

Abstract
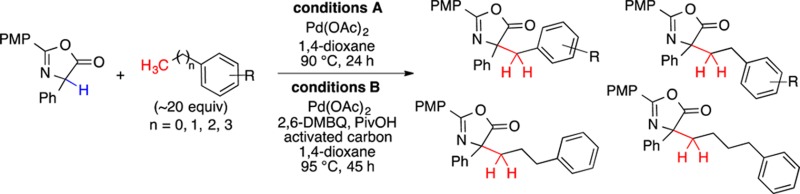
The first selective coupling of a carbon nucleophile with methyl, ethyl, propyl, and butyl arenes in the absence of a directing group is described. Pd(OAc)2 double C–H activation displays remarkable selectivity for the terminal methyl sites in alkyl arenes, rather than the more commonly observed arene sp2 C–H activation. Mechanistic studies indicate the intermediacy of an azlactone dimer, obtained from oxidation with Pd(OAc)2, and are consistent with a Pd-catalyzed C–H activation vs a radical process. The observed reactivity establishes that typical reaction solvents (e.g., toluene) can readily participate in C–H activation chemistry.
The ability of metals, and particularly Pd catalysts, to selectively insert into C–H bonds has provided a host of new, useful methods for constructing organic structures.1 C–H activation is highly significant because additional steps to preactivate a center for bond construction (e.g., halogenation) can be avoided, thereby increasing efficiency by reducing step-count and decreasing waste. Catalytic dehydrogenative cross-coupling (CDC) is the ideal version of this process, where C–H bonds from each of two reacting partners are selectively cleaved, accompanied by an oxidative fragment union.2 In this strategy, coordinating groups play a key role by complexing the substrate with the catalyst and thus lowering the energy barrier for C–H activation.3 Advances in such alkyl C–H activation are considerable, but much less progress has been made in systems lacking coordinating groups.4
Toluene compounds are easy-to-handle, stable, and commercially available, rendering them ideal as benzylation reagents, and far more atom-economical than the corresponding benzyl halides. Even though the benzylic C–H bond in toluene is 20–30 kcal/mol weaker than the arene C–H, Pd displays a remarkable selectivity for arene C–H activation (Figure 1).5 In fact, methyl and other alkyl substituents on arenes are compatible with Pd-catalyzed sp2 C–H functionalization. Presumably, Pd π-coordination positions the metal carboxylate for a favorable deprotonative metalation (Figure 1).6
Figure 1.
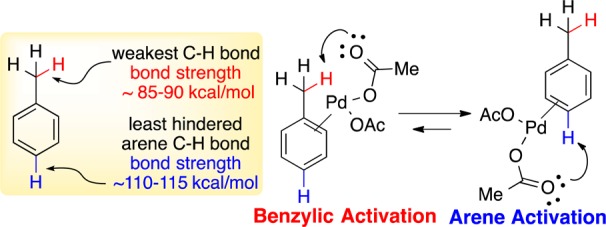
sp3 C–H vs sp2 C–H activation of toluene with Pd.
Of late, radical-mediated processes for activating toluene have shown considerable promise in CDC (Scheme 1, eqs 1 and 2).7−9 A benzylic radical is easily formed at elevated temperatures with di-tert-butyl peroxide and can merge with Pd-catalyzed processes. Cu catalysts have also been reported to form benzylic amines and alcohols from toluene and stabilized radicals.10
Scheme 1. sp3 C–H Activation of Toluene with Pd.
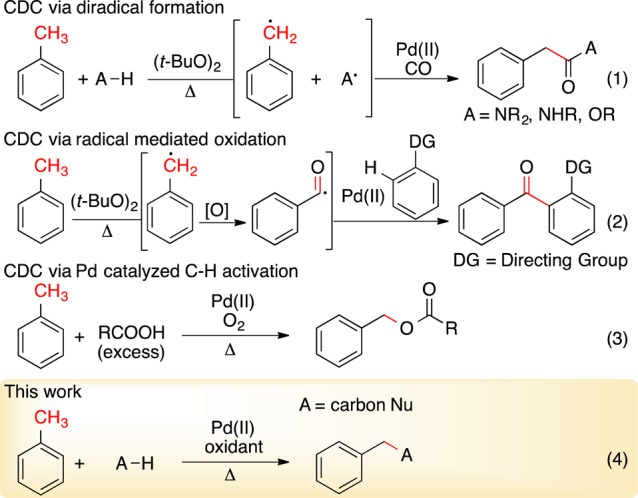
However, activating toluene by nonradical processes is a long-standing challenge.11,12 White and Shi developed an elegant system for the C–H activation of the doubly activated allylbenzene.13 Since the discovery of Pd-catalyzed acetoxylation of toluene in 1968 by Bryant, few advances have been made (Scheme 1, eq 3).14In fact, toluene is considered benign in most Pd-catalyzed processes and is frequently used as solvent. Our ongoing interest15 in C–H activation prompted us to investigate the oxidative coupling of toluene. Herein, we disclose the discovery of selective Pd activation of benzylic and alkyl sp3 C–H bonds relative to arene sp2 C–H bonds to achieve CDC coupling reactions with a carbon nucleophile (Scheme 1, eq 4).
