

Version Changes
Revised. Amendments from Version 1
We have responded to the reviewers suggestions. We have made a small labeling addition to Figure 1 and added a new Figure S2 which is an expanded view of Figure 2B.
Abstract
We are currently faced with a global infectious disease crisis which has been anticipated for decades. While many promising biotherapeutics are being tested, the search for a small molecule has yet to deliver an approved drug or therapeutic for the Ebola or similar filoviruses that cause haemorrhagic fever. Two recent high throughput screens published in 2013 did however identify several hits that progressed to animal studies that are FDA approved drugs used for other indications. The current computational analysis uses these molecules from two different structural classes to construct a common features pharmacophore. This ligand-based pharmacophore implicates a possible common target or mechanism that could be further explored. A recent structure based design project yielded nine co-crystal structures of pyrrolidinone inhibitors bound to the viral protein 35 (VP35). When receptor-ligand pharmacophores based on the analogs of these molecules and the protein structures were constructed, the molecular features partially overlapped with the common features of solely ligand-based pharmacophore models based on FDA approved drugs. These previously identified FDA approved drugs with activity against Ebola were therefore docked into this protein. The antimalarials chloroquine and amodiaquine docked favorably in VP35. We propose that these drugs identified to date as inhibitors of the Ebola virus may be targeting VP35. These computational models may provide preliminary insights into the molecular features that are responsible for their activity against Ebola virus in vitro and in vivo and we propose that this hypothesis could be readily tested.
Keywords: ebola virus, computational models, machine learning
Introduction
The current Ebola virus (EBOV) crisis has demonstrated that globally we are not prepared to respond with therapeutics to treat existing infections or act as prophylactics as there is no Food and Drug Administration (FDA) or European Medicines Agency (EMEA) approved therapeutic. More importantly this suggests we should have been prepared for a pathogen which has been known about for nearly forty years. The current EBOV outbreak is already proving remarkably costly in terms of the mortality and financial ramifications 1, 2. The best approaches to EBOV so far have relied on public health measures for containment 3 which have been used in past outbreaks 4. These lessons with EBOV will undoubtedly be important for the next virus outbreak 5 but they also raise many questions 6 which point to how little we know about these viruses in general, as well as how best to share knowledge openly 7.
There have been a relatively small number of studies that have attempted to identify compounds active against EBOV. Two recent studies utilized high-throughput screens of a subset of FDA approved drugs against different EBOV strains (Zaire and Sudan) in vitro and in vivo. These independent reports suggested the promise of the antimalarials amodiaquine and chloroquine in one study 8, while the selective estrogen receptor modulators (SERMs) clomiphene and toremifene were active in another 9. Chloroquine to date has not progressed beyond the mouse EBOV model used in these studies. We hypothesized that we could use these four molecules to computationally define the features that are important for activity. The previous studies were not exhaustive screens of all FDA drugs and so we have taken this opportunity to suggest additional compounds. Looked at from another perspective “non-antiviral” drugs may be worth following up even though their molecular mechanism is unknown. These compounds may themselves have broad antiviral activity as reports describe modest inhibitory activity against other viruses 10– 13.
Several studies have identified non-FDA approved drugs including an in silico docking approach to identify molecules targeting the viral Nedd4-PPxY interface 14. These molecules were similar to the FDA benzimidazole and aminoquinoline 8, 9 compounds that were active against EBOV. Another good example is the recent in silico docking of 5.4 million drug-like compounds docked in the viral protein VP35 protein 15. This identified multiple pyrrolidinones which inhibit its polymerase cofactor activity 15. The pyrrolidinones bind to an alpha helix which is proposed as important for viral function 16. With the limited knowledge of small molecules and potential targets we have studied whether the FDA-approved drugs that are active in vitro and in vivo versus EBOV could be targeting VP35.
Methods
Common features pharmacophore for EBOV actives
Two papers from 2013 described compounds active as inhibitors of different EBOV strains in vitro and in vivo, namely amodiaquine and chloroquine in one study 8, clomiphene and toremifene in another 9. These active molecules were used as they have both in vitro and in vivo activity to build a common features pharmacophore with Discovery Studio 4.1 (Biovia, San Diego, CA) from 3D conformations of the molecules generated with the CAESAR algorithm. This identified key features. The pharmacophore was then used to search various databases (for which up to 100 molecule conformations with the FAST conformer generation method with the maximum energy threshold of 20 kcal/mol, were created). The pharmacophore was then used to search the Microsource Spectrum database ( http://www.msdiscovery.com/spectrum.html) as well as the CDD FDA drugs dataset ( https://www.collaborativedrug.com/pages/public_access). In both cases over 300 hits were retrieved initially. The van der Waals surface of amodiaquine (which was more potent than chloroquine 8) was added to limit the number of hits retrieved 17– 19.
