

Introduction
Pulmonary arterial hypertension (PAH) is characterized by increased pulmonary vascular resistance due to remodeling of distal pulmonary arterioles that occurs as a consequence of a complex interplay between molecular and genetic factors1. The incidence of PAH is estimated at 7–10 individuals per million people2, with a prevalence of up to 50 cases/million3. The most recent World Health Organization (WHO) clinical classification of pulmonary hypertension (PH)4 distinguishes Group 1 PH from pulmonary vascular disease related to lung disease, left atrial hypertension or venothromboemoblism by including PAH in association with anorexigen exposure, connective tissue disease, Human Immunodeficiency Virus (HIV), or portal hypertension, among other specific co-morbidities. In turn, idiopathic PAH (iPAH) is diagnosed in patients without a hereditary or other identifiable cause of PAH. Owing to the diversity of diseases implicated in the pathogenesis of PAH, a central goal among PH care centers5 is pathophenotyping patients with pulmonary vascular disease to calibrate suitable therapy6.
Prior to “PAH-specific” drug treatment availability, diagnosing PAH functioned principally to inform (a dismal) patient prognosis as treatment was relegated primarily to the careful use of warfarin, digoxin, diuretics, and oxygen7. The discovery of calcium channel blocker efficacy in this disease was a breakthrough, but this therapy was deemed to be appropriate for only a minority of patients and this remains true today8, 9. However, subsequent seminal discoveries of key signaling pathways implicated in the pathogenesis of PAH in some patients exposed for the first time disease-specific treatment targets10. In turn, results from conventional randomized clinical trials (RCTs) validated their translational relevance and introduced four novel drug classes to clinical practice that improve incrementally quality of life and/or longevity in PAH patients8, 11–17. Even in the era of contemporary PAH therapies, progressive heart failure and diminished quality of life remain common and are associated with a one-year mortality rate of 7–17%18, 19. The persistently elevated rates of PAH-associated morbidity and mortality raise speculation that currently used tactics to identify optimal treatments and predict therapeutic responsiveness in PAH are insufficient, and that additional molecular treatment targets remain unidentified20.
With the maturation and enhanced availability of applied clinical genomic- and proteomics-based research, the defining features of PAH biology is in continual flux. Over the previous few years, numerous molecules that contribute to PAH pathophysiology have been identified in at least two experimental animal models of PAH in vivo or in affected patients21–24. Moreover, the pool of potential monogenetic forms of PAH has expanded through the recent identification of novel gene mutations in PAH family clusters25. The ramifications of these advances are not inconsequential: the current methods for clinical diagnosis of PAH, which hinge primarily on achieving hemodynamic metrics without regard to other clinical variables, such as right ventricular function or patients’ molecular pathophenotype, is increasingly recognized as antiquated and insufficient26–28.
The National Institutes of Health announced recently a major funding initiative to stimulate investigations that leverage proteomics and genomics for the characterization of pulmonary vascular disease phenotype29. Collectively, momentum is shifting in the PAH field toward a personalized medicine approach to disease categorization, diagnosis, and, ultimately, treatment implementation30. The barriers to achieving truly individualized care are extensive, complex, and may not be surmountable. Nevertheless, in the spirit of this aim we believe that PAH is a disease model well suited for smaller trial designs that selectively target patients based on pathobiology (rather than general hemodynamic data alone) and maintain adequate statistical fidelity. Additional potential virtues of these alternative clinical research approaches in PAH include maneuverability between therapies to improve the identification of effective drugs or drug combinations31.
The RCT is the principle clinical research method to assess efficacy of novel treatment in PAH, and has been instrumental for identifying the vast majority of Food and Drug Administration-approved therapies for this disease. By recruiting clinical resources from PAH centers of excellence worldwide, RCTs have been successful at providing outcome data relevant to this pulmonary vascular disease patient population despite the (relatively) low prevalence of PAH. However, RCTs in PAH trials generally do not incorporate the totality of clinical, genetic, and molecular data when designating inclusion/exclusion criteria for enrollment20. This, in turn, increases the probability that a study cohort includes a heterogeneous range of PAH substrates, which we believe accounts for inconsistent rates of clinical benefit reported within RCTs, across similarly designed RCTs, and, ultimately limits the translation of clinical trial observations to “real world” practice. One often cited justification for the use of conventional RCT design includes unavailability of suitable alternative study designs. Here, we discuss clinical trial designs for the forthcoming era of advanced molecular and genomic PAH diagnosis that maintain rigorous analysis of outcome despite lower patient volume, which we believe are necessary elements of contemporary clinical research studying this heterogeneous and uncommon disease. Although RCTs will continue to play a vital role in PAH research, we feel that we must pivot and start incorporating other designs that will better answer certain questions when a conventional RCT is unlikely to.
PAH and Randomized Controlled Trials: An Imperfect Strategy to Study a Complex Disease
Applying randomized clinical trial data to patient care in PAH
The traditional RCT design hinges on a reductionist approach to establishing patient appropriateness for study consideration, which often involves 20 or more patient inclusion/exclusion criteria for study enrollment11–14, 16, 32,33. Still, this approach does not appear to offset the heterogeneity of PAH, as poor generalizability of findings from RCT to clinical practice are reported26. Additional factors specific to traditional study design that are likely to contribute to this dilemma include trial duration variability and flawed study end-points34.
