

A novel role for FoxO1 in regulating macrophage phenotypic polarization and therapeutic interventions for chronic inflammatory conditions.
Keywords: monocyte, metabolic stress, M1-like, M2-like, inflammation
Abstract
Macrophages are a heterogeneous population of immune cells that are essential for the initiation and containment inflammation. There are 2 well-established populations of inflammatory macrophages: classically activated M1 and alternatively activated M2 macrophages. The FoxO family of transcription factors plays key roles in a number of cellular processes, including cell growth, metabolism, survival, and inflammation. In this study, we determined whether the expression of FoxO1 contributes polarization of macrophages toward the M2-like phenotype by enhancing IL-10 cytokine expression. We identified that FoxO1 is highly expressed in M-CSF-derived (M2-like) macrophage subsets, and this M2-like macrophages showed a preferential FoxO1 enrichment on the IL-10 promoter but not in GM-CSF-derived (M1-like) macrophages during classic activation by LPS treatment, which suggests that FoxO1 enhances IL-10 by binding directly to the IL-10 promoter, especially in BMMs. In addition, our data show that macrophages in the setting of hyperglycemia contribute to the macrophage-inflammatory phenotype through attenuation of the contribution of FoxO1 to activate IL-10 expression. Our data identify a novel role for FoxO1 in regulating IL-10 secretion during classic activation and highlight the potential for therapeutic interventions for chronic inflammatory conditions, such as atherosclerosis, diabetes, inflammatory bowel disease, and arthritis.
Introduction
Macrophages are essential components of innate immunity, which sense microenvironmental signals and control the nature and extent of a subsequent immunologic reaction. The shaping of the monocyte-macrophage inflammatory phenotype is an important link between microenvironmental signals and the inflammatory reaction, including cytokine secretion and acute and chronic inflammatory cell recruitment. There is growing evidence that suggest that the diverse biologic activity of macrophages can be separated into phenotypically distinct, functional classifications that develop in response to their environmental signals [1, 2]. Mirroring Th cell polarization, there are 2 distinct states of polarized activation for macrophages: the classically activated M1 and alternative activated M2 macrophages that have distinct cytokine/chemokine profiles and unique features of the metabolism of iron, folate, and glucose [3]. The M1/M2 nomenclature is derived from the types of cytokines that are associated with these macrophage phenotypes (e.g., IFN-γ, IL-4, or IL-13). The M1 population is thought to contribute to macrophage-mediated tissue injury [4, 5], and has strong microbicidal activity as a result of abundant generation of reactive oxygen and nitrogen species. In contrast to proinflammatory M1 cells, M2 macrophages suppress inflammation and antitumor immunity, facilitate wound repair, and regulate glucose metabolism [3, 6, 7]. Although these 2 major subpopulations of macrophages have characteristic features, it is generally thought that the macrophages phenotype has bidirectional plasticity that is dependent on the microenvironment. Indeed, many studies have shown flexibility in macrophage phenotype in response to new microenvironmental signals [8–10]. Regardless of how they are characterized, functional dysregulation of macrophages could have beneficial or deleterious consequences, depending on the biologic situation. For example, hyper-responsive M1 macrophages are important for pulmonary host defense but can cause irreversible, “off-target,” inflammatory-mediated tissue damage, whereas overactive M2 macrophages are involved in wound healing but can also promote fibrosis and exacerbate allergic inflammatory responses. Although it is important to determine how the expression of these phenotypes is regulated in a specific environment, the molecular determinants of functional diversity and specifically, the transcription factors that dictate alternative functional outcomes are, to a large extent, unknown.
The FoxO family of transcription factors has an important role in many fundamental cellular processes, including cell growth, metabolism, survival, and inflammation. In mammals, the FoxO subclass consists of 4 members, including FoxO1, FoxO3, FoxO4, and FoxO6 [11]. FoxO1 is the most abundant FoxO isoform in multiple metabolic pathways [12]. FoxO1 and FoxO3 are the main isoforms expressed in the immune cells, including macrophages [11]. Regulation of FoxO transcriptional activity is complex and a tightly orchestrated process, dependent on the status of post-translational modifications, including phosphorylation, acetylation, ubiquitination, and methylation [11]. PI3K/Akt signaling and CBP/p300, which are activated by certain cytokines and growth factors, are well-established upstream regulators of FoxO protein, leading to determine nuclear/cytoplasmic localization [13, 14]. Although several studies have been focused on the fundamental role of FoxO3 in hematopoietic and immune cells, a potential role of FoxO1 in mediating interconversion of the M1- and M2-inflammatory phenotype in macrophages has not been investigated. The published literature has only narrowly pointed to a proinflammatory role of FoxO1 in inflammatory signaling [15, 16]. The purpose of our study is to examine whether FoxO1 has a specialized function for determining macrophage phenotype committing to M2 or M2-like lineage.
It has long been known that macrophage function and metabolism are interconnected [17–19], but whether this is bidirectional or unidirectional is unknown. Macrophages are insulin-sensitive cells [20], and a defective insulin signaling in macrophages seems to predispose to foam-cell formation in insulin-resistant states that are characteristic of atherosclerotic lesions [21, 22]. Polarized macrophages show a distinct regulation of glucose metabolism that is pathologically linked to obesity [23, 24]. Activation of inflammatory pathways by metabolic cues leads to macrophage recruitment, which leads to exposure of glucose- and/or lipid-rich environments [24, 25]. As such, the presence of persistent metabolic stresses could cause dysregulation of macrophage phenotype, resulting in a state of unresolved chronic inflammation [26, 27]. However, the endogenous factors that induce macrophage phenotypic polarization in the glucose- and lipid-rich microenvironments are poorly understood.
In this study, we examined whether the expression of FoxO1 can have profound effects on the BMMs and GM-BMMs during classic activation by LPS treatment. Our data show that FoxO1 has an effect on the production of M2 signature cytokine IL-10, together with published results, indicating a specific role as a critical sensor for microenvironmental inflammatory signaling in macrophages through enhanced TLR4 signaling [15]. Consequently, those macrophages in the setting of hyperglycemia are likely to contribute to the augmented, proinflammatory macrophage phenotype, in addition to the effect of FoxO1 on activation of IL-10 gene expression. This unique role of FoxO1 provides an important clue for understanding the mechanisms involved in macrophage phenotypic polarization, which could have an impact on development of innovative therapeutic interdiction in the molecular pathogenesis of chronic inflammatory conditions, such as atherosclerosis, type 1 diabetes, inflammatory bowel disease, arthritis, and allergic asthma.
MATERIALS AND METHODS
Materials
Unless otherwise stated, all biochemical reagents used in this study were purchased from Sigma (St. Louis, MO, USA). Antibody against FoxO1 was purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against CD11b and MARCO were purchased from BD Biosciences (San Jose, CA, USA) and R&D Systems (Minneapolis, MN, USA), respectively. Anti-CD80 was purchased from eBioscience (San Diego, CA, USA). Anti-actin antibody was purchased from Pierce (Rockford, IL, USA).
Cell cultures
BMDMs from mice were isolated, according to published protocols [28, 29], and grown in DMEM/F12 supplemented with 10% FBS, 1% penicillin/streptomycin, rmM-CSF (20 ng/ml; PeproTech, Rocky Hill, NJ, USA), or rmGM-CSF (10 ng/ml; PeproTech) [30, 31]. After 7 days, adherent cells were washed with PBS and replated and then stimulated LPS (100 ng/ml; Alexis Biochemicals, Farmingdale, NY, USA). Mouse monocyte/macrophage Raw 264.7 cells were cultured in DMEM, supplemented with 10% FBS and 1% penicillin/streptomycin. For studies of the effect of glucose, bone marrow differentiation was done in DMEM/F12 with 5.5 or 25 mM endotoxin-free D-glucose (Sigma) [32]. During the differentiation, glucose was supplemented every 24 h to prevent glucose depletion.
Enriched populations of human monocytes were isolated by Percoll gradient of citrate-treated blood (kind gift of Dr. Jaehyung Cho, University of Illinois at Chicago, Chicago, IL, USA), as described [30, 33]. M1 and M2 macrophages were obtained after 5 days of culture of human monocytes in RPMI-1640 medium, supplemented with rhM-CSF (10 ng/ml; PeproTech) or rhGM-CSF (10 ng/ml; PeproTech).
