

Abstract
Engineered biological circuits are often disturbed by a variety of environmental factors. In batch culture, where the majority of synthetic circuit characterization occurs, environmental conditions vary as the culture matures. Turbidostats are powerful characterization tools that provide static culture environments; however, they are often expensive, especially when purchased in custom configurations, and are difficult to design and construct in a lab. Here, we present a low cost, open source multiplexed turbidostat that can be manufactured and used with minimal experience in electrical or software engineering. We demonstrate the utility of this system to profile synthetic circuit behavior in S. cerevisiae. We also demonstrate the flexibility of the design by showing that a fluorometer can be easily integrated.
Keywords: continuous culture, turbidostat, characterization, fluorescence detection
The characterization of biological building blocks and complex synthetic systems is fundamental to synthetic biology.1,2 Historically, characterization of biological systems has been carried out using batch culture methods.3−9 The low cost, ease of culture propagation, and scalability through use of well plates and culture flasks have contributed to the prevalence of this culture method. However, in batch culture, the chemical environment is continually changing as cells grow, divide, consume nutrients, and excrete waste products.10 These changes associated with growth have substantial effects on cellular physiology11−13 and engineered biological components, which can in turn cause the mischaracterization of a system’s input-output response or require the consideration of uncontrolled disturbances to the system.
To more accurately characterize biological systems, researchers must turn to static environments, which yield lower noise in quantitative phenotyping. A primary tool for culturing cells in a static environment is continuous culture, where inoculated growth medium is continually diluted with fresh medium. At steady state, a continuous culture device will dilute cells and waste products at the same rate that they are being produced leading to an unchanging environment.14,15
Two main categories of continuous culture devices are chemostats and turbidostats. Chemostats dilute liquid culture at a fixed rate and reach steady state when a limiting nutrient has been depleted, or toxins have been accumulated, at which point the growth rate is equal to the dilution rate. Therefore, chemostats are ideal for interrogating the effects of nutrient limitation or where defined growth rates are desired. In contrast, turbidostats use a feedback control loop to keep cell density constant; in a turbidostat, the cell density is continually monitored and used to compute the dilution rate that achieves a prespecified cell density. Thus, turbidostats are ideal for characterization of systems without nutrient limitation, when cells are growing at their maximum rate.
In spite of the advantages, use of chemostats and turbidostats is not commonplace. Factors that limit the use of continuous culture systems include the fact that commercially available bioreactors either lack feedback capability, have such large volumes that dilution rates needed to sustain a log phase culture would be impractical, or are prohibitivly expensive. As a result, researchers often design their own turbidostats, which vary widely in implementation based on the needs of the experimenter.16−23 Unfortunately, the design and construction can be laborious and uncertain. Design decisions that are seemingly innocuous can lead to failures or inflexible devices. Furthermore, development of turbidostats requires specialized knowledge of electrical and computer engineering, which has further limited their popularity.
We have developed an open source turbidostat design (the Flexostat) that contrasts with other publicly available continuous culture devices.18,20,24 Using common hand tools and standard laboratory equipment combined with inexpensive online or university provided fabrication services and 3D printing, our instrument can be built for under $2000 USD. Indeed, many individuals from a wide range of backgrounds have already built turbidostats from our design. Currently, five laboratories have at least one complete instrument (see acknowledgments), some with custom modifications. As part of our commitment to the community, the authors will continue to contribute updated designs to the Web site and ask, but do not require, that all derived designs be submitted so that the community may benefit.
The Flexostat implements eight parallel culture chambers, which enable users to gather replicate data and perform parallel experiments requiring no extra pumps or controllers. Each chamber contains electronics that enable distributed sensing and mixing control allowing users to add new chamber modules with new capabilities with minimal hardware redesign. A platform agnostic Python program collects data from each chamber and computes dilution rates through a user programmable function.
