conditions and experiments were conducted using protocols and conditions approved by the institutional animal care and use committee of the appropriate consortium member. Ten mice (solid tumors) or eight mice (leukemias) were used in each control or treatment group. Tumor volumes (cm3) [solid tumor xenografts] or percentages of human CD45-positive [hCD45] cells [acute lymphoblastic leukemia (ALL) xenografts] were determined as previously described [18] and responses were determined using three activity measures as previously described [19].
Determination of Response
Responses were determined using three activity measures [19]. For individual mice, progressive disease (PD) was defined as <50% regression from initial volume during the study period and >25% increase in initial volume at the end of study period. Stable disease (SD) was defined as <50% regression from initial volume during the study period and ≤25% increase in initial volume at the end of the study. Partial response (PR) was defined as a tumor volume regression ≥50% for at least one time point but with measurable tumor (≥0.10 cm3). Complete response (CR) was defined as a disappearance of measurable tumor mass (<0.10 cm3) for at least one time point. A CR was considered maintained (MCR) if the tumor volume was <0.10 cm3 at the end of the study period. For treatment groups only, if the tumor response was PD, then PD was further classified into PD1 or PD2 based on the tumor growth delay (TGD) value. TGD values were calculated based on the numbers of days to event. For each individual mouse that had PD and had an event in the treatment groups, a TGD value was calculated by dividing the time to event for that mouse by the median time to event in the respective control group. Median times to event were estimated based on the Kaplan–Meier event-free survival distribution. If a mouse had a TGD value ≤1.5, that mouse was considered PD1. If the TGD value was >1.5, the mouse was considered PD2. Mice that had PD but did not have an event at the end of the study were coded as PD2.
Event-Free Survival
An event in the solid tumor xenograft models was defined as a quadrupling of tumor volume from the initial tumor volume. Event-free survival was defined as the time interval from initiation of study to the first event or to the end of the study period for tumors that did not quadruple in volume. The time to event was determined using interpolation based on the formula:
where tx is the interpolated day to event, t1 is the lower observation day bracketing the event, t2 is the upper observation day bracketing the event, V1 is the tumor volume on day t1, V2 is the tumor volume on day t2 and Ve is the event threshold (four times initial tumor volume for solid tumor xenografts).
Response and Event Definitions for Acute Lymphoblastic Leukemia (ALL) Xenograft Models
Individual mice were categorized as PD if their percentage of hCD45 cells never dropped below 1% and they had an event before the end of the study period. An event is defined as hCD45 cells above 25% in the peripheral blood with times to event calculated as above. Individual mice were classified as SD if their percentage of hCD45 cells never dropped below 1% and no event occurred before the end of the study. PR was assigned if the percentage of cells dropped below 1% for any one-time point regardless of whether the percentage eventually reached 25%. A CR was assigned if the percentage of hCD45 cells dropped below 1% for two consecutive weeks of the study and regardless of whether the percentage reached 25% or not. A CR was considered maintained if the percentage of hCD45 was <1% for the last three measurements of the study. For treatment groups, PD was further classified into PD1 and PD2 according to the TGD value.
The time to event was determined using interpolation based on the formula:
where tx is the interpolated day to event, t1 is the lower observation day bracketing the event, t2 is the upper observation day bracketing the event, V1 is the hCD45 percentage on day t1, V2 is the tumor volume (or hCD45 percentage) on day t2 and Ve is the event threshold (25% for ALL xenografts).
Summary Statistics and Analysis Methods
Overall group response
Each individual mouse was assigned a score from 0 to 10 based on their response: PD1 = 0, PD2 = 2, SD = 4, PR = 6, CR = 8, and MCR = 10, and the median for the group determined the overall response. Studies in which toxicity was >25% or in which the control group was not at least SD, were considered in evaluable and were excluded from analysis. Treatment groups with PR, CR, or MCR are considered to have had an objective response. Agents inducing objective responses are considered highly active against the tested line, while agents inducing SD or PD2 are considered to have intermediate activity, and agents producing PD1 are considered to have a low level of activity against the tested line.
Tumor volume T/C value
Relative tumor volumes (RTV) for control (C) and treatment (T) mice were calculated at day 21 or when all mice in the control and treated groups still had measurable tumor volumes(if <21 days). The mean RTV for control and treatment mice for each study were then calculated and the T/C value was the mean RTV for the treatment group divided by the mean RTV for the control group. For the tumor volume T/C response measure, agents producing a T/C of ≤15% are considered highly active, those with a mean tumor volume T/C of ≤45% but >15% are considered to have intermediate activity, and those with mean T/C values >45% are considered to have low levels of activity [20].
EFS T/C value
An EFS T/C value was defined by the ratio of the median time to event of the treatment group and the median time to event of the respective control group. If the treatment group did not have a median time to event, then EFST/C was defined as greater than the ratio of the last day of the study for the treatment group divided by the median time to event for the control group. For the EFS T/C measure, agents are considered highly active if they meet three criteria: (a) an EFS T/C >2; (b) a significant difference in EFS distributions (P ≤ 0.050), and (c) a net reduction in median tumor volume for animals in the treated group at the end of treatment as compared to at treatment initiation. Agents meeting the first two criteria, but not having a net reduction in median tumor volume for treated animals at the end of the study are considered to have intermediate activity. Agents with an EFS T/C <2 are considered to have low levels of activity. Xenografts in which the median EFS for the control line was greater than one-half of the study period or in which the median EFS for the control line did not exist are considered not evaluable for the EFS T/C measure of activity.
Statistical methods
The exact log-rank test, as implemented using Proc StatXact for SAS®, was used to compare event-free survival distributions between treatment and control groups. P-values were two-sided and were not adjusted for multiple comparisons given the exploratory nature of the studies. The Mann–Whitney test was used to test the difference of medians of IC50 values between cell lines of a given histotype compared to the remaining cell lines of the panel.
Western blotting
Western blot analyses were performed as previously described with minor modifications [21]. Primary antibodies to AKR1C3 (Sigma, St. Louis, MO) and GAPDH (Cell Signalling Technologies, Beverly, MA) were used.