Receptor-ligand pharmacophores for VP35
Receptor-ligand pharmacophores for the VP35 protein were generated from crystal structures (4IBB, 4IBC, 4IBD, 4IBE, 4IBF, 4IBG, 4IBI, 4IBJ, 4IBK) in the protein data bank PDB. Pharmacophores were constructed using the receptor-ligand pharmacophore generation protocol in Discovery Studio version 4.1 (Biovia, San Diego, CA) with a maximum number of pharmacophores (10), minimum features (4), and maximum number of features (6) as are described elsewhere 20.
In silico docking of molecules in VP35 structure
PDB 4IBI was used for docking using LibDock in Discovery Studio (Biovia, San Diego CA) 21. The proposed binding site was centered on the ligand and a site sphere created (coordinates 2.14, 20.93, 1.71) with 9.45 Å diameter. The protocol included 10 hotspots and docking tolerance (0.25). The FAST conformation method was also used along with steepest descent minimization with CHARMm. Further parameters followed the default settings. The ligand VPL57 was removed from the binding site and re-docked. The four FDA approved drugs with activity against Ebola were docked in the structure from an sdf file. Molecules were visualized alongside the original ligand VPL57 and the 2D interaction plots generated.
Results
Data was downloaded sourced from Microsource Spectrum and CDD Drugs. Dataset includes sd files used to create the 3D database that was searched. Note that models only run on Discovery Studio.
Common features pharmacophore for EBOV actives
The pharmacophore was generated using the in vivo and in vitro active amodiaquine, chloroquine, clomiphene and toremifene ( Supplemental Table 1) as these represent the most relevant FDA approved drugs to date. This pharmacophore consists of 4 hydrophobic features and a hydrogen bond acceptor feature ( Figure 1). The pharmacophore with van der Waals surface was also used to search FDA drug various libraries ( Supplemental Table 2 and Supplemental Table 3). The most interesting observations from this virtual screen are that various estradiol analogs score well (e.g. estradiol valerate Fit value 4.23). Previously estradiol was suggested to be active in the EBOV pseudotype assay in vitro 8. In addition, dibucaine was also retrieved (Fit value 1.58) which was also active in the EBOV pseudotype assay 8. Amodiaquine, chloroquine, clomiphene and toremifene can be used as positive controls for future screens. Because the original complete sets of FDA approved compounds screened are not publically accessible it is difficult to compare hit rates versus all compounds tested to date.
Figure 1. Pharmacophore based on 4 hits.

A. amodiaquine, B. chloroquine, C. clomiphene D. toremifene and E. Overlap showing all molecules in the van der Waals surface of amodiaquine. Pharmacophore features are Hydrophobic (H, cyan) and Hydrogen bond acceptor (HBA, green).
Receptor-ligand pharmacophores for VP35
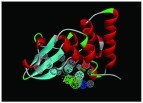
The nine receptor-ligand pharmacophores created all consisted of three to four hydrophobic features and one to two hydrogen bonding features ( Table 1). Eight of these pharmacophores also had a negative ionizable feature. These suggest that the receptor-ligand based approach results in a general similarity across the nine structures, likely indicating the similar binding mode and importance of features for interfering with this generally hydrophobic pocket for protein-protein interactions.
Table 1. Pharmacophores for EBOV VP35 generated from crystal structures in the protein data bank PDB.
