

SUMMARY
Neural circuit formation relies on interactions between axons and cells within the target field. While it is well established that target-derived signals act on axons to regulate circuit assembly, the extent to which axon-derived signals control circuit formation is not known. In the Drosophila visual system, anterograde signals numerically match R1–R6 photoreceptors with their targets by controlling target proliferation and neuronal differentiation. Here we demonstrate that additional axon-derived signals selectively couple target survival with layer-specificity. We show that Jelly belly (Jeb) produced by R1–R6 axons interacts with its receptor, anaplastic lymphoma kinase (Alk), on budding dendrites to control survival of L3 neurons, one of three postsynaptic targets. L3 axons then produce Netrin, which regulates the layer-specific targeting of another neuron within the same circuit. We propose that a cascade of axon-derived signals, regulating diverse cellular processes, provides a strategy for coordinating circuit assembly across different regions of the nervous system.
INTRODUCTION
The nervous system comprises local circuits, containing diverse neuronal cell types interlinked by synaptic connections. These are assembled into networks through additional connections established between neurons located in different local circuits. Both intrinsic mechanisms and intercellular signals specify different cell types and many different intercellular signaling pathways pattern connections between neurons.
Two broad classes of interactions have been characterized between growth cones and their environment. It is well established, for instance, that signals produced by cells influence axons through interactions with receptors on growth cones. This includes the production of signals by intermediate targets (Bate, 1976; Ou and Shen, 2010; Palka et al., 1992), the numerical matching of axons and target neurons through retrograde action of trophic factors (Harrington and Ginty, 2013; Levi-Montalcini, 1987) and topographic map formation through graded expression of targeting cues (Cheng et al., 1995; Flanagan, 2006).
Signals released by axons also regulate circuit assembly. For example, agrin, secreted by motor axons, regulates development of the postsynaptic membrane at the vertebrate neuromuscular junction (Burgess et al., 1999; Gautam et al., 1996; Kummer et al., 2006; Lin et al., 2005; Misgeld et al., 2005; Nitkin et al., 1987), and BDNF, produced by sympathetic axons, induces pruning of less active neighboring sympathetic axons (Singh et al., 2008). In addition, within the Drosophila visual system, anterograde signals regulating cell proliferation and differentiation control the numerical matching of photoreceptors with their synaptic targets (see below). While it is clear that axon-derived signals contribute to circuit assembly and maintenance in some isolated contexts, whether they act in a broader fashion to coordinate neural circuit assembly is not known. Here, we demonstrate that a sequence of signals released by different axons couples neural circuit development between different regions of the Drosophila visual system in a cell type-specific manner. We suggest that integration of such anterograde signaling pathways may provide a general scheme for coordinating circuit assembly.
The fly visual system comprises photoreceptor neurons (R cells) in the compound eye or retina and a large collection of neurons that process visual information in the optic lobe (i.e. in the lamina, medulla, lobula and lobula plate) (see Figure 1). These neurons are organized in a modular fashion. The retina comprises some 750 modules called ommatidia, each containing eight photoreceptor neurons (R1–R8) that fall into three classes (R1–R6, R7 and R8) based on spectral sensitivity and synaptic specificity. R1–R6 neurons form synapses within modules called cartridges aligned in a parallel fashion in the lamina, a thin structure that lies directly below the retina. R7 and R8 neurons project axons through the lamina which terminate in discrete layers within medulla modules referred to as columns. Each column also receives input from R1–R6 neurons indirectly, through the projections of lamina neurons L1–L3. As a consequence of the pattern of connections between neurons, lamina cartridges and medulla columns are topographically matched and process information collected by R1–R8 neurons that “see” the same point in visual space. Within medulla columns, different visual features are computed within specific layers.
Figure 1. The Drosophila visual system.
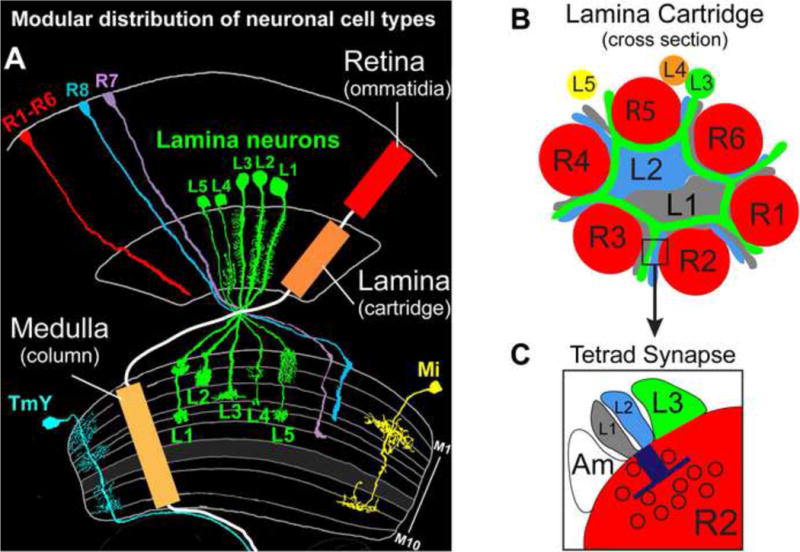
(A) The visual system is organized in a modular fashion with an array of topographically matched units called ommatidia, cartridges, and columns, in the retina, lamina and medulla, respectively. The morphologies of representative cells found within these repeated modules are shown separately. (Adapted from Fischbach and Dittrich 1989.) (B) Schematic of a lamina cartridge shown in cross section. The positions of the neurons within the cartridge are based on an electron microscopy-based reconstruction of a lamina cartridge (Meinertzhagen and O’Neil, 1991; Rivera-Alba et al., 2011). (C) Schematic of a tetrad synapse. The blue “T” represents the T-bar structure characteristic of pre-synaptic specializations in the visual system, and the black circles symbolize synaptic vesicles. Amacrine cells, Am.
A detailed description of the cellular complexity and pattern of synaptic connections between neurons in the lamina and medulla has been determined by a series of studies over the past 100 years, culminating with the recent assembly of a dense connectome of synaptic connections determined by serial electron microscopic reconstruction (Cajal, 1915; Fischbach, 1989; Meinertzhagen and O’Neil, 1991; Rivera-Alba et al., 2011; Takemura et al., 2013; Takemura et al., 2008). In each lamina cartridge the processes of some 19 different neurons are interlinked by thousands of synaptic connections, and each matched medulla column contains the processes of approximately 60 different neurons, which arborize in specific layers and form over 10,000 synaptic connections. As in the vertebrate visual system, neurons in the fly visual system form multiple contact synapses (Dowling JE, 1966; Meinertzhagen and Hanson, 1993). These synapses comprise a single presynaptic release site which juxtaposes multiple postsynaptic elements (i.e. dendritic processes). The best characterized of these are tetrad synapses within the lamina (Meinertzhagen and Hanson, 1993), comprising a pre-synaptic release site on R1–R6 axons and four tightly opposed postsynaptic elements, typically from L1, L2, and L3 neurons within each cartridge, and from an amacrine cell spanning multiple cartridges (see Figure 1) (Meinertzhagen and O’Neil, 1991). L1, L2, and L3 neurons convey information to different layers in medulla columns comprising ON (L1) and OFF (L2) circuits regulating motion detection and what is likely to be a color processing pathway (L3) (Takemura et al., 2013). The developmental strategy underlying the formation of these columnar circuits and the underlying molecular mechanisms remain poorly understood.
Classical genetic studies indicated that the patterning of neuronal processes within the lamina relies on R cell-derived signals (Meyerowitz and Kankel, 1978; Power, 1943). In the late 1990s, Kunes and colleagues identified signals released from R cell growth cones that regulate lamina neuron development. As R cell growth cones enter the developing lamina, they produce Hedgehog, which induces lamina neuron precursor cells to undergo a terminal cell division and express the EGF receptor (Huang and Kunes, 1996; Huang et al., 1998). R2 and R5 growth cones then locally secrete the EGF ligand Spitz (Yogev et al., 2010), which is required for the differentiation of all five lamina neuron subclasses (L1–L5) within each developing cartridge (Huang et al., 1998). Thus, anterograde signals delivered by R cell growth cones to precursors within the developing target field provide a way of matching R1–R6 photoreceptors to the appropriate number of target neurons (Kunes, 2000). These findings raised the possibility that additional anterograde signals might regulate target neuron development.
More recently a third R cell derived signal Jelly belly (Jeb), a secreted protein produced by R cell growth cones, and its receptor anaplastic lymphoma kinase (Alk), a receptor tyrosine kinase expressed in the optic lobe, were shown to be required for the targeting of R cell axons within the lamina and medulla (Bazigou et al., 2007). Disrupting Jeb/Alk signaling reduced the expression of several cell adhesion molecules and it was proposed that the R cell targeting defects were a consequence of these changes rather than abnormal development of neurons within the target region (Bazigou et al., 2007). Here, we demonstrate that anterograde Jeb/Alk signaling is essential for the survival of L3 neurons and that R cell mis-targeting is a consequence of L3 loss. Moreover, we show that Jeb is part of a sequence of axon-derived signals, also involving the secreted protein Netrin, which selectively control assembly of a layer-specific circuit. We consider these findings within the broader context of circuit assembly.
RESULTS
Alk is required cell-autonomously for lamina neuron L3 development
As a step towards understanding intercellular signaling mechanisms regulating the development of L3 neurons, we screened RNAi constructs directed to all genes in the Drosophila genome predicted to encode cell surface and secreted proteins (Pecot et al., 2013). RNAi constructs were expressed in lamina neuron precursor cells and the neurons derived from them, L1–L5, using a specific GAL4 transgene and phenotypes in L3 were assessed using an L3-specific marker expressed by the LexA/LexAop system. Strikingly, no L3 neurons were detected in animals expressing an Alk RNAi construct (Figures 2A, B) which significantly reduced Alk protein in the lamina (Figures S1A, B). To determine if Alk acts in a cell-autonomous fashion, we generated single Alk null mutant lamina neurons (Alk1) (Loren et al., 2003) using mosaic analysis with a repressible cell marker (MARCM) (Lee and Luo, 2001) and assessed L3 morphology in adult animals using an L3-specific marker, different from that used in the RNAi experiments (see Experimental Procedures). In 25 control adult animals, 56 L3 neurons homozygous for a control chromosome were generated and displayed wild type morphology (Figure 2C). By contrast, no L3 neurons homozygous for an Alk null mutation were found in 29 experimental adult animals (Figure 2C). Thus, Alk is required for L3 development in a cell-autonomous fashion.