Immunohistochemistry
Immunohistochemistry was performed on the xenograft tissue microarray using the same antibody as for Western blot analysis and visualized using Cytomation EnVision (DAKO Corp., Carpentaria, CA).
Gene expression analysis
Affymetrix U133 Plus 2.0 array expression data were generated and analyzed as previously described [22].
Pharmacokinetic analysis
The metabolism of PR-104 is shown in Supplemental Figure 1. Plasma levels of PR-104, PR-104A (alcohol), PR-104H (hydroxylamine), PR-104M (amine), PR-104S (semi-mustard), and PR-104G (O-glucuronide) were measured by LC/MS/MS using the method of Gu and Wilson [23]. Three mice were used per time point, and two or three replicate determinations were done for each mouse using procedures described by Gu and Wilson [23]. PR-104H (hydroxylamine) and PR-104M (amine) are the active cytotoxic species. No major nonlinearity with dose has been demonstrated in several species, including humans [24], consequently the pharmacokinetics for PR-104 and its metabolites were determined at a single dose (270 mg/kg; 807 mg/m2) in the current study.
Drugs and formulation
PR-104 and PR-104A were provided to the Pediatric Preclinical Testing Program by Proacta, Inc., through the Cancer Therapy Evaluation Program (NCI). For in vitro testing, stock solutions of PR-104A were prepared in DMSO, with dilutions in culture media. For in vivo testing, PR-104 was dissolved in sterile water, diluted in sterile saline, and immediately administered I.P., once weekly for 6 weeks at a dose of 550, 270, or 110 mg/kg. The schedule for administration was chosen after consultation with Proacta, Inc. PR-104 was provided to each consortium investigator in coded vials for blinded testing.
RESULTS
PR-104A In Vitro Testing
Although the half-life is relatively short in cell culture medium, PR-104A inhibited growth of the majority of the cell lines from the PPTP in vitro panel (Table I) with only two cell lines (Rh18 and CHLA-90) having IC50 values >100 μM. Under the aerobic testing conditions employed, the median IC50 value for all of the cell lines in the panel was 16.5 μM (Table I). IC50 values were significantly lower for the ALL cell lines (median = 2.4 μM) compared to the remaining cell lines (P = 0.001) (Fig. 1, panel A). Typical examples of concentration response curves are shown for the rhabdomyosarcoma line Rh30, and the leukemia cell line, COG-LL-317 (Fig. 1, panel B).
TABLE I.
Activity of PR-104A Against Cell Lines in the PPTP In Vitro Panel Under Aerobic Conditions
| Cell line | Histology | IC50 (μM) |
|---|---|---|
| RD | Rhabdomyosarcoma | 23.1 |
| Rh41 | Rhabdomyosarcoma | 46.6 |
| Rh18 | Rhabdomyosarcoma | >100 |
| Rh30 | Rhabdomyosarcoma | 18.6 |
| BT-12 | Rhabdoid | 39.3 |
| CHLA-266 | Rhabdoid | 26.9 |
| TC-71 | Ewing sarcoma | 6.0 |
| CHLA-9 | Ewing sarcoma | 11.7 |
| CHLA-10 | Ewing sarcoma | 17.4 |
| CHLA-258 | Ewing sarcoma | 16.6 |
| GBM2 | Glioblastoma | 16.5 |
| NB-1643 | Neuroblastoma | 6.4 |
| NB-EBc1 | Neuroblastoma | 5.1 |
| CHLA-90 | Neuroblastoma | >100 |
| CHLA-136 | Neuroblastoma | 18.5 |
| NALM-6 | ALL | 4.6 |
| COG-LL-317 | ALL | 1.7 |
| RS4;11 | ALL | 4.0 |
| MOLT-4 | ALL | 2.4 |
| CCRF-CEM | ALL | 1.4 |
| Kasumi-1 | AML | 27.6 |
| Karpas-299 | ALCL | 24.9 |
| Ramos-RA1 | NHL | 10.1 |
| Median | 16.6 | |
| Minimum | 1.4 | |
| Maximum | >100 |
Fig. 1.

PR-104 in vitro activity. A: Relative sensitivity of the cell lines using the IC50 values displayed by histotype. The black line indicates the median IC50 (16.5 μM) for the panel. Cells were exposed to PR-104 for 96 hr under aerobic conditions at concentrations from 10 nM to 100 μM, and viable cells determined by fluorescein diacetate staining. Concentrations of PR-104 that inhibited cell proliferation by 50% (IC50) are plotted for each cell line of each histotype. B: Typical growth inhibition curves for Rh30 and COG-LL-317. Error bars represent standard deviations for each concentration tested.
PR-104 In Vivo Testing
PR-104 was evaluated in 45 xenograft models using a weekly schedule at a dose of 550 mg/kg, the MTD for the agent using this schedule. Forty-six of 846 mice died during the study (5.4%), with 8 of 419 in the control arms (1.9%) and 38 of 427 in the PR-104 treatment arms (8.9%). Four lines (NB-SD, NB-1691, NB-1643, and ALL-3) were excluded from analysis due to toxicity >25%. A complete summary of results is provided in Supplemental Table I, including total numbers of mice, number of mice that died (or were otherwise excluded), numbers of mice with events and average times to event, TGD, as well as numbers of responses and T/C values.
Antitumor effects were evaluated using the PPTP activity measures for time to event (EFS T/C), TGD (tumor volume T/C), and objective response. PR-104 induced significant differences in EFS distributions compared to controls in 31/34 (91%) solid tumor models and 6/7(86%)ALL models (Table II). Twenty-three out of 34 evaluable lines met the criteria for high activity with EFS T/C values >2 and with final tumor volumes less than the initial volumes (Table II). An additional eight models met criteria for intermediate activity for the EFS T/C activity measure by having EFS T/C values exceeding 2.0 and significant differences in EFS distribution between treated and control groups.
TABLE II.