Optimal therapy duration and ethical consideration of placebo use in PAH trials
The optimal duration of therapy in PAH clinical trials is unresolved. While RCTs completed over the last two decades have demonstrated that a 12-week end-point correlates positively with outcomes assessed in longer extension studies,35 a number of PAH studies have included time points ranging from 8–26 weeks. Moreover, other trials have demonstrated a benefit at 12 weeks only to observe diminished benefit at 9 months36. Data to systemically characterize PAH-specific treatment efficacy as a function of time are unavailable; however, the rapid trajectory of clinical decline in many patients is an important consideration to trial design, especially in the setting of delayed clinical presentation and diagnosis that often characterizes PAH in clinical practice37. Recent estimates indicate that despite the availability of PAH-specific therapy, 1-year mortality rates in untreated PAH7, 38 rival patients with moderate or severe congestive heart failure due to advanced left-sided heart disease (New York Heart Functional Class III/IV)(Figure 1)2, 18, 39. However, clinical trials in systolic heart failure use follow-up periods on a scale of years compared to the much shorter durations commonly used in PAH trials40, 41.
Figure 1. Mortality rates in patients with pulmonary arterial hypertension (PAH), left-sided heart failure with reduced ejection fraction (HFrEF), and left-sided heart failure with preserved ejection fraction (HFpEF).

(A) Kaplan–Meier survival analysis of 6,076 patients hospitalized with left-sided heart failure hospitalized over a 15-year period (1987–2001) at Mayo Clinic Hospital (Olmsted County, Minnesota). Compared to patients with HFpEF (red line), decreased survival was observed in HFrEF (black line) at 5 years (adjusted hazard ratio for death, 0.96; P = 0.03). Adapted with permission from62. (B) Kaplan–Meier analyses compares survival in the contemporary era (2002–2005) for patients with idiopathic, familial, or anorexigen-associated PAH (56 incident and 298 prevalent cases) (solid line) with predicted survival data derived from the original National Institutes of Health (NIH) PAH registry. The original NIH PAH registry included 194 patients diagnosed between July 1981 and December 1985 and followed through August 198838. Adapted with permission from18. (C) Kaplan–Meier analyses from panels A and B were merged using Adobe Illustrator CS5 on Win7 OS to compare mortality rates from HFpEF (purple dotted line), HFrEF (green solid line), and PAH (observed, blue dotted line; predicted, red line). Graph derived from 18, 38, 62.
In light of the progressive (and generally poor) natural history of untreated PAH, concern has been raised regarding the ethical implications of placebo use in RCTs, which adds to the complexity of performing controlled clinical studies in this disease42. Although the association between placebo use and unanticipated mortality during RCTs in PAH is unresolved38, withholding active treatment for the duration of RCTs (12–18 weeks) is associated with a significantly increased short-term risk of morbidity43, including clinical worsening. It is notable, however, that, despite these data, up to 50% of patients randomized to placebo in the most recent PAH RCTs were not on background pulmonary vasodilator therapy at all13, 15.
The clinical outcome dilemma in PAH clinical research
The cornerstone outcome measure to assess intervention efficacy in PAH RCTs has historically been distance achieved on the 6-minute walk test.11 Completion of a RCT powered sufficiently to measure drug effect on other primary end-points, such as survival, is uncommon due to the low prevalence of this disease and high costs associated with extended length studies to achieve sufficient statistical power12–14, 16, 17. Although 6-minute walk distance (6-MWD) is proposed as a marker of global health and baseline 6-MWD is an established predictor of survival in PAH, a consistent relationship has never been observed between change from baseline in 6-MWD and survival, PAH-associated hospitalization, or PAH therapy escalation44. In addition, while most therapies affect mean 6-MWD to a similar, albeit modest magnitude (approximately 20–50 m), studies evaluating the effect of an exercise program on 6-MWD in PAH demonstrate superior improvements in 6-MWD as compared to PAH pharmacotherapy. These findings underscore the potential bias of the training effect on assessing functional capacity as an outcome measure in this (and other) cardiopulmonary diseases and raises the question of the true clinical impact that a small improvement in 6-MWD achieved actually has on PAH outcomes45. For these reasons, many contemporary study designs in PAH have transitioned away from utilizing 6-MWD as the sole primary end-point15, 46. Instead, other clinical endpoints have been introduced in recently published trials, such as time to the first clinical event related to PAH and time to general clinical worsening15.
Time-to-clinical-worsening, however, may conflict with patients’ clinical care goals by illuminating treatment failure as inevitable. In fact, many professional societies are now underscoring the importance of integrating patient-reported outcomes in clinical research47. With this in mind, many PAH trials now emphasize patient reported outcomes measurements (PROMs), such as dyspnea or quality of life. Recently, the Cambridge Pulmonary Hypertension Outcome Review (CAMPHOR) questionnaire was demonstrated to predict clinical deterioration at study enrollment in PAH, even after adjusting for functional class and 6-MWD48. If validated in subsequent studies, the integration of CAMPHOR or similar tools into future trials should be considered.
Investigational endpoints can inform the pathophysiological basis for treatment success or failure. Often, the unavailability of technology and costs limits the widespread use of these endpoints within RCTs when studied across large populations and different centers. While such endpoints are unlikely to serve as the basis for drug approval, utilization of investigational end-points in future trial designs can help further understand the relevance of basic science or pre-clinical observations to iPAH patients. Examples of this might include changes in pulmonary vascular metabolic status using fluorodeoxyglucose (FDG) uptake,49 or distinguishing adaptive from maladaptive RV structural changes using cardiac magnetic resonance imaging (CMRI). Such an approach to including investigational secondary endpoints is not without precedent; for example, many historic trials in myocardial infarction and heart failure were designed to achieve the primary clinical endpoint while also providing information on disease epidemiology through surrogate end-points50.
Clinical Research Designs for PAH: Alternative to the Randomized Clinical Trial
Factorial Design
Factorial studies allow investigators to test multiple hypotheses at once. The simplest example is a 2×2 design where two treatments are studied. For example, if studying drug A and drug B, a factorial design would comprise four groups: (1) active drug A plus placebo drug B, (2) placebo drug A plus placebo drug B, (3) placebo drug A plus active drug B, (4) active drug A plus active drug B (Table 1). When deciding on the various therapies to be tested using a factorial design, it is important to consider the potential for drug-drug interaction(s) between each therapy as a confounder.