Figure 2. Jeb/Alk signaling is required for L3 development.
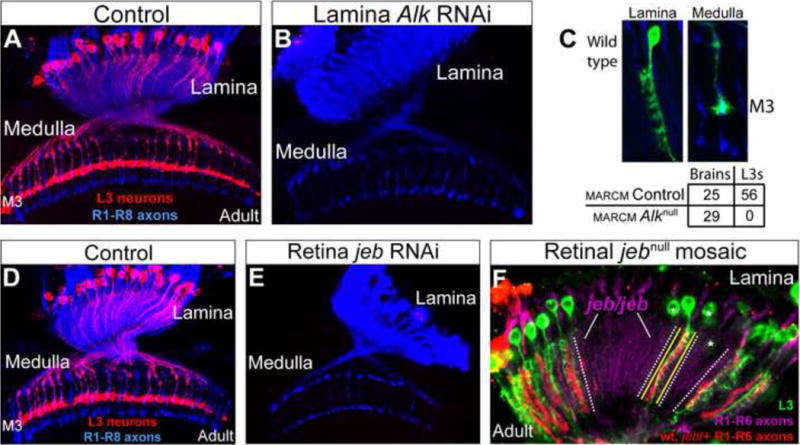
(A–C) Alk is required for L3 development. A. In wild type adult animals the cell bodies and dendritic processes of L3 neurons (red) were visualized in the lamina, and L3 axons terminated within the M3 layer in the medulla. R1–R8 axons (blue) allow visualization of the lamina and medulla. (B) L3 neurons were eliminated by the expression of an Alk RNAi construct in the developing lamina. (C) MARCM experiments demonstrate that Alk regulates L3 development in a cell-autonomous manner. The cell body and dendritic arbor (Top left panel), and the axon terminal (Top right panel) of a single GFP-labeled wild type L3 neuron in an adult brain are shown. Table shows the quantification. (D–F) jeb is required for L3 development. Wild type (i.e. a different “wild type” genotype from (A), as an appropriate control for E). (E) Selective expression of jeb RNAi in the retina eliminated L3 neurons. (F) L3 neurons were missing in regions of the adult lamina innervated by jeb mutant photoreceptor axons in genetically mosaic animals. L3 neurons (green); jeb mutant axons (RFP negative; cartridges innervated by jeb/jeb photoreceptors are demarcated by dotted white lines). Yellow lines demarcate a single cartridge containing a wild type R cell axon(s) associated with an L3 neuron. Neighboring cartridges did not contain L3 neurons (i.e. RFP negative), demonstrating that Jeb acts locally. All R1–R6 axons were labeled with the mAb24B10 antibody (purple). Asterisks indicate portions of L3 neurons (e.g. cell bodies) within RFP positive cartridges in different focal planes. (See also Figure S1)
Jeb is required in R cells for L3 development and acts locally
We next sought to assess whether the Alk ligand Jeb (Englund et al., 2003; Lee et al., 2003) is also required for L3 development. As Jeb is expressed by R cells and not by lamina neurons (Bazigou et al., 2007), we selectively knocked down Jeb protein levels in R cells through the targeted expression of a jeb RNAi construct (Figures S1C, D), and assessed L3 morphology with a specific marker in adult animals. No L3 neurons were seen in laminas innervated by Jeb deficient R cells (Figures 2D, E). L3 neurons were also missing in regions of genetically wild type laminas innervated by jeb mutant R cells generated by mitotic recombination. In these mosaics, axons from wild type and heterozygous R cells expressed RFP, whereas axons from R cells homozygous for a jeb null mutation did not (Figure 2F). Large regions of the lamina and medulla innervated by homozygous mutant R cells were devoid of L3 neurons (Figure 2F). There was a strict correlation between the genotype of the R cells innervating a lamina cartridge and the presence of L3. Indeed, single isolated L3 neurons were invariably associated with RFP expressing (i.e. wild type or heterozygous) R cell axons (Figure 2F). These experiments demonstrate that R cell-derived Jeb acts locally, in a non-autonomous fashion to control L3 development. Moreover, this indicates that, within this context, Jeb interacts with Alk expressed on the surface of L3 neurons.
Jeb/Alk signaling is required for survival of developing L3 neurons
The loss of L3 neurons could be due to a defect in cell fate determination, differentiation, or cell death. When Alk and Jeb levels were knocked down by RNAi constructs, lamina neuron specification was normal, including L3, based on the expression of subclass-specific markers at an early stage of lamina development (Figures S2A–D). Furthermore, the morphology of L3 neurons including their axonal projections into the medulla in early pupa was indistinguishable from wild type (Figure 3A). Therefore, we sought to establish at what stage L3 neurons die in the absence of Alk function. We performed Alk MARCM experiments and assessed L3 morphology at different stages of pupal development, beginning at 20–30h after puparium formation (APF) when L3 neurons have projected axons into the medulla. As lamina neurons are specified in a wave, at this stage there is a gradient of L3 ages across the lamina and medulla (axons). We found that control and homozygous mutant L3 neurons were generated with a similar frequency (Figure 3C). Although most Alk mutant L3 neurons displayed wild type morphology (Figure 3A), about 20% were degenerating, as indicated by fragmentation and abnormal growth cone morphology (Figures 3B and 3C, see below), and these abnormal neurons were preferentially seen in older regions of the lamina and medulla (see below). In MARCM experiments at 40h APF, only one homozygous Alk mutant L3 neuron was seen in 17 experimental animals analyzed, and this cell was degenerating (Figure 3D). By contrast, in three control animals analyzed at this stage, we observed 31 L3 neurons all with normal morphology (Figure 3D). Thus, Alk is required for L3 survival, and L3 neurons lacking Alk function die between 20–40h APF.
Figure 3. Jeb/Alk signaling regulates L3 survival.
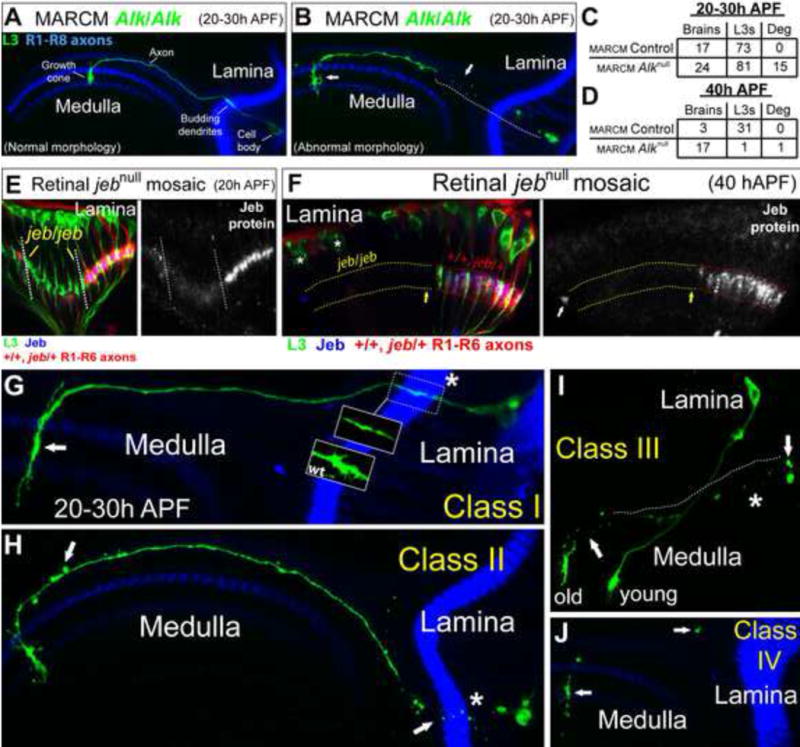
(A–C) Alk MARCM experiments analyzed at 20–30h APF. (A) An Alk/Alk L3 neuron (green) displaying normal morphology. R1–R8 axons (blue) highlight the lamina and medulla neuropils. (B) An Alk/Alk L3 neuron (green) in the process of degenerating. (C) Quantification of Alk MARCM experiments at 20–30h APF. (Deg, degenerating). (D) Quantification of Alk MARCM experiments at 40h APF. (E) jeb genetic mosaic experiments at 20h APF. L3 neurons (green, left panel) were present in all lamina cartridges, even those innervated by jeb/jeb photoreceptors (RFP negative). Patch of cartridges innervated by jeb/jeb photoreceptors demarcated by dotted lines. Jeb protein expression (blue) corresponds with photoreceptor genotype (i.e. jeb/jeb photoreceptors did not express Jeb protein). (Right panel) Jeb protein expression (white) is shown more clearly. (F) As in (E) but at 40h APF. L3 neurons (green, left panel) were lost from cartridges innervated by jeb/jeb photoreceptors (RFP negative; parallel dotted yellow lines). L3 neurons were present in cartridges in which Jeb (blue) was expressed (parallel red dotted lines) by wild type or jeb/+ photoreceptors (RFP positive). L3 neurons were not found within jeb/jeb cartridges (yellow arrow) adjacent to wild type cartridges demonstrating that Jeb acts locally. Asterisks indicate the cell bodies of L3 neurons within wild type cartridges (RFP positive) in a different focal plane. (Right panel) Jeb expression (white) was absent from jeb/jeb cartridges demonstrating that L3 survival correlates with Jeb expression. The white arrow indicates non-specific staining. (G–J) Morphological classification of the progression of L3 degeneration in Alk MARCM experiments at 20–30h APF. Asterisks indicate budding dendritic regions of L3 neurons (green) within the lamina neuropil (blue) (except for Class IV neurons which no longer have even remnants of a lamina spanning region). R1–R8 axons (blue) mark the lamina and medulla neuropils (except in 3I). (G) Class I. The white arrow indicates an abnormal L3 growth cone. The lamina portion of the L3 neuron is outlined with a dotted box. In wild type neurons (wt box) this region comprises filopodia that represent budding dendritic processes. However, in Class I neurons this region was characteristically lacking these filopodia (blown up region within the dotted box). (H) Class II. Degeneration was first observed within budding L3 dendrites (white arrow near asterisk). The white arrow within the medulla highlights blebbing along the axon. (I) Class III. The arrow in the lamina denotes remnants of the cell body associated with the degenerating L3 neuron. The dotted line indicates the path of the axon before it began to degenerate inferred from the line of fluorescent dots remaining. The arrow within the medulla shows where the medulla portion of the axon has started to degenerate. Based on its position within the lamina, the degenerating L3 neuron is clearly older (i.e. more posterior) than an Alk/Alk L3 neuron displaying normal morphology (young) that is also visible in this image. (J) Class IV. Arrows indicate the last few remnants of L3 neurons. Typically, the growth cone region was last to disappear. (See also Figure S2)
To assess whether disrupting jeb in R cells results in L3 death within a similar period, we specifically labeled L3 neurons within laminas innervated by jeb mutant R cells between 20 and 40h APF. At 20h APF, L3 neurons were present within lamina cartridges innervated by jeb mutant R cell axons and were morphologically indistinguishable from L3 neurons within cartridges innervated by wild type or heterozygous R cell axons (Figure 3E). In mosaics analyzed between 20 and 40h APF, degenerating L3 neurons were observed in cartridges innervated by jeb mutant R cell axons (Figure S2E), and at 40h APF, no L3 neurons were seen in these cartridges (Figure 3F). Collectively, these findings demonstrate that disrupting jeb in R cells and Alk in L3 results in L3 death between 20–40h APF. Thus, Jeb/Alk signaling is essential for L3 survival.