Activity for PR-104 Against the PPTP In Vivo Panel
| Xenograft line | Histology | KM estimate of median time to event | P-value | EFS T/C | Median final RTV | T/C | P-value | T/C volume activity | EFS activity | Response activity |
|---|---|---|---|---|---|---|---|---|---|---|
| BT-29 | Rhabdoid | 33.9 | <0.001 | 2.4 | >4 | 0.41 | <0.001 | Int | Int | Int |
| KT-14 | Rhabdoid | >EP | <0.001 | >1.7 | 0.3 | 0.18 | <0.001 | Int | NE | High |
| KT-12 | Rhabdoid | >EP | <0.001 | >4.0 | 0.0 | 0.00 | <0.001 | High | High | High |
| KT-10 | Wilms | >EP | <0.001 | >3.9 | 0.0 | 0.00 | <0.001 | High | High | High |
| KT-11 | Wilms | >EP | <0.001 | >3.7 | 0.0 | 0.31 | 0.004 | Int | High | High |
| KT-13 | Wilms | >EP | <0.001 | >4.5 | 0.0 | 0.22 | <0.001 | Int | High | High |
| SK-NEP-1 | Ewing | >EP | <0.001 | >5.0 | 0.0 | 0.08 | <0.001 | High | High | High |
| EW5 | Ewing | >EP | <0.001 | >6.2 | 0.0 | 0.10 | <0.001 | High | High | High |
| EW8 | Ewing | >EP | <0.001 | >3.9 | 2.2 | 0.34 | <0.001 | Int | Int | Int |
| TC-71 | Ewing | 21.7 | <0.001 | 2.5 | >4 | 0.32 | 0.004 | Int | Int | Int |
| CHLA258 | Ewing | >EP | <0.001 | >6.1 | 0.0 | 0.00 | <0.001 | High | High | High |
| Rh10 | ALV RMS | >EP | 0.002 | >1.6 | 0.0 | 0.15 | <0.001 | High | NE | High |
| Rh28 | ALV RMS | >EP | <0.001 | >2.5 | 0.0 | 0.06 | <0.001 | High | High | High |
| Rh30 | ALV RMS | >EP | <0.001 | >2.4 | 0.0 | 0.07 | <0.001 | High | High | High |
| Rh30R | ALV RMS | >EP | <0.001 | >3.6 | 0.3 | 0.23 | <0.001 | Int | High | High |
| Rh41 | ALV RMS | >EP | <0.001 | >3.8 | 0.0 | 0.19 | <0.001 | Int | High | High |
| Rh18 | EMB RMS | >EP | <0.001 | >2.4 | 0.0 | 0.10 | <0.001 | High | High | High |
| BT-28 | Medulloblastoma | 10.1 | 0.161 | 1.4 | >4 | 0.66 | 0.035 | Low | Low | Low |
| BT-45 | Medulloblastoma | 16.3 | 0.073 | 1.1 | >4 | 0.87 | 0.143 | Low | Low | Low |
| BT-50a | Medulloblastoma | >EP | 1.000 | — | 0.0 | 0.53 | 0.052 | Low | NE | Int |
| BT-41 | Ependymoma | >EP | <0.001 | >1.4 | 1.0 | 0.44 | <0.001 | Int | NE | Int |
| BT-44 | Ependymoma | >EP | <0.001 | >2.1 | 0.0 | 0.04 | <0.001 | High | High | High |
| GBM2 | Glioblastoma | >EP | <0.001 | >3.1 | 1.2 | 0.37 | <0.001 | Int | Int | Int |
| BT-39 | Glioblastoma | >EP | <0.001 | >2.8 | 0.0 | 0.11 | <0.001 | High | High | High |
| D645 | Glioblastoma | >EP | <0.001 | >4.0 | 0.5 | 0.06 | <0.001 | High | High | High |
| D456 | Glioblastoma | >EP | <0.001 | >8.1 | 2.7 | 0.18 | <0.001 | Int | Int | Int |
| NB-1771 | Neuroblastoma | 8.7 | 0.003 | 2.1 | >4 | 0.40 | 0.005 | Int | Int | Int |
| NB-EBc1 | Neuroblastoma | >EP | <0.001 | >8.1 | 2.9 | 0.18 | <0.001 | Int | Int | Int |
| OS-1 | Osteosarcoma | >EP | <0.001 | >1.3 | 0.2 | 0.68 | <0.001 | Low | NE | High |
| OS-2 | Osteosarcoma | >EP | <0.001 | >1.6 | 0.3 | 0.67 | <0.001 | Low | NE | High |
| OS-17 | Osteosarcoma | 35.5 | <0.001 | 2.3 | >4 | 0.48 | 0.002 | Low | Int | Int |
| OS-9 | Osteosarcoma | >EP | <0.001 | >2.0 | 0.2 | 0.54 | 0.036 | Low | High | High |
| OS-33 | Osteosarcoma | >EP | <0.001 | >2.4 | 0.2 | 0.41 | <0.001 | Int | High | High |
| OS-31 | Osteosarcoma | 27.8 | <0.001 | 1.7 | >4 | 0.58 | <0.001 | Low | Low | Int |
| ALL-2 | ALL B-precursor | >EP | 0.002 | >2.6 | 0.0 | — | — | — | High | High |
| ALL-4 | ALL B-precursor | >EP | <0.001 | >4.2 | 0.1 | — | — | — | High | High |
| ALL-7 | ALL B-precursor | >EP | <0.001 | >9.7 | 0.0 | — | — | — | High | High |
| ALL-8 | ALL T-cell | >EP | <0.001 | >12.2 | 0.0 | — | — | — | High | High |
| ALL-16 | ALL T-cell | >EP | 0.061 | >1.9 | 0.0 | — | — | — | NE | High |
| ALL-17 | ALL B-precursor | >EP | <0.001 | >6.7 | 0.0 | — | — | — | High | High |
| ALL-19 | ALL B-precursor | >EP | 0.040 | >3.9 | 0.1 | — | — | — | High | High |
EP, evaluation period.
BT-50 control growth did not reach median RTV >4, so EFS T/C is not evaluable.
The in vivo testing results for the objective response measure of activity are presented in Figure 2 in a “heat-map” format as well as a “COMPARE”-like format, based on the scoring criteria described in the Materials and Methods Section. The latter analysis demonstrates relative tumor sensitivities around the midpoint score of 5 (SD). Objective responses were seen in 21 of 34 solid tumor models with examples of representative solid tumor response shown in Figure 3 (KT-12, KT-10, and OS-33). Maintained CRs were seen in all seven evaluable ALL models with representative examples of ALL responses shown in Figure 4 (ALL-2, ALL-8, and All-19).
