

Abstract
Immune responses must be well restrained in a steady state to avoid excessive inflammation. However, such restraints are quickly removed to exert anti-microbial responses. Here, we report a role of autophagy in an early host anti-fungal response by enhancing NFκB activity through A20 sequestration. Enhancement of NFκB activation is achieved by autophagic depletion of A20, an NFκB inhibitor, in F4/80hi macrophages in the spleen, peritoneum, and kidney. We show that p62, an autophagic adaptor protein, captures A20 to sequester it in the autophagosome. This allows the macrophages to release chemokines to recruit neutrophils. Indeed, mice lacking autophagy in myeloid cells show higher susceptibility to Candida albicans infection due to impairment in neutrophil recruitment. Thus, at least in the specific aforementioned tissues, autophagy appears to break A20-dependent suppression in F4/80hi macrophages, which express abundant A20 and contribute to the initiation of efficient innate immune responses.
Introduction
Autophagy is a highly conserved cellular process in eukaryotes, and is induced by various pathogen-associated molecular patterns (PAMPs) and cytokines to eliminate intracellular microbes through autophagosomal digestion in mammals1, 2, 3. Autophagy can unselectively eliminate cytoplasmic proteins and organelles, and contributes to amino acid recycling within a cell. At the same time, selective degradation of particular proteins is mediated by autophagy adaptor proteins such as p62, also called sequestosome-1, which contributes to maintenance of cellular activity. Recent studies have shown that autophagy eliminates assembled inflammasomes in macrophages4 and BCL10 to limit T cell receptor (TCR) signaling in T cells5, both resulting in downregulation of immune responses. Thus, autophagy is currently known to suppress immune responses to protect hosts from possible collateral damage caused by overly active immunity6.
It is reported that dectin-1, a fungal cell wall component β-glucan receptor, triggers conversion of microtubule-associated protein 1 light chain 3 (LC3)-I to LC3-II7, which is used to monitor autophagy. A recent report demonstrated a protective role of autophagy in Candida infection8, while others showed that autophagy is not critical for host protection against C. albicans9, 10. Thus, the specific role(s) of autophagy during Candida infection may be considered controversial.
Various cell types in the host immune system play a protective role against Candida infection. For example, neutrophils prevent fungal growth and invasion, particularly at the early stage of infection. Therefore, neutropenia is a major causal factor in disseminated candidiasis11, 12. Neutrophils prevent Candida hyphal formation by phagocytosed conidia, although macrophages are destroyed by germinated hyphae of ingested Candida13. As a result, neutrophils are viewed as critical cells for the host’s protection against Candida infection. Because of their short life span (half-life of ~6 h in circulation14), prompt recruitment of neutrophils from BM is critical for host protection. On the other hand, tissue-resident macrophages are considered to be an important source of neutrophil chemoattractants; thus, deletion of tissue-resident macrophages results in the failure of neutrophil recruitment and neutrophil-dependent immune responses15, 16, 17. F4/80hi macrophages are largely tissue-resident18, 19, 20, 21, although it is possible that F4/80hi macrophages from different organs have tissue-specific phenotypes. In contrast, as previously demonstrated with BM chimera mice, 95–100% of F4/80lo macrophages in various organs are replaced by donor BM cells20. It is also known that, during infection, F4/80lo monocytes/macrophages are recruited in the infected sites 22, 23, while the spleen has a number of monocytes even in naïve mice. Distinct gene expression pattern between F4/80lo and F4/80hi macrophages20 suggests different functions between the two populations. Currently, it is unknown which macrophage subset is the main producer of neutrophil-chemoattractants in fungal infection.
Host cells are equipped with various mechanisms that negatively regulate immune responses to avoid hyper-inflammation and autoimmunity. For example, A20 acts as a pivotal NFκB suppressor under Toll-like receptors (TLRs), and tumor necrosis factor receptor (TNFR)24. A20 deficiency causes severe autoimmune inflammation by excessive NFκB activation25, 26, 27. However, such a negative regulatory system, if sustained, hampers host immune responses to act quickly on the clearance of pathogens. In order to solve this dilemma, immune suppression by A20 must be strictly controlled when infections occur. Therefore, timely downregulation of A20 is considered to be important during acute infections. Although NFκB is known to induce A20 expression28, the mechanism underlying the negative regulation of A20 is not clear.
In the current study, we evaluate anti-fungal immunity in mice conditionally lacking autophagy-related protein-7 (ATG7), essential for autophagosome formation29 and LC3-associated phagocytosis (LAP)30, in myeloid cells. Our data demonstrate that autophagy downregulates A20 in F4/80hi macrophages through p62-associated autophagic sequestration and that the resulting NFκB activation induces expression of chemokines at an early stage of Candida infection. Based on these results, we suggest a mechanism by which autophagy protects hosts by enhancing NFκB signaling in tissue-resident macrophages, which indeed play a sentinel role at the early stage of fungal infections.
Results
Lack of ATG7 attenuates host resistance against Candida
To assess the impact of ATG7 on fungal immunity, we generated Atg7fl/fllysozyme M (LysM)cre/+ mice (denoted as “Atg7 CKO mice” hereafter), in which myeloid cells, such as macrophages, DCs and neutrophils, lack autophagy. The mice were confirmed that Atg7 mRNA expression was abolished in peritoneal macrophages (Supplementary Fig. 1). When the mice were intravenously injected with either 106 (Fig. 1a, b) or 2×105 (Fig. 1c, d) conidia of Candida, weights and the survival of Atg7 CKO mice were significantly reduced compared to control LysMcre/+ mice (denoted as “WT mice” hereafter). Importantly, the significant difference in weight reduction was observed on day 1 or 2 (Fig. 1b, d). Therefore, the impact of autophagy in myeloid cells on host protection was apparent at a very early stage (24–48 hrs) of fungal infection. We next compared fungal burdens in WT and Atg7 CKO mice in the kidney and spleen on day 3. Fungal loads in both organs were significantly increased in Atg7 CKO mice (Fig. 1e), and the increased fungal load was also histologically identified in the kidney (Fig. 1f).
Figure 1. Absence of ATG7 in myeloid cells decreases host resistance against Candida infection.

(a–d) Atg7fl/flLysMcre/+ (Atg7 CKO)(n=9) and LysMcre/+ (WT)(n=12) mice were systemically infected by i.v. injection of C. albicans. Mouse survival (a, c) and weight (b, d) were monitored. Innocula of Candida were 106 spores/mouse to Atg7 CKO (n=9) and WT (n=12)(a, b), and 2×105 spores/mouse to Atg7 CKO (n=12) and WT(n=10)(c, d). (e) Fungal burdens in the kidney and the spleen of Atg7 CKO and WT mice 3 days after 106 C. albicans i.v. injection. One circle denotes a result from one mouse. (f) H&E and silver staining of sections from kidneys of WT and Atg7 CKO mice 3 days after 106 C. albicans i.v. injection. Candida is stained with black in silver staining. Magnified views of the panels in the middle (yellow rectangles) are shown on the right. Scale bars in the left and middle panels indicate 200 μm; and scale bars in the panels on the right indicate 60 μm. Data are representative of independent two experiments. Error bars represent mean ± SD. Student’s t-test was used for statistical analysis. *; p < 0.05, **; p <0.01.
ATG7 has no direct influence on fungal killing
In an effort to describe the protection mechanism of autophagy in innate immune responses against fungal pathogen, we first examined whether or not Candida could induce autophagy in innate immune cells. Macrophage cell line (RAW-Blue cell) was co-cultured with Candida conidia, and the induction of autophagy was evaluated by detecting LC3 puncta formation and LC3-II induction. Sixty minutes after the stimulation with conidia, LC3 puncta formation was clearly observed (Fig. 2a). Immunoblotting data also confirmed induction of autophagy as quickly as 5 min following the co-culture set up (Fig. 2b). Autophagy is known to enhance phagocytosis of viral, bacterial, and parasitic pathogens1, 2, 3. We thus next examined whether or not ATG7-mediated phagocytic activity in macrophages and neutrophils. The phagocytic capacity of both bone marrow-derived macrophages (BMDMs) and neutrophils, obtained from Atg7 CKO and WT mice, was assessed by flow cytometry. Unexpectedly, the absence of autophagy did not affect the percentages of host cells showing a fluorescent marker for Candida (Fig. 2c, d) and phagocytic index (the number of Candida detected with single macrophages)(Fig. 2e). No change in Atg7 CKO was found also in thioglycollate-elicited peritoneal and total splenic macrophages (Supplementary Fig. 2).
Figure 2. Autophagy is induced by Candida but does not play a role in Candida uptake by macrophages and neutrophils.