Pharmacophores were generated using the receptor-ligand pharmacophore generation protocol in Discovery Studio version 4.1 (Biovia, San Diego, CA) with minimum features (3) and maximum features (6). Pharmacophore features are Hydrophobic (H, cyan), Hydrogen bond acceptor (HBA, green), hydrogen bond donor (HBD, purple) and 1 negative ionizable (neg, blue). Excluded volumes (grey) were also automatically added. Further details on this approach are described elsewhere 20.
| PDB | Pharmacophore
features |
Pharmacophore with ligand
mapped |
|---|---|---|
| 4IBB | 4H, 1HBD,
1 neg ionizable |
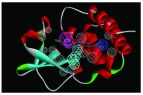
|
| 4IBC | 3H, 2HBA,
1 neg ionizable |
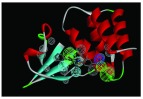
|
| 4IBD | 4H, 1 HBA,
1 neg ionizable |
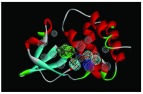
|
| 4IBE | 4H, 1HBA |
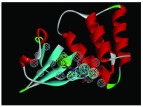
|
| 4IBF | 4H, 1 HBA,
1 neg ionizable |
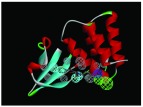
|
| 4IBG | 3H, 2 HBA,
1 neg ionizable |
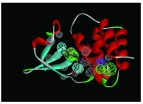
|
| 4IBI | 4H, 1HBA,
1 neg ionizable |
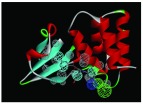
|
| 4IBJ | 4H, 1HBA,
1 neg ionizable |
|
| 4IBK | 4H, 1HBA,
1 neg ionizable |
|
In silico docking of molecules in VP35 structure
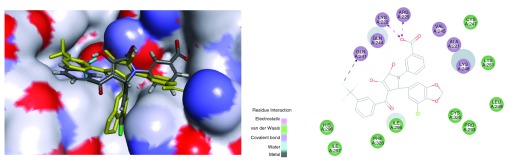
Redocking the 4IBI ligand in the protein resulted in an RMSD of 3.02Å, which generally indicates the difficulty of predicting orientations for compounds binding in what is a relatively hydrophobic and shallow pocket ( Figure S1). This molecule was ranked the 29 th pose and had a LibDock score of 86.62 ( Figure S1 higher scores are better). The four FDA approved drugs were docked into the VP35 structure 4IBI. All compounds docked similarly and overlapped with the co-crystal ligand ( Figure 2). Amodiaquine and chloroquine had higher LibDock scores (> 90) than the 4IBI ligand, while clomiphene and toremifene had LibDock scores less than 70. All four FDA approved drugs bound similarly to the pyrrolidinone ligands in the pocket formed by residues from the α-helical and β-sheet subdomains 15. We have highlighted proposed energetically favorable interactions of the antimalarial candidate binders with ILE295, LYS248 and GLN244, which scored favorably. Previously published studies suggested mutation of ILE295, LYS248 resulted in near-complete loss of binding activity 15.
Figure 2. Docking FDA approved compounds in VP35 protein showing overlap with ligand (yellow) and 2D interaction diagram.

4IBI was used, 4IBI ligand VPL57 shown in yellow. A. Amodiaquine (grey) and 4IBI LibDock score 90.80, B. Chloroquine (grey) LibDock score 97.82, C. Clomiphene (grey) and 4IBI LibDock score 69.77, D. Toremifene (grey) and 4IBI LibDock score 68.11
Discussion
Our previous experience with common feature and quantitative pharmacophore models has demonstrated their value in predicting novel actives from collections of FDA approved drugs 22– 27. Candidate predicted actives may be assessed by their Fit Value to the pharmacophore model. This score can be used to prioritize compounds for eventual testing. In the current study it was hypothesized that two different classes of compounds showing activity against EBOV in vitro and in vivo may share a common pharmacophore. Construction of this pharmacophore ( Figure 1) indicated four hydrophobic features and a hydrogen bond acceptor feature. This pharmacophore (with an added van der Waals surface to limit the number of hits retrieved) was then used to screen and score other FDA drugs from a small database and identified 120 and 124 structures for future evaluation in vitro testing ( Supplemental Table 2 and Supplemental Table 3). Out of these compounds estradiol and dibucaine had been previously described as active in in vitro EBOV assays. This suggested the pharmacophore could retrieve some structurally diverse classes of known hits 8.