Fig. 2.

PR-104 in vivo objective response activity. Left: The colored “heat map” depicts group response scores. A high level of activity is indicated by a score of 6 or more, intermediate activity by a score of ≥2 but <6, and low activity by a score of <2. Right: representation of tumor sensitivity based on the difference of individual tumor lines from the midpoint response (stable disease). Bars to the right of the median represent lines that are more sensitive, and to the left are tumor models that are less sensitive. Red bars indicate lines with a significant difference in EFS distribution between treatment and control groups, while blue bars indicate lines for which the EFS distributions were not significantly different.
Fig. 3.

PR-104 activity against individual solid tumor xenografts. Kaplan–Meier curves for EFS, median relative tumor volume graphs, and individual tumor volume graphs are shown for selected lines. Controls (gray lines); treated (black lines).
Fig. 4.

PR-104 activity against ALL xenografts. Kaplan–Meier curves for EFS and graphs of median and individual percentages of hCD45 cells, are shown for selected lines. Controls (gray lines); treated (black lines).
To determine the dose–response relationship for PR-104, five sensitive tumors (EW-5, Rh30, CHLA258, Rh41, and OS-1) that showed a range of expression of AKR1C3 (see below) were selected. Tumor bearing mice were treated at 550, 270, or 110 mg/kg q 7D. These doses were selected to bracket the dose range at which PR-104A systemic exposures comparable to those tolerated in humans are achieved. Although there were no objective responses in any tumor model at dose levels below the MTD, Table III, growth of CHLA-258 and Rh30 xenografts was significantly retarded at lower doses, as shown for several sarcoma models (Fig. 5).
TABLE III.
Dose–Response Activity for PR-104 Against Selected PPTP In Vivo Lines
| Xenograft line | Histology | Treatment group (mg/kg) | KM estimate of median time to event | P-value | EFS T/C | Median final RTV | T/C | P-value | Median group response |
|---|---|---|---|---|---|---|---|---|---|
| CHLA258 | Ewing | 550 | >EP | <0.001 | >4.9 | 0.8 | 0.27 | <0.001 | SD |
| 270 | 20.3 | <0.001 | 2.4 | >4 | 0.45 | 0.002 | PD2 | ||
| 110 | 14.3 | 0.012 | 1.7 | >4 | 0.61 | 0.023 | PD2 | ||
| EW5 | Ewing | 550 | >EP | <0.001 | >4.1 | 0 | 0.02 | <0.001 | MCR |
| 270 | 25.2 | 0.204 | 2.4 | >4 | 0.58 | 0.165 | PD2 | ||
| 110 | 15 | 0.394 | 1.5 | >4 | 0.64 | 0.029 | PD1 | ||
| Rh30 | ALV RMS | 550 | >EP | <0.001 | >4.1 | 0 | 0.23 | <0.001 | MCR |
| 270 | 20.6 | 0.006 | 2 | >4 | 0.51 | <0.001 | PD2 | ||
| 110 | 9.4 | 0.504 | 0.9 | >4 | 0.85 | 0.274 | PD1 | ||
| Rh41 | ALV RMS | 550 | >EP | <0.001 | >3.4 | 0 | 0.31 | <0.001 | MCR |
| 270 | 17.4 | 0.107 | 1.4 | >4 | 0.62 | 0.004 | PD1 | ||
| 110 | 16.5 | 0.03 | 1.3 | >4 | 0.65 | 0.017 | PD1 | ||
| OS-1 | Osteosarcoma | 550 | >EP | <0.001 | >1.8 | 2.1 | 0.6 | 0.007 | PD2 |
| 270 | 24.9 | 0.432 | 1 | >4 | 1.09 | 0.853 | PD1 | ||
| 110 | 19.9 | 0.888 | 0.8 | >4 | 1.25 | 0.280 | PD1 |
Fig. 5.

Dose–response of sarcoma xenografts treated with PR-104. Tumor bearing mice were treated with PR-104 at 550 mg/kg (■), 270 mg/kg (◆), or 110 mg/kg (▼), or received no treatment (●). Curves show the median growth of 10 tumors per dose group.
Pharmacokinetic and Pharmacodynamic Studies
Nitroreduction of PR-104A is catalyzed either by one-electron reductases in hypoxic cells [10] or by the oxygen-independent two electron reductases (e.g., AKR1C3 in humans) [11]. Levels of PR-104 and its metabolites following a single administration of drug (270 mg/kg; 807 mg/m2) are shown in Figure 6. For parent drug and all metabolites except PR-104G, the Cmax occurred at 15 min with rapid clearance for all metabolites (Table IV). Comparison to human pharmacokinetic parameters at the human MTD (1,100 mg/m2) is also shown in Table IV. Systemic exposure in SCID mice to PR-104A at the 270 mg/kg (807 mg/m2) dose in mice is approximately 30% higher than that for humans at the PR-104 MTD. Consistent with previous reports for CD1 nude mice [9,25], SCID mice show less extensive O-β-glucuronidation of PR-104A (the PR-104G metabolite) compared to humans, while mice showed more extensive N-dealkylation (the PR-104S metabolite) than humans. Systemic exposures for activated metabolites (PR-104H and PR-104M) were also evaluated for SCID mice, and these were much lower than those previously described for CD1 nude mice (2% vs. approximately 20% for the ratio of the summed activated metabolite AUCs, PR-104H and PR-104M, to the PR-104A AUC). This may reflect a strain difference, as blood samples were processed rapidly to prevent degradation of these labile metabolites [25].
Fig. 6.

Plasma concentration–time profiles of PR-104 metabolites in scid−/− mice dosed i.p. at 270 mg/kg (466 μmol/kg).
TABLE IV.