To demonstrate the utility of our device over standard batch culture, we recharacterized part of the auxin plant hormone pathway cloned into S. cerevisiae and grown at various constant cell densities. Previous work has characterized the interactions between the small molecule auxin and the family of F-box proteins in batch culture resulting in a table of key parameters for each interaction.6 Here, we show that one of these parameters, a degredation rate in the presence of auxin that was previously considered a constant, varies by more than 4-fold depending on the cell density.
Finally, we showcase how the modular design and 3D printed nature of the Flexostat allows us to add new capabilities. In particular, we show a proof of concept single chamber, mixture controlled variant that is capable of detecting strongly fluorescent cultures (the Fluorostat) such as E. coli expressing super folder GFP. Fluorescence detection is accomplished through the addition of a single blue excitation source and a pair of low cost, spectrally orthogonal filters. The light sources for the Optical density (OD) measurement and fluorescence measurements alternate in time and share the same photodetector. The dual use of the photosensor allows us to use the same electronics for the standard Flexostat and the Fluorostat, requiring that only software and a single 3D printed part be changed.
Results and Discussion
Theory of Operation
The operation of the Flexostat (Figure 1) consists of a discrete dilution cycle with an adjustable period (typically 1 min). First, the current optical density of each chamber is measured. Then, for each chamber, a user-programmable Python module computes the dilution rate as a function of error between desired OD and measured OD. The dilution rates, which may be different for each chamber, are then sent to the embedded circuitry, which controls the media pump and valve system. As new media is added, it is mixed by magnetic stir bars diluting old media, cells, and waste products. When the media level rises to the mouth of the effluent tube, waste is forced out by positive pressure provided by an aeration input.
Figure 1.

(a) Schematic of the Flexostat. Optical density (OD) is measured through the chamber wall and reported to a control system. A dilution rate is then calculated for each chamber, which is carried out by the pumping system. Valves select the flow source or destination while a single multiplexed syringe pump determines the volume and direction of flow. (b) A photograph of the turbidostat chamber with integrated OD measurement and stirring. (c) A 3D printed syringe pump. (d) A 3D printed four way normally closed pinch-valve with the upper right valve selected.
Specifications and Characterization
Biochemical reactions in cells are highly sensitive to experimental preparation, equipment variation, and environmental changes. Indeed, some systems even have multimodal steady state distributions,25 which are sensitive to initial conditions and require replicates to thoroughly probe. Furthermore, most experimental outcomes must be compared to those of a control experiment or between multiple variants. In a single chamber turbidostat, each replicate and control must be performed serially, which is time-consuming and susceptible to day to day variations in experimental conditions. For the Flexostat, we chose to multiplex eight culture chambers into the same media source and pump, which allows us to perform eight experiments in parallel, all of which are subjected to the same conditions. A single multiplexed pump has the additional advantage of lowering cost relative to buying eight pumps.
In the continuous culture literature, culture volumes can range from a few femtoliters26 to industrial fermenters containing thousands of liters of media. The culture volume has logistical impacts that need to be carefully weighed. Yeast in rich media go through approximately 16 generations in a day if kept at maximal growth rates. Since one volume must be replaced for every generation and there are eight culture chambers, 16 generations/day/chamber × 1 volume/generation × 8 chambers = 128 volumes per day will be consumed, which quickly adds up for large culture volumes. Small volumes also have drawbacks. If sampling is required for characterization, enough effluent must be available for a single measurement. A typical flow cytometer for example requires at least 50 μL for measurement.27 Sampling the 50 μL required for flow cytometry from a 1 mL vessel would require close to 5 min of collection time from effluent. We have found that a culture volume of 15 mL is a good balance, allowing for enough effluent to be collected over a sample period of 2 min while limiting the amount of media used to less than 2 L a day.
For characterization in replicate, all chambers must measure OD uniformly. Commercial OD probes are too large to fit into a 15 mL volume without interfering with other essential processes (e.g., stirring, dilution, etc.), so we developed a customized solution. We measure optical density through the walls of the culture tube using a 650 nm laser diode, similar to those found in common laser pointers, and a pair of photo sensors configured to reject changes in transmitted light intensity caused by temperature variation and electrical noise (Figure 2). Tests with reference medium consisting of black dye, which has an absorbance spectrum similar to E. coli (in the 500–700 nm range), show chamber to chamber variation of less than ±2.5%. For further characterization information including noise, linearity, and detection threshold see Supporting Information section S2.