Recently identified co-crystal structures of the EBOV VP35 protein were used to derive receptor-ligand pharmacophores. These nine receptor-ligand pharmacophores suggested the importance of hydrophobic, hydrogen bonding and negative ionizable interactions to interfere with this protein-protein interaction ( Table 1). Eight out of nine of the pharmacophores had one or more hydrogen bond acceptor feature. These pharmacophores are grossly similar to our ligand based pharmacophore (derived from four FDA approved drugs that inhibit EBOV), as both types of model had multiple hydrophobic features and at least one hydrogen bond acceptor. When we docked the antimalarials and SERMs into a representative VP35 structure these compounds were found to overlap with the X-ray ligand to differing extents. Amodiaquine and chloroquine had LibDock scores greater than 90 and higher than that for the redocked X-ray ligand. This indicated that VP35 may be a potential target for these two distinct classes of compounds. However, it is important to point out that we have not compared docking to other proteins in EBOV and it could also be possible that these molecules are active elsewhere as well as via other mechanisms than by specific binding to proteins 28, 29. Further, VP35 may be a preferred target for the antimalarials while the SERMs are not predicted to bind as well as the X-ray ligand. The use of other docking and scoring methods may produce differences in the pose and predicted binding affinity, which could be of interest for further studies.
A combination of the promising efficacy of chloroquine (EC 50 16 μM 8) and amodiaquine (EC 50 8.4 μM 8) versus EBOV, their availability and likely low cost should prioritize their further laboratory exploration. Mechanistic studies against VP35 and possibly other proteins should also be pursued and may be enlightened by the observation that both of these compounds also have reported activity against other viruses. For example, chloroquine is active against human coronavirus OC43 ( in vitro and in infected mice) as well as SARS ( in vitro) 10, 30, 31, while amodiaquine also inhibits dengue virus 2 replication and infectivity in vitro 11.
Conclusions
In summary, this study has built on the previous publications that identified four FDA approved compounds active against different strains of EBOV 8, 9. Our pharmacophore model for SERMs and aminoquinolines suggests that these compounds share multiple chemical features based on their overlap to the four hydrophobic features and a hydrogen bond acceptor ( Figure 1E) and they may have a common mechanism or target. We suggest that VP35 may be the likely target based on the overlap of receptor-based pharmacophores and docking into the crystal structure. Amodiaquine and chloroquine score particularly well in terms of docking to VP35. If this is the case it could provide a means to follow up with other small molecule analogs and/or additional FDA approved drugs that could target this protein-protein interaction. As with our other tuberculosis-focused research 32, 33, and computational approaches to repositioning compounds 34 we embrace the essentiality for computational predictions to be interrogated through rigorous experimental studies. For example at least two in silico docking studies screened commercially available compounds 14, 15. We propose that docking FDA approved drugs could also be a viable first step to identifying potential compounds that could be used. We are actively seeking collaborators with experience with EBOV assays to enable further translational studies. We believe this computationally inspired approach may be applicable for other known infectious pathogens that do not have current treatments such as other viruses related to Ebola. Ultimately we need to be able to leverage such approaches to provide antivirals for future pathogens.
Data availability
F1000Research: Dataset 1. Pharmacophores, receptor ligand models and docking data for FDA-approved drugs inhibiting the Ebola virus, 10.5256/f1000research.5741.d38449 35.
The ligand-based pharmacophore was previously made available: http://figshare.com/articles/Ebola_active_cpds_pharmacophore/1190902.
The following PDB structures were used in this study ( 4IBB, 4IBC, 4IBD, 4IBE, 4IBF, 4IBG, 4IBI, 4IBJ, 4IBK).
For models and advice please contact Sean Ekins ( ekinssean@yahoo.com).
Acknowledgments
Dr. Christopher D. Southan, Dr. Peter Madrid and Dr. Nadia Litterman are acknowledged for discussions on Ebola. Biovia are kindly acknowledged for providing Discovery Studio. An earlier preliminary version of this pharmacophore was described previously: http://figshare.com/articles/A_pharmacophore_of_ebola_active_compounds/1190787.
Funding Statement
The author(s) declared that no grants were involved in supporting this work.
v2; ref status: indexed
Supplementary materials
Supplemental Table 1. Information for common features pharmacophore generation.
| Maximum Omitted features | Principal | |
|---|---|---|
| chloroquine | 0 | 1 |
| amodiaquine | 0 | 2 |
| clomiphene | 0 | 1 |
| Toremifene | 0 | 2 |
Supplemental Table 2. FDA drugs and common features pharmacophore. The dataset of 2643 molecules was downloaded from the CDD Public Access ( https://www.collaborativedrug.com/pages/public_access) as an sdf and then a 3D database was created in Discovery Studio using FAST conformer generation with up to 255 conformations. The database was searched with the common feature pharmacophore developed from amodiaquine, chloroquine, clomiphene and toremifene. The search 3D database protocol was used with the Fast search method. In some cases the indication for the molecules is not described (ND).