Non-Compartmental Pharmacokinetic Parameters of PR-104 Metabolites in Non-Tumoredscid−/− Mice Administered i.p. at 270 mg/kg
| Species | SCID mice AUC (μM h) | SCID mice Cmax (μM) | SCID mice t½ (min) | Human AUC (μM h) |
|---|---|---|---|---|
| PR-104 | 74.6 | 136.9 | 7.80 | 19.0 |
| PR-104A | 44.5 | 62.0 | 12.8 | 34.9 |
| PR-104S | 10.6 | 11.3 | 19.7 | 0.78 |
| PR-104G | 5.91 | 8.92 | 13.1 | 44.6 |
| PR-104H | 0.534 | 0.723 | 15.5 | 1.75 |
| PR-104M | 0.368 | 0.518 | 16.0 | 0.29 |
Comparison is made to systemic exposures in humans at the PR-104 MTD (1,100 mg/m2 as a 60-min intravenous infusion) [24].
Using the PPTP Affymetrix U133 Plus 2.0 gene expression database, high-level expression of AKR1C3 was demonstrated in many solid tumors and ALL models (Fig. 7A). These data were largely consistent with immunoblot analysis (Fig. 7B) for those xenografts for which both were performed. As in vitro testing was done under aerobic conditions, we performed exploratory analyses to evaluate whether IC50 values correlated with the AKR1C3 expression level (probeset 209160_at of the Affymetrix arrays). No correlation between AKR1C3 expression and cytotoxic activity was observed in vitro, and median expression level was roughly comparable for the ALL cell lines (the histotype most responsive to PR-104A) and for the non-ALL cell lines. For the in vivo panel, there was a trend for higher AKR1C3 expression in solid tumor xenografts with maintained CR compared to those with PD (PD1 or PD2) (Mann–Whitney P = 0.13). We also evaluated the relationship between PR-104 in vivo activity and tumor VEGF expression (Affymetrix probeset 201512_s_at) as a potential measure of tumor hypoxia. Median VEGF expression levels were indistinguishable between xenografts with PD as their best response and those with maintained CR (P = 0.98).
Fig. 7.
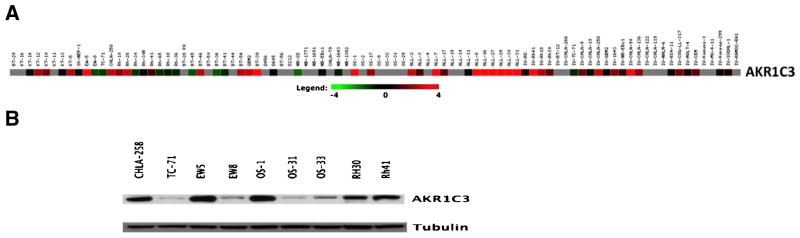
AKR1C3 reductase levels. A: Gene expression data obtained from Affymetrix HG-U133 Plus 2.0 for AKR1C3 for PPTP cell lines and xenografts as visualized using GeneSifter software (VizX Labs, Seattle, WA). Gray indicates an absent call from Affymetrix quality control. B: AKR1C3 protein expression levels as determined by Western blotting in selected xenografts.
DISCUSSION
PR-104 represents a novel preprodrug bifunctional DNA alkylating agent in adult phase 2 trials for treatment of cancer. In vitro, PR-104A (the prodrug form) was tested under standard conditions (21% O2) and exhibited greater potency against ALL cell lines compared to cell lines derived from solid tumors. The ALL cell lines were also more sensitivity than the other cell lines of the PPTP in vitro panel to the alkylating agent melphalan [26]. The median IC50 concentration was 16.5 μM (range 1.4 to >100 μM). The activity of PR-104A under the aerobic conditions employed could be the result of aerobic nitroreduction (e.g., as described for AKR1C3) and subsequent interstrand cross-linking (ICL) by PR-104H or PR-104M, although cytotoxicity in the absence of ICL has been described for several cell lines [27]. The lower IC50 values under aerobic conditions for PR-104A observed in this report compared to previous reports may represent the longer exposure to PR-104A used for the PPTP testing (96 hr vs. 2–4 hr in prior reports) [8,27], which would allow for greater reductive metabolism of PR-104A. Hypoxic sensitivity was not evaluated in this study but PR-104A is reported to be 10- to 100-fold more dose-potent in the absence of oxygen [8,27].
In vivo, PR-104 at its MTD caused maintained CRs in 19/34 solid tumor models and demonstrated high-level activity (by all three activity criteria) against 11 solid tumors and 6 ALL models. Of note was the response of Rh18 xenografts (MCR) whereas this cell line was the most refractory in vitro. The least responsive tumors were neuroblastomas (all six with PD despite toxicity) and medulloblastoma (BT-28 and BT-45). Maintained CRs were obtained in most other tumor panels. While PR-104 and the bifunctional alkylating agent cyclophosphamide [19] showed broad-spectrum activity against both solid tumor and ALL models, their spectra of antitumor activity at the MTD were not identical. For example, PR-104 induced maintained CR in models (KT-11, KT-12, KT-14, Rh18, and Rh41) that responded poorly to cyclophosphamide (PD2), whereas TC-71 and OS-17 achieved maintained CRs in mice treated with cyclophosphamide, but had PD when treated with PR-104. The neuroblastoma panel, derived largely from drug refractory patients, was the least responsive panel for both agents.
The high level of in vivo activity for PR-104 could be related to both hypoxic and oxic bioreduction of the agent. Previous reports have shown that xenografts have regions of hypoxia [28–30], with the extent of hypoxia increasing with tumor volume [28] and with hypoxic tumor regions being distant from blood vessels and areas of perfusion [29]. However, tumor xenograft hypoxia was not explicitly measured in this study. Activation of PR-104 in regions of hypoxia could result in an antitumor activity against adjacent tumor regions through a “bystander” effect [8,27,31], accounting for greater reductions in tumor volume than could be accounted for simply on the basis of regions of hypoxia. PR-104 in vivo activity could also be related to aerobic activation, as illustrated by its activation in humans by the reductase AKR1C3 [11]. Relative levels of AKR1C3 gene expression correlated with protein levels of AKR1C3 in eight of nine tumors for which both were measured, but neither correlated with overall tumor responsiveness to PR-104. Immunohistochemical staining for AKR1C3 in xenograft tumors and normal tissues (human) revealed heterogeneous staining in malignant tissues and intense staining in human liver and kidney (data not shown). Consistent with this observation, AKR1C3 gene expression is observed in liver and kidney as well as other normal tissues [32]. Activation of PR-104 in nude mice (as evidenced by tissue levels of PR-104H and PR-104M) has been documented for several normal tissues, including liver, kidney, small intestine, and bone marrow [9]. While there is evidence for physiologic hypoxia in these tissues, it is likely that a substantial proportion of the activation is through oxygen-insensitive two-electron reductases rather than hypoxia-dependent one-electron reductases [9]. The reductase(s) responsible for oxic activation of PR-104 in mice is not identified, but may be distinct from AKR1C3 [9].