Generation of mice with FoxO1−/− myeloid cells (LysMFoxO1)
To generate myeloid FoxO1−/− mice, FoxO1fl/fl mice [34] were crossed with LysM Cre mice to homozygosity (LysMFoxO1). DNA extraction and genotyping were performed as described previously [34, 35]. To avoid the possibility that results could be influenced by Cre recombinase-induced toxicity, FoxO1wt/wtCreTg mice were used as WT controls. Fox endotoxemia experiments, WT, and LysMFoxO1 mice were injected i.p. with LPS (2 mg/kg). At 3 or 18 h post-LPS injection, mice were killed, and macrophages/blood were harvested.
Hyperglycemic mice
Diabetic db/db mice and nondiabetic db/+ controls on a C57Bl/6 background were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). For the generation of BMDMs, bone marrow of WT or db/db was cultured in DMEM/F12, as described above. In other experiments, diabetes was induced by STZ (mixed anomers No. S130, 160 mg/kg; Sigma), dissolved immediately before use in freshly made citrate buffer (0.1 M, pH 4.5), and single injected i.p. in male C57Bl/6 mice. Diabetic mice with nonfasted blood glucose values >500 mg/dl were measured by a blood glucose monitor (Germaine Laboratories, San Antonio, TX, USA).
Adipose tissue macrophages were isolated according to published protocols [36, 37]. Isolation of CD11b+ cells from stromal vascular fraction isolates was performed by magnetic immunoaffinity isolation with anti-CD11b antibodies conjugated to magnetic beads (MACS; Miltenyi Biotec, Bergisch Gladbach, Germany). Cells were isolated by use of positive-selection columns before preparation of whole-cell lysates.
Measurement of cytokines
Cytokine secretion in culture supernatants was analyzed by ELISA, specific for mIL-10, mIL-12/IL-23 p40, mTNF-α, and mIL-6 (R&D Systems), following the protocols supplied by the manufacturer.
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay lysis buffer (Millipore, Temecula, CA, USA) with 1× protease inhibitor cocktail (Pierce). Cell lysates containing equal amounts of protein were electrophoresed and immunoblotted by use of appropriate antibodies, as described [28].
Flow cytometry
Single-cell suspensions (1 × 104/sample) were washed and incubated on ice for 30 min with appropriate fluorescently labeled antibodies. Cells were analyzed on a CyAn ADP analyzer (Beckman Coulter, Brea, CA, USA), where gating was based on respective unstained cell population and isotype-matching control antibodies. The data were analyzed with FlowJo software (Tree Star, Ashland, OR, USA).
RNA extraction and quantitative real-time RT-PCR
RNA was extracted from cells by use of an RNeasy Mini kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s instruction. cDNA synthesis with RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Pittsburgh, PA, USA) and gene expression was measured by the change-in-threshold method, based on quantitative real-time PCR in a LightCycler 480 system (Roche, Rotkreuz, Switzerland), normalizing to GAPDH expression as an endogenous control.
FoxO1 DNA-binding assay
FoxO1 DNA-binding activity was analyzed by an ELISA-based FoxO1-DNA-binding assay (TransAM FKHR; Active Motif, Carlsbad, CA, USA), according to the manufacturer's instructions. In brief, nuclear extracts were applied to 96-well plates coated with oligonucleotides containing FoxO-DNA-binding elements. Bound (i.e., active) FoxO was then detected by use of an antibody directed against FoxO1, the binding of which was assayed by use of a secondary antibody conjugated with HRP [38].
ChIP
ChIP assays were performed with the ChromaFlash One-Step kit (Epigentek, Farmingdale, NY, USA) or SimpleChIP enzymatic ChIP kit (Cell Signaling Technology) with anti-FoxO1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), as described [39]. The immunoprecipitated DNA subjected to RT-PCR analysis with PerfeCTa SYBR Green FastMix (Quanta BioSciences, Gaithersburg, MD, USA). Data were analyzed with the LightCycler 480 software (Roche).
siRNA knockdown
The ON-TARGETplus siRNA SMARTpool (GE Dharmacon, Lafayette, CO, USA), designed to target FoxO1, was used for siRNA knockdown experiments by use of Amaxa Mouse Macrophage Nuclefector kit (Lonza, Cologne, Germany). After 24 h post-transfection, BMDMs were stimulated by LPS (100 ng/ml) for 24 h.
Adenoviral infection
An adenovirus construct that encoded FoxO1 mutants (FoxO1-TSS) is described elsewhere [40, 41] and was a gift from Dr. Terry Unterman (University of Illinois at Chicago). M2-like macrophages were infected with adenoviruses (1–50 multiplicity of infection) and treated with LPS after 24 h.
Luciferase reporter assay
Relative luciferase reporter assays were performed as described [16]. Transfections were carried out by use of Lipofectamine 2000 or Lipofectamine LTX (Invitrogen, Carlsbad, CA, USA). The thymidine kinase promoter-Renilla luciferase reporter plasmid was used as a control for transfection efficiency in the dual-luciferase system (Promega, Madison, WI, USA). mIL-10 promoter genomic sequences were analyzed for the FoxO1-binding sites (TGTTTGC and TGTTT) [15]. A 1.2 kb of mIL-10 was amplified by PCR from the genomic DNA isolated from the mBMDM by use of the primer pair 5-CCGCTCGAGAGCAGTGTGTCCACACCTAAAACATC-3 (restriction site XhoI) and 5-CCCAAGCTTCAGCTGTTCTATGTACAGAGGCCCTC-3 (restriction site HindIII). The amplified product was purified by use of the PCR Clean-Up kit (Qiagen), digested with XhoI and HindIII, and ligated in pGL3 basic vector (Promega) by use of the Instant Sticky-end Ligase Master Mix (New England BioLabs, Ipswich, MA, USA). The ligation product was transformed in Escherichia coli (NEB 5-alpha Competent cells; New England BioLabs). We selected positive clones by restriction digestion with XhoI and HindIII and labeled as IL-10. The 1.2 kb promoter region was digested with NheI + BglII and BglII + HindIII to generate distal, 700 bp (harboring 4 TGTTT FoxO1-binding sites and 1 TGTTTGC FoxO1-binding site) and proximal, 521 bp (harboring 3 TGTTT FoxO1-binding sites) fragments, respectively. Both of the fragments were cloned in pGL3 basic vector. The distal 700 bp clone was labeled as IL-10-fr1, and the proximal 521 bp was labeled as IL-10-fr2. All constructs were sequenced to confirm identity from the sequencing facility at the DNA Services Facility at the University of Illinois at Chicago, and sequence analysis was performed by use of EditSeq and SeqMan (DNASTAR, Madison, WI, USA).
Statistical analysis
Results are expressed as means ± sem. Statistical analysis of significance was calculated by Student’s t-test. Statistical significance is indicated in figure legends.
RESULTS
Distinct phenotypic features of BMMs and GM-BMMs and high expression of FoxO1 in M2 macrophages
Macrophage polarization is driven by cues in the tissue microenvironment, including cytokines, growth factors, and pathogen-associated molecular patterns [42]. These changes result in activation of a subset of defining transcription factors that are phenotypically characterized by the production of anti-inflammatory and proinflammatory cytokines via distinct transcriptional programs. M-CSF and GM-CSF are mediators involved in regulating the numbers and function of macrophage lineage populations [30, 31, 43].
To examine difference in the macrophage phenotypes, primary mouse bone marrow cells were cultivated for 7 days into M2-like macrophages in the presence of M-CSF (20 ng/ml) or into M1-like macrophages in the presence of GM-CSF (10 ng/ml) [31, 43, 44]. Bacterial product, LPS, was used in this study as a classic activation stimulus to interrogate the distinct macrophage subsets. Macrophages that were polarized to the M1 phenotype were larger with central nucleus or “fried egg shape,” as described previously, whereas M2-polarized macrophages were smaller and elongated with a spindle-like shapes as shown in Fig. 1A [45–47]. To determine the expression of macrophage surface markers, we analyzed the expression of CD11b (monocyte marker), F4/80 (macrophage marker), CD80 (M1 profile marker), CD11c (M1 profile marker), and MARCO (M2 profile marker) antigen on the surface of M-CSF/GM-CSF-differentiated BMDMs by use of flow cytometry (Fig. 1A) [48]. Compared with M1-like GM-BMM, BMM had lower CD80/CD11c but higher expression of MARCO (gray-filled histogram). We found a markedly higher level of intracellular Ym1 (M2 marker) protein in BMM compared with GM-BMM. However, GM-BMM secreted significantly less IL-10 but more IL-12/IL-23p40 in response to LPS compared with BMM (Fig. 1B). In these cultures, we also observed less induction of expression of mRNA for IL-10 but more expression of IL-12p40 mRNA in GM-BMM (Fig. 1C). Surprisingly, we found that the treatment with M-CSF resulted in marked up-regulation of FoxO1 protein expression in response to LPS treatment. along with M2-like phenotypic expression in macrophages, but the treatment with GM-CSF, an M1-differentiating factor, did not (Fig. 1D). To determine the transcriptional activity of FoxO1 in the nuclear extract protein, we performed a transcription factor-binding assay in which transcriptionally active FoxO1 transcription factors binding to the FoxO consensus sequence were detected [38]. Apparently, GM-CSF decreased FoxO1 transactivation potential, possibly by reducing their DNA-binding activity of nuclear FoxO1 (Fig. 1E). Thus, these data illustrates an association of FoxO1 with a M2-like property suggestion that FoxO1 might have greater impact on the function of M-CSF- rather than GM-CSF-dependent macrophages.