Supplemental Table 3. Microsource Spectrum and common features pharmacophore. The dataset of 2311 molecules was provided by Microsource ( http://www.msdiscovery.com/spectrum.html) as an sdf and then a 3D database was created in Discovery Studio using FAST conformer generation with up to 255 conformations. The database was searched with the common feature pharmacophore developed from amodiaquine, chloroquine, clomiphene and toremifene. The search 3D database protocol was used with the Fast search method.
Figure S1. Redocking VPL57 in 4IBI.
The 4IBI ligand was removed from the structure and redocked. The closest pose (grey) was ranked 29 with RMSD 3.02A and LibDock score 86.62 when compared to the actual ligand in 4IBI (yellow).
Figure S2. Docking chloroquine in 4IBI showing complete protein.

4IBI ligand VPL57 shown in yellow. Chloroquine (grey). The compounds bind in the first basic patch of the interferon inhibitory domain which is important for the viral nucleoprotein interaction and replication complex formation 15.
References
- 1.Butler D, Morello L: Ebola by the numbers: The size, spread and cost of an outbreak. Nature. 2014;514(7522):284–5. 10.1038/514284a [DOI] [PubMed] [Google Scholar]
- 2.Piot P: Ebola’s perfect storm. Science. 2014;345(6202):1221. 10.1126/science.1260695 [DOI] [PubMed] [Google Scholar]
- 3.Trad MA, Fisher DA, Tambyah PA: Ebola in west Africa. Lancet Infect Dis. 2014;14(11):1045. 10.1016/S1473-3099(14)70924-7 [DOI] [PubMed] [Google Scholar]
- 4.Dhillon RS, Srikrishna D, Sachs J: Controlling Ebola: next steps. Lancet. 2014;384(9952):1409–1411. 10.1016/S0140-6736(14)61696-2 [DOI] [PubMed] [Google Scholar]
- 5.Anon: Call to action. Nature. 2014;514(7524):535–536. 10.1038/514535b [DOI] [PubMed] [Google Scholar]
- 6.Check Hayden E: The Ebola questions. Nature. 2014;514(7524):554–7. 10.1038/514554a [DOI] [PubMed] [Google Scholar]
- 7.Piot P: The F1000Research: Ebola article collection [v1; ref status: not peer reviewed, http://f1000r.es/4ot]. F1000Res. 2014;3:269 10.12688/f1000research.5685.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madrid PB, Chopra S, Manger ID, et al. : A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS One. 2013;8(4):e60579. 10.1371/journal.pone.0060579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johansen LM, Brannan JM, Delos SE, et al. : FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci Transl Med. 2013;5(190):190ra79. 10.1126/scitranslmed.3005471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keyaerts E, Li S, Vijgen L, et al. : Antiviral activity of chloroquine against human coronavirus OC43 infection in newborn mice. Antimicrob Agents Chemother. 2009;53(8):3416–21. 10.1128/AAC.01509-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boonyasuppayakorn S, Reichert ED, Manzano M, et al. : Amodiaquine, an antimalarial drug, inhibits dengue virus type 2 replication and infectivity. Antiviral Res. 2014;106:125–34. 10.1016/j.antiviral.2014.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Acosta EG, Bruttomesso AC, Bisceglia JA, et al. : Dehydroepiandrosterone, epiandrosterone and synthetic derivatives inhibit Junin virus replication in vitro. Virus Res. 2008;135(2):203–12. 10.1016/j.virusres.2008.03.014 [DOI] [PubMed] [Google Scholar]
- 13.Dyall J, Coleman CM, Hart BJ, et al. : Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother. 2014;58(8):4885–93. 10.1128/AAC.03036-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han Z, Lu J, Liu Y, et al. : Small-molecule probes targeting the viral PPxY-host Nedd4 interface block egress of a broad range of RNA viruses. J Virol. 2014;88(13):7294–306. 10.1128/JVI.00591-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown CS, Lee MS, Leung DW, et al. : In silico derived small molecules bind the filovirus VP35 protein and inhibit its polymerase cofactor activity. J Mol Biol. 2014;426(10):2045–58. 10.1016/j.jmb.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chakraborty S, Rao B, Asgeirsson B, et al. : Characterizing alpha helical properties of Ebola viral proteins as potential targets for inhibition of alpha-helix mediated protein-protein interactions [v1; ref status: approved with reservations 1, http://f1000r.es/4lg]. F1000Res. 2014;3:251 10.12688/f1000research.5573.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamichhane G, Freundlich JS, Ekins S, et al. : Essential metabolites of Mycobacterium tuberculosis and their mimics. MBio. 2011;2(1):e00301–10. 10.1128/mBio.00301-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ekins S, Bradford J, Dole K, et al. : A collaborative database and computational models for tuberculosis drug discovery. Mol Biosyst. 2010;6(5):840–851. 10.1039/b917766c [DOI] [PubMed] [Google Scholar]