The high level of activity observed in ALL xenografts could initially be interpreted as a consequence of the tissue distribution of the rapidly proliferating leukemia cell precursors, since oxygen tension levels in the bone marrow are low [33,34], but as treatments began when engraftment was well established and leukemia cells have already reached other organs this explanation is insufficient. The pronounced effects observed in vitro on the ALL cell lines, under the same oxic conditions used for other cell lines, also points to some particular feature of the histotype which may underpin chemosensitivity. As activated PR-104 metabolites are present in mice treated with PR-104 [9,25], the leukemia panel in vivo activity may reflect the general sensitivity of ALL cells to alkylating agents.
Because the antitumor activity of PR-104 was so profound across multiple tumor models, we investigated the dose range over which this agent demonstrated activity. Five tumor models that were sensitive in the initial screen and that showed a range of AKR1C3 expression were chosen to evaluate lower dose levels (270 and 110 mg/kg) using the same weekly schedule of administration. Reduction of the PR-104 dose from 550 to 270 mg/kg resulted in significant loss of antitumor activity in each solid tumor model, although lower doses still significantly inhibited growth of several sarcoma models.
For a drug that has a narrow therapeutic dose range, it is imperative to ascertain the systemic drug exposures associated with tumor regression in mice and to compare these to drug exposures tolerated in humans [35–37]. Detailed analysis of pharmacokinetic parameters have been described for PR-104 metabolites in CD1 nude mice receiving PR-104 at 326 mg/kg and for the same metabolites in humans at the PR-104 MTD (1,100 mg/m2) [25]. The total exposure (AUC0–inf) to circulating reduced metabolites (i.e., PR-104H and PR-104M) was approximately 6-fold higher in nude mice versus humans under these conditions, and the PR-104A exposure was 2.3-fold higher in mice compared to humans [25]. These results suggest that the relevant PR-104 dose for efficacy testing in CD1 nude mice (based on achievable exposures in humans to either PR-104A or to the reduced metabolites of PR-104) is well below 326 mg/kg. We conducted similar pharmacokinetic analyses for SCID mice and demonstrated that PR-104A systemic exposure at the 270 mg/kg dose is similar to that observed for nude mice (when adjusted for dose) and exceeds PR-104A systemic exposure in humans at the PR-104 MTD (Table IV). These results suggest that the human-relevant SCID mouse dose for efficacy testing is <270 mg/kg, a dose that was ineffective in causing tumor regression in the solid tumor models evaluated. Comparing course duration between mice and humans is challenging, since tumors double or quadruple as quickly as 7–10 days in the former and perhaps increase by 25% to 50% in 3 weeks in the latter. Arguments can be made for either a literal translation of time from mice to man or for adjusting for the more rapid pace of tumor growth in mice versus man. For other cytotoxic agents such as topotecan, the plasma exposure (AUC) in mice yielding antitumor activity appears to correlate well with that yielding a similar response rate in patients with neuroblastoma [36,38] or for melphalan in patients with rhabdomyosarcoma at diagnosis [39]. In our study, the 270 mg/kg PR-104 dose produced PR-104A systemic exposures that were 3.8-fold greater than those achieved at the MTD in humans per 21-day course of treatment when taking into account that three doses of PR-104 were given to mice during each 21-day period. Conversion of PR-104 to PR-104A has demonstrated approximate dose-linear pharmacokinetics in several species including human [24]. Assuming such dose-proportionality for PR-104A systemic exposures, a 7.6-fold increase in PR-104A systemic exposure beyond that achieved at the adult MTD would be required for consistent tumor regressing activity. Consequently, using this approach for translating dose between species suggests that the agent is highly unlikely to reach the plasma exposures in children that were associated with high-level activity in pediatric preclinical models representing solid tumors. Using an alternative approach that compares the systemic exposures at the active dose used in mice “per tumor volume doubling” (i.e., 550 mg/kg) and again assuming dose-proportionality for systemic exposure to PR-104A, the systemic exposure of PR-104A at the human MTD is 2.6-fold below the systemic exposure associated with tumor regressing activity in mice. Even this more favorable analysis suggests that it is unlikely that PR-104 can achieve adequate levels in humans for robust tumor regressing activity. A similar conclusion was reached by Patel et al. [24], who estimated the exposure in humans at doses causing dose-limiting myelosuppression was approximately 25% of that achievable in mice.
An additional issue that needs addressing with respect to toxicity is the potential long-term deleterious effects on the hematopoietic stem cell compartment, as these cells have been shown to benefit from a hypoxic niche in the bone marrow [40]. Normally these cells are refractory to chemotherapy and playa crucial role inpatient recovery from chemotherapy-induced hematologic toxicity. While this study does not establish whether reductive activation of PR-104 in murine bone marrow, or other tissues, is due to oxic or hypoxic reductase(s) [9], it is probable that the hematopoietic toxicity observed in humans is independent of hypothetical risks related to the hypoxic marrow niche. In support of this conjecture is the observation that hematologic recovery following administration of PR-104 in patients is relatively rapid [12]. In contrast, agents that are cytotoxic to bone marrow stem cells (e.g., chloroethylating nitrosoureas) induce prolonged marrow suppression.
It is apparent that PR-104 shows substantial activation in normal tissues and that this may contribute to the preclinical activity described in this report as well as the clinical toxicity observed in the PR-104 phase 1 trial [12]. Achieving monotherapy activity with hypoxia-activated agents likely requires unrealistic circumstances (e.g., 100% hypoxic fraction or a dramatic effective bystander effect) or off-target effects (e.g., aerobic activation). The present study does not establish whether PR-104 kills hypoxic cells in pediatric xenografts at human-equivalent doses; if it does, it could well have utility in combination with chemotherapy agents that are able to kill tumor cells in non-hypoxic tumor regions. The concept of using prodrugs that are selectively activated in tumors, either because of tumor hypoxia or because of cancer-specific reductases, is an attractive one, and further research in this area is clearly warranted.