Figure 1. M-CSF drives BMM into M2-like phenotype and induces high FoxO1 expression.
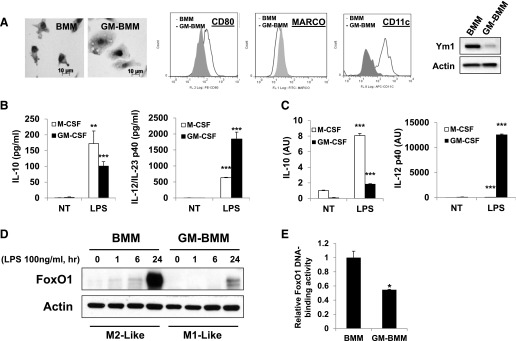
(A) Representative morphologic images and flow cytometric expression of CD80, MARCO, and CD11c of BMDMs differentiated for 7 days into M2-like macrophages with M-CSF (BMM) or into M1-like macrophages with GM-CSF (GM-BMM). Immunoblot analysis of Ym1 from BMM and GM-BMM. FL 1/2/8, Fluorescence 1/2/8; APC, allophycocyanin. (B) ELISA of the secretion of IL-10 and IL-12/IL-23 by BMM/GM-BMM stimulated with LPS (100 ng/ml). NT, nontreated. (C) RT-PCR analysis of IL-10 and IL-12p40 mRNAs by BMM/GM-BMM stimulated with LPS (100 ng/ml). (D) Immunoblot analysis of FoxO1 from BMM/GM-BMM; extracts of adherent cells were probed with antibody to FoxO1. (E) FoxO1-DNA-binding activity was analyzed by an ELISA-based (TransAM) FoxO1-DNA-binding assay from nuclear extracts BMM/GM-BMM. *P < 0.05, **P < 0.01, and ***P < 0.001 versus nontreated (Student’s t-test). Results are from at least 3 independent experiments.
We next examined FoxO1 expression in primary human monocyte-derived macrophages that were polarized to M2 or M1 phase by treatment with M-CSF or GM-CSF, respectively [10, 46]. In this experiment, the macrophage phenotypes were characterized by evaluating cytokine production (IL-10 for M2, IL-12/IL-23 p40 for M1). As can be seen in Fig. 2, primary human monocyte-derived M2 macrophages, differentiated by M-CSF for 5 days, are characterized by secretion of bioactive IL-10, followed by stimulation with LPS. Treatment with GM-CSF resulted in up-regulation of IL-12/IL-23 (Fig. 2A). Similar with their mouse counterparts, macrophages that were polarized to the M1 phenotype were larger with a central nucleus, whereas M2-polarized macrophages were smaller and elongated with spindle-like shapes, as shown in Fig. 1A [45, 46]. FoxO1 protein expression is much higher in primary human monocyte-derived M2 macrophages than M1 macrophages (Fig. 2B), suggesting that it might be involved in M2 polarization or the M2 phenotype. Based on these data, we will refer to FoxO1 as "M-CSF-dependent macrophages associated transcription factor."
Figure 2. Phenotypic features of M1 and M2 human macrophages.

(A) ELISA measurement of the secretion of IL-10 and IL-12 stimulated for 24 h with LPS (100 ng/ml) and representative morphologic images of human (h) PBMCs differentiated for 5 days into M1 macrophages with hGM-CSF (hGM-BMM) or into M2 macrophages with hM-CSF (hBMM). (B) Immunoblot analysis of FoxO1 from human PBMCs differentiated for 5 days into hGM-BMM or into M2 macrophages with hBMM; extracts of adherent cells were probed with the antibody to FoxO1. **P < 0.01, and ***P < 0.001 (Student’s t-test) versus nontreated. Results are from a different donor or from 3 independent experiments.
FoxO1 influences the production of macrophage lineage-specific IL-10 cytokines
We next asked whether the loss of FoxO1 function influenced the M-CSF-dependent macrophage phenotype, resulting in greater production of IL-10 relative to IL-12/IL-23 in response to LPS treatment. BMMs, cultivated for 7 days in the presence of M-CSF (20 ng/ml), were used for this experiment. We transfected BMM with siRNA constructs targeting FoxO1, and this was controlled by transfection with a scRNA nonsense control construct. After transfection of scRNA and siFoxO1, an immunoblot demonstrated that siFoxO1 effectively decreased FoxO1 protein levels by ∼80% compared with control (Fig. 3A). In the FoxO1 knockdown BMM, there was a blunted rise of IL-10 in response to LPS, whereas the increase was only one-half of that in the scRNA-transfected group of cells. In contrast, there was no significant difference in IL-12/IL-23 production, even proinflammatory IL-6 and TNF-α cytokine production by the scRNA- and siRNA-transfected groups (Fig. 3B).
Figure 3. Knock down of FoxO1 expression attenuates the M2-like phenotype of BMMs, which were transfected with siFoxO1 or scRNA.
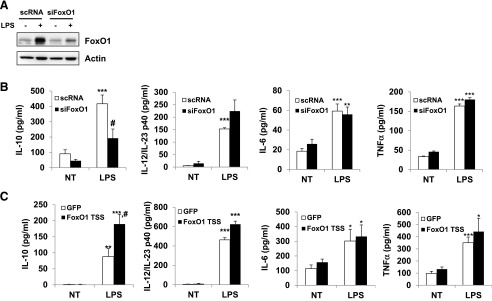
(A) Immunoblot of FoxO1 levels after treatment with siFoxO1. (B) ELISA of the secretion of IL-10, IL-12/IL-23, and proinflammatory cytokines IL-6 and TNF-α by BMM transfected with siFoxO1 and stimulated for 24 h with LPS (100 ng/ml). (C) ELISA of the secretion of IL-10, IL-12/IL-23, and proinflammatory cytokines IL-6 and TNF-α by BMM infected with FoxO1 TS and stimulated for 24 h with LPS (100 ng/ml). *P < 0.05, **P < 0.01, and ***P < 0.001 versus nontreated; #P < 0.05 versus scRNA or GFP (Student’s t-test). Results are from at least 3 independent experiments.
We next performed a reciprocal "gain-of-function" experiment, where we overexpressed a constitutive, active form of FoxO1 with an adenoviral expression construct in BMM, which resulted in a significant increase of IL-10 release (Fig. 3C). In this experiment, BMMs were transfected with an adenovirus expressing GFP or FoxO1 TSS and stimulated with LPS (100 ng/ml) for 24 h. In spite of no significant difference in IL-12/IL-23 release, there was a significant increase M2 signature IL-10 cytokine secretion in the FoxO1 TSS-infected macrophages compared with GFP-infected macrophages following LPS treatment. Further evidence for the role of FoxO1 in shaping M2-like macrophages was obtained pharmacologically through the use of AS1842856 (FoxO1 inhibitor; Calbiochem, San Diego, CA, USA). This selective FoxO1 inhibitor is newly discovered and according to a published report [49], potently inhibits the transcription activity of FoxO1 compared with inhibition of functionally related family members, including FoxO3a and FoxO4 (70%, 20%, and 3% inhibition, respectively) via direct binding of the active FoxO1 but not the Ser256-phosphorylated/inactive form of FoxO1. Like siFoxO1 knockdown, our in vitro experiment with BMM treated with AS1842856 resulted dramatically in inhibition of IL-10 release and IL-10 mRNA expression but not in those with IL-12/IL-23 (Fig. 4A and B). Thus, the combined data in Figs. 1–4 are consistent with a cause and effect relationship between FoxO1 and M2-like macrophages, resulting in equipping the cells with an IL-10 (high):IL-12/IL-23 (low) cytokine production profile.