Fragmentation of L3 is initiated in budding L3 dendrites
MARCM analyses at 20–30h APF indicated that L3 neurons lacking Alk function degenerated in a characteristic manner. Based on morphology, we divided the degeneration phenotypes observed in these experiments into four classes with increasing severity (Figures 3G–J): 1. Class I neurons lack budding dendrites and form elongated growth cones in the medulla (Figure 3G, arrow; note that wild type L3 dendrites emerge from the proximal region of the axon, as is typical of most neurons in Drosophila); 2. In Class II neurons fragmentation was observed in the lamina, within the budding dendritic region (Figure 3H, arrow and asterisk), and blebbing was seen along the axons projecting into the medulla although they remained intact (Figure 3H, arrow); 3. Class III neurons lacked intact axons, although the cell body and portions of the growth cone remained (Figure 3I); and 4. In Class IV neurons only remnants of the cell remain (Figure 3J, arrows). The growth cone was consistently the last region to degenerate. More severe phenotypes were seen in older L3 neurons (Figure 3I, old and young neurons). In summary, when Jeb/Alk signaling is disrupted, L3 neurons degenerate within a discrete time window (20–40h APF) and in a characteristic fashion. Fragmentation is initiated within the budding dendritic region in the lamina neuropil, followed by degeneration of the cell body and axon. Based on previous descriptions of L3 development, cell death occurs before the formation of mature dendrites and prior to the segregation of L3 growth cones into the M3 layer (Chen et al., 2014; Pecot et al., 2013).
Expression of the anti-apoptotic protein p35 suppresses L3 death
The cell death phenotype seen in jeb and Alk mutants could reflect a direct role for this signaling pathway in providing trophic support for L3, or alternatively this pathway may control cell survival indirectly by regulating a specific step in L3 development, such as dendritogenesis, which may, in turn, be essential for L3 survival. To address this question we blocked cell death, induced by disrupting Alk function in the lamina, through targeted expression of the caspase inhibitor, baculovirus p35 protein (Chen et al., 1996; Grether et al., 1995; Hay et al., 1994; White et al., 1996; Xue and Horvitz, 1995) in lamina neurons. P35 expression suppressed the death of most, if not all, mutant L3 neurons. Furthermore, the morphology of these “rescued” neurons in the adult, including dendritic arbors and axon terminals, was indistinguishable from wild type (Figures 4A–C). The simplest interpretation of these data is that Jeb/Alk signaling directly regulates L3 survival.
Figure 4. Expression of p35 suppresses L3 death and R cell mis-targeting in the absence of Jeb/Alk signaling.
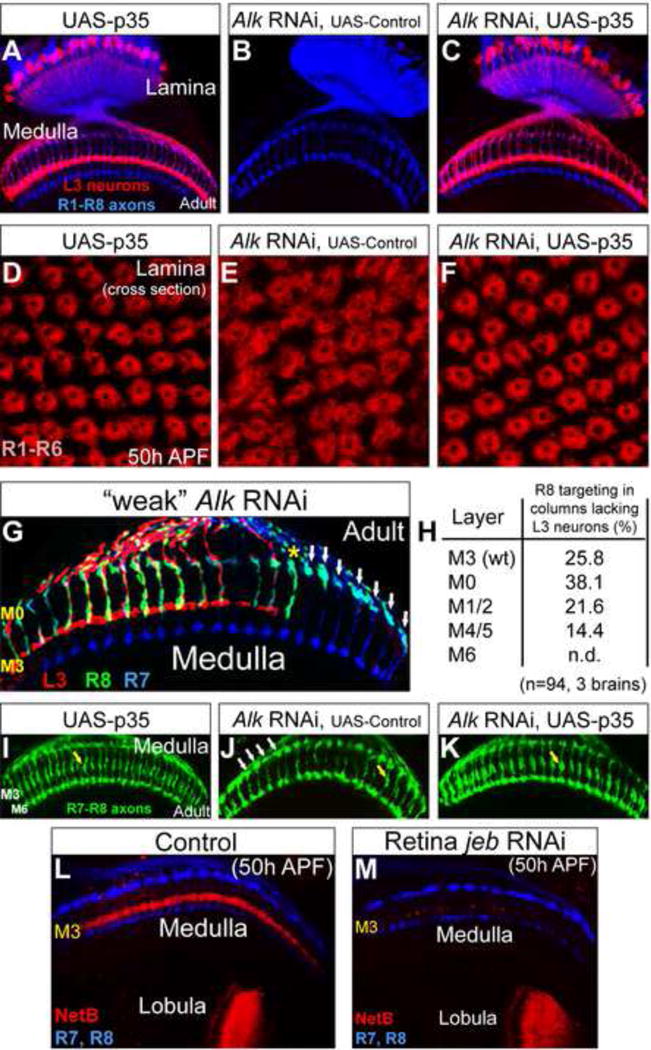
(A) As a control, expression of p35 in lamina neurons did not affect the morphology of L3 neurons (red) in adult animals. R1–R8 axons (blue) highlight the lamina and medulla neuropils. (B) L3 neurons were lost in Alk RNAi experiments in the presence of a gratuitious UAS transgene (CD8-GFP) included here as a control for UAS-P35. (C) The expression of p35 in lamina neurons rescued L3 death when Alk was disrupted in the lamina through RNAi. The morphology of L3 neurons (red) under these conditions was indistinguishable from wild type L3 neurons. (D–F) Cross-section of lamina cartridges at 50h APF. (D) Based on the organization of R cell axons (red, mAb24B10), lamina cartridges appeared qualitatively normal when p35 was expressed in lamina neurons. (E) Disrupting Alk in lamina neurons through RNAi caused mild disorganization of lamina cartridges that was corrected by rescuing L3 death through the expression of p35 in the lamina (F). (G) An RNAi construct specific for Alk was weakly expressed in lamina neurons (see Experimental Procedures) to remove some but not all L3 neurons. The targeting of R8 axons (green) to the M3 layer correlated with the presence of L3 neurons (red) within the same or directly adjacent columns (yellow asterisk), indicating that L3 acts locally to control R8 layer specificity. White arrows indicate mis-targeting R8 axons in columns lacking L3 neurons that were not adjacent to L3-containing columns. The layer specificity of R7 neurons (blue) was not affected by the loss of L3 neurons. (H) Quantitation of (G). Mis-targeting to M6 was not determined (n.d.) as a small subset of R7 neurons (which terminate in M6) were labeled with the R8 marker (see Experimental Procedures). The targeting of R8 neurons within columns not containing L3 neurons that were also adjacent to L3-lacking columns (n=94, 3 brains) was assessed. (I–K) Images are from (A–C). R7 and R8 axons are shown more clearly in green and in the absence of L3 labeling. (I) R8 targeting to the M3 layer was normal (yellow arrow) in UAS-p35 control animals. (J) In the absence of L3 neurons, induced by disrupting Alk in the lamina, R8 axons mis-targeted and were observed near the top of the medulla (white arrows). The yellow arrow indicates an R8 axon targeting correctly to M3. (K) R8 targeting was restored (yellow arrow) in the absence of Alk function when L3 death was rescued by expressing p35 in lamina neurons. (L) In wild type animals at 50h APF, Netrin (red) was concentrated within the developing M3 layer and the lobula. R7 and R8 axons (blue) outline the medulla neuropil. (M) Netrin expression within the M3 layer but not the lobula was strongly reduced when L3 neurons were lost due to the disruption of jeb in the retina through RNAi. Some residual Netrin was seen indicating other neurons may weakly express Netrin within the medulla or remnants of L3 axons may remain. (see also Figure S3)
Jeb/Alk signaling indirectly regulates R cell targeting by controlling L3 survival
In a previous study, Salecker and colleagues demonstrated that loss of function mutations in jeb and Alk disrupted the targeting of R1–R6 and R8 axons within the lamina and medulla (Bazigou et al., 2007). They argued that this was due to a requirement for Jeb and Alk to regulate expression of cell adhesion molecules in target neurons, thereby shaping “their environment for target recognition”. In light of our findings, we sought to assess whether the R cell targeting defects result solely from the loss of L3 neurons. If so, rescuing L3 death in the absence of Jeb/Alk signaling would abrogate R cell mis-targeting. To test this hypothesis, we expressed p35 in lamina neurons lacking Alk function due to RNAi and assessed the organization of R1–R6 axons in lamina cartridges and the targeting of R8 axons in the medulla.