Figure 2.
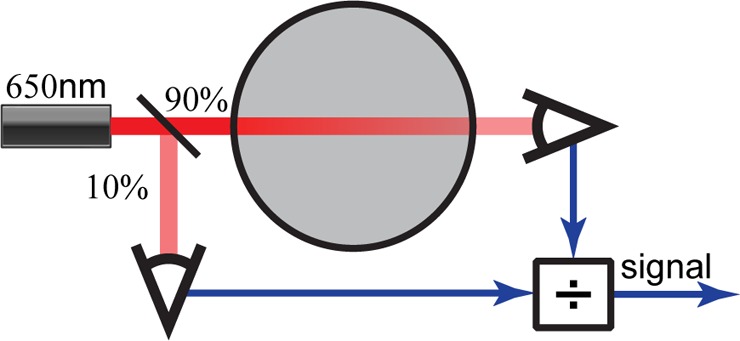
In each chamber, a laser diode emits a 650 nm beam that is then split two ways. Approximately 10% of the light is sent to a photosensor used to measure noise while the remaining 90% is used to measure light transmitted through the cell culture. The ratio of these two signals is a linear function of the transmitted light, which is then normalized to a blank measurement and log transformed to obtain the optical density.
A large component of the cost of a continuous culture device is the pump. In the Flexostat, we multiplexed a single syringe pump that feeds all eight chambers (Figure 1). A multiplexed pump saves the cost of seven additional pumps and reduces the number of valves needed from 16 to 9. To further reduce the cost, we have designed a 3D printed syringe pump that may be used in place of a commercial syringe pump. These two strategies have reduced pumping costs by 96% compared to designs that utilize commercial peristaltic pumps.
Modeling and Programming
A continuous culture device can be modeled by a simple set of ordinary differential equations (ODEs),14
| 1 |
where x(t) is the cell mass, s(t) is the mass of substrate, s0 is the substrate concentration of the feedstock, φ(s) is the growth rate, γ is the growth yield in biomass per substrate mass, and u is the dilution rate. It is worth noting that the units of x can be substituted by OD, which only changes the units of γ to OD per substrate mass.
If a turbidostat’s set point is too close to the maximal cell density, γs0, then it is possible that slight variations in media, conditions, or cell morphology may cause large changes in dilution rate, or even prevent dilution entirely. More formally, if we assume the growth rate as a function of substrate concentration is a Hill function without cooperativity10
with max growth rate φmax and half-maximum constant k, we can examine the sensitivity to substrate concentration
| 2 |
which rises sharply as the substrate mass goes to zero. The high sensitivity to low substrate concentrations makes running a turbidostat in nutrient limited regimes impractical. Difficulty maintaining set points near the maximal OD is analogous to the difficulty chemostats have maintaining set points near the washout rate.
Assuming cultures will be run sufficiently far from maximal densities, where the substrate is not limiting, we can assume a nearly constant growth rate allowing us to simplify the model further
| 3 |
Furthermore, because a turbidostat’s culture is always near its reference point r, the bilinear model (eq 3) can be further simplified to a linear model around the operating point (x*, u*) = (r, φ):
| 4 |
As stated earlier, turbidostats maintain cell density through feedback control. We have chosen the proportional plus integral feedback controller28
| 5 |
where kp and ki are proportional and integral gains, respectively. This controller eliminates any steady state error in optical density and yields the system
| 6 |
System 6 can oscillate when the condition rkp2/(4ki) > 1 is not met. To prevent oscillations we assume that the minimal OD set point will be 0.1 (r > 0.1) OD units and pick kp and ki so that rkp/(4ki) = 5 > 1. Indeed, gains so chosen have yielded robust, nonoscillatory OD signals with the Flexostat.