Table 1.
Characteristics of proposed and established study designs in pulmonary arterial hypertension (PAH).
| Trial type | Design | Advantage | Limitation | Example in PAH |
|---|---|---|---|---|
| Randomized Controlled Trial |
|
|
|
Reference 11–17. |
| Factorial Design | ≥2 Factors, each with ≥2 levels: 2 × 2 Factorial Design Drug A + Placebo B Placebo A + Placebo B Placebo A + Active B Active A + Active B |
|
|
Reference 51. |
| Crossover Study |
|
|
|
Reference 52. |
| Randomized Discontinuation Trial |
|
|
|
Reference 54. |
| N-of-1 Clinical Trial |
|
|
|
None reported. |
Using this design, Kawut and investigators conducted a randomized, double-blind, placebo-controlled 2×2 factorial clinical trial of simvastatin and aspirin in PAH patients receiving background PAH therapy (Figure 2)51. Subjects were randomly assigned in a 1:1:1:1 ratio to aspirin 81 mg once daily/simvastatin 40 mg once daily, aspirin 81 mg once daily/simvastatin placebo once daily, aspirin placebo once daily/simvastatin 40 mg once daily, or aspirin placebo once daily/simvastatin placebo once daily. Subjects were then evaluated at baseline, week 6, month 3 and month 6. The study was both informative and instructive: despite demonstrating no significant benefit from either aspirin or statin therapy on 6-MWD at 6 months, findings highlighted the feasibility and role of performing a factorial study in PAH, particularly when different mechanistic pathways are under investigation.
Figure 2. Factorial study design used in the ASA-STAT trial involving patients with pulmonary arterial hypertension (PAH).

Patients were randomly assigned in a 1:1:1:1 ratio by a Web-based computerized system to: (1) aspirin 81 mg once daily plus simvastatin 40 mg once daily, (2) aspirin 81 mg once daily plus placebo simvastatin once daily, (3) placebo aspirin placebo once daily plus simvastatin 40 mg once daily, or (4) placebo aspirin once daily plus placebo simvastatin once daily. NSAID, nonsteroidal anti-inflammatory drug; PFT, pulmonary function test; and ASA, aspirin. Reproduced with permission from51.
Crossover study
The crossover study design is divided into specific phases. In phase I, the dependent variable (i.e., end-point) is assessed at baseline and following randomization to treatment with study drug or placebo for a pre-determined duration of time. In phase II, patients are administered therapy opposite to Phase I and the end-point is re-assessed at the completion of the study (Figure 3). Within-subject analyses are performed to compare differences in outcome between the study drug and placebo. Advantages of this trial design include blinding and use of a smaller sample size compared to parallel trial design20.
Figure 3. Schematic representation of a crossover study design.
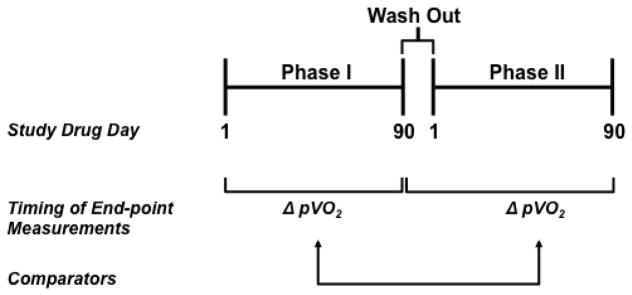
In Phase I, patients are randomized to treatment with placebo or study drug and testing relevant to the study end-points, such as peak volume of oxygen consumption (pVO2), occurs at study drug day 1 (i.e., baseline) and day 90. Following a 21-day drug wash out period, subjects enter Phase II of the trial, which is characterized by cross-over to therapy opposite of Phase I. Repeat end-point assessment will be performed at study drug day 90 of Phase II. Change in performance on end-points from study drug day 1 at study drug day 90 (Phase I) are compared to change in performance from study drug day 1 at study drug day 90 (Phase II), using a 2-sided, paired Student’s t test.
As discussed previously, PAH is often characterized by rapid clinical deterioration and symptom transition across various stages of disease natural history (i.e. exertional intolerance at early stages versus syncope and progressive right heart failure at advanced stages). Thus, crossover studies, in which a proportion of patients are randomized to upfront placebo generally involve patients with moderate symptom burden and do not control for timing of drug initiation. Additionally, owing to the observation that PAH-specific therapies appear more efficacious in patients with more severe disease, delayed drug therapy may be a confounding factor in the interpretation of cross-over study design results in demonstrating drug efficacy in PAH.
Singh and colleagues randomized patients with PAH (n=10) and Eisenmenger syndrome (n=10) to receive sildenafil or placebo for 6 weeks and then crossed over to opposite therapy after a washout period of 2 weeks52. Sample size calculation was based on a predetermined definition of improvement in 6-MWD by 50 m as clinically relevant, and the primary outcome was compared using repeated-measures analysis of variance. The authors also recorded and compared cardiopulmonary hemodynamic changes with Friedman tests. Individual changes in mean pulmonary artery pressure, New York Heart Association functional status, and metabolic equivalents achieved during exercise were analyzed using the Wilcoxon signed rank test. A benefit for sildenafil use was observed in the studied patient populations, which was later confirmed in larger RCTs12.
