

Abstract
Lipid-derived electrophiles (LDEs) that can directly modify proteins have emerged as important small-molecule cues in cellular decision-making. However, because these diffusible LDEs can modify many targets [e.g., >700 cysteines are modified by the well-known LDE 4-hydroxynonenal (HNE)], establishing the functional consequences of LDE modification on individual targets remains devilishly difficult. Whether LDE modifications on a single protein are biologically sufficient to activate discrete redox signaling response downstream also remains untested. Herein, using T-REX (targetable reactive electrophiles and oxidants), an approach aimed at selectively flipping a single redox switch in cells at a precise time, we show that a modest level (∼34%) of HNEylation on a single target is sufficient to elicit the pharmaceutically important antioxidant response element (ARE) activation, and the resultant strength of ARE induction recapitulates that observed from whole-cell electrophilic perturbation. These data provide the first evidence that single-target LDE modifications are important individual events in mammalian physiology.
This communication shows that a single-target chemical modification by a small-molecule signaling electrophile, 4-hydroxynonenal (HNE) is sufficient to regulate cellular antioxidant response. This finding is made possible through the application of a unique chemical tool named T-REX (targetable reactive electrophiles and oxidants) with which we are able to read out a downstream response specifically elicited by a single-target HNEylation event in living cells.
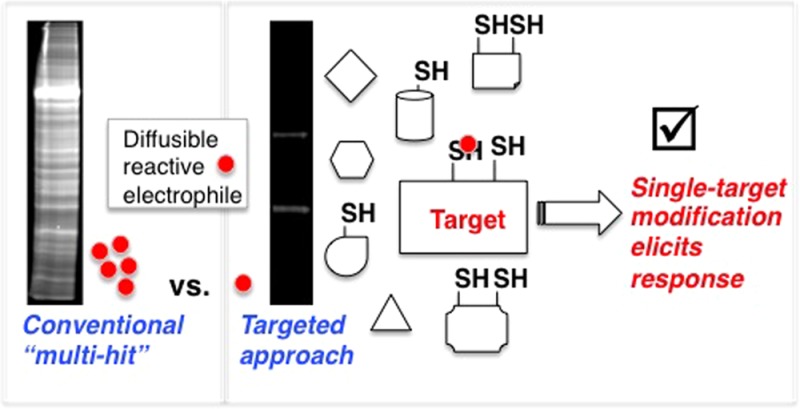
Lipid-derived electrophiles (LDEs), such as HNE, are central to redox-dependent cell signaling.1 However, the lability of the LDE adducts2a and toxic nature of LDEs above physiological concentrations and after prolonged exposure2b,2c render the consequences of nonenzyme-assisted LDE modifications largely intractable. The only general way to study the impacts of LDE modifications on specific proteins is with “overload” approaches in which the entire cell is treated with LDE in excess (Figure 1a).1−3
Figure 1.

(a) Whole-cell reactive electrophile (red circle) bathing turns on multiple stress responses. The T-REX approach interrogates importance of specific redox events. Inset: HaloTag system for T-REX. Structure of inert precursor (HtPHA) is shown in the inset in panel c. Blue spheres designate intracellular proteins. (b) Whole-cell HNE flooding elicits ARE activation but many upstream proteins (e.g., Akt, PTEN, PKC, GSK3, and Keap1) are HNE-sensitive ARE regulators.4a,5,11 (c) Binding of the chloroalkane-functionalized caged precursor to HNE-alkyne (HtPHA) to HaloTag (PDB: 1BN6) and subsequent energy minimization using MacroModel (Schrödinger, Inc.) showed that the cage motif is solvent-exposed. Low-energy light activation liberates HNE-alkyne efficiently.12
“Multi-hit” approaches have yielded important information about various stress-associated pathways.1−4 Proteomics-based innovations involving global treatment with electrophilic probes have enabled reactivity ranking of cysteines (Cys’s).4c For instance, 790 Cys’s have been quantitatively profiled as HNE-sensitive targets against >1000 Cys’s assayed from soluble fractions of HNE-treated human cell lysates.5 Phenotypic outputs resulting from whole-cell HNEylation are also well annotated for numerous physiologic processes such as anti-inflammatory, heat shock, metabolic, antioxidant, and immune responses.1,2c,3
It has thus been proposed that even low-stoichiometry HNEylation may induce signaling responses.1b,5 However, whether such substoichiometric modifications, or even modifications on one target alone, are sufficient to elicit downstream responses remain untested.