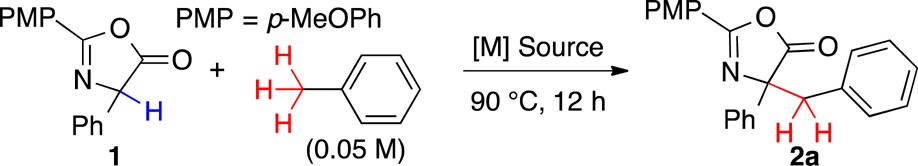
We initially explored the coupling of phenylglycine azlactone and toluene with late-transition metals based on their propensity for C–H activation (Table 1). Notably, only Pd(II) carboxylates provided any cross-coupled product (Table 1, entries 6 and 7). PdCl2 provided the azlactone dimer, but all other Pd sources were inactive (Table 1, entries 8–12). Surprisingly, the coupling product 2a arose from deprotonative metalation into the sp3 C–H bond rather than the typical sp2 C–H activation.5,6
Table 1. Metal Sources for C–H Activation of Toluene.
| entry | [M] source (100 mol%) |
yield (%)a |
entry | [M] source (100 mol%) |
yield (%)a |
|---|---|---|---|---|---|
| 1 | Au(OAc)3b | 0 | 7 | Pd(TFA)2 | 81 |
| 2 | Cu(OAc)2b | 0 | 8 | PdCl2 | 0 |
| 3 | Rh2(OAc)4b | 0 | 9 | PdCl2(MeCN)2 | 0 |
| 4 | Ni(OAc)2b | 0 | 10 | PdCl2(COD)2 | 0 |
| 5 | [Pt4(OAc)8]HOAcb | 0 | 11 | [Pd(allyl)Cl2]2 | 0 |
| 6 | Pd(OAc)2 | 77 | 12 | Pd2dba3 | 0 |
Isolated yield.
Azlactone dimer as substrate, 20 mol% metal source, 5 h.
With this rare example of selective C–H activation of the benzylic position of toluene,12 we attempted to minimize the amount of toluene. A screen of cosolvents16 revealed that 1,4-dixoane was optimal to reduce toluene from 80 to 20 equiv while efficiently providing the benzylated product (Scheme 2, 2a). These conditions also proved successful for several toluene derivatives. In contrast to Bryant’s results with acetoxylation of xylenes, the monobenzylated product was the only product observed (Scheme 2, 2b–2d).14a Although m-xylene (2c) was highly effective, mesitylene proceeded in only 14% yield; increased temperature and longer reaction times did not improve the outcome. We speculate that the three alkyls of mesitylene hinder the coordination of the Pd to the π-system needed to initiate C–H activation (Figure 1).
Scheme 2. C–H Activation of Tolyl Analogues.