(a) RAW-Blue cells (macrophage cell line) were co-cultured with Candida labeled with Alexa Fluor-647 (AF-647)(pink) for 0 or 60 min at the 1:1 ratio of Raw-Blue and spore cell. LC3 and nuclei were shown with green and blue, respectively. Corresponding Nomarski images are shown below. Scale bars = 5 μm. (b) Western blotting detection of LC3II in RAW-Blue cells stimulated with C. albicans (1:1 cell ratio) at the indicated time points. (c–e) BMDMs (c) and neutrophils (d) obtained from Atg7 CKO and WT mice were fluorescently labeled and co-cultured with AF-647-labeled Candida (1:5 ratio of host cells and Candida spores) for indicated duration. AF-647-positive cells were determined by flow cytometry. Intensity of AF-647 (Candida) in BMDMs at 30 min was assessed by flow cytometry analysis (e). The histogram is pre-gated on F4/80+. (f) To determine macrophage ROS production, dihydroethidium (DHE) staining in BMDMs was evaluated by flow cytometry 2 hrs after co-culture with Candida (1:1 ratio of BMDMs and spores). (g–i) Inhibition of Candida expansion by BMDM (g, h) and neutrophils (i) was evaluated by the method described in the METHODS section. Candida expansion with or without BMDMs was shown at indicated time points (g), and inhibition of Candida expansion was evaluated at 12 hrs after co-culture with host cell culture. (j) Intensity of LC3 signals (green) in a Candida (pink)-engulfed RAW-Blue cell at 60 min after starting co-culture was analyzed along with the yellow line in the panel at right bottom. Scale bar = 2.5 μm. Shown in (a) and (j) are typical examples from 100 cells observed in four independent experiments. Results in (b-i) are representatives of at least two experiments with more than three mice/group. Error bars represent mean ± SD. Student’s t-test was used for statistical analysis. N.S.; not significant. **; p <0.01. ***; p < 0.001.
Because flow cytometry does not distinguish Candida-engulfed host cells from those simply attaching to Candida, we eye-counted Candida-engulfed macrophages from at least 100 cells randomly captured by confocal microscopic images. The results also indicated that WT and Atg7 CKO macrophages had similar abilities to phagocytose Candida (Supplementary Fig. 3). In addition, the generation of reactive oxygen species (ROS), toxic for ingested microbes including Candida, was also unaltered in Atg7-deficient macrophages (Fig. 2f). Our ex vivo assay to evaluate host cell inhibition of Candida growth showed comparable results between WT and Atg7 CKO with BMDMs, thioglycollate-elicited peritoneal macrophages, total splenic macrophages, and neutrophils (Fig. 2g–i; Supplementary Fig. 2b, d). These results suggested that ATG7 likely does not play a role either in host cell-intrinsic functions to phagocytose, or in inhibiting expansion of Candida both in macrophages and neutrophils. This is in clear contrast to the role of autophagy in infections by viruses, bacteria, and parasites.
We then tried to evaluate the time course of live Candida engulfment in macrophages, particularly within LC3 cargos. A few conidia were phagocytosed at 5 and 10 min, and we could not evaluate the presence of Candida-containing LC3 cargos. Yet, complete phagocytosis of conidia was observed at 30, 45 and 60 min (Supplementary Fig. 4a). Unexpectedly, no conidia were found within LC3 cargos, but LC3 protein was rather randomly distributed (Fig. 2j; Supplementary Fig. 4a; Supplementary Table 1). Spores contained within LC3 cargos were sought in at least 100 macrophages at each time point, but we found no spores contained in LC3 cargos. As a positive control, we could confirm LC3 recruitment to zymosan particles (Supplementary Fig. 4b, c; Supplementary Table 1), as previously reported7. These data suggest that live Candida was not cleared in autophagosomes; and this is also a clear contrast to the role of autophagy against viruses, bacteria, and parasites.
Autophagy increases neutrophil recruitment
Atg7 CKO mice showed severe weight reduction as early as day 1 (Fig. 1b). As neutrophils are responsible for Candida clearance at the early stage of infection12, 31, we investigated if the lack of autophagy affected neutrophil recruitment to the kidney and spleen at 4 and 24 hrs post infection (hpi). The four-hour time point was too early to statistically evaluate neutrophil infiltration; but, the result at 24-hpi using WT mice showed a sizeable increase in numbers of neutrophils (Supplementary Fig. 5). At 24-hpi, the cellularity of total splenocytes in Atg7 CKO mice was comparable to that in WT mice (Supplementary Fig. 6a); however, Atg7 CKO mice showed significantly reduced neutrophil recruitment in both the kidney and spleen (Fig. 3a, b). Interestingly, WT mice showed a more drastic decrease in BM neutrophil population at 24-hpi than did Atg7 CKO mice after infection (Fig. 3c), while WT and Atg7 CKO naïve mice had similar sizes of the BM neutrophil population in an uninfected condition (Fig. 3d, e). This suggests that Atg7 CKO mice had less neutrophil egress from the BM than did WT mice. To further evaluate the neutrophil recruitment to infected sites, we injected Candida to the peritoneal cavity of Atg7 CKO and WT mice. The lack of Atg7 significantly reduced recruitment of neutrophils in the peritoneal cavity (Fig. 3f). Collectively, these results suggest the general and noteworthy contribution of autophagy in neutrophil recruitment to inflammatory sites.
Figure 3. Autophagy enhances neutrophil recruitment and clearance of Candida.

Indicated mouse groups received i.v. injection of 106 Candida spores (except for (f)), and neutrophils (CD11b+Ly6G+) were enumerated in indicated organs 24 hrs post infection by flow cytometry. (a, b) Proportions and numbers of neutrophils in the kidney from WT and Atg7 CKO mice (a); and in the spleen from naïve or infected WT mice (b, n=4). (c–e) Proportions of neutrophils in BM 24 hrs after Candida infection (c). Representative flow cytometry CD11b/Ly6G staining patterns (d) and proportions (e) of neutrophils in BM in naïve mice. WT and Atg7 CKO mice were compared. (f) Proportions and numbers of neutrophils infiltrated into the peritoneal cavity at 18 hrs after i.p. injection of 5×106 Candida spores. One circle denotes a result from one mouse (a, e, f). Horizontal lines indicate mean values. Data are representative of two independent experiments. Error bars represent mean ± SD. Student’s t-test was used for statistical analysis for (a), (c), (e) and (f) and ANOVA was used for (b). N.S.; Not significant. *; p < 0.05. **; p <0.01. ***; p < 0.001.
We evaluated immune cells other than neutrophils in the spleen at 24-hpi. Proportions and numbers of splenic conventional dendritic cells (cDCs) were slightly less in Atg7 CKO mice than in WT mice (Supplementary Fig. 6b), suggesting that recruitment of cDCs was somewhat affected by autophagy. Proportions and numbers of plasmacytoid DCs (pDCs), B cells, CD8+ and CD4+ T cells did not show a significant difference between WT and Atg7 CKO mice either before or after infection (Supplementary Fig. 6c). Numbers of F4/80lo and F4/80hi macrophage populations (gating strategy in Supplementary Fig. 6d), were also unaltered (Supplementary Fig. 6e). In sum, increased numbers of neutrophils recruited to inflammatory sites appear to be important in autophagy-mediated host resistance during the early stage of Candida infection.
Autophagy enhances production of neutrophil chemoattractants
CXCR2-deficiency in mice reduces neutrophil influx into Candida-infected sites, and increases susceptibility to gastric candidiasis and acute systemic candidiasis32 We first found that expression of CXCR2 was comparable between WT and Atg7-deficient neutrophils in the BM, peripheral blood, and the spleen (Fig. 4a). Chemotactic ability toward CXCL1 and CXCL2, CXCR2 ligands, was also comparable between WT and Atg7-deficient neutrophils (Fig. 4b). These results suggested that Atg7-deficient neutrophils have normal migration ability. We next assessed levels of major neutrophil chemoattractants in the serum and kidney at 24-hpi. Atg7 CKO mice showed significantly lower concentrations of both CXCL1 and/or CXCL2 in both the serum and kidney (Fig. 4c, d). These results suggest that autophagy increases neutrophil recruitment by enhancing production of chemoattractants at the site of inflammation, but not by enhancing the migratory ability of neutrophils.
Figure 4. Autophagy induced levels of CXCL1 and CXCL2 in serum and the kidney.

(a) Histogram and MFI of CXCR2 staining determined by flow cytometry. Neutrophils were obtained from the BM, blood, and spleen from WT and Atg7 CKO mice at 24 hrs after i.v. injection of 106 Candida spores. (b) Transwell migration assay of neutrophils towards CXCL1 and CXCL2. Neutrophils were FACS-sorted as CD11b+Ly6G+ from the BM of naïve mice, and plated in the upper chamber of transwell. Indicated concentrations of CXCL1 and CXCL2 were added in the lower chamber. Cells were cultured for 1 hr and the transmigration of neutrophils was assessed by flow cytometry. Data are representative of two independent experiments with at least three mice per each group (a, b). Error bars represent mean ± SD. (c, d) Levels of chemokines in the serum (c) and the kidney (d) from from WT and Atg7 CKO mice. Naïve mice and infected mice at 24 hrs after i.v. injection of 106 Candida spores were analyzed. One circle denotes a result from one mouse. Horizontal lines indicate values of the mean. Error bars represent mean ± SD. ANOVA was used for statistical analysis of (a, b) and Student’s t-test was used for (c, d). N.S., not significant. *; p < 0.05. **; p <0.01.