To measure error and validate the controller in the implemented system, we grew S. cerevisiae in duplicate at four different ODs and quantified the average, maximum, and 95th percentile tracking error (difference between set OD and measured OD) over a period of 22 h. Average tracking error was no more than 3.14 × 10–4 OD units between all eight chambers while the maximum error observed at any time point was ±4.8 × 10–3 OD units with 95% of the error inside of ±2.3 × 10–3 OD units. Thus, accurate maintenance of a desired OD set point is achievable over experimentally relevant time periods.
Since the Flexostat is programmable through a Python application program interface (API), it can also be easily reprogrammed for different objectives (see Supporting Information section S1 for details). Dilution and feedback can be turned off allowing for the measurement of growth curves in the same device (Supporting Figure S3), which can be useful for determining batch growth rates and growth yields. It can also be programmed to follow arbitrary reference trajectories so long as they are physiologically feasible (Figure 3).
Figure 3.

Experimental data collected from parallel experiments. S. cerevisiae was grown in synthetic complete media and made to track four sinusoidal reference trajectories (red). Measured OD values are shown in blue while the normalized dilution rate is shown in green. The maximum positive rate of change in OD is determined by the growth rate, while the maximum negative rate of change in OD is determined by the device’s maximum dilution rate.
Gene Network Parameter Dependence on Cell Density
In previous work,6 we studied the auxin plant hormone pathway cloned into S. cerevisiae. In our engineered yeast strain, as in planta, indole-3-acetic acid (auxin) promotes the interaction between a member of the F-box family of plant proteins (AFBs) and a member of the Aux/IAA family of plant proteins. AFBs target our engineered EYFP-Aux/IAA proteins to promote degradation via the proteasome (Figure 4a). In yeast transformed with a member of each family of AFB and EYFP-Aux/IAA proteins, strong YFP fluorescence can be measured through flow cytometery. With the addition of auxin, the EYFP-Aux/IAA fusion protein is degraded and within 2 h only background fluorescence is detectable. The exact degradation rates were characterized and shown to be dependent on the combination of AFBs and Aux/IAAs.
Figure 4.
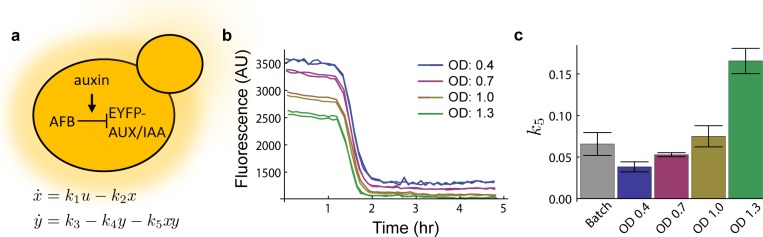
(a) Top: In the presence of auxin, AFB degrades the fusion protein EYFP-AUX/IAA. Bottom: A model from previous work6 of the network shown. State x represents auxin bound to AFB with input u being the auxin concentration, parameter k1 the association rate of auxin and AFB, and k2 the natural degradation rate of AFB. State y represents the concentration of EYFP-AUX/IAA where k3 is the production rate, k4 is the natural degradation rate, and k5 is the degradation rate in the presence of auxin bound to AFB. (b) YKL73 characterized in a turbidostat at four different ODs. At t = 1.25 h auxin was added to the culture chamber and media source to reach a concentration of 10 μM, and the response was measured by sampling effluent and reading mean fluorescence in a flow cytometer at a period of roughly 6 min. The cytometery data was gated to only include singlet (nonbudding) yeast in a healthy size range. An untreated time course is available as Supporting Information Figure S9 and shows no singnifigant change in fluorescence. (c) Model parameter k5 was fit to the data collected in part b (See Havens et al.6 Supplemental Data for detailed methods) and compared to previous work (gray).6
In previous work, each member of a combinatorial AFB Aux/IAA library was grown in synthetic complete medium (SC) under batch conditions at 30 °C in a shaker incubator overnight. The following day, the cultures were diluted and allowed to reenter exponential growth phase before induction at a cell count roughly equivalent to OD 0.4 in the Flexostat. Over the course of three or more hours, cells were sampled from batch and fluorescence was measured in a flow cytometer. These data were then fit to an ODE model (Figure 4a) where the parameter k5 represents the degradation rate of EYFP-Aux/IAA in the presence of auxin bound to AFB.