Randomized discontinuation trial and Withdrawal studies
A randomized discontinuation trial (RDT) is optimal for studying long-term, non-curative therapies, especially when the use of placebo is deemed unethical53. The RDT consists of two-phases. In the first phase, all patients are treated with the study drug, and in the second phase, drug therapy responders are randomly assigned to switch to placebo or continue the same treatment53. Predictive enrichment techniques are used to select subjects for study who have the greatest chance of benefit, as medication non-adherent patients or those reporting adverse events are generally not considered for study enrollment20. Withdrawal studies, which are similar to RDTs in principle, aim to determine if patients may be transitioned safely to an alternative form of therapy. Such a randomized, placebo-controlled withdrawal trial was performed by Rubenfire and colleagues, in which clinically stable PAH patients on epoprostenol (PGI2) therapy were randomized to transition to subcutaneous treprostinil (PGI2) or placebo in a 2:1 manner over a period of up to 14 days (Figure 4)54. In this study, of the 8 patients withdrawn to placebo, seven (88%) had clinical deterioration, while only 1 of 14 patients (7%) withdrawn to treprostinil deteriorated (p<0.001).
Figure 4. Discontinuation and Transition study design in pulmonary arterial hypertension (PAH).
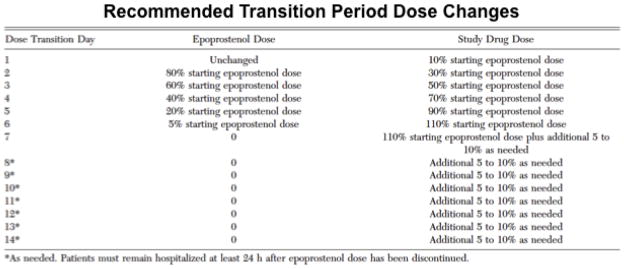
In this study, PAH patients stable on intravenous epoprostenol therapy were transitioned to study drug (subcutaneous treprostinil or placebo, 2:1) over a period of ≤14 days. Patients were hospitalized during the transition period and for at least 24 hr after the epoprostenol infusions were stopped. Patients who did not complete the transition to study drug or who had clinical deterioration were returned to continuous intravenous epoprostenol. Assessments were conducted at baseline, prior to discharge after the transition period, and at weeks 4 and 8. Reproduced with permission from 54.
More recently, Channick and colleagues used a RDT to assess outcomes following transition from parenteral prostacyclin to inhaled iloprost55 (Figure 5). In this study of 37 consecutive patients, the transition period began on the first day of inhaled iloprost with intent of discontinuing parenteral prostacyclin, and completed on the first day of treatment with inhaled iloprost free of parenteral prostacyclin. Almost 92% of patients had an overlapping transition with a mean transition period of 10.5 ± 13.9 days. At one year follow up, 78% of the patients remained on inhaled iloprost alone, and 81% were free of clinical worsening. It should be noted, however, that successful transition in this study appeared related to concomitant oral medication use, which must be considered during RDT planning.
Figure 5. Study design of an 8-week, multicenter, randomized, placebo-controlled withdrawal trial in pulmonary arterial hypertension (PAH).
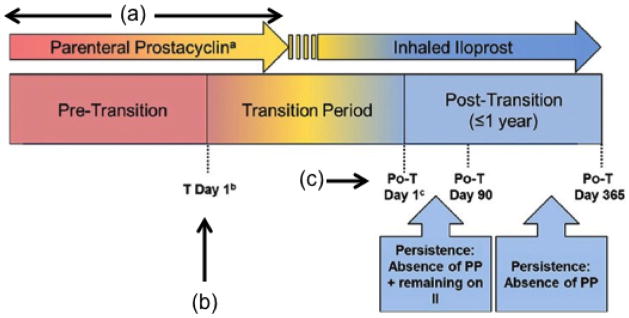
In this randomized discontinuation trial, patients were transitioned from parenteral to inhaled illoprost and the effects of this on safety and outcome was assessed. During time period (a) parenteral prostacyclins are administered comprising of intravenous epoprostenol and intravenous/subcutaneous treprostinil. At time point (b) transition Day 1 is defined as the start day of inhaled iloprost with intent of discontinuing parenteral prostacyclin therapy. At time point (c) post-transition Day 1 is defined as the first day on inhaled iloprost free of parenteral prostacyclin therapy. Depending on the clinical site and/or patient, there may be no period of concurrent (overlapping) administration of inhaled iloprost and parenteral prostacyclin therapy and, therefore, no transition period. In such cases, transition Day 1 is synonymous as post-transition Day 1. Po-T: post-transition; PP: parenteral prostacyclin; II: inhaled iloprost. Reproduced with permission from 55.
An important consideration of this study design in PAH is the possibility for adverse events to occur upon therapy withdrawal. Therefore, the RDT planning phase requires particular consideration to the individual patient’s clinical profile, particularly disease severity, when determining appropriateness for RDT trial enrollment.
The N-of-1 Clinical Trial
A common N-of-1 trial design involves multiple crossover experiments performed over pre-defined time periods to compare the effects of different treatments on outcome measure(s) within an individual patient (Figure 6). Although under-represented in the cardiovascular disease literature, Gabler and colleagues identified 108 N-of-1 trials involving 2,154 patients published between 1985–201056, which include chronic diseases such as insomnia, attention deficit hyperactivity disorder, chronic obstructive pulmonary disease, and sleep disordered breathing. Generally, following informed consent, a patient enrolled in an N-of-1 trial undergoes baseline measurement of a specific outcome measure. The patient is randomized to receive placebo or a therapeutic intervention for a pre-specified time period, after which performance on the outcome measure(s) is reassessed. Following a drug washout period, the same experimental design is repeated to measure the effect of a second therapy on the same outcome measure(s). Ultimately, a comparison of the effect of each treatment on outcome is performed to characterize drug efficacy. Similar to RCTs, clinicians and patients are generally blinded to the therapeutic agent (or placebo) during the study to avoid the introduction of bias on outcomes. Various permutations in study design involving the number of therapy cycles, duration of therapy, role of blinding, sequence of randomization, and potential for co-therapy is considered according to the disease process and pharmacokinetics of the drug(s) under investigation. Overall, a favorable cost value of an N-of-1 trial compared to RCT is likely, but hinges on the complexity of the selected end-points and scale of the comparator RCT (Table 2).