Source of CXCL1 and CXCL2 early after C. albicans infection
To determine the cellular source of CXCL1 and CXCL2, we sorted splenic F4/80hi macrophages, F4/80lo macrophages, cDCs, pDCs, NK cells, T cells and B cells, by FACS from spleens of WT mice at 24-hpi, and then cultured the cells for 24 hrs. F4/80hi macrophages were the only cell type that produced high levels of CXCL1 and CXCL2 (Fig. 5a). When F4/80hi macrophages were depleted from total splenocytes, the levels were significantly reduced (Fig. 5b). Thus, F4/80hi macrophages appear to be critical producers of CXCL1 and CXCL2.
Figure 5. F4/80hi macrophages are a main source of CXCL1 and CXCL2 early after Candida infection.
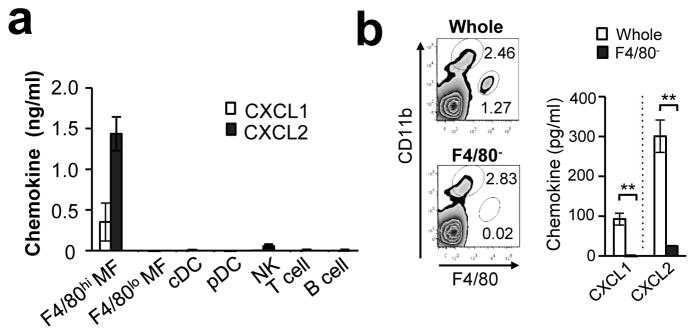
(a) CXCL1 and CXCL2 production by various splenic cell populations. Indicated splenic cell populations were purified by FACS from the spleen of mice 24 hrs after 106 Candida i.v. inoculation, and each population was cultured for 24 hrs (106 cells/ml). The chemokine production in the supernatants was assessed by ELISA. n=3. (b) Splenocytes were obtained from mice 24 hrs after i.v. injection of 106 Candida spores. F4/80hi macrophage population was depleted by magnetic beads, and the rest of splenocytes were confirmed to be lacking the F4/80hi population by flow cytometry (left panels). Whole splenocytes and F4/80hi macrophages-removed splenocytes (F4/80−) were cultured (5×107/ml) for 24 hrs and chemokine production in the supernatants was evaluated by ELISA. n=3. Error bars represent mean ± SD. Student’s t-test was used for statistical analysis. **; p <0.01.
Autophagy enhances chemokine production
We next examined the potential impact of Atg7-deficiency in the production of CXCL1 and CXCL2 in vivo. At 24-hpi, splenic WT F4/80hi macrophages expressed significantly higher levels of Cxcl1 and Cxcl2 mRNA than did Atg7-deficient F4/80hi macrophages and F4/80lo macrophages of WT and Atg7−/− backgrounds (Fig. 6a). High levels of CXCL2 protein expression was also identified in splenic WT F4/80hi macrophages, but not in Atg7-deficient, or F4/80lo macrophages in infected mice (Fig. 6b). Similarly, gene expression of Ccl3 and Ccl4 was highly expressed in splenic WT F4/80hi macrophages from infected mice (Supplementary Fig. 7a). (Note that CCL3 and CCL4 are ligands of C-C chemokine receptor 5 (CCR5), and act as chemoattractants for monocyte-derived macrophages and DCs33.)
Figure 6. ATG7-deficiency decreased chemokine production by F4/80hi peritoneal-resident macrophages.

(a) Levels of Cxcl1 and Cxcl2 mRNA in splenic F4/80lo and F4/80hi macrophages obtained from WT and Atg7 CKO mice 24 hpi with 106 Candida spores. (b) Levels of CXCL2 protein in supernatants of macrophages subsets indicated in (a). n=3. (c) CXCL2 levels in culture supernatants of renal F4/80hi macrophages from naïve mice. Cells were stimulated with zymosan (100 μg/ml) for 24 hrs. n=3. (d) Expression of Cxcl1 and Cxcl2 mRNA in peritoneal F4/80hi macrophages obtained from naïve mice. Cells were stimulated with zymosan (Zym; 100 μg/ml) or heat-killed Candida (HKCA; 5×105/ml) for 3 hrs. (e) Levels of CXCL1 and CXCL2 proteins in culture supernatants of F4/80hi peritoneal macrophages stimulated with Pam3CSK4 (Pam; 100 ng/ml), zymosan (100 μg/ml), or curdlan (100 μg/ml) for 24 hrs. n=3. (f) Peritoneal F4/80hi macrophages were pretreated with 3-MA (10 mM), then stimulated 2 hrs later with Pam3CSK4 (100 ng/ml) for additional 24 hrs. Supernatants were analyzed by ELISA to measure CXCL1 (left panel) and CXCL2 (right panel) production. (g) Peritoneal F4/80hi macrophages were pretreated with rapamycin (1 μg/ml) for 1 hr, then zymosan (100 μg/ml) was added and harvested 3 hrs later. Cxcl1 and Cxcl2 mRNA expression was assessed by real-time PCR analysis. n=3. (h, i) A million F4/80hi peritoneal macrophages of WT or Atg7 CKO F4/80hi were transferred into the peritoneal cavity of Atg7 CKO mice; then 5×106 Candida spores were i.p. injected to the mice. Recruitment of neutrophils (h) and fungal burden (i) in the peritoneal cavity was evaluated 20 hpi. One circle denotes a result from one mouse. In all the experiments in Fig. 6, except for (h) and (i), cells were pooled from at least three mice for one group to get enough numbers of macrophages. Data are representative of at least two independent experiments. Error bars for mRNA data denote RQ-Max/Min as described in the Method section. Error bars for protein data represent mean ± SD. ANOVA and Student’s t-test were used for (e, f) and (b, c), respectively. N.S.; not significantly different. *; p < 0.05 and **; p <0.01.
We have shown, importantly, that ATG7-sufficient splenic F4/80hi macrophages produce high levels of chemokines both by in vivo and ex vivo. As Candida is accumulated in the kidney, we FACS-sorted renal F4/80hi macrophages, and stimulated the cells with zymosan ex vivo. Again, the absence of ATG7 in the cells reduced CXCL2 production (Fig. 6c). We next tested chemokine expression in F4/80hi peritoneal macrophages, a dominant cell population in the peritoneal cavity of naïve mice21. We obtained peritoneal-resident F4/80hi macrophages, and then stimulated the cells ex vivo with either zymosan (a cell wall component from Saccharomyces cerevisiae), heat killed C. albicans (HKCA), Pam3CSK4, or curdlan. Atg7-deficient peritoneal F4/80hi macrophages again showed reduced expression of CXCL1, CXCL2, CCL3, and CCL4 at the level of mRNA and/or protein after cell stimulation, compared to WT cells (Fig. 6d, e; Supplementary Fig. 7b). As expected, WT peritoneal F4/80hi macrophages expressed significantly higher levels of chemokines compared to WT peritoneal F4/80lo (Supplementary Fig. 8a), as found in the splenic macrophages. The absence of ATG7 had no impact on CXCL2 production in F4/80lo peritoneal macrophages (Supplementary Fig. 8b). Importantly, inhibition of autophagy with 3-methyladenine (3-MA) significantly reduced CXCL1 and CXCL2 production by WT F4/80hi peritoneal macrophages stimulated with Pam3CSK4 (Fig. 6f). Indeed, induction of autophagy by rapamycin treatment enhanced the gene expression of Cxcl1 and Cxcl2 induced by zymosan (Fig. 6g). As an additional representative of F4/80lo macrophages, BMDMs were also tested. Atg7-deficient BMDMs did not alter chemokine expression in CCL1, CCL2, CCL3, and CCL4 upon zymosan stimulation (Supplementary Fig. 9a). Further, treatment with rapamycin did not alter chemokine production by BMDMs (Supplementary Fig. 9b). Therefore, autophagy induces expression of tested chemokines in F4/80hi macrophages but not in F4/80lo macrophages.
We then tested whether mice, intraperitoneally transferred with Atg7-deficient F4/80hi macrophages, have less ability to recruit neutrophils and clear Candida, compared to mice transferred with WT F4/80hi macrophages. Less neutrophil recruitment and more fungal burden were identified in the peritoneal cavity of the recipients transferred with Atg7-deficient F4/80hi macrophages compared to those with WT F4/80hi macrophages (Fig. 6h, i), although Atg7-deficiency in BMDMs transferred to peritoneal cavity of recipients did not affect neutrophil recruitment and Candida clearance (Supplementary Fig. 10). Taken together, results using various sources of macrophages suggested that ATG7 works through autophagy to induce chemokine production by F4/80hi macrophages, and plays a critical role in neutrophil-mediated fungal clearance.