We hypothesize that culture density has an effect on degradation rates either directly or indirectly through changes in nutrients, signaling molecules, and waste products that correspond with cell density in a given media. To test our hypothesis, we selected a strain YKL73 from previous work that coexpresses AFB2 and EYFP-Aux/IAA6. YKL73 was grown in SC in our turbidostat inside of a 30 °C incubator at four different ODs: 0.4, 0.7, 1.0, and 1.3 in duplicate. Although we chose ODs ranging from 0.4 to 1.3, we did not observe a significant difference in growth rate between cultures, which is typical for growth at nonlimiting nutrient concentrations. To measure fluorescence in each chamber, 200 μL of effluent was sampled every 6 min and measured in the same flow cytometer as previous work (Figure 4b). After fitting the new degradation curves we found k5 to vary from 0.036 ± 0.006, an average degrader, to 0.171 ± 0.015 (Figure 4c and Supporting Information Table S2), which is 35% faster than the fastest pair previously reported6 from batch data.
Another apparent feature of the data are the initial AUX/IAA levels (quantity k3/k4 in the model). The initial expression levels also show an inverse relationship between OD and untreated expression of AUX/IAA. Figure 4c and Supporting Information Figures S10 and S11 illustrate the relationship between OD, initial expression, and degradation rates though the exact cause of the relationships is unknown.
In continuous culture, steady state levels of waste products and other exportable chemicals are proportional to cell density. Through the use of cytometer gating, we controlled for cell size and morphology (singlets vs budding cells). This leads us to hypothesize that either the rate k5 and the ratio k3/k4 are highly sensitive to differences in growth rate (within the error margin of our measurements), or that the cells’ internal state was affected by the density dependent effects in environment. Since k5 is modeled as a constant, the density dependent effects, which could not have been measured in batch, highlight the utility of continuous culture in model validation and the need, in this case, for a higher order model to explain the data when density cannot be controlled. Further study is required to determine the exact nature and source of the physiological changes that influence expression and degradation rates.
Fluorescence Detecting Turbidostat
A key capability in synthetic and systems biology is the ability to read fluorescent reporters. In the previous section, we used a flow cytometer, which resulted in individual cell data but was labor intensive. An alternative method to collecting cytometry data is to obtain bulk fluorescence in situ, which can be collected without user intervention.
We modified the original chamber design to include a blue excitation light source at a right angle to the absorbance photo sensor (Figure 5a). The new design multiplexes the original light sensor in time to serve the dual purpose of measuring absorbance and fluorescence. For OD measurements, the chamber works as described in earlier sections. When fluorescence measurements are made, the 600 nm LED is turned off and the 470 nm LED excitation source is turned on. To prevent bleed over from the excitation source into the emissions wavelengths, we added a band-pass optical filter (see Methods and Supporting Information section S4). Similarly, to prevent the excitation source from being detected by the photo sensor, a long-pass optical filter was added.
Figure 5.
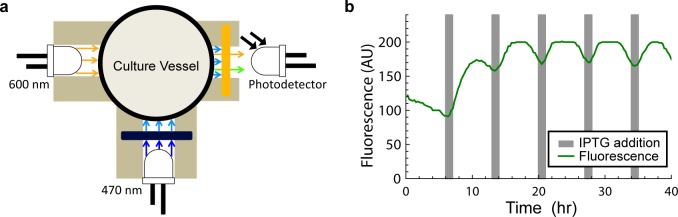
(a) Diagram representing the layout of the modified fluorostat culture chamber. The light sensor is time divison multiplexed between light from the absorbance source (600 nm) and fluorescence excited by the excitation source (470 nm). A filter set ensures that only light generated by fluorescence is detected. (b) An IPTG inducible GFP expressing strain of E. coli was grown at OD 0.6 in alternating M9 media with (gray area) and without IPTG.