Supplementary Material
Acknowledgments
Grant sponsor: National Cancer Institute; Grant number: NO1-CM-42216; Grant number: CA21765; Grant number: CA108786; Grant sponsor: Health Research Council of New Zealand; Grant number: HRC08/103.
This work was supported by NO1-CM-42216, CA21765, and CA108786 from the National Cancer Institute and the Health Research Council of New Zealand, grant HRC08/103. PR-104 and PR-104A were supplied by Proacta, Inc. In addition to the authors represents work contributed by the following: Sherry Ansher, Catherine A. Billups, Ingrid Boehm, Joshua Courtright, Mila Dolotin, Edward Favours, Henry S. Friedman, Debbie Payne-Turner, Charles Stopford, Chandra Tucker, Jianrong Wu, Joe Zeidner, Ellen Zhang, and Jian Zhang. Children’s Cancer Institute Australia for Medical Research is affiliated with the University of New South Wales and Sydney Children’s Hospital. We appreciate the valuable discussions with Proacta, Inc. in planning and undertaking this study.
Footnotes
Conflict of interest: William R. Wilson is a shareholder in Proacta, Inc. Yongchuan Gu is an employee of Proacta. The other authors consider that there are no actual or perceived conflicts of interest.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 2.Gillies RJ, Gatenby RA. Hypoxia and adaptive landscapes in the evolution of carcinogenesis. Cancer Metastasis Rev. 2007;26:311–317. doi: 10.1007/s10555-007-9065-z. [DOI] [PubMed] [Google Scholar]
- 3.Sullivan R, Graham CH. Hypoxia-driven selection of the metastatic phenotype. Cancer Metastasis Rev. 2007;26:319–331. doi: 10.1007/s10555-007-9062-2. [DOI] [PubMed] [Google Scholar]
- 4.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 5.Graeber TG, Osmanian C, Jacks T, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- 6.Denny WA. Tumor-activated prodrugs—A new approach to cancer therapy. Cancer Invest. 2004;22:604–619. doi: 10.1081/cnv-200027148. [DOI] [PubMed] [Google Scholar]
- 7.Denny WA. Hypoxia-activated prodrugs in cancer therapy: Progress to the clinic. Future Oncol. 2010;6:419–428. doi: 10.2217/fon.10.1. [DOI] [PubMed] [Google Scholar]
- 8.Patterson AV, Ferry DM, Edmunds SJ, et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin Cancer Res. 2007;13:3922–3932. doi: 10.1158/1078-0432.CCR-07-0478. [DOI] [PubMed] [Google Scholar]
- 9.Gu Y, Guise CP, Patel K, et al. Reductive metabolism of the dinitrobenzamide mustard anticancer prodrug PR-104 in mice. Cancer Chemother Pharmacol. 2010 doi: 10.1007/s00280-010-1354-5. [DOI] [PubMed] [Google Scholar]
- 10.Guise CP, Wang AT, Theil A, et al. Identification of human reductases that activate the dinitrobenzamide mustard prodrug PR-104A: A role for NADPH:cytochrome P450 oxidoreductase under hypoxia. Biochem Pharmacol. 2007;74:810–820. doi: 10.1016/j.bcp.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 11.Guise CP, Abbattista MR, Singleton RS, et al. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res. 2010;70:1573–1584. doi: 10.1158/0008-5472.CAN-09-3237. [DOI] [PubMed] [Google Scholar]
- 12.Jameson MB, Rischin D, Pegram M, et al. A phase I trial of PR-104, a nitrogen mustard prodrug activated by both hypoxia and aldo-keto reductase 1C3, in patients with solid tumors. Cancer Chemother Pharmacol. 2010;65:791–801. doi: 10.1007/s00280-009-1188-1. [DOI] [PubMed] [Google Scholar]
- 13.Frgala T, Kalous O, Proffitt RT, et al. A fluorescence microplate cytotoxicity assay with a 4-log dynamic range that identifies synergistic drug combinations. Mol Cancer Ther. 2007;6:886–897. doi: 10.1158/1535-7163.MCT-04-0331. [DOI] [PubMed] [Google Scholar]
- 14.Houghton PJ, Morton CL, Kolb EA, et al. Initial testing (stage 1) of the proteasome inhibitor bortezomib by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:37–45. doi: 10.1002/pbc.21214. [DOI] [PubMed] [Google Scholar]
- 15.Friedman HS, Colvin OM, Skapek SX, et al. Experimental chemotherapy of human medulloblastoma cell lines and transplantable xenografts with bifunctional alkylating agents. Cancer Res. 1988;48:4189–4195. [PubMed] [Google Scholar]
- 16.Graham C, Tucker C, Creech J, et al. Evaluation of the antitumor efficacy, pharmacokinetics, and pharmacodynamics of the histone deacetylase inhibitor depsipeptide in childhood cancer models in vivo. Clin Cancer Res. 2006;12:223–234. doi: 10.1158/1078-0432.CCR-05-1225. [DOI] [PubMed] [Google Scholar]
- 17.Peterson JK, Tucker C, Favours E, et al. In vivo evaluation of ixabepilone (BMS247550), a novel epothilone B derivative, against pediatric cancer models. Clin Cancer Res. 2005;11:6950–6958. doi: 10.1158/1078-0432.CCR-05-0740. [DOI] [PubMed] [Google Scholar]
- 18.Liem NL, Papa RA, Milross CG, et al. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood. 2004;103:3905–3914. doi: 10.1182/blood-2003-08-2911. [DOI] [PubMed] [Google Scholar]
- 19.Houghton PJ, Morton CL, Tucker C, et al. The Pediatric Preclinical Testing Program: Description of models and early testing results. Pediatr Blood Cancer. 2007;49:928–940. doi: 10.1002/pbc.21078. [DOI] [PubMed] [Google Scholar]
- 20.Plowman JCR, Alley M, Sausville E, et al. US-NCI testing procedures. In: Feibig HH, editor. Relevance of tumor models for anticancer drug development. Basel: Karger; 1999. pp. 121–135. [Google Scholar]