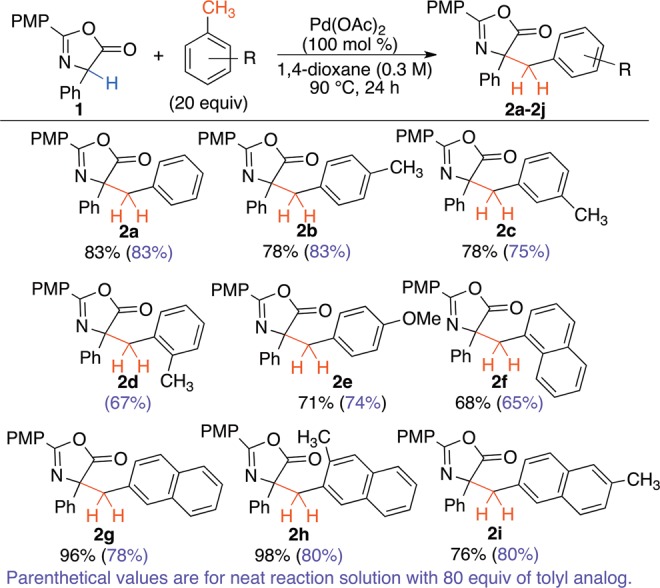
Naphthalene analogues of benzyl bromide are relatively unstable for SN2 transformations and are not widely available. Use of 1- and 2-methylnaphthalene in CDC via radical-mediated processes is uncommon because the tolyl analogue is typically used neat, a difficult proposition with these solids. With the optimal conditions, naphthyl derivatives of phenylglycine azlactone were formed in good yield (Scheme 2). Hydrolysis of these compounds permits facile access to unnatural α,α-disubstituted α-amino acids.17
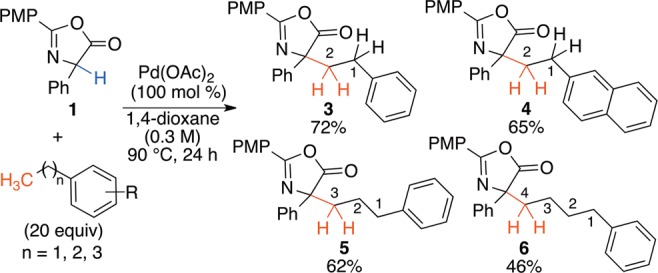
Success with the selective sp3 C–H bond activation of primary benzylic sites prompted exploration of secondary benzylic compounds (Scheme 3).18 Unexpectedly, the secondary benzylic product was not observed; rather, with each substrate, substitution occurred at the primary methyl.19 Most surprisingly, propylbenzene and butylbenzene gave rise to products 5 and 6, respectively, while <5% of the other cross-coupling isomers were observed. This chemoselectivity provides access to chemical space that would not be feasible via a radical-mediated process.
Scheme 3. Selective Primary C–H Activation.
Furthermore, the novel activation of benzylic, homobenzylic, and bis/tris-homobenzylic sp3 C–H bonds by Pd(OAc)2 without concomitant arene sp2 C–H reaction represents a paradigm shift in the behavior of Pd catalysts. We initially reasoned that a catalytic cycle might involve Pd(II) deprotonative carbopalladation of the benzylic component, preceded or followed by ligand exchange with the azlactone. Subsequent reductive elimination would yield the product and Pd(0), consistent with the observed formation of Pd black.20 With this reasoning, we turned our attention to identifying a suitable oxidant to allow turnover.
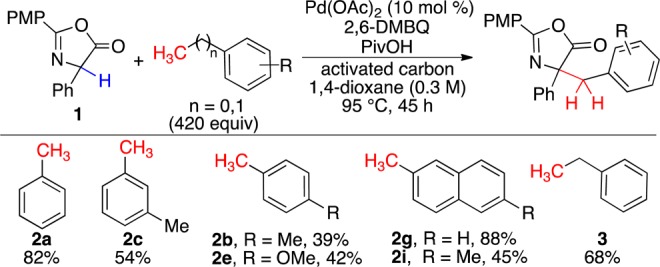
A PME screen21 with 23 diverse oxidants (2 equiv) and 2 Pd carboxylates (30 mol%)16 revealed that 2,6-dimethylbenzoquinone (2,6-DMBQ) was superior. 2,6-DMBQ loading could be reduced with a co-oxidant, MnO2, but the most successful additive was PivOH,16 which presumably promotes dissociation of the hydroquinone anion from Pd(II).22 Benzylation of phenylglycine azlactone with a series of tolyl and methylnaphthyl derivatives under the optimal conditions for Pd catalysis revealed that the transformation was substrate dependent. Further studies on additives and cosolvents found that dioxane (cf. Scheme 2) as a solvent permitted the use of smaller amounts of the benzylic compound, even when catalytic Pd was employed. Among known Pd(0) stablilizers (DMSO, DMA, White’s sulfoxide ligand,13b phenanthroline, BIPY, and cyclohexene), activated carbon was found to improve upon the initial catalytic findings (Scheme 4). These conditions were also explored in a homobenzylic system and provided the ethylated phenylglycine azlactone product in good yield.