- 19.Zheng X, Ekins S, Raufman JP, et al. : Computational models for drug inhibition of the human apical sodium-dependent bile acid transporter. Mol Pharm. 2009;6(5):1591–1603. 10.1021/mp900163d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meslamani J, Li J, Sutter J, et al. : Protein-ligand-based pharmacophores: generation and utility assessment in computational ligand profiling. J Chem Inf Model. 2012;52(4):943–55. 10.1021/ci300083r [DOI] [PubMed] [Google Scholar]
- 21.Rao SN, Head MS, Kulkarni A, et al. : Validation studies of the site-directed docking program LibDock. J Chem Inf Model. 2007;47(6):2159–71. 10.1021/ci6004299 [DOI] [PubMed] [Google Scholar]
- 22.Diao L, Ekins S, Polli JE: Novel inhibitors of human organic cation/carnitine transporter (hOCTN2) via computational modeling and in vitro testing. Pharm Res. 2009;26(8):1890–1900. 10.1007/s11095-009-9905-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng X, Ekins S, Raufman JP, et al. : Computational models for drug inhibition of the human apical sodium-dependent bile acid transporter. Mol Pharm. 2009;6(5):1591–603. 10.1021/mp900163d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diao L, Ekins S, Polli JE: Quantitative structure activity relationship for inhibition of human organic cation/carnitine transporter. Mol Pharm. 2010;7(6):2120–2131. 10.1021/mp100226q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ekins S, Diao L, Polli JE: A substrate pharmacophore for the human organic cation/carnitine transporter identifies compounds associated with rhabdomyolysis. Mol Pharm. 2012;9(4):905–913. 10.1021/mp200438v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong Z, Ekins S, Polli JE: Structure-activity relationship for FDA approved drugs as inhibitors of the human sodium taurocholate cotransporting polypeptide (NTCP). Mol Pharm. 2013;10(3):1008–19. 10.1021/mp300453k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong Z, Ekins S, Polli JE: Quantitative NTCP pharmacophore and lack of association between DILI and NTCP Inhibition. Eur J Pharm Sci. 2014;66C:1–9. 10.1016/j.ejps.2014.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kazmi F, Hensley T, Pope C, et al. : Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab Dispos. 2013;41(4):897–905. 10.1124/dmd.112.050054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nadanaciva S, Lu S, Gebhard DF, et al. : A high content screening assay for identifying lysosomotropic compounds. Toxicol In Vitro. 2011;25(3):715–23. 10.1016/j.tiv.2010.12.010 [DOI] [PubMed] [Google Scholar]
- 30.de Wilde AH, Jochmans D, Posthuma CC, et al. : Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob Agents Chemother. 2014;58(8):4875–84. 10.1128/AAC.03011-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vincent MJ, Bergeron E, Benjannet S, et al. : Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. 2005;2:69. 10.1186/1743-422X-2-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ekins S, Freundlich JS: Computational models for tuberculosis drug discovery. Methods Mol Biol. 2013;993:245–62. 10.1007/978-1-62703-342-8_16 [DOI] [PubMed] [Google Scholar]
- 33.Ekins S, Freundlich JS, Choi I, et al. : Computational databases, pathway and cheminformatics tools for tuberculosis drug discovery. Trends Microbiol. 2011;19(2):65–74. 10.1016/j.tim.2010.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ekins S, Williams AJ, Krasowski MD, et al. : In silico repositioning of approved drugs for rare and neglected diseases. Drug Disc Today. 2011;16(7–8):298–310. 10.1016/j.drudis.2011.02.016 [DOI] [PubMed] [Google Scholar]
- 35.Ekins S, Freundlich JS, Coffee M: Dataset 1 in: A common feature pharmacophore for FDA-approved drugs inhibiting the Ebola virus. F1000Research. 2014. Data Source [DOI] [PMC free article] [PubMed] [Google Scholar]