To test the fluorescence detection capability of our fluorescence detecting turbidostat (or Fluorostat for short), we measured a range of fluorescent cultures. Wild type E. coli (BL21) and BL21 containing a high expression super folder green fluorescent protein (sfGFP) plasmid were cultured for use in quantification. The cultures were then mixed at various ratios and measured in the Fluorostat, which showed a linear relationship between mix ratio and measured fluorescence (Supporting Information Figure S13).
Next, we tested the Fluorostat under more experimentally realistic conditions. We cultured an IPTG inducible sfGFP expressing strain of E. coli in the Fluorostat. At periods of 1 and 6 h, media containing 1 mM and 0 mM IPTG, respectively, were alternated and the resulting fluorescence signal was measured (Figure 5b). The results of this experiment demonstrate the ability of an LED based fluorimeter to read sfGFP reported outputs during the course of a turbidostat experiment. These results also highlight the adaptability of our design to work with evolving experimental needs.
While linear within its dynamic range and capable of measuring changes in expression levels, we discovered that only cultures bright enough to be visibly fluorescent under blue light transillumination were capable of being detected. The YFP expressing strain of yeast measured using cytometry in the previous section was too dimly fluorescent to measure in the Fluorostat. While its low sensitivity precludes the use of the Fluorostat in some experiments, we believe that through further refinement, the use of a more sensitive fluorometer, or an automatic cytometer sampling system, all experiments requiring fluorescence measurement should be possible without manual sampling.
Conclusion
With the advent of 3D printing and other inexpensive custom fabrication processes, laboratories are now more capable of building their own equipment from open source designs than ever before. These community driven designs have the benefit of open design elements that allow laboratories to customize software and hardware for their own unique experiments rather than using designs optimized for industrial purposes. We presented a new 3D printable and customizable turbidostat for the next generation of “3D printed labs”.
To demonstrate its utility, we have shown an example system with previously thought to be constant parameters that turned out to be dependent on cell density. We also demonstrated that our design can be adapted to accommodate fluorescence measurement, enabling the automatic collection of reporter expression data within the device.
Methods
All data fitting methods, yeast methods, strains, media, and flow cytometery are as in previous work,6 unless otherwise stated below.
Yeast Methods
Diploid S. cerevisiae strain YKL73 was obtained from previous work6 and expresses AFB2 and IAA6 with EYFP fused to its N-terminus. All yeast cultures were grown at 30 °C in Synthetic Complete medium according to standard protocols. Batch cultures were grown from colonies in 15 mL tubes in a shaker incubator and used to inoculate turbidostat cultures. Auxin was dosed from a stock solution of 100 mM auxin in 95% ethanol to a working concentration of 10 μM into each turbidostat chamber while simultaneously dosing the media reservoir.
Fabrication
Complete construction and assembly instructions, design files (including 3D CAD and circuit boards), and a full bill of materials can be found on our Web site http://klavinslab.org. All ABS plastic parts were 3D printed with OEM natural colored filament on the UP! 3D printer from http://www.pp3dp.com/, which can be purchased for $1500 USD.
Delrin parts were laser cut from 5 mm delrin sheet by https://www.ponoko.com/.
Circuit boards were manufactured by http://www.seeedstudio.com/ and assembled by hand in the laboratory. Surface-mount soldering was done on a standard laboratory hot plate using CHIPQUIK Sn96.5Ag3.0Cu0.5 solder paste at a temperature of 230 °C and through hole was done by hand using a Weller W60p soldering iron.