Figure 4. Effect of FoxO1 inhibitor AS1842856 on IL-10/IL-12 cytokine releases.

(A) ELISA of the secretion of IL-10 and IL-12/IL-23 from BMM. Cells were preincubated with 0.5–5 μM FoxO1 inhibitor (AS1842856) for 1 h before stimulation with 100 ng/ml LPS (w/ LPS) or left unstimulated (w/o LPS). (B) RT-PCR analysis of IL-10 and IL-12p40 by BMM pretreated with FoxO1 inhibitor and stimulated for 24 h with LPS (100 ng/ml; n = 3 or more). **P < 0.01, and ***P < 0.001 versus nontreated; #P < 0.05, and ###P < 0.001 versus LPS with noninhibitor (Student’s t-test). Results are from at least 3 independent experiments.
FoxO1 is involved in transcriptional regulation of IL-10
Next, we investigated the LPS-induced recruitment of FoxO1 to the promoter loci of the IL-10 genes. We designed primers encompassing these FoxO1-binding sites on each cytokine gene and used them in a quantitative ChIP assay along with LPS. With the use of the ChIP assay, after LPS stimulation, we observed the enrichment of FoxO1 at the promoter regions of IL-10 up to 24 h in BMM (Fig. 5A), but it did not occur at the promoter region of IL-12 gene (data not shown). In this assay. we also observed that putative FoxO1-binding sites on distal Region #1 were displaying highly variable and no significant difference in response to LPS, although FoxO1 was detected at all regions. These data indicate that FoxO1 is recruited in the promoter region of the gene exclusively encoding IL-10 through preferential binding to specific Regions #2 and #3. To ascertain whether the identified FoxO1-binding sites are transcriptionally active, a luciferase reporter assay was performed by use of the IL-10 luciferase reporter plasmid plus a construct encoding FoxO1. Expression of FoxO1 caused higher IL-10 luciferase activity in response to LPS at Raw 264.7 macrophage cells (Fig. 5B). Moreover, we observed highly enriched FoxO1 on the IL-10 promoter in BMM but less in GM-BMM, consistent with data obtained with IL-10 expression (Fig. 5C). To evaluate the importance of the binding of FoxO1 to the IL-10 promoter, we generated a mutant of FoxO1 lacking the DNA-binding domain. This mutant domain induced much less luciferase activity (Fig. 5D), which indicates that FoxO1 enhances IL-10 by binding directly to the IL-10 promoter.
Figure 5. FoxO1 is a recruited IL-10 promoter in macrophages.

(A) The upstream locations of putative FoxO1-binding sites in IL-10 gene. The primers were designed for each region of a putative binding site of FoxO1 and used for amplification. The fold enrichment following LPS stimulation was measured, as described in Materials and Methods, by use of a FoxO1-specific antibody. The binding of FoxO1 to these 3 regions of the IL-10 promoter, as determined by a ChIP assay in BMM (n = 5), is described in Materials and Methods. (B) The reporter plasmid containing the upstream region of the IL-10 gene was constructed by use of the dual luciferase systems. The reporter construct was transfected into Raw macrophage cells for 24 h, and the cells were treated with LPS (n = 4). (C) Binding activity of FoxO1 to the promoter Region #2 of the IL-10 gene was determined in BMM/GM-BMM by ChIP assay (n = 4 or more). (D) Reporter luciferase activity of the upstream promoter region of IL-10 in Raw macrophage cells. The cells were transfected with a WT FoxO1-encoding IL-10 promoter reporter (IL10) or 2 mutants of FoxO1 lacking the DNA-binding domain (IL10-fr1 or -fr2) and then stimulated with LPS (n = 3 or more). *P < 0.05, and **P < 0.01 versus nontreated; ##P < 0.01 versus M-CSF-polarizing BMDMs (Student’s t-test). Results are from at least 3 independent experiments.
We next generated a mouse model with myeloid-specific FoxO1−/− (Fig. 6A), which is referred to as the LysMFoxO1 mice. A deletion efficiency of ∼80% was determined in LysMFoxO1 BMDMs compared with WT control (Fig. 6B and C). To establish whether macrophage FoxO1 plays a role in regulation of IL-10 in an in vivo model, LysMFoxO1 and littermate WT were injected with a low dose of LPS (2 mg/kg). At 3 h post-LPS, LysMFoxO1 mice had decreased significantly the serum IL-10 level compared with controls. The numbers of macrophages recruited into the peritoneal cavity of LPS-challenged mice were similar between LysMFoxO1 and WT control, but IL-10 mRNA levels were reduced markedly in LysMFoxO1 mice (Fig. 6D). BMM obtained from LysMFoxO1 secreted significantly less IL-10 and reduced expression of IL-10 mRNA, indicative of decreased M2-like cells (Fig. 6E). The expression of genes encoding M2 macrophage markers (IL-10, Arg1, Fizz1, and IL-13ra1) was significantly less in LysMFoxO1 mice (Fig. 6F); however, M1 markers (iNOS, IL-12a, IL-12b and CCR2) in these cells were significantly higher or did not follow the expected pattern of FoxO1 dependence, which possibly reflects earlier works showing a FoxO1-Tlr4 or FoxO1-CCR2 axis [15, 50].
Figure 6. Myeloid FoxO1-dependent induction of IL-10 production in vivo and in vitro.
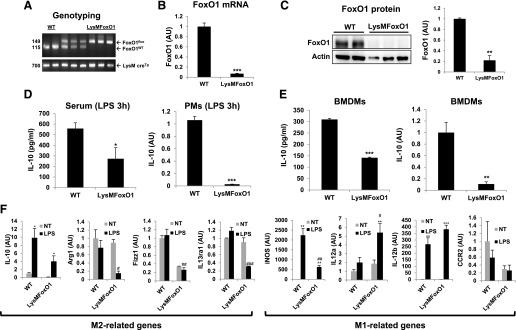
(A) FoxO1 PCR yields a 149 bp for the LoxP allele and 115 bp for the WT FoxO1 allele (upper) and a 700 bp for the LysM cre recombinase allele (lower). (B) Loss of FoxO1 mRNA in peritoneal macrophages from LysMFoxO1 mice. (C) Reduced FoxO1 protein in peritoneal macrophages from LysMFoxO1 mice. (D) ELISA (IL-10) of the serum concentrations of cytokine in LysMFoxO1 mice and mRNA of IL-10 by peritoneal macrophages (PMs; n = 4–6) injected i.p. with LPS (2 mg/kg) assessed 3 h later. (E) ELISA and mRNA of IL-10 by BMM obtained from LysMFoxO1 mice and their WT littermate (n = 4–6) and stimulated for 24 h or 3 h with LPS (100 ng/ml). (F) Expression of mRNA for M1 and M2 markers in LPS-stimulated peritoneal macrophages from LysMFoxO1. *P < 0.05, **P < 0.01, and ***P < 0.001 versus nontreated; #P < 0.05, ##P < 0.01, and ###P < 0.001 versus WT-LPS (Student’s t-test).
Down-regulation of FoxO1 and IL-10 secretion is correlated in hyperglycemic M1-like macrophages
There is growing evidence that indicates that macrophages are able to sense the metabolic stresses and are activated to a certain phenotype that could play a key role in metabolic dysregulation. We investigated a potential link between FoxO1 and M2 activation of macrophages in glucose-enriched microenvironments. For this study, bone marrow differentiation and activation were attained in a 5.5 (normal) or 25 mM (high) D-glucose condition. Previous studies showed that macrophages that differentiated in the presence of a high-glucose concentration exhibited elevated levels of proinflammatory cytokines [32, 51]. Our work expands the implications of the skewed classically activated M1-like macrophages by suggesting M1/M2 cytokine profiles and FoxO1 expression under high-glucose conditions. Interestingly, macrophages differentiated in the presence of high-glucose concentration exhibited a lower level of FoxO1 protein than did a normal glucose condition (Fig. 7A), and this decrease was associated with direct binding of FoxO1 to the promoter region of IL-10 in the high-glucose condition (Fig. 7B). By ChIP assay, we observed LPS-induced enrichment of FoxO1 at the promoter region of IL-10 (Fig. 7B), which matched, more or less, recruitment of FoxO1 in BMM and GM-BMM in Fig. 1E, possibly because of the decreased M2-related cytokine IL-10 under high-glucose condition (Fig. 7C) [32].
Figure 7. Differential activation of FoxO1 in hyperglycemic M1-like macrophages.