R1–R6 Targeting
Jeb and Alk mutations cause defects in lamina cartridge organization that result from the abnormal targeting of R1–R6 axons (Bazigou et al., 2007). These defects can be readily appreciated in top down views of the cartridge array. P35 expression suppressed defects in cartridge assembly caused by the lack of Alk function, leading to a cartridge array indistinguishable from wild type (Figures 4D–F). Thus, R cell targeting defects in the lamina are an indirect effect of the loss of L3. L3 neurons may regulate cartridge assembly by directly interacting with R1–R6 growth cones, by interacting with other lamina neurons within the cartridge, or by participating in both types of interactions.
R8 Targeting
Both R8 and L3 axons target to the M3 layer of the medulla. Previous studies established that L3 neurons target to M3 prior to R8 (Nern et al., 2008; Pecot et al., 2013; Timofeev et al., 2012). R8 initially terminates in a temporary region at the outer surface of the medulla and extends to M3 between 45 and 55 APF (Ting et al., 2005). When jeb or Alk is disrupted, R8 axons fail to terminate within M3 with most remaining at the temporary layer (Bazigou et al., 2007). To determine if R8 mis-targeting under these conditions correlated with the loss of L3 neurons within medulla columns, we used RNAi under suboptimal conditions to generate an optic lobe in which only some L3 neurons were missing (see Experimental Procedures). In these optic lobes, loss of L3 occurred in a patchy fashion and the targeting of R8 axons to the M3 layer correlated with the presence of L3 axons within the same or an immediately neighboring column (Figure 4G, yellow asterisk). When L3 neurons were not present within the same and neighboring columns (Figure 4G, white arrows), about 75% of the R8 axons failed to terminate within M3, with 80% of these terminating in more superficial layers (e.g. M0–M2) and 20% terminating in deeper layers (e.g. M4–M5) (Figure 4H). Rescuing L3 death through p35 expression suppressed this phenotype (Figures 4I–K). This supports the view that L3 neurons are required for R8 targeting.
How does L3 regulate R8 targeting?
Salecker and colleagues reported that Netrin is required for R8 targeting to the M3 layer, provided evidence that L3 neurons express Netrin, and demonstrated that expression of a Netrin transgene in L3 neurons is sufficient to rescue R8 mis-targeting in animals lacking Netrin (Timofeev et al., 2012). As shown in the previous section, we established that L3 neurons are required for R8 targeting (Figures 4G–K). Together, these findings led us to predict that the loss of L3 would result in a marked decrease in Netrin expression within the M3 layer. Furthermore, since Jeb/Alk signaling indirectly regulates R8 targeting through control of L3 survival, we anticipated that Netrin expression would be restored by preventing L3 death through p35 expression. Indeed, we found that netrin expression in M3 correlated with the presence of L3 terminals within this layer (Figures 4L–M, and S3). Thus, together with findings from Salecker and co-workers, these results are consistent with Netrin expression in L3 as both necessary and sufficient for R8 targeting.
In summary, we show that Jeb/Alk signaling controls R cell targeting, only indirectly, through the regulation of L3 survival.
Jeb expression in R1–R6 growth cones in the lamina is sufficient for L3 survival
Two lines of evidence support the view that Jeb/Alk signaling within the lamina neuropil acts locally to provide trophic support for L3. First, the fragmentation of L3 neurons lacking Alk function is initially observed in the lamina neuropil within budding L3 dendrites (see Figure 3H), and then spreads to the cell body and axon. Second, Jeb and Alk proteins are localized in a complementary fashion within this region (Figure 5A), with Jeb expressed in R1–R6 growth cones (Figure 5B) and Alk localized to budding L3 dendrites (Figure 5C). The more restricted localization of Jeb than the R cell specific membrane marker used in Figure 5B may reflect localization of Jeb to only restricted regions of the growth cone or alternatively selective expression in only a subset of them. To test whether Jeb expression within the developing lamina neuropil is sufficient for L3 survival, we selectively removed R cell-derived Jeb protein from the medulla. This was done using a genetic background in which expression of the senseless transcription factor (Nolo et al., 2000) is selectively removed from the eye using a senseless transgene, lacking an eye-specific enhancer (Pepple et al., 2008), to rescue lethality caused by a senseless null mutation. In these flies, R8 neurons are inappropriately specified as R2/R5-like cells (Frankfort et al., 2001) and their growth cones terminate within the lamina. As R7 neurons rely on an inductive signal from R8 (Reinke and Zipursky, 1988), they do not form. Although R1–R6 neurons are reduced in number (Frankfort et al., 2001) and their axons are disorganized, they terminate within the lamina neuropil (Figures 5D and 5E). As a consequence, these growth cones are the only source of R cell-derived Jeb (Figures 5D′ and 5E′). Despite these abnormalities, many L3 neurons survive (Figures 5F and 5G). Interestingly, L3 neurons did not project axons into the medulla under these conditions, suggesting that L3 axons may extend along the surface of R7 and R8 axons from the lamina into the medulla. In summary, these data support a model in which Jeb, released from R1–R6 growth cones in the lamina, binds to Alk on budding L3 dendrites to promote survival.
Figure 5. L3 survival relies on Jeb/Alk signaling in the lamina.
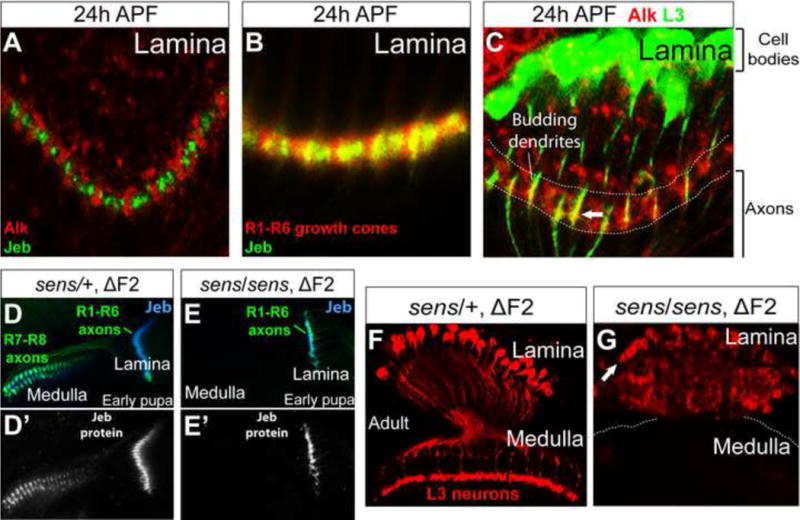
(A) Endogenous Alk (red) and Jeb (green) were expressed in complementary patterns within the lamina neuropil at 24h APF. (B) Endogenous Jeb (green) colocalized with R1-R6 growth cones (red) within the lamina neuropil at 24h APF. (C) Endogenous Alk (red) was localized on budding L3 dendrites (green) within the lamina neuropil at 24h APF (white arrow). Alk was also localized on the processes of other neurons within the neuropil. (D) In early pupae heterozygous for senseless (sensE2/+) (Nolo et al., 2000) carrying a sens transgene lacking the F2 fragment (ΔF2) required for expression in the eye, R1–R6 axons (green) projected into the lamina and R7 and R8 neurons projected axons (green) into the medulla. Jeb expression (blue) was observed in the lamina and medulla. (D’) Jeb expression (white) more clearly shown than in (D) was found in both the lamina and medulla neuropils associated with R cell growth cones. (E) In early sens/sens mutant pupae, R7 and R8 neurons were not specified and R1–R6 neurons, the only remaining R cell source of Jeb (blue), projected axons into the lamina. (E’) Jeb expression (white) is more clearly visualized. (F) L3 neurons (red) were morphologically normal in adult animals heterozygous for sensE2 (same genotype as (D) and (D’)). (G) In adult animals lacking Jeb expression in the medulla (same genotype as (E) and (E’)) L3 neurons are present (white arrow indicates cell bodies), although the lamina was disorganized (see text).
Jeb/Alk signaling selectively regulates L3 development
As each cartridge comprises five lamina monopolar neurons (L1–L5), we sought to determine whether Jeb/Alk signaling was selective for L3 or whether it also provides trophic support for the remaining neurons. The survival of L1 and L2 neurons was not affected by the knockdown of jeb mRNA in the retina (Figures 6A–D), although some subtle differences in the morphology of some L2 axon terminals were observed. The knockdown of Alk mRNA in the lamina resulted in a mild decrease in the number of L4 neurons and some morphological defects (Figures 6E, F). As L3 and L4 axons occupy neighboring positions within lamina cartridges (see Figure 1B) (Meinertzhagen and O’Neil, 1991; Rivera-Alba et al., 2011), however, these effects may result non-autonomously from the loss of neighboring L3 neurons. We have not been able to distinguish between an autonomous or non-autonomous role for Alk in L4 using MARCM experiments due to technical limitations. And finally, to determine if Alk is required in L5 neurons, we performed MARCM analysis using an L5-specific GAL4 marker to identify single homozygous Alk null mutant L5 neurons in adults. A similar number of control and homozygous Alk mutant L5 neurons were generated in these experiments, and the Alk mutant L5 neurons were morphologically indistinguishable from wild type (Figure 6G).
Figure 6. Jeb/Alk signaling does not control the survival of other lamina neurons.