Figure 6. One potential N-of-1 clinical trial design for idiopathic pulmonary arterial hypertension (iPAH).

This schematic depicts one potential N-of-1 clinical trial, designed to test the efficacy of drug therapy on change in peak volume of oxygen consumption (VO2) from baseline as the outcome measure for a 34-year old woman with iPAH. This trial model designation is A-B-C by virtue of the three experimental phases involving Drug A (placebo), followed by Drug B (endothelin receptor antagonist), followed by a period of therapy with Drug C (phosphodiesterase type-V inhibitor). Each drug therapy period is separated by a drug washout phase in order to avoid potential residual effects of prior therapy on outcome. The time period for assessment of drug efficacy is 6 weeks, which is within the time frame of previously published randomized clinical trials demonstrating drug efficacy in iPAH.
Table 2. Sample cost-analysis for a placebo-controlled, randomized clinical trial in idiopathic pulmonary arterial hypertension (iPAH).
Published cost estimates for a typical randomized clinical trial (RCT) in iPAH vary substantially based on several key factors, including study duration, complexity of selected end-points, enrollment, and study drug price. Differences in Institutional Review Board (IRB) and study drug fees for an N-of-1 trial compared to a RCT reflect the single center nature of the design and differences in drug treatment duration.
| Randomized Clinical Trial | N-of-1 Trial | ||||
|---|---|---|---|---|---|
| Study Element | Baseline Cost (USD) | Iterations/Study (N) | Total Cost Per Element (USD) | Iterations/Study (N) | Total Cost Per Element (USD) |
| Informed Consent Processing | 150.00 | 1 | 150.00 | 1 | 150.00 |
| History and PE | 500.00 | 3 | 1,500.00 | 4 | 2,000.00 |
| Vital sign assessment | 50.00 | 3 | 150.00 | 4 | 150.00 |
| Outcomes questionnaires | 100.00 | 3 | 300.00 | 4 | 400.00 |
| Drug Compliance Assessment | 50.00 | 3 | 150.00 | 4 | 200.00 |
| Study Personnel | 700.00 | 1 | 700.00 | 0 | 0.00 |
| 6-MWT | 550.00 | 1 | 550.00 | 4 | 2,200.00 |
| Cardiopulmonary Exercise Test | 1,100.00 | 2 | 2,200.00 | 4 | 4,400.00 |
| Echocardiography | 300.00 | 1 | 300.00 | 4 | 1,200.00 |
| IRB fees | 4,000.00 | 1 | 4,000.00 | 1 | 1,000.00 |
| Study Drug* | |||||
| ERA (12 wk) High Dose |
12,100.00 | 1 | 12,100.00 | - | - |
| ERA (12 wk) Low Dose |
12,100.00 | 1 | 12,100.00 | - | - |
| ERA (6 wk) | - | - | - | 1 | 6,050.00 |
| PDE-V (6 wk) | - | - | - | 1 | 2,700.00 |
| ERA+PDE-V (6 wk) | - | - | - | 1 | 8,750.00 |
| Subtotal Costs | 34,200/patient | 29,200/patient | |||
| Total Costs For Trial | 2,052,000.00 | 29,200 | |||
Value for Study Drug Costs reflects mean costs for daily study drug use and placebo, based on published costs associated with phosphodiesterase type V inhibitor class medication.9 USD, United States Dollars ($); PE, physical examination; 6-MWT, 6-minute walk test.
A limitation of the N-of-1 trial in PAH is the potential rapid nature of disease progression as well as the perils of drug withdrawal. Indeed, N-of-1 trials may be suited better for chronic, progressive diseases characterized by a predictable mortality and event rate, as demonstrated in a recent N-of-1 analysis of statin therapy57. Nevertheless, certain PAH patients may warrant consideration for N-of-1 trial protocols when the disease pathophenotype is known in order to characterize individualized response to therapy. Consider these three patients with PAH (i) a loss of function BMPR-2 mutation that promotes angioproliferative pulmonary vascular injury58, (ii) a loss of function KCNK3 mutation that impairs potassium channel function to promote pulmonary vascular dysfunction21, or (iii) pulmonary vascular inflammation and fibrosis in the setting of scleroderma-associated pulmonary arterial hypertension. Despite an overlapping histopathology between these three patients, interchangeability of directed therapy to restore BMPR-2-dependent signaling is unlikely to abrogate pulmonary hypertension in the latter two patients, and vice versa for drugs that target pulmonary vascular inflammation to treat scleroderma-associated PAH in the first two cases. Therefore a trial of individualized therapy in an N-of-1 setting may play a role in these patients. The N-of-1 clinical trial design is well-positioned to identify therapies that are beneficial for specific PAH sub-pathophenotypes, but is unlikely to lend insight to the management of PAH patients broadly, which is where investigational endpoints play a role, i.e., by correlating the clinical improvements with changes in novel markers of disease59. A thorough discussion of the various statistical methods used to analyze data from N-of-1 trials is reviewed in reference60.
Limitations of novel trials in PAH
There are important characteristics of PAH that may influence use or success of novel trial designs. PAH is a progressive disease with a variable clinical trajectory, which may confound drug efficacy within a single patient to generate both false positive and false negative results. Along these lines, since currently available therapies for PAH have never been shown to reverse disease pathobiology, the assessment of drug efficacy within a patient across different clinical stages of PAH is challenging. Thus, the timing of PAH-specific therapy initiation, which is controversial among experts, is unlikely to be resolved by these alternative trial designs. Additionally, drug withdrawal is associated with acute clinical worsening in some PAH patients, and, therefore, RDTs, withdrawal studies, and N-of-1 trial planning should be well within the framework of expert consensus guidelines for good clinical practice in PAH, including access to expert PAH care providers and specialized clinical PAH systems61.