Fluorescence Detection
Fluorescence characterization was done with E. coli strain BL21 transformed with a plasmid containing the pLac promotor driving sfGFP. All growth of E. coli in the turbidostat was done in M9-1% glucose at an OD600 2 cm of 0.6 and a temperature of 37 °C. Batch growth was done in M9-1% glucose media at 37 °C in a baffled shaker flask to stationary phase and diluted to OD600 1 cm 1.0 in M9 salts. For fluorescent strains a saturating level of 100 μM IPTG was used to induce sfGFP expression.
The following filter set and excitation source was used for fluorescence measurement: Thorlabs part number FGB25, Thorlabs part number FGL530, http://www.ledsupply.com part number L1-0-B5TH30-1.
Flow Cytometry
Samples ≥200 μL were collected over 3 min sampling periods into 2 mL 96 deep well plates. The plates were then transferred to an Accuri C6 flow cytometer with CSampler attachment for immediate measurement. All settings and analysis were done as in previous work.6
Acknowledgments
This work was supported by grants to E.K. from the National Science Foundation (NSF) (1317653) and the Paul G. Allen Family Foundation and grants to M.J.D. from the NSF (1120425) and the National Institute of General Medical Sciences (P41 GM103533) from the National Institutes of Health. A.W.M. was supported by T32 HG00035. M.J.D. is a Rita Allen Foundation Scholar and Fellow in the Genetic Networks program at the Canadian Institute for Advanced Research. The authors thank the Murray lab at the California Institute of Technology, the Tabor lab at Rice University, and the Savage lab at the University of California, Berkeley, for their becoming early adopters as well as for their input on our design and assembly instructions.
Supporting Information Available
Additional characterization data, Python interface specifications, schematics, and 3D renderings. This material is available free of charge via the Internet at http://pubs.acs.org. All assembly instructions are available through our Web site http://klavinslab.org. Computer source code and design files are available through our Web site or directly from the public github repository https://github.com/klavinslab/.
The authors declare no competing financial interest.
Supplementary Material
References
- Canton B.; Labno A.; Endy D. (2008) Refinement and standardization of synthetic biological parts and devices. Nat. Biotechnol. 26, 787–793. [DOI] [PubMed] [Google Scholar]
- Alper H.; Fischer C.; Nevoigt E.; Stephanopoulos G. (2005) Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. U.S.A. 102, 12678–12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson E.; Hartsough L.; Landry B.; Shroff R.; Tabor J. (2014) Characterizing bacterial gene circuit dynamics with optically programmed gene expression signals. Nat. Methods 11, 449–455. [DOI] [PubMed] [Google Scholar]
- Ceroni F.; Furini S.; Stefan A.; Hochkoeppler A.; Giordano E. (2012) A synthetic post-transcriptional controller to explore the modular design of gene circuits. ACS Synth. Biol. 1, 163–171. [DOI] [PubMed] [Google Scholar]
- Kelly J. R.; Rubin A. J.; Davis J. H.; Ajo-Franklin C. M.; Cumbers J.; Czar M. J.; de Mora K.; Glieberman A. L.; Monie D. D.; Endy D. (2009) Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havens K. A.; Guseman J. M.; Jang S. S.; Pierre-Jerome E.; Bolten N.; Klavins E.; Nemhauser J. L. (2012) A synthetic approach reveals extensive tunability of auxin signaling. Plant Physiol. 160, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. C.; Voigt C. A.; Arkin A. P. (2007) Environmental signal integration by a modular AND gate. Mol. Syst. Biol. 3, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutalik V. K.; Guimaraes J. C.; Cambray G.; Mai Q.-A.; Christoffersen M. J.; Martin L.; Yu A.; Lam C.; Rodriguez C.; Bennett G.; et al. (2013) Quantitative estimation of activity and quality for collections of functional genetic elements. Nat. Methods 10, 347–353. [DOI] [PubMed] [Google Scholar]