Autophagy does not affect cell death or PRR expression
We next sought to identify the mechanism for the autophagy-mediated induction of chemokine expression by F4/80hi macrophages. First, there was no difference in cell survival between WT and Atg7-deficient F4/80hi macrophages (Supplementary Fig. 11a). Inhibition of autophagy with 3-MA in RAW-Blue cells also did not alter the proportion of live cells, compared to untreated cells (Supplementary Fig. 11b). Absence of autophagy did not seem to affect the expression levels of PRRs (pattern-recognition receptors) that detect Candida, such as TLR2, TLR4, dectin-1, and dectin-2, in F4/80hi macrophages from the spleen, kidney, and peritoneal cavity following Candida infection (Supplementary Fig. 11c–e). While F4/80hi macrophages produce more chemokines than do F4/80lo macrophages, this phenomenon was also not explained by expression levels of TLR2, TLR4, dectin-1, and dectin-2 (Supplementary Fig. 12). These results suggested that autophagy contributes to enhancement of chemokine secretion in F4/80hi macrophages without inhibiting cell death or upregulation of PRR expression.
Here, we have evaluated the ability of host cells to phagocytose Candida and to inhibit Candida growth, as shown in Fig. 2 and Supplementary Fig. 2 and 3, by using peritoneal F4/80hi macrophages. Again, the absence of ATG7 in F4/80hi macrophages did not alter Candida phagocytosis nor inhibited Candida growth, although LC3 puncta were clearly produced in WT F4/80hi macrophages (Supplementary Fig. 13a–d). Interestingly, F4/80hi macrophages were more susceptible than were F4/80lo macrophages to intracellular germination of Candida (Supplementary Fig. 13e–g), suggesting that F4/80hi macrophages are easily killed by Candida after completing their job as sentinels to recruit mainly neutrophils.
Autophagy enhances NFκB activation in F4/80hi macrophages
A previous report showed that knockdown of either Atg5 or Atg7 inhibits NFκB signaling through TNFR34. Therefore, we sought to identify the mechanism in activation of NFκB, which induces gene expression of Cxcl1, Cxcl2, Ccl3, and Ccl435, 36, 37. We first confirmed that an NFκB inhibitor strongly suppressed CXCL1 and CXCL2 production by F4/80hi peritoneal macrophages, stimulated with zymosan or HKCA (Fig. 7a). Indeed, Atg7-deficient F4/80hi macrophages showed lower levels of NFκB activity than did WT F4/80hi macrophages, as demonstrated by less IκB phosphorylation, greater IκB levels (i.e., less IκB degradation)(Fig. 7b), impaired NFκB p65 nuclear translocation (Fig. 7c; Supplementary Table 2), and impaired NFκB p65 binding to a target DNA promoter sequence (Fig. 7d). Inhibiting autophagy with 3-MA also downregulated NFκB activity in RAW-Blue cells detected with a secreted embryonic alkaline phosphatase (SEAP) reporter, and nuclear translocation assays (Supplementary Fig. 14a, b). Taken together, these results strongly suggest that autophagy positively regulates NFκB activation in F4/80hi macrophages.
Figure 7. Autophagy enhances NFκB activation in F4/80hi macrophages.

(a) Production of CXCL1 and CXCL2 by F4/80lo and F4/80hi peritoneal macrophages. Cells were stimulated with zymosan (100 μg/ml) or (HKCA; 5×105 spores/ml) for 24 hrs with or without an NFκB inhibitor, QNZ. Culture supernatants were analyzed by ELISA. n=3. (b) Western blotting analysis of p-IκB and total IκB in F4/80hi peritoneal macrophages stimulated with zymosan (100 μg/ml) for 30 min. (c) Translocation of NFκB p65 (green) to the nucleus (DAPI, blue) in F4/80hi peritoneal macrophages stimulated with Pam3CSK4 (100 ng/ml) for 30 min. NFκB localization was analyzed by confocal microscopy. Yellow broken lines in enlarged photos indicate borders of the nucleus. Shown are representative images from at least 50 cells. Scale bars indicate 5 μm. (d) Relative activity of NFκB in WT and Atg7 CKO F4/80hi peritoneal macrophages was evaluated. Cells were stimulated with zymosan (100 μg/ml) for 30 min, and p62 binding to a target NF-κB response element in DNA was evaluated by colorimetric reading. Values were calculated as a fold increase of O.D. values of stimulated cells from those of unstimulated cells. n=3. Data are representative of two independent experiments. Error bars represent mean ± SD. ANOVA and Student’s t-test were used for statistical analysis of (a) and (d), respectively. N.S.; not significant. ***; p < 0.001.
Autophagy sequesters A20 to induce NFκB activation
Since autophagy is known to specifically sequester certain proteins, we hypothesized that it also eliminates an NFκB signaling inhibitor to induce NFκB activity. For example, A20 is known to inhibit NFκB signaling25, 38 by deubiquitinating IκB kinase gamma (IKKγ; also known as NFκB essential modifier, NEMO) and TRAF639. A20-deficient mice spontaneously develop multi-organ inflammation and die prematurely26. Here, we sought to show whether or not autophagy sequesters A20, particularly because A20 was shown to colocalize with lysosomes subsequent to TNF-α stimulation40. Stimulated Atg7-deficient peritoneal F4/80hi macrophages indeed showed higher levels of A20 protein than did WT F4/80hi macrophages (Fig. 8a; Supplementary Fig. 15). Our time-course results of A20 levels showed a peak at 1 hr after Pam3CSK4 stimulation for WT and Atg7 CKO macrophages; but, the initial increase in Atg7-deficient F4/80hi macrophages were much higher than that in WT cells (Fig. 8a, b). Even after the peak, Atg7-deficient F4/80hi cells maintained A20 levels significantly higher than those of WT F4/80hi cells (Fig. 8a, b). As expected from previous studies26, the degree of NFκB activation showed a reverse correlation with A20 levels -- weak NFκB activation in Atg7 CKO macrophages than WT macrophages (Fig. 8a, b). Here, we confirmed highly K63 de-ubiquitinated IKKγ, suggesting the increased impact of A20 in Atg7 CKO F4/80hi macrophages (Fig. 8c; negative controls in Supplementary Fig. 16a. b). ATG7-specific downregulation of A20 did not occur at the mRNA level (Fig. 8d), suggesting post-translational downregulation of A20 in WT F4/80hi macrophages. Indeed, autophagy negatively regulated A20 protein levels as evidenced by 3-MA treatment on WT F4/80hi macrophage increasing A20 expression 1 hr after Pam3CSK4 stimulation (Fig. 8e).
Figure 8. Absence of ATG7 decreases A20 levels in F4/80hi macrophages.

(a, b) Turnover of A20, pIκB and total IκB protein levels in F4/80hi peritoneal macrophages (WT vs. Atg7 CKO) stimulated with Pam3CSK4 (100 ng/ml). Cell lysates were harvested at indicated time points and submitted for Western detection (a). Abundance of proteins evaluated by densitometry was plotted for A20, p-IκB normalized, total IκB normalized with β-actin, total IκB, and β-actin, respectively. (c) Detection of K63 ubiquitination of IKKγ. F4/80hi peritoneal macrophages were stimulated with zymosan (100 μg/ml) for 30 min. IKKγ was immunoprecipitated and K63 ubiquitination of IKKγ was detected by immunoblotting with K63-Ub antibody. Results of negative control using unstimulated cells and control IgG are shown in Suppl. Fig. 15a, b. (d) Expression of Tnfaip3 mRNA in F4/80hi peritoneal macrophages (WT vs. Atg7 CKO) stimulated with Pam3CSK4 (100 ng/ml) for indicated duration. Evaluated by qPCR. Error bars for mRNA data denote RQ-Max/Min as described in the Method section. (e) Expression of A20 in peritoneal F4/80hi macrophages stimulated by Pam3CSK4 (100 ng/ml) for 1 hr in the absence or presence of 3-MA (5 μM). (f) Confocal microscopy analysis to localize A20 and LC3. NIH-3T3 cells were transfected with V5-tagged A20 expression vector, and stimulated with Pam3CSK4 (100 ng/ml) for 60 min. LC3 and V5 antibodies were used to detected LC3 and A20, respectively. Scale bars in unmagnified and magnified panels indicate 10 μm and 4 μm, respectively. A20, LC3, and nuclei were shown with green, pink, and blue, respectively. White arrows indicate co-localization of A20 and LC3. (g) Western blotting detection of A20 in naïve peritoneal F4/80hi macrophages obtained from WT and Tnfaip3+/− mice. (h) Levels of CXCL1 in culture supernatants of WT and Tnfaip3+/− F4/80hi macrophages stimulated with Pam3CSK4 (100 ng/ml) for 24 hrs. Indicated groups were treated with 3-MA (3 μM) starting an hour before the addition of Pam3CSK4. CXCL1 production was evaluated by ELISA. n=3. Data shown in all the panels are representatives of at least two independent experiments. Error bars represent mean ± SD. Student’s t-test was used. N.S.; not significant. *; p < 0.05.