Scheme 4. Pd(OAc)2-Catalyzed sp3 C–H Activation.
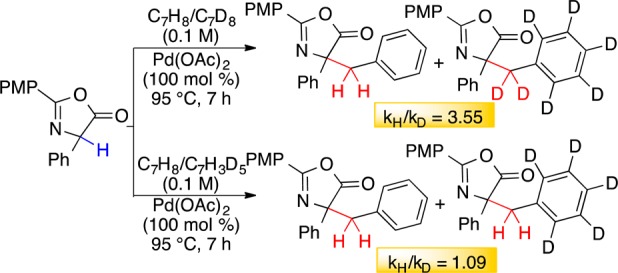
With these novel findings in hand, the mechanism of C–H activation was investigated. An equimolar mixture of toluene and d8-toluene with phenylglycine azlactone and Pd(OAc)2 provided kH/kD = 3.5. Parallel experiments23 with the two substrates also revealed that that deuterated analogue was 2–4 times slower, suggesting a metal-catalyzed C–H activation step as rate-determining (Scheme 5).13a,14b Radical-mediated processes (e.g., Scheme 1, eqs 1 and 2) typically exhibit a more significant isotopic effect >5.7a,10b The absence of an isotope effect and deuterium scrambling with d5-toluene indicates that initial Pd metalation of an arene C–H is unlikely.
Scheme 5. Kinetic Isotope Studies.
In the course of these studies, the dimer of the azlactone was observed frequently, prompting further studies to determine if it was necessary for benzylation or was a side product. Dimer formation can be initiated with a metal oxidant (e.g., NiO2, MnO2) or with air in a polar solvent (DMSO).24 We thus developed mild conditions to form the azlactone dimer 8 in 83% yield with Pd(OAc)2 (5 mol%) using Ag2O (100 mol%) at room temperature.16 When this dimer was subjected to catalytic Pd(OAc)2 and toluene (no added oxidant), the benzylated product was formed in 72% yield (Scheme 6, top).25 The high yield in the absence of additional oxidant suggests that, once the azlactone dimer is formed, the benzylation is redox neutral, unlike other processes involving dimeric Pd.26
Scheme 6. Involvement of the Azlactone Dimer.
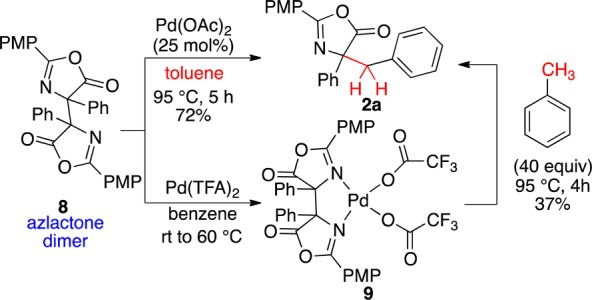
The azlactone dimer was discovered to form metal complex 9 upon treatment with Pd(TFA)2 (Scheme 6, bottom), as judged by a downfield 1H NMR shift of the C-4 phenyl o-H (7.31–9.47 ppm) and X-ray crystallography.16 Treatment of this complex with toluene at elevated temperatures provided the benzyl product (Scheme 6, bottom), although in lesser yield (cf. Table 1, entry 7). Monitoring complex 9 at room temperature with d8-toluene revealed dissociation of the complex within 30 min. From these results, it appears that complex 9 is not a reactive intermediate.
Phenylalanine azlactone dimer is known to undergo homolytic cleavage above 115 °C.24b Thus, the possibility of such an event being closely coupled to C–H activation was considered. Evidence found against such a path includes the transformation proceeding below the dimer homolysis temperature and not proceeding in the absence of Pd(II). Further, studies with the PMP-phenylglycine dimer 8 and Ph-phenylglycine dimer at 85–95 °C with and without Pd(OAc)2 reveal no dimer recombination characteristic of such a homolytic cleavage.16
Although the full mechanistic details of this transformation remain to be elucidated, the following mechanism accounts for the observations to date (Figure 2a). 1H NMR spectroscopic monitoring reveals that the phenylglycine azlactone 1 is converted to the azlactone dimer 8 in the first 30 min when exposed to Pd(OAc)2 and heat. Toluene can undergo C–H activation with Pd(OAc)2 at elevated temperatures to generate benzylic Pd(II) A.12,14 The electron-rich A may undergo metathesis with the azlactone dimer (path a or c). Displacement of the better acetate leaving group (path a) would generate B, which would provide product upon reductive elimination. However, the C-acetoxy byproduct (C) of this event was not observed. Pd(IV) intermediate D may instead form by oxidative addition of the labile C–C bond of the azlactone dimer to A (path b). Reductive elimination would yield the benzylated azlactone product and E, both of which could also form via metathesis path c. Pd(OAc)2 would be regenerated from E in the presence of AcOH. Re-forming phenylglycine azlactone consumes the remaining Pd(OAc)2, accounting for the 72% yield observed commencing from the azlactone dimer in the absence of oxidant (Scheme 6).