(A) FoxO1 immunoblot analysis from BMDMs, differentiated in 5.5 (normal) or 25 (high) mM D-glucose and stimulated with LPS (100 ng/ml). (B) Binding of FoxO1 to Region #2 of the IL-10 promoter, as determined by ChIP assay in BMM differentiated in 5.5 (normal) or 25 (high) mM D-glucose and stimulated with LPS (100 ng/ml; n = 3 or more). (C) ELISA of the secretion of IL-10 from BMM differentiated in 5.5 or 25 mM D-glucose and stimulated with LPS (100 ng/ml). (D) Macrophages cultured from diabetic db/db mice have M1-like phenotypic characteristics, as well as the suppressed FoxO1 expression. Representative morphologic images of BMM. The immunoblots against FoxO1 and iNOS of BMM cultured from db/db mice and their WT counterpart BMM were analyzed. The cells were stimulated for 24 h with LPS (100 ng/ml). (E) The binding of FoxO1 to the putative FoxO1-binding Region #2 of the IL-10 gene was determined by ChIP assay in LPS-stimulated, M-CSF-polarizing BMM, which was derived from db/db and their WT counterpart and stimulated for 24 h with LPS (100 ng/ml; n = 3 or more). (F) RT-PCR analysis of IL-10 mRNA expression and ELISA of IL-10 from BMM cultured from db/db and their WT counterpart. The cells were stimulated for indicated times with LPS (100 ng/ml). Results are from at least 3 independent experiments. (G) Hyperglycemia was induced by STZ (n = 5). Representative morphologic images of peritoneal macrophages. Loss of FoxO1 mRNA and reduced FoxO1 protein in peritoneal macrophages from hyperglycemic mice. (H) ELISA of IL-10 and IL-12/IL-23 by peritoneal macrophages obtained from STZ-hyperglycemic mice and their control (n = 5) and stimulated for 24 h with LPS (100 ng/ml). (I) RT-PCR analysis of FoxO1, Ym1, IL-10, IL-12b, and iNOS mRNA expression in adipose tissue macrophages from STZ-hyperglycemic mice (n = 5–6). *P < 0.05, **P < 0.01, ***P < 0.001 versus nontreated, and ##P < 0.01 versus 5.5 mM D-glucose (Student’s t-test).
To investigate if these results could be confirmed in an in vivo metabolic stress condition, we obtained BMDMs from obese db/db mice. Because of the lack of a leptin receptor, db/db mice are genetically obese, showing metabolic abnormalities, such as insulin-resistant and impaired glucose tolerance [16]. Similar to the data on macrophages from a high-glucose condition, BMDMs obtained from db/db mice exhibited less expression of FoxO1 protein than their WT counterpart (Fig. 7D). Consistent with the less FoxO1 expression in macrophages from db/db mice, we observed reduced FoxO1 enrichment at the promoter regions of IL-10 in obese db/db mice compared with the WT counterpart (Fig. 7E), suggesting a probable causal relationship between less FoxO1 and IL-10 in metabolic stress conditions. Consequently, BMM from db/db mice expressed less IL-10 gene encoding and a low level of IL-10 in response to stimulation with LPS than did WT cells (Fig. 7F); consistent with recent findings, the setting of diabetes is likely to contribute to the augmented, proinflammatory M1 macrophage phenotype [32].
In addition, freshly isolated peritoneal macrophages from STZ hyperglycemic mice displayed an M1-like phenotype and reduced FoxO1 expression (Fig. 7G), similar to what we observed in macrophages from db/db mice. We found that macrophages from hyperglycemic mice showed a pancake-like shape, which exhibits an M1 phenotype [47], and expressed a low level of FoxO1 and IL-10 but not IL-12/IL-23 (Fig. 7G and H) . We also observed FoxO1 dysregulation of the M1-like (Ym1low-IL-10low-IL-12bhigh-iNOShigh) state in relevant target adipose tissue macrophages in a STZ-induced hyperglycemic condition. Together, these results strongly suggest that FoxO1 is important in establishing an alternative M2-like macrophage skewing as a result of augmented IL-10 release, which could be a key molecular adaptor integrating an inflammatory response in the context of a hyperglycemic condition.
DISCUSSION
Macrophages are key regulators of the innate immune response that originates from bone marrow precursors, and functional differentiation and polarization are hallmarks of macrophages that result in the phenotypic diversity of the macrophage population [3, 30]. Interestingly, macrophages have pro- and anti-inflammatory properties. Classically (M1) and alternatively (M2) activated macrophages have distinct cytokine/chemokine profiles, phagocytic activity, expression of cell-surface receptors, and homeostatic functions [52]. However, M1 and M2 diversity subsets are not firmly differentiated, as are TH1 and TH2 lymphocyte subsets, implying that macrophages undergo dynamic transition between differential states [42]. This plasticity in the dual-functional properties of M1/M2 macrophages is determined by the immunologic microenvironment. The molecular determinants of functional diversity and specifically, the transcription factors that dictate alternative functional outcomes are, to a large extent, unknown. With the use of molecular biologic analyses of phenotype-specific transcription factors, this study shed new light on the transcriptional mechanisms of macrophage development and activation. We determined a novel role for FoxO1 in macrophages as a key regulator of the inflammatory process, where it contributes to regulation of IL-10 gene expression during classic activation by LPS treatment. Our data also show a link between FoxO1-derived macrophage phenotype and hyperglycemia and hyperlipidemia, as these are critical for the development of a metabolic syndrome. In the presence of high-glucose concentration, macrophages skewed an M1-like phenotype with an attenuated level of IL-10, and this decrease was associated with FoxO1.
Treatment with M-CSF generates highly purified mature macrophages from murine bone marrow cells in vitro, called BMDMs, which are a widely used population to study macrophage biology [31, 53]; on the other hand, with a similar protocol, GM-CSF also generates a population (GM-BMM) of cells that has macrophage properties [43, 44, 54]. GM-BMM and BMM adopt distinct cytokine profiles following TLR stimulation, and it was proposed that they can be viewed as being in "M1-like" and "M2-like" polarization states, respectively [43]. In the context of this study, we have used M-CSF to drive macrophage differentiation to the M2-like phenotype, whereas GM-CSF drives macrophages to the M1-like phenotype.
FoxO proteins participate in a wide range of fundamental cellular processes, including apoptosis, autophagy, repair of DNA, response to oxidative stress, stress resistance, and regulation of metabolism [55–57]. FoxOs belong to the forkhead box family of transcription factors having a winged helix motif in the protein structure domain [58]. FoxOs are known to be subject to extensive post-translational modifications affecting their functions, including activity and localization [59]. The phosphorylation by PI3K/Akt and p300-mediated acetylation on a specific site of FoxOs is a major pathway and results in the control of their transcriptional activity. Interestingly, several transcription factors can directly affect the expression of FoxO genes, including FoxO1 and FoxO3 themselves [59, 60]. Among the FoxO isoforms, FoxO1 and FoxO3 are mainly expressed in the immune system [11]. A majority of studies has been focused on the role of FoxO3 in the immune system, whereas only few pointed to a role of FoxO1. Recent works have shown that FoxOs regulate specialized characteristics of lymphocyte and myeloid homeostasis [34, 61–63]. More recently published literature showed that FoxO1 stimulates expression of the proinflammatory IL-1β and enhances TLR4 signaling in mature macrophages, resulting in enhanced inflammatory responses [15, 16]. This correlates with transcriptional activity of FoxO1 as responsible for enhanced TLR4-meidated signaling, supporting that FoxO1 enhances innate immunity in macrophages. Indeed, FoxO1 has also been shown to regulate M1-like macrophage recruitment into adipose tissues [50]. Overexpression of FoxO1 promoted transcription of CCR2 in adipose tissue macrophages. These findings imply that FoxO1 is underlying the progress of inflammation in response to metabolic demand or stress. Conversely, FoxO1 displayed anti-inflammatory responses by blunting the NF-κB pathway [64–66], pinpointing a dual function of FoxO1 as a key transcription factor of macrophage inflammation, and this dual role warrants additional investigations. In this current study, the expression of mRNA for M2 markers (IL-10, Arg1, Fizz1, and IL-13ra1) was considerably reduced in LysMFoxO1 macrophages, whereas some M1-related genes (iNOS and CCR2) did not follow the expected pattern of FoxO1 dependence. This possibly reflects the impact of LPS on macrophage gene expression, as reported earlier studies show a FoxO1-TLR4 pathway [15] and/or FoxO1-CCR2 axis [50] that emphasizes the importance of a dual function for FoxO1 in this context.