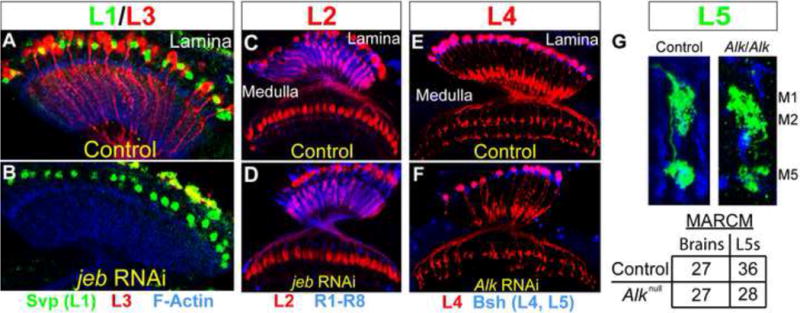
(A) In wild type adult animals, L1 neurons (green nuclei) were visualized in the lamina via anti-Svp staining (Z.C. and C.D., unpublished). L3 neurons are shown in red and F-Actin (blue) highlights the lamina. (B) Selectively disrupting jeb in the retina through RNAi, resulted in the loss of L3 neurons, but not L1 neurons. (C) L2 neurons (red) in adult wild type animals. R1–R8 axons (blue) mark the lamina and medulla neuropils. (D) Disrupting jeb function in the retina through RNAi did not affect L2 survival, although the morphology of some L2 axon terminals was slightly abnormal. (E) L4 neurons (red) in adult wild type animals. L4 and L5 nuclei (blue) were labeled with anti-Bsh. (F) When Alk function was disrupted in lamina neurons through RNAi, some L4 neurons were lost although the vast majority survive, and moderate morphological defects were seen (see text). (G) Alk MARCM experiments in L5 neurons. In adult animals, morphologically normal L5 neurons (green) homozygous for a control chromosome or an Alk null mutation were observed with similar frequencies. L5 neurons project axon terminals into the M5 layer and also elaborate branches that span the M1 and M2 layers.
In conclusion, Jeb acts as an anterograde signal produced by R1–R6 growth cones to specifically regulate, through the Alk receptor, the survival of L3 neurons.
DISCUSSION
Here we demonstrate that Jeb/Alk signaling regulates the survival of L3 neurons, one of several post-synaptic targets of R1–R6 neurons. Jeb is expressed in R1–R6 growth cones and acts at short-range, prior to synapse formation (see below), through the Alk receptor tyrosine kinase localized on budding L3 dendrites within the lamina neuropil. Jeb/Alk signaling is highly selective, as the survival of other R1–R6 postsynaptic targets (i.e. L1 and L2) is not affected when signaling is disrupted. We also show that, at a later stage of development, L3 growth cones produce Netrin within the medulla, which is required for the targeting of R8 growth cones to the M3 layer. We speculate that a cascade of growth cone-derived signals acting across different brain regions provides a general strategy for the assembly of neural circuits.
Anterograde Jeb/Alk signaling controls the survival of a subset of synaptic targets
In many regions of the developing nervous system neurons are produced in excess, and significant cell death occurs after axons innervate their targets (Oppenheim, 1991). In vertebrates, it is well established that target-derived neurotrophins, such as nerve growth factor, regulate neuronal numbers (Levi-Montalcini, 1987). These factors are produced by target neurons in limiting amounts, and locally promote survival in a retrograde manner through receptors localized on axon terminals, providing a mechanism for matching the number of axons to targets (Levi-Montalcini, 1987; Lewin and Barde, 1996). In recent years, diverse classes of molecules have been shown to control neuronal survival during development (de Araujo et al., 2009; Vanderhaeghen and Cheng, 2010). Anterograde sources of trophic factors may also regulate survival, as denervation has been shown to induce excessive target neuron cell death (Linden, 1994). Indeed, several signals, including BDNF, are transported, in some contexts, in an anterograde manner within axons (Altar et al., 1997; Caleo and Cenni, 2004; Caleo et al., 2000). In addition, the overexpression of BDNF in afferents can rescue cell death within the target field (Alonso-Vanegas et al., 1999; Caleo et al., 2000; Spalding et al., 2002), and the disruption of BDNF through function blocking antibodies has been reported to decrease the number of target neurons within the rat superior colliculus (Caleo et al., 2000). As BDNF may be produced by both axons and cells within the superior colliculus, it remains unclear whether endogenous axon derived BDNF, and thus anterograde signaling, is required to regulate neuron survival.
Although a role for target-derived retrograde trophic factors in vertebrate neural development was established many decades ago, trophic factors have only recently been shown to regulate neuronal development in Drosophila. Three Drosophila proteins Neurotrophins 1 and 2 (DNT1 and 2) and Spatzle (Spz) are distantly related to vertebrate neurotrophins (Mizuguchi et al., 1998; Zhu et al., 2008), and it has been shown that, like their vertebrate counterparts, they function as target-derived retrograde survival signals (Zhu et al., 2008). Unlike their vertebrate homologs, however, which act through receptor tyrosine kinases, fly neurotrophins promote cell survival through Toll-like receptors (McIlroy et al., 2013; Morisato and Anderson, 1994).
Although Jeb bears no significant homology to fly or vertebrate neurotrophins, Jeb acts through a receptor tyrosine kinase, Alk, which is distantly related to vertebrate neurotrophin receptors or Trks (Englund et al., 2003; Iwahara et al., 1997; Loren et al., 2003; Morris et al., 1997). Alk was originally identified as part of a fusion protein associated with large cell anaplastic lymphoma (Morris et al., 1994). Its role in mammals remains poorly understood. Drosophila Alk was initially found to regulate visceral mesoderm development through interaction with Jeb (Englund et al., 2003; Lee et al., 2003; Loren et al., 2003; Loren et al., 2001), and subsequently, Jeb/Alk signaling has been shown to regulate diverse cellular processes. Recent studies in vertebrates and Drosophila demonstrated that disrupting Alk function causes a decrease in the number of neurons (Cheng et al., 2011; Weiss et al., 2012; Yao et al., 2013). While in the vertebrate studies, Alk’s mechanism of action was not established, in Drosophila Alk was shown to antagonize pathways that restrict neurogenesis under conditions of nutrient deprivation (Cheng et al., 2011). Whether Jeb and Alk regulate neuronal survival in contexts outside of L3 development is not known, although Alk is widely expressed in the developing visual system (Bazigou et al., 2007) and Jeb is expressed by several populations of neurons, in addition to photoreceptors (not shown).
The cellular specificity of the Jeb/Alk requirement is particularly surprising. Indeed, at all R1–R6 synapses containing L3 postsynaptic elements, L1 and L2 neurons each contribute a single postsynaptic element juxtaposing the same presynaptic site on R cell axons (Meinertzhagen and O’Neil, 1991). In the absence of Jeb/Alk signaling, however, only L3 neurons die. The mechanisms that underlie this selectivity are not known. Alk is broadly expressed in the lamina (see Figure S1A) suggesting specificity may be controlled at the level of downstream signaling, or that other trophic signals act redundantly with Jeb to control L1 and L2 survival. Collectively, the findings reported here demonstrate that anterograde Jeb/Alk signaling acts selectively to control L3 survival, providing, to our knowledge, the first evidence that anterograde signaling directly regulates target neuron survival in vivo.
Jeb/Alk signaling is required for L3 survival during a discrete step in circuit assembly
Several lines of evidence indicate that signaling between Jeb, expressed by R1–R6 growth cones, and Alk, localized to budding L3 dendrites, controls L3 survival between 20–40h APF. First, Alk mutant L3 neurons, or wild-type L3 neurons innervated by jeb mutant R1–R6 axons, die between 20–40h APF (see Figure 3). Second, R cell populations containing only R1–R6 neurons, are sufficient for L3 survival (see Figures 5D–G). Third, Alk and Jeb are expressed in a complementary fashion at the appropriate time on budding L3 dendrites and R1–R6 growth cones, respectively (see Figures 5A–C). And finally, L3 degeneration begins within budding L3 dendrites juxtaposed to R1–R6 growth cones (see Figure 3H). The temporal requirement for Alk/Jeb signaling corresponds to a critical and fascinating phase of lamina circuit assembly.
R1–R6 growth cones form connections with lamina neurons in three discrete steps. First, R1–R6 growth cones from the same ommatidium associate with a single cartridge of differentiating lamina neurons. Second, through a highly stereotyped re-assortment process occurring between 24–38h APF (Clandinin and Zipursky, 2000), these six growth cones diverge from one another and project locally to 6 different developing cartridges (Braitenberg, 1967; Meinertzhagen and Hanson, 1993; Trujillo-Cenoz, 1965). As a consequence of this rearrangement, the R1–R6 cells which “see” the same point in space form connections with L1, L2 and L3 neurons within the same cartridge. And third, R1–R6 then commence synapse formation at 45 hours APF and this process continues until eclosion (~96 h) (Chen et al., 2014). Thus, L3 death in jeb and Alk mutants occurs prior to synapse formation, during the process of R1–R6 growth cone rearrangement. The suppression of L3 death by expression of the caspase inhibitor p35 argues that during normal development Jeb/Alk signaling acts to inhibit caspase activity. Which caspases contribute to L3 death, and whether caspases antagonize other cellular processes necessary for wiring is not known. Regardless of how Jeb/Alk signaling functions at the molecular level, it acts to ensure that visual input from R1–R6 neurons is transmitted to the L3 pathway.
A cascade of intercellular interactions regulates circuit assembly in the fly visual system
Our findings and the work of others (Kunes, 2000; Salecker et al., 1998) suggest a logic underlying neural circuit assembly within the Drosophila visual system. The retina, lamina and medulla are distinct yet interconnected regions comprising columnar modules (i.e. ommatidia, cartridges and columns, respectively) that are matched topographically between each region. Within each module, intrinsic mechanisms and intercellular interactions control cell fate determination. For instance, R8 neurons provide a discrete locally acting signal to induce R7 development in the developing retina (Reinke and Zipursky, 1988), while in the medulla Notch/Delta interactions between daughter cells generated from the same ganglion mother cell promote acquisition of distinct cell fates (Li et al., 2013). Superimposed upon these interactions are axon-derived signals that coordinate development between matched modules from different regions (Figure 7). Together, these mechanisms organize the assembly of columnar units in multiple regions (i.e. super-columns), each processing visual information captured from a discrete region of the visual field. Indeed, the modular assembly of these super-columns spanning different regions of the visual system reflects the function of these circuits in the parallel processing of visual information.
Figure 7. Signals released by growth cones coordinate development between different regions of the visual system.