Here, we hypothesized that autophagy sequesters A20, and sought to first identify LC3 and A20 co-localization. (In this regard, we had to overexpress A20 in 3T3 cells, since detection of endogenous A20 by confocal microscopy was difficult.) Although A20 was not colocalized with LC3 in unstimulated cells (Supplementary Fig. 16c), its colocalization was identified after cell stimulation (Fig. 8f; Supplementary Table 3). To further evaluate the impact of autophagy-mediated A20 downregulation in CXCL1 expression, we used Tnfaip3+/− (A20 heterozygous knock-out) peritoneal F4/80hi macrophages, which have reduced levels of A20 expression compared to WT macrophages (Fig. 8g). (Knocking-down A20 and using Tnfaip3 homozygous knock-out were not technically optional here because of (1) poor ex vivo viability of F4/80hi macrophages, and (2) pre-mature death in Tnfaip3−/− mice by multi-organ inflammation25, 26, respectively.) When autophagy was inhibited by 3-MA, CXCL1 production in WT cells was reduced, but no corresponding reduction was found in Tnfaip3+/− cells (Fig. 8h), suggesting that autophagic removal of abundant A20 in WT cells played a role in high CXCL1 production. Impact of inhibiting autophagy in Tnfaip3+/− macrophages was minimal due most likely to the fact that A20 was not sufficiently abundant in the Tnfaip3+/− cells to make a difference with or without autophagy (Fig. 8g). In summary, these results strongly suggest that NFκB is activated by autophagic sequestration of A20 in F4/80hi macrophages.
p62 mediates the autophagic sequestration of A20
p62 is known as an adaptor protein which has a role in the selective delivery of target proteins to autophagosome41. p62 was colocalized with A20 (Fig. 9a; Supplementary Fig. 16d; Supplementary Table 4), and co-immunoprecipitated with A20 (Fig. 9b; Supplementary Fig. 16e, f) in naïve and stimulated cells. These results strongly suggest an interaction between p62 and A20. In addition, a deficiency of Sqstm1, encoding p62, increased protein levels of A20 in F4/80hi macrophages (Fig. 9c; Supplementary Fig. 17a), in which chemokine production was subsequently reduced (Fig. 9d; Supplementary Fig. 17b). Collectively, it was thus suggested that p62 scavenges A20 to autophagic degradation and that the downregulation of A20 enhances NFκB activity for chemokine production.
Figure 9. p62 mediates A20 sequestration by autophagy.

(a) Co-localization of A20 and p62 was evaluated by confocal microscopy. NIH-3T3 cells expressing exogenous A20-V5 and p62-HA were stimulated with Pam3CSK4 (100 ng/ml) for 60 min, and A20 (pink) and p62 (green) were detected by V5 and HA antibodies, respectively. White arrows indicate co-localization of A20 and p62. Scale bar = 10 μm. (b) Co-immunoprecipitation of A20 and p62. Peritoneal F4/80hi macrophages were stimulated with Pam3CSK4 (100 ng/ml) for 60 min. Cell lysates were immunoprecipitated with A20 antibody, then p62 was detected with p62 antibody by Western. Results for negative controls using unstimulated cells and control IgG is shown in Suppl. Fig. 15e, f. (c) Expression of A20 in WT and p62 (Sqstm1)-deficient peritoneal F4/80hi macrophages, which were unstimulated or stimulated with Pam3CSK4 (100 ng/ml) for 1 hr. (d) CXCL1 and CXCL2 production by WT and p62 (Sqstm1)-deficient peritoneal F4/80hi macrophages. Macrophages were stimulated with Pam3CSK4 (100 ng/ml) for 24 hrs, and chemokine levels in the culture supernatants were evaluated by ELISA. n=3. Data in all the panels are representative of independent at least two experiments. Error bars represent mean ± SD. ANOVA was used for statistical analysis. N.S.; not significant. *; p < 0.05.
F4/80hi macrophages express high levels of A20
We then asked why the autophagy-mediated NFκB activation is specific for F4/80hi macrophages. F4/80hi macrophages expressed higher levels of Tnfaip3 (A20) mRNA than do F4/80lo macrophages and BMDMs both with and without cell stimulation (Fig. 10a). Further, Western blotting showed high levels of A20 protein, particularly in Atg7-deficient F4/80hi macrophages after zymosan stimulation, while levels of A20 in F4/80lo macrophages were too weak to be detected regardless of Atg7 deficiency (Fig. 10b). Even without stimulation, F4/80hi macrophages expressed A20 protein abundant enough to be detected in by Western blotting (Supplementary Fig. 16e, f). These results suggest that F4/80hi macrophages express higher levels of A20 than do F4/80lo macrophages regardless of cell stimulation. In addition, A20 and p62 appeared to constitutively interact (Supplementary Fig. 16e), suggesting that A20 is ready to be sequestered in autophagsomes with p62 as soon as autophagosomes appear. Sequestration of A20 in F4/80hi macrophages by autophagy thus appears to be a critical event for robust NFκB activation upon fungal infection. Deficiency of autophagy allows accumulation of A20 in F4/80hi macrophages, presumably contributing to the defect in their chemokine expression. Our results here suggest that tissue-resident F4/80hi macrophages take advantage of autophagy to sequester intrinsically abundant A20 for the activation of NFκB in an effort to quickly respond to fungal infection (Fig. 10c).
Figure 10. A20 is highly expressed in F4/80hi peritoneal macrophages, but not in F4/80lo peritoneal macrophages.

(a) Comparison of Tnfaip3 mRNA levels among F4/80lo and F4/80hi peritoneal macrophages, and BMDMs. Unstimulated and stimulated cells with HKCA (5x105/ml, 1:1 ratio) for 4 hrs were analyzed by real-time PCR. Error bars denote RQ-Max/Min as described in the Method section. (b) Western blotting analysis of A20 and LC3 expression in F4/80lo and F4/80hi peritoneal macrophages stimulated with zymosan (100 μg/ml) for 30 min. Data are representative of independent two experiments (a, b). (c) Schematic illustration of autophagy-mediated NFκB activation in F4/80hi macrophages and the resulting induction of resistance to Candida infection. F4/80hi macrophages induce autophagy upon Candida detection. Autophagy then sequesters abundant A20 through interaction between p62 and A20, then unleash NFκB activity in F4/80hi macrophages. As a result, the macrophages secrete high levels of CXCL1 and CXCL2 to recruit neutrophils for fungal clearance.
Discussion
In viral, bacterial, and parasitic infections, autophagy contributes to host protection by containing pathogens in the autophagosome for pathogen clearance1, 2, 3. Autophagy also promotes phagocytosis and antigen processing in these microbial infections42, 43, 44. Based on these findings, we initially expected the involvement of autophagy in the direct killing of fungal pathogens. However, the data in this study demonstrated that ATG7 did not affect Candida phagocytosis or the inhibition of Candida expansion by macrophages and neutrophils. Unexpectedly, our intensive search did not identify LC3 cargos containing live Candida spores, although zymosan particles successfully recruited LC3. In line with our findings, an in vivo study using zebrafish also demonstrated that very few conidia of C. albicans were contained in LC3 cargos45. In addition, a recent study showed that live Candida did not induce clear LC3 staining surrounding conidia, while HKCA and β-1,3-glucan-coated polystyrene beads were clearly engulfed by LC cargos46, suggesting the exposure of β-glucans is not sufficient on live Candida for being recruited in LC3 cargos. Although the possibility of transient inclusion of live Candida in LC3 cargos has not been ruled out, host cells do not appear to directly kill Candida in autophagosomes and LC3-associated phagosomes.
A recent article reported that disruption of host autophagy ex vivo by Atg5 shRNA in a macrophage cell line decreased the phagocytic index of Candida, as well as the level of Candida-killing activity8. This result is not quite the same as what we found here; but, multiple differences exist between the earlier study’s experimental conditions and ours, as follows: The study8 used the SC5314 Candida strain, Atg5 knock-down in J774.16 cell lines, and evaluated phagocytosis and Candida-killing activity at 30 min. On the other hand, we used Atg7 CKO in BMDMs, splenic macrophages, thioglycollate-elicited peritoneal macrophages, and neutrophils (Fig. 2c, d, e; Supplementary Fig. 2, 3). We also evaluated phagocytosis at 0.5, 1, 2, and 3 hrs, as well as the inhibition of Candida expansion at 1.5, 3, 6, and 12 hrs. Despite these differences, a key conclusion of our study is the same as the previous report8 in terms of the involvement of autophagy in host protection against Candida. We should also note another recent research effort that found only slightly worse survival from Candida infection in Atg7 CKO (LysM-Cre) mice9. One possible explanation of the latter is that the recent study9 used another different Candida strain, because Candida strains are indeed known to have a significant impact on host responses47, 48.