We identified that FoxO1 is highly expressed in BMM and induced characteristic cytokine IL-10 secretion. Depletion of FoxO1 by siRNA in BMM resulted in reduction of IL-10 secretion, whereas the IL-12/IL-23 level is not responsible. We have extended these findings by testing a newly developed pharmacological inhibitor of FoxO1, detecting distinct phenotypic cytokine secretions. This inhibitor is believed to interfere with FoxO1 interaction by binding the dephosphorylated FoxO1, thereby repressing FoxO1-mediated activation [49]. These latter experiments are consistent with the idea that IL-10 secretion is dependent on FoxO1 activation in response to LPS. Interestingly, this inhibitor resulted unexpectedly in increased IL-12/IL-23 secretion, suggesting potent reciprocal regulation by FoxO1. From this point of view, the role of FoxO1 would be greater in BMM than in GM-BMM during classic activation with LPS treatment. It should be noted that an interesting aspect of our data is that ablation of FoxO1 expression is accentuated in response to LPS treatment in M1-like macrophages, which is consistent with a counter-regulatory role of FoxO1, presumably though enhancement of the M2-like phenotype. Thus, these data, in combination with those shown above, support the conclusion that endogenous FoxO1 has profound effects on BMM, as BMMs express abundant FoxO1, whereas GM-BMMs have decreased FoxO1 compared with control values when treated in vitro with endotoxin. Many studies confirmed that the polarization status of macrophages correlates well with the immune system and metabolism in disease [67–70]. In addition to the proinflammatory cytokines, anti-inflammatory cytokines, such as IL-10, IL-1Ra, and secreted frizzled-related protein 5, increased in metabolic dysfunction [71, 72]. The observation that elevated anti-inflammatory cytokines in metabolic dysfunction support the validity of homeostatic balance between pro- and anti-inflammatory cytokines defines the magnitude of inflammation [67]. Thus, M1 and M2 character coexistence during progression of an inflammatory response enables the dual role of macrophages in orchestrating the onset of inflammation. This indicates that there could be different collaborating transcription factors controlling M2- versus M1-specific gene expression separately, and these reciprocal factors might be associated with FoxO1 on phenotypic differences of macrophages.
It is becoming increasingly clear that macrophages have an important, central role in metabolic stress-associated inflammation [34, 50, 65]. In our study, we showed that in vitro exposure of macrophage to hyperglycemia reduced FoxO1 gene expression. We also showed that BMDMs from obese db/db mice had a reduced level of FoxO1 protein, suggesting that hyperglycemia may impact FoxO1 gene expression. Of interest, macrophages in these hyperglycemic conditions showed an inferior FoxO1 enrichment on the IL-10 promoter following challenge with LPS, but not in the control animal. These findings do add to evidence that links association of low IL-10 levels with the metabolic syndrome [73–75]. Our observations indicate that positive interactions between FoxO1 and IL-10 promoter DNA in a high-glucose condition were less than a normal condition, which suggests that loss of recruitment of FoxO1 to the IL-10 does mimic the effect of the M1 inflammatory phenotype during glucose-enriched conditions. We confirmed the phenotypic change of tissue-resident macrophages in in vivo exposure to hyperglycemia, as we detected low IL-10 and FoxO1 expressions in db/db mice. Thus, macrophages in the setting of hyperglycemia are likely to contribute to the augmented proinflammatory macrophage phenotype through attenuation of the contribution of FoxO1 to activate IL-10 expression. We propose that during hyperglycemia, FoxO1 is required to regulate the macrophage phenotype by promoting IL-10 genes. Our work expands the implications of the increased IL-10 levels associated with improved insulin sensitivity [76] by suggesting that FoxO1 is a key enhancer of macrophage IL-10 secretion in patients with persistent hyperglycemia.
A recent study shows that PDK1-FoxO1 signaling regulated M1 macrophage recruitment into adipose tissue during a high-fat diet via CCR2 expression [50]. In that study, they suggested adipose tissue macrophage autonomous PDK-FoxO1 signaling regulates adipose tissue inflammation and insulin sensitivity in vivo. In addition to the above ability of FoxO1, myeloid FoxO knockout mice develop more severe atherosclerosis by increased oxidative stress and iNOS-derived NO overproduction in macrophages, possibly M1-like macrophages [34]. Therefore, it is plausible to explain that FoxO function in myeloid cells plays an important role in response to metabolic stress, raising the possibility of FoxO1-dependent expansion of IL-10 secretion, as proinflammatory M1-like macrophages are linked to obesity, diabetes mellitus, and atherosclerosis with no doubt. In the present study, the distinct role of FoxO1 in the concept of M2-like macrophages seems to be specific: it only had high expression in M2-like BMM and promoted robust IL-10 expression during classic activation. Nevertheless, regulation of FoxO1 is an important issue, as this protein may be a key molecular adaptor integrating proinflammatory response in the context of insulin resistance and hyperglycemia. As the concept of pathobiology of a metabolic syndrome, a specific biologic output of the FoxO1 signaling pathway that engages macrophages to polarize has become a major consideration in exploring the potential role of insulin signaling to FoxO1 in macrophages.
In summary, our data show that FoxO1 is associated with the M2-like macrophages, as IL-10 production by BMM is disrupted by genetic ablation and overexpression of FoxO1 in a highly predictable manner. Although results of biologic output of the new myeloid-specific, FoxO1−/− mice remain to be defined in response to metabolic stress, this study provides a novel role for FoxO1 in M2-like macrophages. We also have shown that loss of FoxO1 expression contributes to skew the macrophage phenotype as M1 like, following metabolic stress condition. Together, these data suggest that FoxO1 has transcriptional influence on polarization of macrophages toward the M2-like phenotype, suggesting that interdiction could lead to innovative treatments for inflammation as a link between the hyperglycemia and metabolic syndrome.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health Grants 2R01HL075557-08 and 5R01HL103643-03 and U.S. Department of Veterans Affairs Merit Review Grant 5I01BX000108. The authors thank Dr. Jaehyung Cho for support with human PBMC experiments. The authors gratefully acknowledge Drs. Timothy Koh and Aaron J. Trask for providing bone marrow from the db/db mice.
Glossary
- −/−
deficient
- Arg1
arginase 1
- BMDM
bone marrow-derived macrophage
- BMM
M-CSF-derived macrophage
- ChIP
chromatin immunoprecipitation
- Fizz1
found in inflammatory zone 1
- FoxO
forkhead box protein O
- GM-BMM
GM-CSF-derived macrophage
- LysMFoxO1
forkhead box protein Oflox/floxCreTg
- m
mouse
- MARCO
macrophage receptor with collagenous structure
- PDK
phosphoinositide-dependent protein kinase
- rh
recombinant human
- rm
recombinant mouse
- scRNA
scrambled control small interfering RNA
- siFoxO1
forkhead box protein O 1 small interfering RNA
- siRNA
small interfering RNA
- STZ
streptozotocin
- TSS
constitutively active mutant containing the mutations at T24A, S256A and S319A
- WT
wild-type
AUTHORSHIP
S.C. designed, performed, and analyzed all experiments and wrote the manuscript. R.R., Y.G.L, J.D., L.X., M.K., J.Y.K., T.G.U., and G.Y.P. provided significant assistance in design, analysis, and editing. J.W.C. provided the funds, resources, and mentorship in experimental design, analysis, and editing of the manuscript.
DISCLOSURES
The authors declare no competing financial interests.