- Mutalik V. K.; Guimaraes J. C.; Cambray G.; Lam C.; Christoffersen M. J.; Mai Q.-A.; Tran A. B.; Paull M.; Keasling J. D.; Arkin A. P.; et al. (2013) Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods 10, 354–360. [DOI] [PubMed] [Google Scholar]
- Monod J. (1949) The growth of bacterial cultures. Annu. Rev. Microbiol. 3, 371–394. [Google Scholar]
- Brauer M. J.; Huttenhower C.; Airoldi E. M.; Rosenstein R.; Matese J. C.; Gresham D.; Boer V. M.; Troyanskaya O. G.; Botstein D. (2008) Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol. Biol. Cell 19, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valgepea K.; Adamberg K.; Seiman A.; Vilu R. (2013) Escherichia coli achieves faster growth by increasing catalytic and translation rates of proteins. Mol. BioSyst. 9, 2344. [DOI] [PubMed] [Google Scholar]
- Scott M.; Gunderson C. W.; Mateescu E. M.; Zhang Z.; Hwa T. (2010) Interdependence of cell growth and gene expression: Origins and consequences. Science 330, 1099–1102. [DOI] [PubMed] [Google Scholar]
- Monod J. (1950) La technique de culture continue, theorie et applications. Ann. Inst. Pasteur 79, 390–410. [Google Scholar]
- Saldanha A. J.; Brauer M. J.; Botstein D. (2004) Nutritional homeostasis in batch and steady-state culture of yeast. Mol. Biol. Cell 15, 4089–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomson K.; Barber J.; Vanatalu K. (2006) Adaptastat—A new method for optimizing of bacterial growth conditions in continuous culture: Interactive substrate limitation based on dissolved oxygen measurement. J. Microbiol. Methods 64, 380–390. [DOI] [PubMed] [Google Scholar]
- Toprak E.; Veres A.; Michel J. B.; Chait R.; Hartl D. L.; Kishony R. (2012) Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat. Genet. 44, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt K. M.; Carlson J. C.; Liu D. R. (2011) A system for the continuous directed evolution of biomolecules. Nature 472, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovitt R. W.; Wimpenny J. W. T. (1981) The gradostat: A bidirectional compound chemostat and its application in microbiological research. Microbiology 127, 261–268. [DOI] [PubMed] [Google Scholar]
- Miller A. W.; Befort C.; Kerr E. O.; Dunham M. J. (2013) Design and use of multiplexed chemostat arrays. J. Vis. Exp. e50262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markx G. H.; Davey C. L.; Kell D. B. (1991) The permittistat: A novel type of turbidostat. J. Gen. Microbiol. 137, 735–743. [Google Scholar]
- Larsson G.; Enfors S.-O.; Pham H. (1990) The pH-auxostat as a tool for studying microbial dynamics in continuous fermentation. Biotechnol. Bioeng. 36, 224–232. [DOI] [PubMed] [Google Scholar]
- Tappe W.; Tomaschewski C.; Rittershaus S.; Groeneweg J. (1996) Cultivation of nitrifying bacteria in the retentostat, a simple fermenter with internal biomass retention. FEMS Microbiol. Ecol. 19, 47–52. [Google Scholar]
- Toprak E.; Veres A.; Yildiz S.; Pedraza J. M.; Chait R.; Paulsson J.; Kishony R. (2013) Building a morbidostat: An automated continuous-culture device for studying bacterial drug resistance under dynamically sustained drug inhibition. Nat. Protoc. 8, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins J. J.; Gardner T. S.; Cantor C. R. (2000) Construction of a genetic toggle switch in Escherichia coli.. Nature 403, 339–342. [DOI] [PubMed] [Google Scholar]
- Wang P.; Robert L.; Pelletier J.; Dang W. L.; Taddei F.; Wright A.; Jun S. (2010) Robust growth of Escherichia coli. Curr. Biol. 20, 1099–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BD Accuri C6 Flow Cytometer Technical Specifications (2012) BD Biosciences, Piscataway, NJ.
- Dorf R. C. and Bishop R. H. (2005) Modern Control Systems, 10th ed.; Prentice Hall, Upper Saddle River, NJ. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.