Here, we demonstrated autophagy-mediated induction of chemokine expression by sequestering an NFκB inhibitor, A20, in a fashion specific to the F4/80himacrophage population at the least in spleens, the peritoneal cavity, and the kidney during systemic Candida infection. A20 is also known to act as a negative regulator of autophagy49, suggesting that fine-tuning of inflammatory responses by feedback between A20 and autophagy is also possible in F4/80hi tissue-resident macrophages. A20 works in TLR and TNFR signaling pathways, and protects hosts from collateral damage due to excessive inflammation initiated by viral and bacterial infections25, 26, 27, 50. However, the role of A20 in fungal infection has not yet been reported. Here, we suggest that abundant A20 in F4/80hi macrophages is an obstacle to jump-start early innate immune responses in fungal infections. Thus, F4/80hi macrophages are able to take advantage of autophagy and remove A20, since the cell type already has high levels of A20. The removal of A20 by autophagy in F4/80hi macrophages subsequent to infection is thus an effective approach to address infection, and prepare for the next step, i.e., recruitment of neutrophils. Given that Atg7 CKO mice are susceptible to Candida infection at an early stage, autophagy in F4/80hi macrophages appears to be a critical and effective intervention, particularly at these earlier stages.
A previous report demonstrated that autophagy eliminates BCL10, and inhibits NFκB activity in T cells upon T cell receptor stimulation5. On the other hand, our results clearly indicate that autophagy enhances NFκB signaling by eliminating A20 in F4/80hi macrophages. Since BCL10 is also expressed in macrophages, it is possible that BCL10 is also sequestered by autophagy in macrophages upon PRR stimulation; however, the autophagy-mediated depletion of BCL10 in macrophages may not have the impact it does in T cells, because signaling pathways inhibited by A20 have a significant impact on responses of macrophages. It is therefore likely that outcomes of autophagy are not the same between different cell types because of their distinct signal transduction networks.
We have shown that autophagy removes abundant A20 in F4/80hi macrophages for copious production of CXCL1/2. Here, F4/80lo macrophages also perform autophagy and do not express abundant A20 as F4/80hi macrophages do. Nevertheless, F4/80lo macrophages express low levels of CXCL1/2. Although further investigation is necessary to better explain why F4/80lo macrophages do not produce CXCL1/2 as well as F4/80hi macrophages, mechanistic machineries to produce the chemokines between the subsets appear to differ. For example, it is possible that expression of Cxcl1 and Cxcl2 genes in F4/80lo macrophages may require a higher threshold of PRR signaling intensity than that in F4/80hi macrophages. A possible impact of negative regulators on chemokine expression, other than A20 in F4/80lo macrophages, cannot be ruled out as well. At the least, the ability to express high levels of CXCL1/2 by F4/80hi macrophages as tissue-resident sentinels in the kidney, spleen, and peritoneum is biologically relevant for early anti-fungal immunity; and, F4/80hi macrophages may have further developed a mechanism for quickly secreting high levels of chemokines during the course of evolution.
Hosts are protected from hyper-inflammation and autoimmunity by the inhibitory effect of A20. At the same time, A20 becomes an obstruction when activation of immune responses is necessary during infections. For exerting rapid and strong immune responses, autophagy quickly sequesters inhibitory molecule A20, which is bound by p62 even before cell stimulation, in F4/80hi macrophages for them to enhance NFκB activation for producing neutrophil chemoattractant (Fig. 10c). Autophagy plays a crucial role in early anti-fungal immunity by jump-starting the innate immune system in F4/80hi macrophages, which are largely tissue-resident macrophages18, 19, 20, 21. It would be helpful if future research efforts focus on whether or not targeted manipulation limited to tissue-resident macrophages is possible. This would allow researchers to further dissect the biology of tissue-resident macrophages in vivo. Results from the current study suggest that F4/80hi macrophages do, indeed, play the role of a sentinel of immunity at the front lines of infection by utilizing autophagy.
Methods
Mice and cell preparation
All the mice used here are on the C57BL/6 background. Atg7fl/fl, Tnfaip3−/−, and Sqstm1fl/fl mice were described previously51, 52, 53. LysMcre/cre mice were purchased from Jackson Laboratories. Six-to-eight week old sex-matched WT, Atg7 CKO, Tnfaip3+/−, or Sqstm1CKO mice were used for all experiments. All the experiments were performed as approved by the Institutional Animal Care and Use Committee (IACUC). Complete RPMI medium (10% (vol/vol) FBS, penicillium/streptomycin (Sigma), 2 mM of L-glutamine)(Life technologies)) was used for host cell culture. RAW-Blue cells were purchased from Invivogen. F4/80hi and F4/80lo macrophages were isolated from kidney or peritoneal cavity by FACS isolation as CD11b+Ly6G−F4/80hi and CD11b+ Ly6G−F4/80lo, respectively. Splenic F4/80hi and F4/80lo macrophages were isolated as AF+Ly6G−F4/80hi and CD11b+Ly6G−F4/80lo after gating out SSChi (eosinophils). BMDMs were derived by culturing BM cells with L929 conditioned medium (15 %, vol/vol) for 7 days as previously described54. Thioglycollate-elicited macrophages and neutrophils were obtained from the peritoneal cavity 3 days and 1 day, respectively, after intraperitoneal (i.p.) injection of thioglycollate (3%, 2.5 ml/mouse). For some experiments, thioglycollate-elicited macrophages and neutrophils were purified by MACS beads (Myltenyi) as CD11b+ or Ly6G+ cells, respectively. Total splenic CD11b+ macrophages were purified from the spleen of naïve mice with CD11c-MACS beads (Myltenyi). For chemotaxis assay, neutrophils were FACS-purified from BM of naïve mice as CD11b+Ly6G+. On the other hand, thioglycollate-elicited peritoneal neutrophils were used for phagocytosis and Candida inhibition assay. In some experiments, macrophages were stimulated with Pam3CSK4 (100 ng/ml), Curdlan (100 μg/ml), zymosan (100 μg/ml) or heat-killed C. albicans (HKCA)(5×106/ml).
Antibodies, reagents, and major machines for analyses
Antibodies against CD11b (clone: M1/70), F4/80 (BM8), TLR4 (MTS510), TLR2 (T2.5), CXCR2 (TG11), CD4 (GK1.5), CD3 (17A2), CD11c (N418) and β-actin were purchased from BioLegend and used at a 1/200 dilution. Antibodies against CD8 (53-6.7), Ly6G (1A8) and B220 (RA3-6B2) were from BD Bioscience (1/200 dilution). Dectin-1 (2A11) and dectin-2 (D2.11E4) antibodies were from AbD Serotec (1/100 dilution). IκB (L35A5), phosphorylated IκB (14D4), p62 (5114S), K63-ubiquitin (5621S) and LC3A/B (4108S)(1/1000 dilution for western blotting) antibodies were purchased from Cell Signaling Technology. LC3 (ab51520)(1/200 dilution for immunofluorescence), IKKγ (ab7890)(1/1000 dilution for Western blotting) antibodies were purchased from Abcam. Antibody against His (HIS.H8)(1/200 dilution) was purchased from Thermo Scientific. V5 antibody (PAB11383) (1/5000 dilution) and HA antibody (HA-7)(1/1000 dilution) were purchased from Abnova and Sigma, respectively. A20 monoclonal antibody (A-12)(1/50 dilution for immunoprecipitation and western blotting) and IKKγ antibody (FL-419)(2 μg for 200 μg proteins, for immunoprecipitation), NFκB p65 polyclonal antibody (sc-109)(1/50 dilution), 3-MA, rapamycin and NFκB inhibitor (QNZ) were purchased from Santa Cruz. Recombinant CXCL1 was purchased from BioLegend. Pam3CSK4 and curdlan were purchased from Invivogen. Zymosan was purchased from MP Biomedicals. FACS-sorting with MoFlo Legacy (Beckman Coulter) was used for some experiments. Sorted cell populations we used were confirmed to be >95 % of purity. FACS CantoTM II (BD) was used for flow cytometry. Mastercycler realplex2 and Mastercycler proS (Eppendorf) were used for real-time PCR and conventional PCR, respectively. Zeiss 710 confocal microscopy and the Zeiss Efficient Navigation (ZEN; Carl Zeiss) software were used for data acquisition and analysis, respectively. Fiji software (http://fiji.sc/Fiji) was used to determine the intensity of fluorescence.