REFERENCES
- 1.Laskin D. L. (2009) Macrophages and inflammatory mediators in chemical toxicity: a battle of forces. Chem. Res. Toxicol. 22, 1376–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laskin D. L., Sunil V. R., Gardner C. R., Laskin J. D. (2011) Macrophages and tissue injury: agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 51, 267–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biswas S. K., Mantovani A. (2010) Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol. 11, 889–896. [DOI] [PubMed] [Google Scholar]
- 4.Holt M. P., Cheng L., Ju C. (2008) Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J. Leukoc. Biol. 84, 1410–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trujillo G., O’Connor E. C., Kunkel S. L., Hogaboam C. M. (2008) A novel mechanism for CCR4 in the regulation of macrophage activation in bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 172, 1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramalingam T. R., Pesce J. T., Mentink-Kane M. M., Madala S., Cheever A. W., Comeau M. R., Ziegler S. F., Wynn T. A. (2009) Regulation of helminth-induced Th2 responses by thymic stromal lymphopoietin. J. Immunol. 182, 6452–6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galli S. J., Borregaard N., Wynn T. A. (2011) Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat. Immunol. 12, 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagemann T., Lawrence T., McNeish I., Charles K. A., Kulbe H., Thompson R. G., Robinson S. C., Balkwill F. R. (2008) “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 205, 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawanishi N., Yano H., Yokogawa Y., Suzuki K. (2010) Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc. Immunol. Rev. 16, 105–118. [PubMed] [Google Scholar]
- 10.Krausgruber T., Blazek K., Smallie T., Alzabin S., Lockstone H., Sahgal N., Hussell T., Feldmann M., Udalova I. A. (2011) IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238. [DOI] [PubMed] [Google Scholar]
- 11.Dejean A. S., Hedrick S. M., Kerdiles Y. M. (2011) Highly specialized role of forkhead box O transcription factors in the immune system. Antioxid. Redox Signal. 14, 663–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobs F. M., van der Heide L. P., Wijchers P. J., Burbach J. P., Hoekman M. F., Smidt M. P. (2003) FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J. Biol. Chem. 278, 35959–35967. [DOI] [PubMed] [Google Scholar]
- 13.van der Heide L. P., Smidt M. P. (2005) Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem. Sci. 30, 81–86. [DOI] [PubMed] [Google Scholar]
- 14.Matsuzaki H., Daitoku H., Hatta M., Tanaka K., Fukamizu A. (2003) Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc. Natl. Acad. Sci. USA 100, 11285–11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan W., Morinaga H., Kim J. J., Bae E., Spann N. J., Heinz S., Glass C. K., Olefsky J. M. (2010) FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J. 29, 4223–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su D., Coudriet G. M., Hyun Kim D., Lu Y., Perdomo G., Qu S., Slusher S., Tse H. M., Piganelli J., Giannoukakis N., Zhang, J., Dong, H. H. (2009) FoxO1 links insulin resistance to proinflammatory cytokine IL-1beta production in macrophages. Diabetes 58, 2624–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mauer J., Chaurasia B., Goldau J., Vogt M. C., Ruud J., Nguyen K. D., Theurich S., Hausen A. C., Schmitz J., Brönneke H. S., Estevez E., Allen T. L., Mesaros A., Partridge L., Febbraio M. A., Chawla A., Wunderlich F. T., Brüning J. C. (2014) Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat. Immunol. 15, 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lumeng C. N., Bodzin J. L., Saltiel A. R. (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Odegaard J. I., Ricardo-Gonzalez R. R., Goforth M. H., Morel C. R., Subramanian V., Mukundan L., Red Eagle A., Vats D., Brombacher F., Ferrante A. W., Chawla A. (2007) Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447, 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tabas I., Tall A., Accili D. (2010) The impact of macrophage insulin resistance on advanced atherosclerotic plaque progression. Circ. Res. 106, 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vats D., Mukundan L., Odegaard J. I., Zhang L., Smith K. L., Morel C. R., Wagner R. A., Greaves D. R., Murray P. J., Chawla A. (2006) Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 4, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang C. P., Han S., Okamoto H., Carnemolla R., Tabas I., Accili D., Tall A. R. (2004) Increased CD36 protein as a response to defective insulin signaling in macrophages. J. Clin. Invest. 113, 764–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biswas S. K., Mantovani A. (2012) Orchestration of metabolism by macrophages. Cell Metab. 15, 432–437. [DOI] [PubMed] [Google Scholar]
- 24.Freemerman A. J., Johnson A. R., Sacks G. N., Milner J. J., Kirk E. L., Troester M. A., Macintyre A. N., Goraksha-Hicks P., Rathmell J. C., Makowski L. (2014) Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 289, 7884–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hotamisligil G. S., Erbay E. (2008) Nutrient sensing and inflammation in metabolic diseases. Nat. Rev. Immunol. 8, 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhargava P., Lee C. H. (2012) Role and function of macrophages in the metabolic syndrome. Biochem. J. 442, 253–262. [DOI] [PubMed] [Google Scholar]
- 27.Tannahill G. M., Curtis A. M., Adamik J., Palsson-McDermott E. M., McGettrick A. F., Goel G., Frezza C., Bernard N. J., Kelly B., Foley N. H., Zheng L., Gardet A., Tong Z., Jany S. S., Corr S. C., Haneklaus M., Caffrey B. E., Pierce K., Walmsley S., Beasley F. C., Cummins E., Nizet V., Whyte M., Taylor C. T., Lin H., Masters S. L., Gottlieb E., Kelly V. P., Clish C., Auron P. E., Xavier R. J., O'Neill L. A. (2013) Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karpurapu M., Wang X., Deng J., Park H., Xiao L., Sadikot R. T., Frey R. S., Maus U. A., Park G. Y., Scott E. W., Christman J. W. (2011) Functional PU.1 in macrophages has a pivotal role in NF-κB activation and neutrophilic lung inflammation during endotoxemia. Blood 118, 5255–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng J., Wang X., Qian F., Vogel S., Xiao L., Ranjan R., Park H., Karpurapu M., Ye R. D., Park G. Y., Christman J. W. (2012) Protective role of reactive oxygen species in endotoxin-induced lung inflammation through modulation of IL-10 expression. J. Immunol. 188, 5734–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krausgruber T., Saliba D., Ryzhakov G., Lanfrancotti A., Blazek K., Udalova I. A. (2010) IRF5 is required for late-phase TNF secretion by human dendritic cells. Blood 115, 4421–4430. [DOI] [PubMed] [Google Scholar]
- 31.Fleetwood A. J., Dinh H., Cook A. D., Hertzog P. J., Hamilton J. A. (2009) GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J. Leukoc. Biol. 86, 411–421. [DOI] [PubMed] [Google Scholar]
- 32.Kanter J. E., Kramer F., Barnhart S., Averill M. M., Vivekanandan-Giri A., Vickery T., Li L. O., Becker L., Yuan W., Chait A., Braun K. R., Potter-Perigo S., Sanda S., Wight T. N., Pennathur S., Serhan C. N., Heinecke J. W., Coleman R. A., Bornfeldt K. E. (2012) Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc. Natl. Acad. Sci. USA 109, E715–E724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hahm E., Li J., Kim K., Huh S., Rogelj S., Cho J. (2013) Extracellular protein disulfide isomerase regulates ligand-binding activity of αMβ2 integrin and neutrophil recruitment during vascular inflammation. Blood 121, 3789–3800, S1–S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsuchiya K., Westerterp M., Murphy A. J., Subramanian V., Ferrante A. W. Jr, Tall A. R., Accili D. (2013) Expanded granulocyte/monocyte compartment in myeloid-specific triple FoxO knockout increases oxidative stress and accelerates atherosclerosis in mice. Circ. Res. 112, 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng X., Zhang W., O-Sullivan I., Williams J. B., Dong Q., Park E. A., Raghow R., Unterman T. G., Elam M. B. (2012) FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J. Biol. Chem. 287, 20132–20143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho K. W., Morris D. L., Lumeng C. N. (2014) Flow cytometry analyses of adipose tissue macrophages. Methods Enzymol. 537, 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramkhelawon B., Hennessy E. J., Ménager M., Ray T. D., Sheedy F. J., Hutchison S., Wanschel A., Oldebeken S., Geoffrion M., Spiro W., Miller G., McPherson R., Rayner K. J., Moore K. J. (2014) Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat. Med. 20, 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seiler F., Hellberg J., Lepper P. M., Kamyschnikow A., Herr C., Bischoff M., Langer F., Schäfers H. J., Lammert F., Menger M. D., Bals R., Beisswenger C. (2013) FOXO transcription factors regulate innate immune mechanisms in respiratory epithelial cells. J. Immunol. 190, 1603–1613. [DOI] [PubMed] [Google Scholar]