Figure 2.
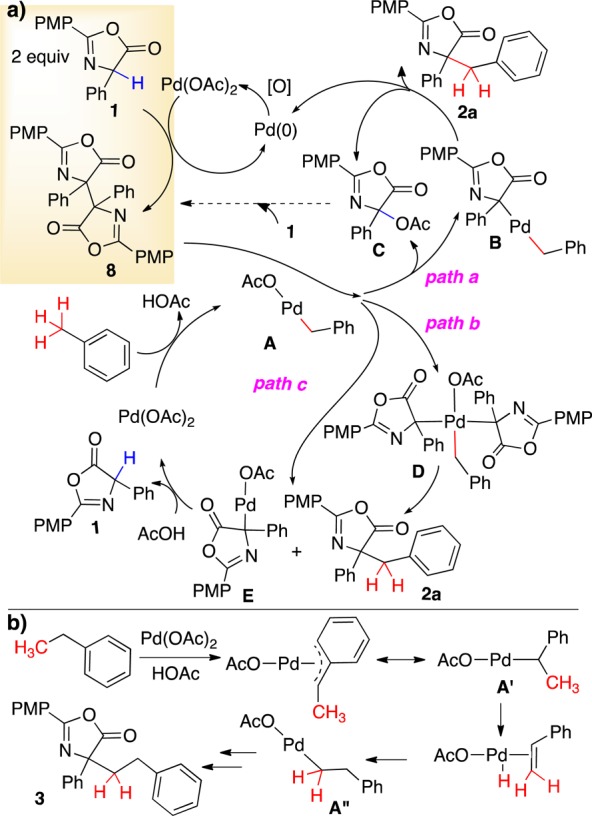
Proposed mechanism for tolyl C–H activation.
A deuterium labeling study conducted with d2-ethylbenzene revealed positional deuterium scrambling (eq 5), supporting initial Pd metalation of the benzylic C–H bond (Figure 2b). Subsequent β-hydride elimination to a styrene, re-addition of Pd onto the terminal C, and cross-coupling with the azlactone would account for the ethylated product (Figure 2b); however, styrenes added to the reaction did not incorporate into the product.
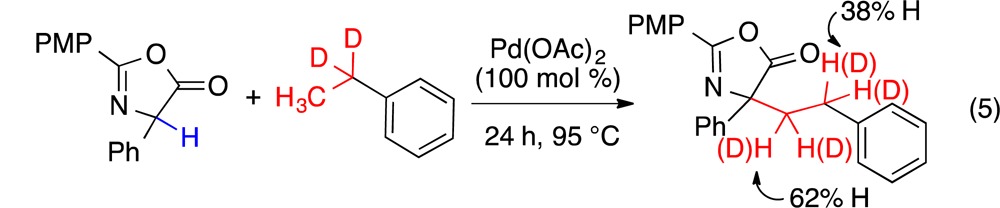 |
5 |
Selective sp3 C–H activation of ethylbenzene provided a unique opportunity to interrogate potential radical pathways. When phenylglycine azlactone was treated with ethylbenzene at 90 °C with Pd(OAc)2 and (t-BuO)2, only reaction at the terminal site (3) was observed (Scheme 7, entry 1). In the absence of Pd, but above the homolysis temperature of (t-BuO)2,27 only the benzylated product was seen (entry 2).28 With Pd(OAc)2 and (t-BuO)2 together at this higher temperature, products from both pathways were seen (entry 3). Radical scavengers also had no effect on the toluene reaction.29 Altogether, these results contraindicate a radical mechanism for this process.