Candida infection, evaluating fungal burdens and histology
Sex-matched mice of age 6–8 week were infected by intravenous (i.v.) injection of 2 × 105 or 106 spores, as described in the figure legends, of Candida albicans (ATCC 18804). In some experiments, the same strain of Candida (5 × 106 spores/mouse) was intraperitoneally injected. Weights of mice were measured, and survival was evaluated every 24 hrs until whichever comes first either 18 days post infection or reaching at the human end-point approved by the mouse protocol. To evaluate in vivo fungal burdens, tissue lysates from kidneys and spleens (or fluids from peritoneal lavage) were first treated with H2O to lyse host cells (but C. albicans is not lysed by water), then plated on YPD agar plates and incubated overnight to determine Candida CFUs. For histological analysis, tissues were harvested 3 days after Candida infection (106 i.v.) and submitted for H&E and silver staining.
Evaluating Candida uptake and intracellular germination
Macrophages and neutrophils were stained with antibodies against CD11b, F4/80 (for macrophages) or Ly6G (for neutrophils), and co-cultured with same number of Alexa Fluor 647 (AF647)-labeled C. albicans (host cell vs. Candida spore numbers as the 1:1 ratio). Cells were fixed with 2 % (wt/vol) paraformaldehyde and submitted for flow cytometry. Proportions of AF647-positive host cells were evaluated for macrophages (CD11b+F4/80+) and neutrophils (CD11b+Ly6G+). Since this method also picks up host cells simply attaching to Candida without phagocytosis, we also evaluated cells by confocal microscopy and eye-counted the cells that completely engulfed Candida. As expected, percentages of AF647-positive cells were larger than those of Candida-engulfed cells evaluated by eye-counting; however, the differences in the percentages were proportional and flow cytometry analysis could be used as a substitute to evaluate phagocytosis between groups used in this study. To evaluate Candida germination, co-culture of Candida and host cells were set up as described above for the uptake analysis, except that cells were cultured and fixed directly cover glasses for confocal microscopic analysis. To evaluate host cell death, live/dead® Fixable Violet Dead Cell Stain Kit (Life technologies) was used and dead cells were enumerated by flow cytometry.
Evaluating inhibition of Candida expansion
Macrophages (BMDMs, thioglycollate-elicited macrophages, splenic CD11b+ macrophages, peritoneal F4/80hi macrophages) and thioglycollate-elicited neutrophils were used for the analyses. To evaluate host cells to inhibit Candida expansion, live Candida was co-cultured with host cells at the 1:10 (conidia : host) ratio (termed “test samples”). Negative control culture had Candida alone. Twelve hours after co-culture, host cells were lysed with water and Candida CFU was determined with YPD plates. Results were obtained with the following formula: Candida Expansion (fold) = [CFU at 12 hrs]/[CFU at 0-h]; Inhibition of Candida expansion (%) = (1 – [CFU of test samples at 12 hrs]/[CFU of negative control at 12 hrs]) × 100.
Evaluation of NFκB activity
NFκB activity was evaluated by either one or combination of the following methods. First RAW-Blue cell line (Invivogen), harboring an NFκB reporter to express secreted embryonic alkaline phosphatase (SEAP), was used with QUANTI-Blue (Invivogen) to detect SEAP activity with a plate reader (TECAN), as indicated by the manufacturer. RAW-Blue cells express PRRs, such as TLRs and dectin-1. Second, functional NFκB activity was carried out by evaluating the binding of p65 to a target DNA fragment wtih NFκB p65 Transcription Factor Assay kit (Thermo scientific). Third, nuclear translocation of NFκB p65 was evaluated by confocal microscopy. Fourth, biochemical analyses to detect phosphorylation of IκB and reduction of total IκB were also carried out by immunoblotting detection.
Neutrophil chemotaxis assay
Neutrophils were FACS-sorted from the BM of naïve mice as CD11b+Ly6G+. Neutrophils (5 × 105) were plated in the upper chamber of transwell (3 μm pore size; Corning), and CXCL1 was added in the lower chamber. After 1 hr of incubation at 37 °C, numbers of migrated cells were determined. Proportion of migrated cells was calculated as follows: Chemotaxis (%) = {[cell number in the lower chamber with CXCL1] – [cell number in the lower chamber without CXCL1] / (5 × 105)} × 100.
Quantitative PCR analysis
mRNA expression levels were determined by real-time PCR using the ΔΔCt method with KAPA SYBR fast qPCR Master Mix (KAPA Biosystems) and the following primer sets: Cxcl1 (forward: 5′-TGGGATTCACCTCAAGAACA-3′, reverse: 5′-TTTCTGAACCAAGGGAGCTT-3′), Cxcl2 (forward: 5′-CCACCAACCACCAGGCTAC-3′, reverse: 5′-GCTTCAGGGTCAAGGGCAAA-3′), Ccl3 (forward: 5′-ACTGCCTGCTGCTTCTCCTACA-3′, reverse: 5′-AGGAAAATGACACCTGGCTGG-3′), Ccl4 (forward: 5′-AAACCTAACCCCGAGCAACA-3′, reverse: 5′-CCATTGGTGCTGAGAACCCT), Tnfaip3 (forward: 5′-TTTGCTACGACACTCGGAAC-3′, reverse: 5′-TTCTGAGGATGTTGCTGAGG-3′), Atg7 (forward: CAGGACAGAGACCATCAGCTCCAC, reverse: TGGCTGCTACTTCTGCAATGATGT) and Actb (forward: 5′-TGTTACCAACTGGGACGACA-3′, reverse: 5′-CTGGGTCATCTTTTCACGGT-3′). Actb expression was used as an internal control. Results shown are representatives from multiple independent experiments with similar results. Error bars calculated were based on the calculation of RQ-Min = 2− (ΔΔCt + T * SD(ΔCt)) and RQ-Max = 2− (ΔΔCt − T * SD(ΔCt)) from triplicate wells as suggested by a manufacturer of PCR machines (Applied Biosystems). Error bars we had indicate RQ-MIN and RQ-MAX, which constitute the acceptable error for a 95% confidence limit according to Student’s t test.
Western blotting and immunoprecipitation
Cells were stimulated with Pam3CSK4 (100 ng/ml) or zymosan (100 μg/ml) and lysed with RIPA buffer and NP-40 (for immunoprecipitation) containing protease inhibitor cocktail (P8340, Sigma). Phosphatase inhibitor cocktail II was added for p-IκB detection. To examine the association between A20 and p62, NIH-3T3 cells were co-transfected with expression vectors for A20 and p62 (pcDNA5-A20-V5/His and pMI-p62-HA, respectively) by using Lipofectamine LTX (Invitrogen), as recommended by the supplier. The expression vectors for A20 and p62 were made in our laboratory, as fusions of the V5/His tag in the pcDNA5 backbone (Invitrogen) and the HA tag in the pMX backbone (Cosmobio), respectively. Immunoprecipitation was carried out by using Dynabeads Protein G Immunoprecipitation system (Invitrogen) with specific antibodies, followed by immunoblotting with indicated antibodies as recommended by the supplier. Signal intensity of blotting was determined by densitometry using the NIH-Image J software (http://rsbweb.nih.gov/ij/). Full blot data are shown in Supplementary Fig. 18.
Cell staining for confocal microscopy analyses
Cells were cultured on covered glasses. For staining, cells were washed with TBS containing 0.05% (vol/vol) Tween (TBS-T), and permeabilized with 0.25% (vol/vol) Triton in TBS, then incubated with a primary antibody at 4 °C overnight, followed by staining with secondary antibody conjugated with fluorophore. Cells on cover glass were embedded to slide glass with mounting medium including DAPI (Prolong Gold, Invitrogen), and analyzed for confocal microscopy. Fiji software (http://fiji.sc/Fiji) was used to determine the intensity of fluorescence.
Statistical analysis
The two-tailed Student’s t-test was used for statistical analyses of two-group comparisons. Multigroup comparisons were performed by a one-way analysis of variance (ANOVA) followed by Tukey–Kramer multiple comparisons test.
Supplementary Material
Acknowledgments
We thank Drs. W. Jia, M. He and I. McLeod for their help setting up the Atg7fl/flLysMcre/+ mouse line and experimental techniques related to autophagy; Dr. T. Yanagawa (University of Tsukuba, Japan) for providing p62fl/flLysM-Cre mice, and Dr. J. Heitman (Department of Molecular Genetics and Microbiology, Duke University) for providing C. albicans. This work was supported by the National Institutes of Health (R01AI088100 to M.L.S.) and by the Uehara Memorial Foundation Fellowship (to M.K.).