- 39.Chung S., Sundar I. K., Hwang J. W., Yull F. E., Blackwell T. S., Kinnula V. L., Bulger M., Yao H., Rahman I. (2011) NF-κB inducing kinase, NIK mediates cigarette smoke/TNFα-induced histone acetylation and inflammation through differential activation of IKKs. PLoS ONE 6, e23488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun Z., Miller R. A., Patel R. T., Chen J., Dhir R., Wang H., Zhang D., Graham M. J., Unterman T. G., Shulman G. I., Sztalryd C., Bennett M. J., Ahima R. S., Birnbaum M. J., Lazar M. A. (2012) Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med. 18, 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shin D. J., Osborne T. F. (2009) FGF15/FGFR4 integrates growth factor signaling with hepatic bile acid metabolism and insulin action. J. Biol. Chem. 284, 11110–11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lawrence T., Natoli G. (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761. [DOI] [PubMed] [Google Scholar]
- 43.Fleetwood A. J., Lawrence T., Hamilton J. A., Cook A. D. (2007) Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J. Immunol. 178, 5245–5252. [DOI] [PubMed] [Google Scholar]
- 44.Lari R., Fleetwood A. J., Kitchener P. D., Cook A. D., Pavasovic D., Hertzog P. J., Hamilton J. A. (2007) Macrophage lineage phenotypes and osteoclastogenesis—complexity in the control by GM-CSF and TGF-beta. Bone 40, 323–336. [DOI] [PubMed] [Google Scholar]
- 45.Wang T., Ge Y., Xiao M., Lopez-Coral A., Azuma R., Somasundaram R., Zhang G., Wei Z., Xu X., Rauscher F. J. III, Herlyn M., Kaurman R. E. (2012) Melanoma-derived conditioned media efficiently induce the differentiation of monocytes to macrophages that display a highly invasive gene signature. Pigment Cell Melanoma Res. 25, 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svensson J., Jenmalm M. C., Matussek A., Geffers R., Berg G., Ernerudh J. (2011) Macrophages at the fetal-maternal interface express markers of alternative activation and are induced by M-CSF and IL-10. J. Immunol. 187, 3671–3682. [DOI] [PubMed] [Google Scholar]
- 47.McWhorter F. Y., Wang T., Nguyen P., Chung T., Liu W. F. (2013) Modulation of macrophage phenotype by cell shape. Proc. Natl. Acad. Sci. USA 110, 17253–17258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han M. S., Jung D. Y., Morel C., Lakhani S. A., Kim J. K., Flavell R. A., Davis R. J. (2013) JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagashima T., Shigematsu N., Maruki R., Urano Y., Tanaka H., Shimaya A., Shimokawa T., Shibasaki M. (2010) Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: improvement of fasting glycemia in diabetic db/db mice. Mol. Pharmacol. 78, 961–970. [DOI] [PubMed] [Google Scholar]
- 50.Kawano Y., Nakae J., Watanabe N., Fujisaka S., Iskandar K., Sekioka R., Hayashi Y., Tobe K., Kasuga M., Noda T., Yoshimura A., Itoh H. (2012) Loss of Pdk1-Foxo1 signaling in myeloid cells predisposes to adipose tissue inflammation and insulin resistance. Diabetes 61, 1935–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shanmugam N., Reddy M. A., Guha M., Natarajan R. (2003) High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes 52, 1256–1264. [DOI] [PubMed] [Google Scholar]
- 52.Mantovani A. (2008) From phagocyte diversity and activation to probiotics: back to Metchnikoff. Eur. J. Immunol. 38, 3269–3273. [DOI] [PubMed] [Google Scholar]
- 53.Hamilton J. A., Stanley E. R., Burgess A. W., Shadduck R. K. (1980) Stimulation of macrophage plasminogen activator activity by colony-stimulating factors. J. Cell. Physiol. 103, 435–445. [DOI] [PubMed] [Google Scholar]
- 54.Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R. M. (1992) Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176, 1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Battiprolu P. K., Hojayev B., Jiang N., Wang Z. V., Luo X., Iglewski M., Shelton J. M., Gerard R. D., Rothermel B. A., Gillette T. G., Lavandero S., Hill J. A. (2012) Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Invest. 122, 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barthel A., Schmoll D., Unterman T. G. (2005) FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 16, 183–189. [DOI] [PubMed] [Google Scholar]
- 57.Sengupta A., Molkentin J. D., Yutzey K. E. (2009) FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 284, 28319–28331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lehmann O. J., Sowden J. C., Carlsson P., Jordan T., Bhattacharya S. S. (2003) Fox’s in development and disease. Trends Genet. 19, 339–344. [DOI] [PubMed] [Google Scholar]
- 59.Hedrick S. M., Hess Michelini R., Doedens A. L., Goldrath A. W., Stone E. L. (2012) FOXO transcription factors throughout T cell biology. Nat. Rev. Immunol. 12, 649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Essaghir A., Dif N., Marbehant C. Y., Coffer P. J., Demoulin J. B. (2009) The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J. Biol. Chem. 284, 10334–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hedrick S. M. (2009) The cunning little vixen: Foxo and the cycle of life and death. Nat. Immunol. 10, 1057–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin L., Hron J. D., Peng S. L. (2004) Regulation of NF-kappaB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity 21, 203–213. [DOI] [PubMed] [Google Scholar]
- 63.Ouyang W., Li M. O. (2011) Foxo: in command of T lymphocyte homeostasis and tolerance. Trends Immunol. 32, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsuchiya K., Banks A. S., Liang C. P., Tabas I., Tall A. R., Accili D. (2011) Homozygosity for an allele encoding deacetylated FoxO1 protects macrophages from cholesterol-induced inflammation without increasing apoptosis. Arterioscler. Thromb. Vasc. Biol. 31, 2920–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Senokuchi T., Liang C. P., Seimon T. A., Han S., Matsumoto M., Banks A. S., Paik J. H., DePinho R. A., Accili D., Tabas I., Tall A. R. (2008) Forkhead transcription factors (FoxOs) promote apoptosis of insulin-resistant macrophages during cholesterol-induced endoplasmic reticulum stress. Diabetes 57, 2967–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baumgartl J., Baudler S., Scherner M., Babaev V., Makowski L., Suttles J., McDuffie M., Tobe K., Kadowaki T., Fazio S., Kahn C. R., Hotamisligil G. S., Krone W., Linton M., Brüning J. C. (2006) Myeloid lineage cell-restricted insulin resistance protects apolipoproteinE-deficient mice against atherosclerosis. Cell Metab. 3, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Osborn O., Olefsky J. M. (2012) The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 18, 363–374. [DOI] [PubMed] [Google Scholar]
- 68.Fujisaka S., Usui I., Bukhari A., Ikutani M., Oya T., Kanatani Y., Tsuneyama K., Nagai Y., Takatsu K., Urakaze M., Kobayashi M., Tobe K. (2009) Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes 58, 2574–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li P., Lu M., Nguyen M. T., Bae E. J., Chapman J., Feng D., Hawkins M., Pessin J. E., Sears D. D., Nguyen A. K., Olefsky J. M. (2010) Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. J. Biol. Chem. 285, 15333–15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patsouris D., Li P. P., Thapar D., Chapman J., Olefsky J. M., Neels J. G. (2008) Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 8, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Juge-Aubry C. E., Somm E., Giusti V., Pernin A., Chicheportiche R., Verdumo C., Rohner-Jeanrenaud F., Burger D., Dayer J. M., Meier C. A. (2003) Adipose tissue is a major source of interleukin-1 receptor antagonist: upregulation in obesity and inflammation. Diabetes 52, 1104–1110. [DOI] [PubMed] [Google Scholar]
- 72.Ouchi N., Higuchi A., Ohashi K., Oshima Y., Gokce N., Shibata R., Akasaki Y., Shimono A., Walsh K. (2010) Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science 329, 454–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Esposito K., Pontillo A., Giugliano F., Giugliano G., Marfella R., Nicoletti G., Giugliano D. (2003) Association of low interleukin-10 levels with the metabolic syndrome in obese women. J. Clin. Endocrinol. Metab. 88, 1055–1058. [DOI] [PubMed] [Google Scholar]
- 74.Calcaterra V., De Amici M., Klersy C., Torre C., Brizzi V., Scaglia F., Albanesi M., Albertini R., Allais B., Larizza D. (2009) Adiponectin, IL-10 and metabolic syndrome in obese children and adolescents. Acta Biomed. 80, 117–123. [PubMed] [Google Scholar]
- 75.Hong E. G., Ko H. J., Cho Y. R., Kim H. J., Ma Z., Yu T. Y., Friedline R. H., Kurt-Jones E., Finberg R., Fischer M. A., Granger E. L., Norbury C. C., Hauschka S. D., Philbrick W. M., Lee C. G., Elias J. A., Kim, J. K. (2009) Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 58, 2525–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grant L., Shearer K. D., Czopek A., Lees E. K., Owen C., Agouni A., Workman J., Martin-Granados C., Forrester J. V., Wilson H. M., Mody N., Delibegovic M. (2014) Myeloid-cell protein tyrosine phosphatase-1B deficiency in mice protects against high-fat diet and lipopolysaccharide-induced inflammation, hyperinsulinemia, and endotoxemia through an IL-10 STAT3-dependent mechanism. Diabetes 63, 456–470. [DOI] [PubMed] [Google Scholar]