Scheme 7. Probing Radical Pathways with Ethylbenzene.
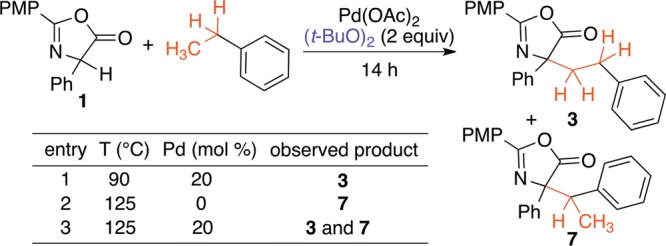
In summary, a novel reactivity mode for alkylarene derivatives has been discovered. With a simple system consisting of Pd(OAc)2 and pivalic acid, CDC with a carbon nucleophile occurs readily for the terminal methyl positions of methyl, ethyl, propyl, and butyl arenes. The resultant azlactone products are masked α-amino acids, with hindered α,α-disubstitution patterns that are difficult to achieve via other means.30 Notably, selective sp3 C–H activation is observed in benzylic systems, even though Pd(OAc)2 typically causes arene C–H activation. Further studies to understand the mechanism and, in particular, the role of an observed azlactone dimer on the sp3 vs sp2 C–H activation selectivity are underway. These studies provide a cautionary tale against use of methyl arene solvents, such as toluene and xylenes, in Pd-catalyzed C–H activation chemistry.
Acknowledgments
We thank the NIH (GM-087605) and the NSF (CHE1213230) for financial support. We thank Dr. Simon Berritt (Penn Merck HTE Lab) for helpful discussions.
Supporting Information Available
Experimental details and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Alberico D.; Scott M. E.; Lautens M. Chem. Rev. 2007, 107, 174. [DOI] [PubMed] [Google Scholar]; b Kuhl N.; Hopkinson M. N.; Wencel-Delord J.; Glorius F. Angew. Chem., Int. Ed. 2012, 51, 10236. [DOI] [PubMed] [Google Scholar]
- a Yeung C. S.; Dong V. M. Chem. Rev. 2011, 111, 1215. [DOI] [PubMed] [Google Scholar]; b Liu C.; Zhang H.; Shi W.; Lei A. Chem. Rev. 2011, 111, 1780. [DOI] [PubMed] [Google Scholar]
- a Lyons T. W.; Sanford M. S. Chem. Rev. 2010, 110, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li B.; Dixneuf P. H. Chem. Soc. Rev. 2013, 42, 5744. [DOI] [PubMed] [Google Scholar]
- a Davies H. M. L.; Hansen T. J. Am. Chem. Soc. 1997, 119, 9075. [Google Scholar]; b Waltz K. M.; Hartwig J. F. J. Am. Chem. Soc. 2000, 122, 11358. [Google Scholar]; c Chen M. S.; White M. C. Science 2007, 318, 783. [DOI] [PubMed] [Google Scholar]
- Blanksby S. J.; Ellison G. B. Acc. Chem. Res. 2003, 36, 255. [DOI] [PubMed] [Google Scholar]
- a Lapointe D.; Fagnou K. Chem. Lett. 2010, 39, 1118. [Google Scholar]; b Balcells D.; Clot E.; Eisenstein O. Chem. Rev. 2010, 110, 749. [DOI] [PubMed] [Google Scholar]; c Ackerman L. Chem. Rev. 2011, 111, 1315. [DOI] [PubMed] [Google Scholar]
- a Xie P.; Xie Y.; Qian B.; Zhou H.; Xia C.; Huang H. J. Am. Chem. Soc. 2012, 134, 9902. [DOI] [PubMed] [Google Scholar]; b Xie P.; Xia C.; Huang H. Org. Lett. 2013, 15, 3370. [DOI] [PubMed] [Google Scholar]; c Liu H.; Laurenczy G.; Yan N.; Dyson P. J. Chem. Commun. 2014, 50, 341. [DOI] [PubMed] [Google Scholar]
- Yin Z.; Sun P. J. Org. Chem. 2012, 77, 11339. [DOI] [PubMed] [Google Scholar]
- a Wu Y.; Choy P. Y.; Mao F.; Kwong F. Y. Chem. Commun. 2013, 49, 689. [DOI] [PubMed] [Google Scholar]; b Xiong F.; Qian C.; Lin D.; Zeng W.; Lu X. Org. Lett. 2013, 15, 5444. [DOI] [PubMed] [Google Scholar]; c Xu Z.; Xiang B.; Sun P. RSC Adv. 2013, 3, 1679. [Google Scholar]