Footnotes
Author contributions
M.K. performed the experiments with assistance from M.I. and K.D. Y.H. and G.H. participated in data interpretation and critical discussions. M.K. and M.L.S. designed experiments, analyzed data, and wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy. 2011;7:341–345. doi: 10.4161/auto.7.3.14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. The Journal of clinical investigation. 2006;116:2366–2377. doi: 10.1172/JCI28796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orvedahl A, MacPherson S, Sumpter R, Jr, Talloczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell host & microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi CS, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nature immunology. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paul S, Kashyap AK, Jia W, He YW, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kappaB. Immunity. 2012;36:947–958. doi: 10.1016/j.immuni.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castillo EF, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E3168–3176. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma J, Becker C, Lowell CA, Underhill DM. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. The Journal of biological chemistry. 2012;287:34149–34156. doi: 10.1074/jbc.M112.382812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicola AM, et al. Macrophage autophagy in immunity to Cryptococcus neoformans and Candida albicans. Infection and immunity. 2012;80:3065–3076. doi: 10.1128/IAI.00358-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smeekens SP, et al. Autophagy is redundant for the host defense against systemic Candida albicans infections. European journal of clinical microbiology & infectious diseases : official publication of the European Society of Clinical Microbiology. 2014;33:711–722. doi: 10.1007/s10096-013-2002-x. [DOI] [PubMed] [Google Scholar]
- 10.Rosentul DC, et al. Role of autophagy genetic variants for the risk of Candida infections. Medical mycology. 2014;52:333–341. doi: 10.1093/mmy/myt035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lortholary O, Dupont B. Antifungal prophylaxis during neutropenia and immunodeficiency. Clinical microbiology reviews. 1997;10:477–504. doi: 10.1128/cmr.10.3.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koh AY, Kohler JR, Coggshall KT, Van Rooijen N, Pier GB. Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog. 2008;4:e35. doi: 10.1371/journal.ppat.0040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rubin-Bejerano I, Fraser I, Grisafi P, Fink GR. Phagocytosis by neutrophils induces an amino acid deprivation response in Saccharomyces cerevisiae and Candida albicans. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:11007–11012. doi: 10.1073/pnas.1834481100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Eeden SF, Bicknell S, Walker BA, Hogg JC. Polymorphonuclear leukocytes L-selectin expression decreases as they age in circulation. The American journal of physiology. 1997;272:H401–408. doi: 10.1152/ajpheart.1997.272.1.H401. [DOI] [PubMed] [Google Scholar]
- 15.Cailhier JF, et al. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. Journal of immunology. 2005;174:2336–2342. doi: 10.4049/jimmunol.174.4.2336. [DOI] [PubMed] [Google Scholar]
- 16.Sun Y, et al. TLR4 and TLR5 on corneal macrophages regulate Pseudomonas aeruginosa keratitis by signaling through MyD88-dependent and -independent pathways. Journal of immunology. 2010;185:4272–4283. doi: 10.4049/jimmunol.1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kubota Y, et al. Role of alveolar macrophages in Candida-induced acute lung injury. Clin Diagn Lab Immunol. 2001;8:1258–1262. doi: 10.1128/CDLI.8.6.1258-1262.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157:832–844. doi: 10.1016/j.cell.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nature immunology. 2013;14:986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 21.Ghosn EE, et al. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:2568–2573. doi: 10.1073/pnas.0915000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mordue DG, Sibley LD. A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. Journal of leukocyte biology. 2003;74:1015–1025. doi: 10.1189/jlb.0403164. [DOI] [PubMed] [Google Scholar]
- 23.Peters W, et al. CCR2-dependent trafficking of F4/80dim macrophages and CD11cdim/intermediate dendritic cells is crucial for T cell recruitment to lungs infected with Mycobacterium tuberculosis. Journal of immunology. 2004;172:7647–7653. doi: 10.4049/jimmunol.172.12.7647. [DOI] [PubMed] [Google Scholar]
- 24.Shembade N, Harhaj EW. Regulation of NF-kappaB signaling by the A20 deubiquitinase. Cellular & molecular immunology. 2012;9:123–130. doi: 10.1038/cmi.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boone DL, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nature immunology. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 26.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammer GE, et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nature immunology. 2011;12:1184–1193. doi: 10.1038/ni.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harhaj EW, Dixit VM. Regulation of NF-kappaB by deubiquitinases. Immunological reviews. 2012;246:107–124. doi: 10.1111/j.1600-065X.2012.01100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komatsu M, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. The Journal of cell biology. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez J, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Fulurija A, Ashman RB, Papadimitriou JM. Neutrophil depletion increases susceptibility to systemic and vaginal candidiasis in mice, and reveals differences between brain and kidney in mechanisms of host resistance. Microbiology. 1996;142 (Pt 12):3487–3496. doi: 10.1099/13500872-142-12-3487. [DOI] [PubMed] [Google Scholar]
- 32.Balish E, Wagner RD, Vazquez-Torres A, Jones-Carson J, Pierson C, Warner T. Mucosal and systemic candidiasis in IL-8Rh-/- BALB/c mice. Journal of leukocyte biology. 1999;66:144–150. doi: 10.1002/jlb.66.1.144. [DOI] [PubMed] [Google Scholar]
- 33.Cravens PD, Lipsky PE. Dendritic cells, chemokine receptors and autoimmune inflammatory diseases. Immunology and cell biology. 2002;80:497–505. doi: 10.1046/j.1440-1711.2002.01118.x. [DOI] [PubMed] [Google Scholar]
- 34.Criollo A, et al. Autophagy is required for the activation of NFkappaB. Cell cycle. 2012;11:194–199. doi: 10.4161/cc.11.1.18669. [DOI] [PubMed] [Google Scholar]
- 35.Li M, et al. An essential role of the NF-kappa B/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. Journal of immunology. 2001;166:7128–7135. doi: 10.4049/jimmunol.166.12.7128. [DOI] [PubMed] [Google Scholar]
- 36.Grove M, Plumb M. C/EBP, NF-kappa B, and c-Ets family members and transcriptional regulation of the cell-specific and inducible macrophage inflammatory protein 1 alpha immediate-early gene. Molecular and cellular biology. 1993;13:5276–5289. doi: 10.1128/mcb.13.9.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z, Bryan JL, DeLassus E, Chang LW, Liao W, Sandell LJ. CCAAT/enhancer-binding protein beta and NF-kappaB mediate high level expression of chemokine genes CCL3 and CCL4 by human chondrocytes in response to IL-1beta. The Journal of biological chemistry. 2010;285:33092–33103. doi: 10.1074/jbc.M110.130377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nature immunology. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 39.Skaug B, Chen J, Du F, He J, Ma A, Chen ZJ. Direct, noncatalytic mechanism of IKK inhibition by A20. Molecular cell. 2011;44:559–571. doi: 10.1016/j.molcel.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li L, et al. Localization of A20 to a lysosome-associated compartment and its role in NFkappaB signaling. Biochimica et biophysica acta. 2008;1783:1140–1149. doi: 10.1016/j.bbamcr.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pankiv S, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. The Journal of biological chemistry. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 42.Paludan C, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 43.English L, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nature immunology. 2009;10:480–487. doi: 10.1038/ni.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinet W, Schrijvers DM, Timmermans JP, Herman AG, De Meyer GR. Phagocytosis of bacteria is enhanced in macrophages undergoing nutrient deprivation. The FEBS journal. 2009;276:2227–2240. doi: 10.1111/j.1742-4658.2009.06951.x. [DOI] [PubMed] [Google Scholar]
- 45.Brothers KM, Gratacap RL, Barker SE, Newman ZR, Norum A, Wheeler RT. NADPH oxidase-driven phagocyte recruitment controls Candida albicans filamentous growth and prevents mortality. PLoS Pathog. 2013;9:e1003634. doi: 10.1371/journal.ppat.1003634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tam JM, et al. Dectin-1-Dependent LC3 Recruitment to Phagosomes Enhances Fungicidal Activity in Macrophages. The Journal of infectious diseases. 2014 doi: 10.1093/infdis/jiu290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saijo S, et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nature immunology. 2007;8:39–46. doi: 10.1038/ni1425. [DOI] [PubMed] [Google Scholar]
- 48.Taylor PR, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nature immunology. 2007;8:31–38. doi: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Science signaling. 2010;3:ra42. doi: 10.1126/scisignal.2000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maelfait J, et al. A20 (Tnfaip3) deficiency in myeloid cells protects against influenza A virus infection. PLoS Pathog. 2012;8:e1002570. doi: 10.1371/journal.ppat.1002570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol. 2011;186:1564–1574. doi: 10.4049/jimmunol.1001822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tavares RM, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010;33:181–191. doi: 10.1016/j.immuni.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harada H, et al. Deficiency of p62/Sequestosome 1 causes hyperphagia due to leptin resistance in the brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:14767–14777. doi: 10.1523/JNEUROSCI.2954-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inoue M, et al. T cells down-regulate macrophage TNF production by IRAK1-mediated IL-10 expression and control innate hyperinflammation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:5295–5300. doi: 10.1073/pnas.1321427111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.