- 21.Kurmasheva RT, Harwood FC, Houghton PJ. Differential regulation of vascular endothelial growth factor by Akt and mammalian target of rapamycin inhibitors in cell lines derived from childhood solid tumors. Mol Cancer Ther. 2007;6:1620–1628. doi: 10.1158/1535-7163.MCT-06-0646. [DOI] [PubMed] [Google Scholar]
- 22.Neale G, Su X, Morton CL, et al. Molecular characterization of the pediatric preclinical testing panel. Clin Cancer Res. 2008;14:4572–4583. doi: 10.1158/1078-0432.CCR-07-5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu Y, Wilson WR. Rapid and sensitive ultra-high-pressure liquid chromatography-tandem mass spectrometry analysis of the novel anticancer agent PR-104 and its major metabolites in human plasma: Application to a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3181–3186. doi: 10.1016/j.jchromb.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 24.Patel K, Choy SS, Hicks KO, et al. A combined pharmacokinetic model for the hypoxia-targeted prodrug PR-104A in humans, dogs, rats and mice predicts species differences in clearance and toxicity. Cancer Chemother Pharmacol. 2010 doi: 10.1007/s00280-010-1412-z. [DOI] [PubMed] [Google Scholar]
- 25.Gu Y, Atwell GJ, Wilson WR. Metabolism and excretion of the novel bioreductive prodrug PR-104 in mice, rats, dogs, and humans. Drug Metab Dispos. 2010;38:498–508. doi: 10.1124/dmd.109.030973. [DOI] [PubMed] [Google Scholar]
- 26.Kang MH, Smith MA, Morton CL, et al. National cancer institute pediatric preclinical testing program: Model description for in vitro cytotoxicity testing. Pediatr Blood Cancer. 2010;74:810–820. doi: 10.1002/pbc.22801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singleton RS, Guise CP, Ferry DM, et al. DNA cross-links in human tumor cells exposed to the prodrug PR-104A: Relationships to hypoxia, bioreductive metabolism, and cytotoxicity. Cancer Res. 2009;69:3884–3891. doi: 10.1158/0008-5472.CAN-08-4023. [DOI] [PubMed] [Google Scholar]
- 28.Saitoh J, Sakurai H, Suzuki Y, et al. Correlations between in vivo tumor weight, oxygen pressure, 31P NMR spectroscopy, hypoxic microenvironment marking by beta-D-iodinated azomycin galactopyranoside (beta-D-IAZGP), and radiation sensitivity. Int J Radiat Oncol Biol Phys. 2002;54:903–909. doi: 10.1016/s0360-3016(02)03013-4. [DOI] [PubMed] [Google Scholar]
- 29.He F, Deng X, Wen B, et al. Noninvasive molecular imaging of hypoxia in human xenografts: Comparing hypoxia-induced gene expression with endogenous and exogenous hypoxia markers. Cancer Res. 2008;68:8597–8606. doi: 10.1158/0008-5472.CAN-08-0677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawrentschuk N, Lee FT, Jones G, et al. Investigation of hypoxia and carbonic anhydrase IX expression in a renal cell carcinoma xenograft model with oxygen tension measurements and (124)I-cG250 PET/CT. Urol Oncol. 2009 doi: 10.1016/j.urolonc.2009.03.028. [DOI] [PubMed] [Google Scholar]
- 31.Hicks KO, Myint H, Patterson AV, et al. Oxygen dependence and extravascular transport of hypoxia-activated prodrugs: Comparison of the dinitrobenzamide mustard PR-104A and tirapazamine. Int J Radiat Oncol Biol Phys. 2007;69:560–571. doi: 10.1016/j.ijrobp.2007.05.049. [DOI] [PubMed] [Google Scholar]
- 32.Lin HK, Jez JM, Schlegel BP, et al. Expression and characterization of recombinant type 2 3 alpha-hydroxysteroid dehydrogenase (HSD) from human prostate: Demonstration of bifunctional 3 alpha/17 beta-HSD activity and cellular distribution. Mol Endocrinol. 1997;11:1971–1984. doi: 10.1210/mend.11.13.0026. [DOI] [PubMed] [Google Scholar]
- 33.Chow DC, Wenning LA, Miller WM, et al. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. II. Modified Kroghian models. Biophys J. 2001;81:685–696. doi: 10.1016/S0006-3495(01)75733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chow DC, Wenning LA, Miller WM, et al. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. I. Krogh’s model. Biophys J. 2001;81:675–684. doi: 10.1016/S0006-3495(01)75732-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peterson JK, Houghton PJ. Integrating pharmacology and in vivo cancer models in preclinical and clinical drug development. Eur J Cancer. 2004;40:837–844. doi: 10.1016/j.ejca.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 36.Zamboni WC, Stewart CF, Thompson J, et al. Relationship between topotecan systemic exposure and tumor response in human neuroblastoma xenografts. J Natl Cancer Inst. 1998;90:505–511. doi: 10.1093/jnci/90.7.505. [DOI] [PubMed] [Google Scholar]
- 37.Leggas M, Stewart CF, Woo MH, et al. Relation between Irofulven (MGI-114) systemic exposure and tumor response in human solid tumor xenografts. Clin Cancer Res. 2002;8:3000–3007. [PubMed] [Google Scholar]
- 38.Santana VM, Furman WL, Billups CA, et al. Improved response in high-risk neuroblastoma with protracted topotecan administration using a pharmacokinetically guided dosing approach. J Clin Oncol. 2005;23:4039–4047. doi: 10.1200/JCO.2005.02.097. [DOI] [PubMed] [Google Scholar]
- 39.Horowitz ME, Etcubanas E, Christensen ML, et al. Phase II testing of melphalan in children with newly diagnosed rhabdomyosarcoma: A model for anticancer drug development. J Clin Oncol. 1988;6:308–314. doi: 10.1200/JCO.1988.6.2.308. [DOI] [PubMed] [Google Scholar]
- 40.Eliasson P, Jonsson JI. The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J Cell Physiol. 2010;222:17–22. doi: 10.1002/jcp.21908. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.