Conclusions
In summary, PAH is a rare and heterogeneous disease characterized by elevated rates of mortality and heart failure-associated morbidity. Variability in PAH pathophenotype is a likely contributing factor to difficulty generalizing RCT findings to patients in clinical practice44. By contrast, we believe that crossover, RDT, and N-of-1 study designs are well positioned to study outcomes in selected PAH patient cohorts defined by converging genetic or molecular PAH pathophenotypes, and provide hypothesis-generating data for future study in large RCTs (Table 1). We anticipate that achieving individualized treatment strategies in PAH ultimately hinges on the application of the novel clinical trial strategies discussed. Furthermore, we believe these strategies are necessary for developing cost-effective methods that identify PAH patients likely to benefit from disease-specific pharmacotherapies
Acknowledgments
Funding Sources
This work was supported, in part, by the US NIH (1K08HL111207-01A1), the Center for Integration and Innovative Technology (CIMIT), Pulmonary Hypertension Association, and Lerner and Klarman Foundations at Brigham and Women’s Hospital to B.A.M..
The authors thank Dr. Stephen Archer, Queen’s University, Kingston, Ontario and Dr. Stuart Rich, University of Chicago, for their review of this article. The authors also thank Barbara J. Stephan, Hallside Gallery curator at the University of Utah for assistance with the figures.
Footnotes
Disclosures
Dr. Maron is an awardee of the Gilead Research Scholars Program from Gilead Sciences Inc. to study pulmonary hypertension.
References
- 1.Maron BA, Loscalzo J. Pulmonary hypertension: pathophysiology and signaling pathways. Handbook of experimental pharmacology. 2013;218:31–58. doi: 10.1007/978-3-642-38664-0_2. [DOI] [PubMed] [Google Scholar]
- 2.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E, Simonneau G. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–30. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 3.Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30:104–9. doi: 10.1183/09031936.00092306. [DOI] [PubMed] [Google Scholar]
- 4.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 5.http://www.phassociation.org/PHCareCenters
- 6.Deano RC, Glassner-Kolmin C, Rubenfire M, Frost A, Visovatti S, McLaughlin VV, Gomberg-Maitland M. Referral of patients with pulmonary hypertension diagnoses to tertiary pulmonary hypertension centers: the multicenter RePHerral study. JAMA internal medicine. 2013;173:887–93. doi: 10.1001/jamainternmed.2013.319. [DOI] [PubMed] [Google Scholar]
- 7.Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Koerner SK, et al. Primary pulmonary hypertension. A national prospective study. Annals of internal medicine. 1987;107:216–23. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- 8.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76–81. doi: 10.1056/NEJM199207093270203. [DOI] [PubMed] [Google Scholar]
- 9.Sitbon O, Humbert M, Jais X, Ioos V, Hamid AM, Provencher S, Garcia G, Parent F, Herve P, Simonneau G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111:3105–11. doi: 10.1161/CIRCULATIONAHA.104.488486. [DOI] [PubMed] [Google Scholar]
- 10.Galie N, Corris PA, Frost A, Girgis RE, Granton J, Jing ZC, Klepetko W, McGoon MD, McLaughlin VV, Preston IR, Rubin LJ, Sandoval J, Seeger W, Keogh A. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D60–72. doi: 10.1016/j.jacc.2013.10.031. [DOI] [PubMed] [Google Scholar]
- 11.Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, Groves BM, Tapson VF, Bourge RC, Brundage BH, Koerner SK, Langleben D, Keller CA, Murali S, Uretsky BF, Clayton LM, Jobsis MM, Blackburn SD, Shortino D, Crow JW. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296–301. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 12.Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, Fleming T, Parpia T, Burgess G, Branzi A, Grimminger F, Kurzyna M, Simonneau G. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–57. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 13.Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, Keogh AM, Langleben D, Kilama MO, Fritsch A, Neuser D, Rubin LJ. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330–40. doi: 10.1056/NEJMoa1209655. [DOI] [PubMed] [Google Scholar]
- 14.Galie N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, Shapiro S, White RJ, Chan M, Beardsworth A, Frumkin L, Barst RJ. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119:2894–903. doi: 10.1161/CIRCULATIONAHA.108.839274. [DOI] [PubMed] [Google Scholar]
- 15.Pulido T, Adzerikho I, Channick RN, Delcroix M, Galie N, Ghofrani HA, Jansa P, Jing ZC, Le Brun FO, Mehta S, Mittelholzer CM, Perchenet L, Sastry BK, Sitbon O, Souza R, Torbicki A, Zeng X, Rubin LJ, Simonneau G. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–18. doi: 10.1056/NEJMoa1213917. [DOI] [PubMed] [Google Scholar]
- 16.Galie N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, Badesch DB, McGoon MD, McLaughlin VV, Roecker EB, Gerber MJ, Dufton C, Wiens BL, Rubin LJ. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117:3010–9. doi: 10.1161/CIRCULATIONAHA.107.742510. [DOI] [PubMed] [Google Scholar]
- 17.Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, Landzberg M, Simonneau G. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- 18.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E, Cottin V, Degano B, Jais X, Montani D, Souza R, Simonneau G. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–63. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 19.Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, Barst RJ, Benza RL, Liou TG, Turner M, Giles S, Feldkircher K, Miller DP, McGoon MD. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137:376–87. doi: 10.1378/chest.09-1140. [DOI] [PubMed] [Google Scholar]