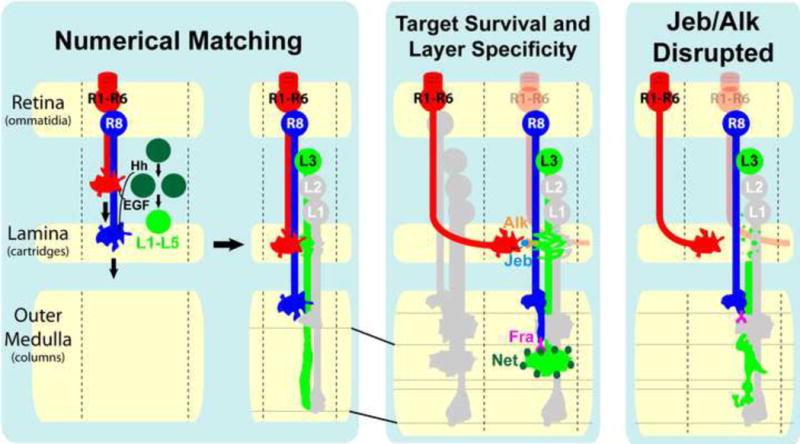
(Left panel, left side) R cell-derived Hedgehog (Hh) and Spitz (EGF) regulate the proliferation of lamina precursor cells (dark green circles) and the differentiation of lamina neurons (light green circle, L1–L5), respectively, thereby matching R cells within each ommatidium to the appropriate number of target neurons within each developing lamina cartridge. For simplicity, we have not shown R7 neurons. While it is clear that R2 and R5 produce EGF, it is not clear whether R1–R6 or R8 produce Hh. (Left panel, right side) After they are specified lamina neurons project axons into the outer medulla. L3 growth cones are initially spear-like and span several developing layers within the outer medulla. (Center panel) Jeb and Netrin act sequentially to couple target neuron survival in the lamina with layer specificity in the medulla in a cell type-specific manner. As the growth cones of R1–R6 neurons that “see” the same point in visual space converge upon the same set of target neurons within each lamina cartridge (see Discussion), they release Jeb which interacts with Alk, localized on budding dendrites in the lamina, to control the survival of L3, but not L1 or L2 neurons. While it is clear that, in the absence of Jeb/Alk signaling, L3 neurons die during growth cone rearrangement it remains possible that Jeb released from R cell growth cones prior to this step may contribute to L3 survival. Subsequently, L3 growth cones, which have segregated into the developing M3 layer, secrete Netrin (Net, dark circles), which becomes concentrated within the layer. Interaction between Netrin within M3 and Frazzled (Fra) expressed in R8 growth cones is required for the targeting of R8 axons to the M3 layer. (Right panel) When Jeb/Alk signaling is disrupted, L3 neurons degenerate in a characteristic manner with fragmentation initiated within the budding dendritic region, and then spreading to the axon and cell body. L3 neurons die before secreting Netrin, which in turn causes defects in the layer-specific targeting of R8 axons.
R cell growth cones produce signals that regulate diverse cellular processes in the developing lamina. Hedgehog drives lamina neuronal precursors through their final division (Huang and Kunes, 1996), cell adhesion proteins promote the association of columns of lamina neurons with R cell axon fascicles (Sugie et al., 2010), EGF induces lamina neuron differentiation (Huang et al., 1998), a yet to be identified signal regulates the development of lamina glia (Suh et al., 2002) and Jeb selectively regulates L3 survival. Thus, axon-derived signals act at multiple levels and in a cell type-specific manner to regulate target development.
Axon-derived signals also coordinate circuit assembly across topographically matched modules. Within medulla columns, L3 growth cones produce Netrin in the M3 layer, which controls the targeting of R8 growth cones to M3. Importantly, Netrin production by L3 occurs after Jeb, released from R1–R6 cells in topographically corresponding lamina cartridges, promotes L3 survival. Thus, Netrin indirectly relies upon prior Jeb signaling. As the L3 and R8 axon terminals within each medulla column transmit information captured from the same point in space to the same layer (M3) and share several postsynaptic targets (Gao et al., 2008; Takemura et al., 2013), the developmental mechanisms giving rise to this circuit may reflect functional relationships between these neurons. Thus, signals produced by axons coordinate assembly of circuits between different brain regions.
We envision that intercellular signaling cascades, analogous to what we have described here, organize other circuit modules in the fly visual system (e.g. ON (L1) and OFF (L2) circuits) (Takemura et al., 2013) comprising different cell types. As many regions of the vertebrate nervous system, including the neocortex, spinal chord, and retina also are arranged in a hierarchically repetitive fashion, this raises the intriguing possibility that similar strategies may coordinate the development of these structures.
EXPERIMENTAL PROCEDURES
General
Unless otherwise indicated flies were grown at 25°C and newly eclosed flies were sacrificed. For developmental analyses, white prepupae were collected and incubated for the indicated number of hours at 25°C. Fly brains were fixed with PLP (4% paraformaldehyde, 75 mM lysine, 37 mM sodium phosphate buffer pH 7.4) for 25′ at RT. Samples were incubated with primary and secondary antibodies for 2 days each at 4°C. Brains were mounted in Slow Fade Gold anti-Fade Reagent (Molecular Probes).
Reagents
Antibodies: Primary antibodies. Chicken pAb α-GFP (abcam; 1:1000); mAb24B10 (Van Vactor et al., 1988) (1:20); rabbit pAb α-DsRed (Clontech; 1:200); guinea pig anti-Jeb (Englund et al., 2003) (1:1000); rabbit anti-Alk (Loren et al., 2003) (1:1000); rabbit anti-NetrinB1 and rabbit anti-NetrinB3 (kindly provided by Dr. Benjamin Altenhein, ICGEB Trieste) (Timofeev et al., 2012) (1:50); guinea pig anti-Bsh (Hasegawa et al., 2011) (1:200); mAb anti-Svp (Kanai et al., 2005) (1:50). Secondary antibodies: Goat α-rabbit 568 (Molecular Probes; 1:500); goat α-mouse 647 (Molecular Probes; 1:500); goat α-chicken 488 (Molecular Probes; 1:500); goat anti-guinea pig 647 (Molecular Probes 1:500). Phalloidin-647 (protein, Molecular Probes; 1:100).
Lamina neuron GAL4 and LexA drivers
R9B08-GAL4: Drives GAL4 expression in lamina neurons L1–L5 and their precursors (Pecot et al., 2013) and was identified by screening a database of GAL4-line expression patterns (Jenett et al., 2012; Pfeiffer et al., 2008).
9-9-GAL4: Was previously shown to specifically drive expression in L3 neurons in the lamina throughout development (Nern et al., 2008; Pecot et al., 2013).
6-60-GAL4: Was previously shown to specifically drive expression in L5 neurons in the lamina throughout development (Nern et al., 2008; Pecot et al., 2013).
R22E09-nlsLexAGADfl: Drives LexA expression specifically in L3 neurons in adult animals (Pecot et al., 2013) and was constructed as previously described (Pfeiffer et al., 2010).
R31C06- nlsLexAGADfl: Drives LexA expression specifically in L4 neurons in adult animals and was generated as described previously (Pfeiffer et al., 2010).
R39D12-nlsLexAGADfl: Drives LexA expression in L2 neurons in adults and was generated as previously described (Pfeiffer et al., 2010).
Alk RNAi experiments
Experimental genotype (L3) Figures 2A–B, S1A–B, and S2D:
UAS-Dcr2/+(Y) ; R22E09-nlsLexAGADfl, LexAop-myrtdTOM/+ ; R9B08-GAL4/UAS-Alk RNAi (Control animals did not have the Alk RNAi transgene)
Experimental genotype (UAS-p35 rescue) Figures 4A–F, I–K, and S3:
UAS-Dcr2/+(Y) ; R22E09-nlsLexAGADfl, LexAop-myrtdTOM/UAS-p35(UAS-CD8-GFP) ; R9B08-GAL4/UAS-Alk RNAi (Control UAS-P35 only animals did not have the Alk RNAi transgene)
Experimental Genotype (L4) Figures 6E and 6F:
UAS-Dcr2/+(Y) ; LexAop-myrtdTOM/+ ; R9B08-GAL4, R31C06-nlsLexAGADfl/UAS-Alk RNAi
Weak Alk RNAi experiments
Experimental genotype Figures 4G and 4H:
sens-FLPg5d/+; GMR-GAL4, UAS-FRT-STOP-FRT-UtrnCHGFP/R22E09-nlsLexAGADfl, LexAop-myrtdTOM ; R9B08-GAL4/UAS-AlkRNAi
sens-FLP drives expression of FLP recombinase in R8 neurons. UAS-UtrnCHGFP, which comprises the calponin homology domain of Utrophin fused to GFP, labels F-Actin.
Jeb RNAi experiments
Experimental genotype (L3) Figures 2D–E, 4L–M, 6A–B, S1, and S2C:
UAS-Dcr2/+(Y) ; R22E09-nlsLexAGADfl, LexAop-myrtdTOM/UAS-jeb RNAi (VDRC v103047) ; GMR-GAL4/+
Experimental genotype (L2) Figures 6C and 6D:
UAS-Dcr2/+(Y) ; LexAop-myrtdTOM/UAS-jeb RNAi ; GMR-GAL4, R39D12-nlsLexAGADfl (L2)/+
MARCM experiments
Experimental genotype (L3) Figures 2C, 3A–D, and 3G–J:
R27G05-FLP1(attP18)/+(Y) ; FRT42D, Tub-GAL80/FRT42D, Alk1 ; 9-9-GAL4, UAS-CD8-GFP/UAS-myrGFP
R27G05 is expressed in the developing lamina (Pecot et al., 2013; Riddiford et al., 2010).
Experimental genotype (L5) Figure 6G:
R27G05-FLP1(attP18)/+(Y) ; FRT42D, Tub-GAL80/FRT42D, Alk1 ; 6-60-GAL4, UAS-CD8-GFP/UAS-myrGFP
Jeb genetic mosaic experiments
Experimental genotype Figures 2F, 3E–F, and S2E:
Ey3.5 FLPg5d/+(Y) ; FRT42D, GMR-RFP/FRT42D, jebl(2)SH0422 ; 9-9-GAL4, UAS-CD8-GFP/+
Co-labeling of L3 neurons axons and endogenous Alk
Figure 5C: 9-9-GAL4 expressed UAS-CD8GFP in L3 neurons and Alk protein was visualized though antibody staining.