- a Powell D. A.; Fan H. J. Org. Chem. 2010, 75, 2726. [DOI] [PubMed] [Google Scholar]; b Rout S. K.; Guin S.; Ghara K. K.; Banerjee A.; Patel B. K. Org. Lett. 2012, 14, 3982. [DOI] [PubMed] [Google Scholar]
- a Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Baudoin O. Chem. Soc. Rev. 2011, 40, 4902. [DOI] [PubMed] [Google Scholar]; c Li H.; Li B.-J.; Shi Z.-J. Catal. Sci. Technol. 2011, 1, 191. [Google Scholar]
- For toluene self-coupling to a 6:94 mixture of benzylic–arene and arene–arene product with Pd(OAc)2 and TFA, see:Rong Y.; Li R.; Lu W. Organometallics 2007, 26, 4376. [Google Scholar]
- a Lin S.; Song C.-X.; Cai G.-X.; Wang W.-H.; Shi Z.-J. J. Am. Chem. Soc. 2008, 130, 12901. [DOI] [PubMed] [Google Scholar]; b Young A. J.; White M. C. J. Am. Chem. Soc. 2008, 130, 14090. [DOI] [PubMed] [Google Scholar]
- a Bryant D. R.; McKeon J. E.; Ream B. C. J. Org. Chem. 1968, 33, 4123. [Google Scholar]; b Liu H.; Shi G.; Pan S.; Jiang Y.; Zhang Y. Org. Lett. 2013, 15, 4098. [DOI] [PubMed] [Google Scholar]
- Lee Y. E.; Cao T.; Toruellas C.; Kozlowski M. C. J. Am. Chem. Soc. 2014, 136, 6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supporting Information.
- Trost B. M.; Xavier A. J. Am. Chem. Soc. 1999, 121, 10727. [Google Scholar]
- Diphenylmethane and cumene were explored with the optimal conditions but did not provide the benzyl product.
- In ref (14a), acetoxylation of ethylbenzene at the terminal methyl gave a complex product mixture in 8.3% overall yield.
- Pun D.; Diao T.; Stahl S. S. J. Am. Chem. Soc. 2013, 135, 8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmink J. R.; Bellomo A.; Berritt S. Aldrichimica Acta 2013, 46, 71. [Google Scholar]
- Grennberg H.; Gogoll A.; Backvall J.-E. Organometallics 1993, 12, 1790. [Google Scholar]
- Simmons E. M.; Hartwig J. F. Angew. Chem., Int. Ed. 2012, 3066. [DOI] [PubMed] [Google Scholar]
- a Rodriguez H.; Marquez A.; Chuaqui C. A.; Gomez B. Tetrahedron 1991, 47, 5681. [Google Scholar]; b Andersen K. K.; Gloster D. F.; Bray D. D.; Shoja M. J. Heteroatom. Chem. 1998, 35, 317. [Google Scholar]
- Pd(0) sources did not provide 2a from dimer and toluene.
- Powers D. C.; Ritter T. Nature 2009, 1, 302. [DOI] [PubMed] [Google Scholar]
- Pryor W. A.; Lee A.; Witt C. E. J. Am. Chem. Soc. 1964, 86, 4229. [Google Scholar]
- Six diastereomers of C- and O-benzylation were observed:Regalado E. L.; Kozlowski M. C.; Curto J. M.; Ritter T.; Campbell M. G.; Mazzotti A. R.; Hamper B. C.; Spilling C. D.; Mannino M. P.; Wan L.; Yu J.-Q.; Liu J.; Welch C. J. Org. Biomol. Chem. 2014, 12, 2161. [DOI] [PubMed] [Google Scholar]
- 2a was obtained in 54, 85, and 59% yield with TEMPO, 1,1-diphenylethylene, and BHT, respectively.
- a Curto J. M.; Dickstein J. S.; Berritt S.; Kozlowski M. C. Org. Lett. 2014, 16, 1948. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dickstein J. S.; Fennie M. W.; Norman A. L.; Paulose B. J.; Kozlowski M. C. J. Am. Chem. Soc. 2008, 130, 15794. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.