- 20.Gomberg-Maitland M, Bull TM, Saggar R, Barst RJ, Elgazayerly A, Fleming TR, Grimminger F, Rainisio M, Stewart DJ, Stockbridge N, Ventura C, Ghofrani AH, Rubin LJ. New trial designs and potential therapies for pulmonary artery hypertension. J Am Coll Cardiol. 2013;62:D82–91. doi: 10.1016/j.jacc.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma L, Roman-Campos D, Austin ED, Eyries M, Sampson KS, Soubrier F, Germain M, Tregouet DA, Borczuk A, Rosenzweig EB, Girerd B, Montani D, Humbert M, Loyd JE, Kass RS, Chung WK. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med. 2013;369:351–61. doi: 10.1056/NEJMoa1211097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA, 3rd, Palomero T, Sumazin P, Kim HR, Talati MH, West J, Loyd JE, Chung WK. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5:336–43. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shintani M, Yagi H, Nakayama T, Saji T, Matsuoka R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet. 2009;46:331–7. doi: 10.1136/jmg.2008.062703. [DOI] [PubMed] [Google Scholar]
- 24.Courboulin A, Paulin R, Giguere NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Cote J, Simard MJ, Bonnet S. Role for miR-204 in human pulmonary arterial hypertension. The Journal of experimental medicine. 2011;208:535–48. doi: 10.1084/jem.20101812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soubrier F, Chung WK, Machado R, Grunig E, Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH, Humbert M. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D13–21. doi: 10.1016/j.jacc.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 26.Ryan JJ, Butrous G, Maron BA. The Heterogeneity of Clinical Practice Patterns among an International Cohort of Pulmonary Arterial Hypertension Experts. Pulmonary circulation. 2014 doi: 10.1086/677357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soubrier F, Chung WK, Machado R, Grunig E, Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH, Humbert M. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D13–21. doi: 10.1016/j.jacc.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 28.Dweik RA, Rounds S, Erzurum SC, Archer S, Fagan K, Hassoun PM, Hill NS, Humbert M, Kawut SM, Krowka M, Michelakis E, Morrell NW, Stenmark K, Tuder RM, Newman J Phenotypes ATSCoPH. An official American Thoracic Society Statement: pulmonary hypertension phenotypes. Am J Respir Crit Care Med. 2014;189:345–55. doi: 10.1164/rccm.201311-1954ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.http://grants.nih.gov/grants/guide/rfa-files/RFA-HL-14-030.html
- 30.Dweik RA, Rounds S, Erzurum SC, Archer S, Fagan K, Hassoun PM, Hill NS, Humbert M, Kawut SM, Krowka M, Michelakis E, Morrell NW, Stenmark K, Tuder RM, Newman J. An official American Thoracic Society Statement: pulmonary hypertension phenotypes. Am J Respir Crit Care Med. 2014;189:345–55. doi: 10.1164/rccm.201311-1954ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grieve AP, Chow SC, Curram J, Dawe S, Harnisch LO, Henig NR, Hung HM, Ivy DD, Kawut SM, Rahbar MH, Xiao S, Wilkins MR. Advancing clinical trial design in pulmonary hypertension. Pulmonary circulation. 2013;3:217–25. doi: 10.4103/2045-8932.109933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukumoto Y, Yamada N, Matsubara H, Mizoguchi M, Uchino K, Yao A, Kihara Y, Kawano M, Watanabe H, Takeda Y, Adachi T, Osanai S, Tanabe N, Inoue T, Kubo A, Ota Y, Fukuda K, Nakano T, Shimokawa H. Double-Blind, Placebo-Controlled Clinical Trial With a Rho-Kinase Inhibitor in Pulmonary Arterial Hypertension. Circulation journal: official journal of the Japanese Circulation Society. 2013 doi: 10.1253/circj.cj-13-0443. [DOI] [PubMed] [Google Scholar]
- 33.Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, Gomez-Sanchez MA, Jondeau G, Klepetko W, Opitz C, Peacock A, Rubin L, Zellweger M, Simonneau G. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT) European heart journal. 2009;30:2493–537. doi: 10.1093/eurheartj/ehp297. [DOI] [PubMed] [Google Scholar]
- 34.McLaughlin VV. Has the 6-min walk distance run its course? Chest. 2012;142:1363–4. doi: 10.1378/chest.12-1110. [DOI] [PubMed] [Google Scholar]
- 35.Oudiz RJ, Brundage BH, Galie N, Ghofrani HA, Simonneau G, Botros FT, Chan M, Beardsworth A, Barst RJ. Tadalafil for the treatment of pulmonary arterial hypertension: a double-blind 52-week uncontrolled extension study. J Am Coll Cardiol. 2012;60:768–74. doi: 10.1016/j.jacc.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 36.Galie N, Humbert M, Vachiery JL, Vizza CD, Kneussl M, Manes A, Sitbon O, Torbicki A, Delcroix M, Naeije R, Hoeper M, Chaouat A, Morand S, Besse B, Simonneau G. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39:1496–502. doi: 10.1016/s0735-1097(02)01786-2. [DOI] [PubMed] [Google Scholar]
- 37.Strange G, Gabbay E, Kermeen F, Williams T, Carrington M, Stewart S, Keogh A. Time from symptoms to definitive diagnosis of idiopathic pulmonary arterial hypertension: The delay study. Pulmonary circulation. 2013;3:89–94. doi: 10.4103/2045-8932.109919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Kernis JT, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Annals of internal medicine. 1991;115:343–9. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 39.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122:164–72. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 40.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–17. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 41.Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, Shusterman NH. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N Engl J Med. 1996;334:1349–55. doi: 10.1056/NEJM199605233342101. [DOI] [PubMed] [Google Scholar]