For a more detailed description of experiments see Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We would like to thank; Iris Salecker for providing jeb and Alk null lines, Graeme Mardon for providing sens ΔF2 flies, Ruth Palmer for providing Jeb and Alk antibodies, and Benjamin Altenhein for providing Netrin antibodies. For critical reading of the manuscript, we would like to thank Sean Millard, Iris Salecker, and members of the S.L.Z. laboratory. We would especially like to thank Barret Pfeiffer, Aljoscha Nern, and Gerald Rubin for generating, annotating and sharing lamina neuron-specific drivers, without which many of our experiments would not have been feasible. M.Y.P. was supported by the Jane Coffin Childs Medical Memorial Research Fund and S.L.Z. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alonso-Vanegas MA, Fawcett JP, Causing CG, Miller FD, Sadikot AF. Characterization of dopaminergic midbrain neurons in a DBH:BDNF transgenic mouse. The Journal of comparative neurology. 1999;413:449–462. doi: 10.1002/(sici)1096-9861(19991025)413:3<449::aid-cne7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Bate CM. Pioneer neurones in an insect embryo. Nature. 1976;260:54–56. doi: 10.1038/260054a0. [DOI] [PubMed] [Google Scholar]
- Bazigou E, Apitz H, Johansson J, Loren CE, Hirst EM, Chen PL, Palmer RH, Salecker I. Anterograde Jelly belly and Alk receptor tyrosine kinase signaling mediates retinal axon targeting in Drosophila. Cell. 2007;128:961–975. doi: 10.1016/j.cell.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Braitenberg V. Patterns of projection in the visual system of the fly. I. Retina-lamina projections. Experimental brain research Experimentelle Hirnforschung Experimentation cerebrale. 1967;3:271–298. doi: 10.1007/BF00235589. [DOI] [PubMed] [Google Scholar]
- Burgess RW, Nguyen QT, Son YJ, Lichtman JW, Sanes JR. Alternatively spliced isoforms of nerve- and muscle-derived agrin: their roles at the neuromuscular junction. Neuron. 1999;23:33–44. doi: 10.1016/s0896-6273(00)80751-5. [DOI] [PubMed] [Google Scholar]
- Cajal SRy. Contribución al conocimiento de los centros nerviosos de los insectos. Parte I, Retina y centros ópticos (Con la colaboración de don D. Sánchez) Trab del Lab de Inv biol. 1915;XIII [Google Scholar]
- Caleo M, Cenni MC. Anterograde transport of neurotrophic factors: possible therapeutic implications. Molecular neurobiology. 2004;29:179–196. doi: 10.1385/mn:29:2:179. [DOI] [PubMed] [Google Scholar]
- Caleo M, Menna E, Chierzi S, Cenni MC, Maffei L. Brain-derived neurotrophic factor is an anterograde survival factor in the rat visual system. Current biology : CB. 2000;10:1155–1161. doi: 10.1016/s0960-9822(00)00713-2. [DOI] [PubMed] [Google Scholar]
- Chen P, Nordstrom W, Gish B, Abrams JM. grim, a novel cell death gene in Drosophila. Genes & development. 1996;10:1773–1782. doi: 10.1101/gad.10.14.1773. [DOI] [PubMed] [Google Scholar]
- Chen Y, Akin O, Nern A, Tsui CY, Pecot MY, Zipursky SL. Cell-type-Specific Labeling of Synapses In Vivo through Synaptic Tagging with Recombination. Neuron. 2014;81:280–293. doi: 10.1016/j.neuron.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HJ, Nakamoto M, Bergemann AD, Flanagan JG. Complementary gradients in expression and binding of ELF-1 and Mek4 in development of the topographic retinotectal projection map. Cell. 1995;82:371–381. doi: 10.1016/0092-8674(95)90426-3. [DOI] [PubMed] [Google Scholar]
- Cheng LY, Bailey AP, Leevers SJ, Ragan TJ, Driscoll PC, Gould AP. Anaplastic lymphoma kinase spares organ growth during nutrient restriction in Drosophila. Cell. 2011;146:435–447. doi: 10.1016/j.cell.2011.06.040. [DOI] [PubMed] [Google Scholar]
- Clandinin TR, Zipursky SL. Afferent growth cone interactions control synaptic specificity in the Drosophila visual system. Neuron. 2000;28:427–436. doi: 10.1016/s0896-6273(00)00122-7. [DOI] [PubMed] [Google Scholar]
- de Araujo EG, da Silva GM, Dos Santos AA. Neuronal cell survival: the role of interleukins. Ann N Y Acad Sci. 2009;1153:57–64. doi: 10.1111/j.1749-6632.2008.03974.x. [DOI] [PubMed] [Google Scholar]
- Dowling JEBB. Organization of the primate retina: electron microscopy. Proc R Soc Lond B Biol Sci. 1966;166:80–111. doi: 10.1098/rspb.1966.0086. [DOI] [PubMed] [Google Scholar]
- Englund C, Loren CE, Grabbe C, Varshney GK, Deleuil F, Hallberg B, Palmer RH. Jeb signals through the Alk receptor tyrosine kinase to drive visceral muscle fusion. Nature. 2003;425:512–516. doi: 10.1038/nature01950. [DOI] [PubMed] [Google Scholar]
- Fischbach KF, Dittrich APM. The optic lobe of Drosophila melanogaster. I. A Golgi analysis of wild-type structure. Cell and Tissue Research. 1989;258:441–475. [Google Scholar]
- Flanagan JG. Neural map specification by gradients. Curr Opin Neurobiol. 2006;16:59–66. doi: 10.1016/j.conb.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Frankfort BJ, Nolo R, Zhang Z, Bellen H, Mardon G. senseless repression of rough is required for R8 photoreceptor differentiation in the developing Drosophila eye. Neuron. 2001;32:403–414. doi: 10.1016/s0896-6273(01)00480-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Takemura SY, Ting CY, Huang S, Lu Z, Luan H, Rister J, Thum AS, Yang M, Hong ST, et al. The neural substrate of spectral preference in Drosophila. Neuron. 2008;60:328–342. doi: 10.1016/j.neuron.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- Grether ME, Abrams JM, Agapite J, White K, Steller H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes & development. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- Harrington AW, Ginty DD. Long-distance retrograde neurotrophic factor signalling in neurons. Nature reviews Neuroscience. 2013;14:177–187. doi: 10.1038/nrn3253. [DOI] [PubMed] [Google Scholar]
- Hasegawa E, Kitada Y, Kaido M, Takayama R, Awasaki T, Tabata T, Sato M. Concentric zones, cell migration and neuronal circuits in the Drosophila visual center. Development. 2011;138:983–993. doi: 10.1242/dev.058370. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- Huang Z, Kunes S. Hedgehog, transmitted along retinal axons, triggers neurogenesis in the developing visual centers of the Drosophila brain. Cell. 1996;86:411–422. doi: 10.1016/s0092-8674(00)80114-2. [DOI] [PubMed] [Google Scholar]
- Huang Z, Shilo BZ, Kunes S. A retinal axon fascicle uses spitz, an EGF receptor ligand, to construct a synaptic cartridge in the brain of Drosophila. Cell. 1998;95:693–703. doi: 10.1016/s0092-8674(00)81639-6. [DOI] [PubMed] [Google Scholar]
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14:439–449. doi: 10.1038/sj.onc.1200849. [DOI] [PubMed] [Google Scholar]
- Jenett A, Rubin GM, Ngo TT, Shepherd D, Murphy C, Dionne H, Pfeiffer BD, Cavallaro A, Hall D, Jeter J, et al. A GAL4-driver line resource for Drosophila neurobiology. Cell reports. 2012;2:991–1001. doi: 10.1016/j.celrep.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai MI, Okabe M, Hiromi Y. seven-up Controls switching of transcription factors that specify temporal identities of Drosophila neuroblasts. Developmental cell. 2005;8:203–213. doi: 10.1016/j.devcel.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Kummer TT, Misgeld T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr Opin Neurobiol. 2006;16:74–82. doi: 10.1016/j.conb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Kunes S. Axonal signals in the assembly of neural circuitry. Curr Opin Neurobiol. 2000;10:58–62. doi: 10.1016/s0959-4388(99)00051-3. [DOI] [PubMed] [Google Scholar]
- Lee HH, Norris A, Weiss JB, Frasch M. Jelly belly protein activates the receptor tyrosine kinase Alk to specify visceral muscle pioneers. Nature. 2003;425:507–512. doi: 10.1038/nature01916. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends in neurosciences. 2001;24:251–254. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Barde YA. Physiology of the neurotrophins. Annual review of neuroscience. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- Li X, Erclik T, Bertet C, Chen Z, Voutev R, Venkatesh S, Morante J, Celik A, Desplan C. Temporal patterning of Drosophila medulla neuroblasts controls neural fates. Nature. 2013;498:456–462. doi: 10.1038/nature12319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Dominguez B, Yang J, Aryal P, Brandon EP, Gage FH, Lee KF. Neurotransmitter acetylcholine negatively regulates neuromuscular synapse formation by a Cdk5-dependent mechanism. Neuron. 2005;46:569–579. doi: 10.1016/j.neuron.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Linden R. The survival of developing neurons: a review of afferent control. Neuroscience. 1994;58:671–682. doi: 10.1016/0306-4522(94)90447-2. [DOI] [PubMed] [Google Scholar]
- Loren CE, Englund C, Grabbe C, Hallberg B, Hunter T, Palmer RH. A crucial role for the Anaplastic lymphoma kinase receptor tyrosine kinase in gut development in Drosophila melanogaster. EMBO reports. 2003;4:781–786. doi: 10.1038/sj.embor.embor897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loren CE, Scully A, Grabbe C, Edeen PT, Thomas J, McKeown M, Hunter T, Palmer RH. Identification and characterization of DAlk: a novel Drosophila melanogaster RTK which drives ERK activation in vivo. Genes to cells : devoted to molecular & cellular mechanisms. 2001;6:531–544. doi: 10.1046/j.1365-2443.2001.00440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlroy G, Foldi I, Aurikko J, Wentzell JS, Lim MA, Fenton JC, Gay NJ, Hidalgo A. Toll-6 and Toll-7 function as neurotrophin receptors in the Drosophila melanogaster CNS. Nature neuroscience. 2013;16:1248–1256. doi: 10.1038/nn.3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinertzhagen IA, Hanson TE. The development of the opticlobe. 1993. pp. 1363–1491. [Google Scholar]
- Meinertzhagen IA, O’Neil SD. Synaptic organization of columnar elements in the lamina of the wild type in Drosophila melanogaster. The Journal of comparative neurology. 1991;305:232–263. doi: 10.1002/cne.903050206. [DOI] [PubMed] [Google Scholar]
- Meyerowitz EM, Kankel DR. A genetic analysis of visual system development in Drosophilia melanogaster. Developmental biology. 1978;62:112–142. doi: 10.1016/0012-1606(78)90096-9. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kummer TT, Lichtman JW, Sanes JR. Agrin promotes synaptic differentiation by counteracting an inhibitory effect of neurotransmitter. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:11088–11093. doi: 10.1073/pnas.0504806102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi K, Parker JS, Blundell TL, Gay NJ. Getting knotted: a model for the structure and activation of Spatzle. Trends in biochemical sciences. 1998;23:239–242. doi: 10.1016/s0968-0004(98)01216-x. [DOI] [PubMed] [Google Scholar]
- Morisato D, Anderson KV. The spatzle gene encodes a component of the extracellular signaling pathway establishing the dorsal-ventral pattern of the Drosophila embryo. Cell. 1994;76:677–688. doi: 10.1016/0092-8674(94)90507-x. [DOI] [PubMed] [Google Scholar]
- Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, Witte DP. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK) Oncogene. 1997;14:2175–2188. doi: 10.1038/sj.onc.1201062. [DOI] [PubMed] [Google Scholar]
- Nern A, Zhu Y, Zipursky SL. Local N-cadherin interactions mediate distinct steps in the targeting of lamina neurons. Neuron. 2008;58:34–41. doi: 10.1016/j.neuron.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitkin RM, Smith MA, Magill C, Fallon JR, Yao YM, Wallace BG, McMahan UJ. Identification of agrin, a synaptic organizing protein from Torpedo electric organ. The Journal of cell biology. 1987;105:2471–2478. doi: 10.1083/jcb.105.6.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolo R, Abbott LA, Bellen HJ. Senseless, a Zn finger transcription factor, is necessary and sufficient for sensory organ development in Drosophila. Cell. 2000;102:349–362. doi: 10.1016/s0092-8674(00)00040-4. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annual review of neuroscience. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Ou CY, Shen K. Setting up presynaptic structures at specific positions. Curr Opin Neurobiol. 2010;20:489–493. doi: 10.1016/j.conb.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palka J, Whitlock KE, Murray MA. Guidepost cells. Curr Opin Neurobiol. 1992;2:48–54. doi: 10.1016/0959-4388(92)90161-d. [DOI] [PubMed] [Google Scholar]
- Pecot MY, Tadros W, Nern A, Bader M, Chen Y, Zipursky SL. Multiple interactions control synaptic layer specificity in the Drosophila visual system. Neuron. 2013;77:299–310. doi: 10.1016/j.neuron.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepple KL, Atkins M, Venken K, Wellnitz K, Harding M, Frankfort B, Mardon G. Two-step selection of a single R8 photoreceptor: a bistable loop between senseless and rough locks in R8 fate. Development. 2008;135:4071–4079. doi: 10.1242/dev.028951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BD, Jenett A, Hammonds AS, Ngo TT, Misra S, Murphy C, Scully A, Carlson JW, Wan KH, Laverty TR, et al. Tools for neuroanatomy and neurogenetics in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9715–9720. doi: 10.1073/pnas.0803697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BD, Ngo TT, Hibbard KL, Murphy C, Jenett A, Truman JW, Rubin GM. Refinement of tools for targeted gene expression in Drosophila. Genetics. 2010;186:735–755. doi: 10.1534/genetics.110.119917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power ME. The effect of reduction in numbers of ommatidia upon the brain of Drosophila melanogaster. Journal of Experimental Zoology. 1943;94:33–71. [Google Scholar]
- Reinke R, Zipursky SL. Cell-cell interaction in the Drosophila retina: the bride of sevenless gene is required in photoreceptor cell R8 for R7 cell development. Cell. 1988;55:321–330. doi: 10.1016/0092-8674(88)90055-4. [DOI] [PubMed] [Google Scholar]
- Riddiford LM, Truman JW, Mirth CK, Shen YC. A role for juvenile hormone in the prepupal development of Drosophila melanogaster. Development. 2010;137:1117–1126. doi: 10.1242/dev.037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Alba M, Vitaladevuni SN, Mishchenko Y, Lu Z, Takemura SY, Scheffer L, Meinertzhagen IA, Chklovskii DB, de Polavieja GG. Wiring economy and volume exclusion determine neuronal placement in the Drosophila brain. Current biology : CB. 2011;21:2000–2005. doi: 10.1016/j.cub.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salecker I, Clandinin TR, Zipursky SL. Hedgehog and Spitz: making a match between photoreceptor axons and their targets. Cell. 1998;95:587–590. doi: 10.1016/s0092-8674(00)81627-x. [DOI] [PubMed] [Google Scholar]
- Singh KK, Park KJ, Hong EJ, Kramer BM, Greenberg ME, Kaplan DR, Miller FD. Developmental axon pruning mediated by BDNF-p75NTR-dependent axon degeneration. Nature neuroscience. 2008;11:649–658. doi: 10.1038/nn.2114. [DOI] [PubMed] [Google Scholar]
- Spalding KL, Tan MM, Hendry IA, Harvey AR. Anterograde transport and trophic actions of BDNF and NT-4/5 in the developing rat visual system. Molecular and cellular neurosciences. 2002;19:485–500. doi: 10.1006/mcne.2001.1097. [DOI] [PubMed] [Google Scholar]
- Sugie A, Umetsu D, Yasugi T, Fischbach KF, Tabata T. Recognition of pre- and postsynaptic neurons via nephrin/NEPH1 homologs is a basis for the formation of the Drosophila retinotopic map. Development. 2010;137:3303–3313. doi: 10.1242/dev.047332. [DOI] [PubMed] [Google Scholar]
- Suh GS, Poeck B, Chouard T, Oron E, Segal D, Chamovitz DA, Zipursky SL. Drosophila JAB1/CSN5 acts in photoreceptor cells to induce glial cells. Neuron. 2002;33:35–46. doi: 10.1016/s0896-6273(01)00576-1. [DOI] [PubMed] [Google Scholar]
- Takemura SY, Bharioke A, Lu Z, Nern A, Vitaladevuni S, Rivlin PK, Katz WT, Olbris DJ, Plaza SM, Winston P, et al. A visual motion detection circuit suggested by Drosophila connectomics. Nature. 2013;500:175–181. doi: 10.1038/nature12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura SY, Lu Z, Meinertzhagen IA. Synaptic circuits of the Drosophila optic lobe: the input terminals to the medulla. The Journal of comparative neurology. 2008;509:493–513. doi: 10.1002/cne.21757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev K, Joly W, Hadjieconomou D, Salecker I. Localized netrins act as positional cues to control layer-specific targeting of photoreceptor axons in Drosophila. Neuron. 2012;75:80–93. doi: 10.1016/j.neuron.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting CY, Yonekura S, Chung P, Hsu SN, Robertson HM, Chiba A, Lee CH. Drosophila N-cadherin functions in the first stage of the two-stage layer-selection process of R7 photoreceptor afferents. Development. 2005;132:953–963. doi: 10.1242/dev.01661. [DOI] [PubMed] [Google Scholar]
- Trujillo-Cenoz O. Some aspects of the structural organization of the intermediate retina of dipterans. Journal of ultrastructure research. 1965;13:1–33. doi: 10.1016/s0022-5320(65)80086-7. [DOI] [PubMed] [Google Scholar]
- Van Vactor D, Jr, Krantz DE, Reinke R, Zipursky SL. Analysis of mutants in chaoptin, a photoreceptor cell-specific glycoprotein in Drosophila, reveals its role in cellular morphogenesis. Cell. 1988;52:281–290. doi: 10.1016/0092-8674(88)90517-x. [DOI] [PubMed] [Google Scholar]
- Vanderhaeghen P, Cheng HJ. Guidance molecules in axon pruning and cell death. Cold Spring Harb Perspect Biol. 2010;2:a001859. doi: 10.1101/cshperspect.a001859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JB, Xue C, Benice T, Xue L, Morris SW, Raber J. Anaplastic lymphoma kinase and leukocyte tyrosine kinase: functions and genetic interactions in learning, memory and adult neurogenesis. Pharmacology, biochemistry, and behavior. 2012;100:566–574. doi: 10.1016/j.pbb.2011.10.024. [DOI] [PubMed] [Google Scholar]
- White K, Tahaoglu E, Steller H. Cell killing by the Drosophila gene reaper. Science. 1996;271:805–807. doi: 10.1126/science.271.5250.805. [DOI] [PubMed] [Google Scholar]
- Xue D, Horvitz HR. Inhibition of the Caenorhabditis elegans cell-death protease CED-3 by a CED-3 cleavage site in baculovirus p35 protein. Nature. 1995;377:248–251. doi: 10.1038/377248a0. [DOI] [PubMed] [Google Scholar]
- Yao S, Cheng M, Zhang Q, Wasik M, Kelsh R, Winkler C. Anaplastic lymphoma kinase is required for neurogenesis in the developing central nervous system of zebrafish. PloS one. 2013;8:e63757. doi: 10.1371/journal.pone.0063757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogev S, Schejter ED, Shilo BZ. Polarized secretion of Drosophila EGFR ligand from photoreceptor neurons is controlled by ER localization of the ligand-processing machinery. PLoS biology. 2010;8 doi: 10.1371/journal.pbio.1000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Pennack JA, McQuilton P, Forero MG, Mizuguchi K, Sutcliffe B, Gu CJ, Fenton JC, Hidalgo A. Drosophila neurotrophins reveal a common mechanism for nervous system formation. PLoS biology. 2008;6:e284. doi: 10.1371/journal.pbio.0060284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.