- 42.Halpern SD, Doyle R, Kawut SM. The ethics of randomized clinical trials in pulmonary arterial hypertension. Proceedings of the American Thoracic Society. 2008;5:631–5. doi: 10.1513/pats.200802-019SK. [DOI] [PubMed] [Google Scholar]
- 43.Helman DL, Jr, Brown AW, Jackson JL, Shorr AF. Analyzing the short-term effect of placebo therapy in pulmonary arterial hypertension: potential implications for the design of future clinical trials. Chest. 2007;132:764–72. doi: 10.1378/chest.07-0236. [DOI] [PubMed] [Google Scholar]
- 44.Savarese G, Paolillo S, Costanzo P, D’Amore C, Cecere M, Losco T, Musella F, Gargiulo P, Marciano C, Perrone-Filardi P. Do changes of 6-minute walk distance predict clinical events in patients with pulmonary arterial hypertension? A meta-analysis of 22 randomized trials. J Am Coll Cardiol. 2012;60:1192–201. doi: 10.1016/j.jacc.2012.01.083. [DOI] [PubMed] [Google Scholar]
- 45.Grunig E, Lichtblau M, Ehlken N, Ghofrani HA, Reichenberger F, Staehler G, Halank M, Fischer C, Seyfarth HJ, Klose H, Meyer A, Sorichter S, Wilkens H, Rosenkranz S, Opitz C, Leuchte H, Karger G, Speich R, Nagel C. Safety and efficacy of exercise training in various forms of pulmonary hypertension. Eur Respir J. 2012;40:84–92. doi: 10.1183/09031936.00123711. [DOI] [PubMed] [Google Scholar]
- 46.http://clinicaltrials.gov/show/NCT01106014
- 47.Anker SD, Agewall S, Borggrefe M, Calvert M, Jaime Caro J, Cowie MR, Ford I, Paty JA, Riley JP, Swedberg K, Tavazzi L, Wiklund I, Kirchhof P. The importance of patient-reported outcomes: a call for their comprehensive integration in cardiovascular clinical trials. European heart journal. 2014;35:2001–9. doi: 10.1093/eurheartj/ehu205. [DOI] [PubMed] [Google Scholar]
- 48.McCabe C, Bennett M, Doughty N, MacKenzie Ross R, Sharples L, Pepke-Zaba J. Patient-reported outcomes assessed by the CAMPHOR questionnaire predict clinical deterioration in idiopathic pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Chest. 2013;144:522–30. doi: 10.1378/chest.12-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hagan G, Southwood M, Treacy C, Ross RM, Soon E, Coulson J, Sheares K, Screaton N, Pepke-Zaba J, Morrell NW, Rudd JH. (18)FDG PET imaging can quantify increased cellular metabolism in pulmonary arterial hypertension: A proof-of-principle study. Pulmonary circulation. 2011;1:448–55. doi: 10.4103/2045-8932.93543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tavazzi L, Maggioni AP, Tognoni G, Gissi Participation versus education: the GISSI story and beyond. American heart journal. 2004;148:222–9. doi: 10.1016/j.ahj.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 51.Kawut SM, Bagiella E, Lederer DJ, Shimbo D, Horn EM, Roberts KE, Hill NS, Barr RG, Rosenzweig EB, Post W, Tracy RP, Palevsky HI, Hassoun PM, Girgis RE. Randomized clinical trial of aspirin and simvastatin for pulmonary arterial hypertension: ASA-STAT. Circulation. 2011;123:2985–93. doi: 10.1161/CIRCULATIONAHA.110.015693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singh TP, Rohit M, Grover A, Malhotra S, Vijayvergiya R. A randomized, placebo-controlled, double-blind, crossover study to evaluate the efficacy of oral sildenafil therapy in severe pulmonary artery hypertension. American heart journal. 2006;151:851 e1–5. doi: 10.1016/j.ahj.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 53.Kopec JA, Abrahamowicz M, Esdaile JM. Randomized discontinuation trials: utility and efficiency. Journal of clinical epidemiology. 1993;46:959–71. doi: 10.1016/0895-4356(93)90163-u. [DOI] [PubMed] [Google Scholar]
- 54.Rubenfire M, McLaughlin VV, Allen RP, Elliott G, Park MH, Wade M, Schilz R. Transition from IV epoprostenol to subcutaneous treprostinil in pulmonary arterial hypertension: a controlled trial. Chest. 2007;132:757–63. doi: 10.1378/chest.06-2118. [DOI] [PubMed] [Google Scholar]
- 55.Channick RN, Frantz RP, Kawut SM, Palevsky H, Tumuluri R, Sulica R, Lauto PO, Benton WW, de Boisblanc B. A multicenter, retrospective study of patients with pulmonary arterial hypertension transitioned from parenteral prostacyclin therapy to inhaled iloprost. Pulmonary circulation. 2013;3:381–8. doi: 10.4103/2045-8932.114768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gabler NB, Duan N, Vohra S, Kravitz RL. N-of-1 trials in the medical literature: a systematic review. Medical care. 2011;49:761–8. doi: 10.1097/MLR.0b013e318215d90d. [DOI] [PubMed] [Google Scholar]
- 57.Joy TR, Monjed A, Zou GY, Hegele RA, McDonald CG, Mahon JL. N-of-1 (Single-Patient) Trials for Statin-Related Myalgia. Ann Intern Med. 2014;160:301–310. doi: 10.7326/M13-1921. [DOI] [PubMed] [Google Scholar]
- 58.Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA, 3rd, Loyd JE. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345:319–24. doi: 10.1056/NEJM200108023450502. [DOI] [PubMed] [Google Scholar]
- 59.Larson EB. N-of-1 clinical trials. A technique for improving medical therapeutics. The Western journal of medicine. 1990;152:52–6. [PMC free article] [PubMed] [Google Scholar]
- 60.Lillie EO, Patay B, Diamant J, Issell B, Topol EJ, Schork NJ. The n-of-1 clinical trial: the ultimate strategy for individualizing medicine? Personalized medicine. 2011;8:161–173. doi: 10.2217/pme.11.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH, Rosenson RS, Rubin LJ, Tapson VF, Varga J. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53:1573–619. doi: 10.1016/j.jacc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 62.Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–9. doi: 10.1056/NEJMoa052256. [DOI] [PubMed] [Google Scholar]