The poor understanding of the mechanistic underpinnings of LDE-modulated phenotypic responses is best exemplified by the debate surrounding the “nuclear factor-erythroid 2 p45-related factor 2–antioxidant response element (Nrf2–ARE) activation, a major mammalian antioxidant signaling axis (Figure 1b).6 The Nrf2 transcription factor regulates transactivation of ∼200 ARE-driven genes essential for antioxidant defense and cellular detoxification.6b ARE inducers are diverse, comprising clinically relevant electrophilic small molecules such as bardoxolone-methyl (CDDO-Me)7 and the recently FDA-approved drug dimethyl fumarate (BG-12),8 as well as innate LDEs such as HNE.1c Because the Kelch-like ECH-associated protein 1 (Keap 1) is the cytosolic anchor of Nrf2,6b the long-standing model of redox-dependent ARE activation involves reactive electrophilic Michael acceptor LDEs such as HNE modifying Cys residue(s) on Keap1, disrupting Keap1–Nrf2 association, allowing Nrf2 to enter the nucleus, and activate numerous ARE-driven cytoprotective genes.9
However, consistent with the >700 known HNE-sensitive targets,4a,5 recent studies have contradicted the above model.6a,10 Indeed, various alternative mechanisms are proposed, including, HNE modification of Nrf2,11a,11b and HNEylation of redox-sensitive kinases such as PKC and GSK3 that can phosphorylate Nrf2.11c−11f The Nrf2–ARE axis is also known to be regulated by (among others) PTEN and Akt, both of which are modified by HNE during overload,11c,11g and recently profiled within the 790 HNE-sensitive targets.5 Similar uncertainties persist in many unrelated redox-dependent signaling pathways.1−3 Because conventional multi-hit methods could trigger (or suppress/nullify) a phenotypic response, a mechanism linking modification of a specific target protein to the downstream ARE response cannot be unequivocally ascertained (Figure 1b). Mechanistic understanding of such a system is therapeutically relevant because CDDO-Me and BG-12 are thought to function by upregulating ARE.7,8 However, these compounds likely react promiscuously with many proteins, reflecting our poor knowledge of the ARE response. The ability to unambiguously pin down a major regulator sufficient for activating a pharmaceutically beneficial response is important for targeted drug design and optimization.
We envisaged that our newly developed temporally controlled targeted HNEylation of specific proteins in cells12 would be ideal to challenge whether single-protein HNEylation is sufficient to elicit a phenotypic response. Because the T-REX approach uses HaloTag13 fused to the target protein to enable directed HNEylation (Figure 1c), we first showed that HaloTagging does not interfere with the ability of Keap1 to homodimerize and bind Nrf2, in vitro and in live HEK-293 cells (Figures 2a and S1–S2 and Table S1). These results agree with previous reports on the nonHaloTagged Keap1–Nrf2 complex.6b,9e,10f Using T-REX, we showed that targeted HNEylation of the Halo-Keap1–Nrf2 complex transfers HNE, within the detection limit, only to Keap1, either in an isolated system (Figure S2c) or in living cells expressing both Halo-Keap1 and Nrf2 (Figure 2b). When compared to global HNE treatment that led to nonspecific HNEylation (Figure 2b), a result in line with the recent quantitative proteomics data on profiling >700 HNE-sensitive targets,5 T-REX constitutes a vast improvement in terms of target selectivity over multi-hit approaches. Furthermore, because HaloTagging in this instance is noninvasive, T-REX is powerfully suited to interrogate the biological effects of HNEylation on Keap1 alone. These data also downplay the functional significance of the previously postulated direct HNE modification of Nrf2.6a,11a,11b
Figure 2.
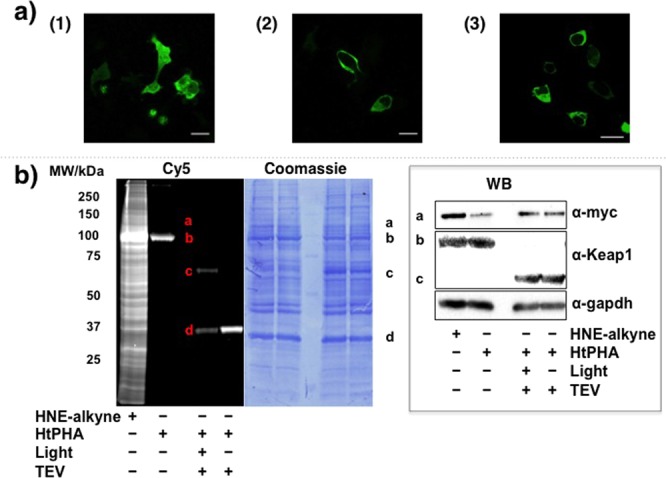
T-REX enables substoichiometric HNEylation of Keap1 alone. (a) The cytosolic protein Halo-Keap16a,6b,12 renders GFP-Nrf2 cytosolic. Confocal images of live HEK-293 cells transiently expressing (1) GFP-Nrf2 alone and (2) GFP-Nrf2 as well as Halo-Keap1. (3) Live HEK-293 cells stably expressing Halo-Keap1 (Figure S1) were transiently transfected with GFP-Nrf2. Scale bars, 20 μm. (b) Keap1-specific HNEylation in HEK-293 cells expressing Halo-Keap1 and myc-Nrf2 enabled by T-REX. Global HNE-alkyne treatment (25 μM) is nonspecific (left-most lane of the “Cy5” gel).5 a, b, c, and d markers designate myc-Nrf2, Halo-Keap1, Keap1, and Halo, respectively. Fluorescence (resulting from Click coupling with Cy5-azide12) allows tracking of any proteins covalently linked to HtPHA (Figure 1c, inset) or adducted by the liberated HNE-alkyne. Keap1-specific targeting efficiency12 in this representative data set is 34%. Coomassie-stained PVDF and western blot (inset) are also shown. TEV, Tobacco Etch Virus cysteine protease, enables separation of Halo and Keap1 domains.
In unstimulated cells, Nrf2 has a short half-life (t1/2 ≈ 15 min–3 h),6 due to Keap1 binding. Keap1 is an adaptor protein for Cul3-based ubiquitin E3 ligase complex, allowing continuous proteasomal degradation of Nrf2.6,9,10 In HEK-293 cells expressing Halo-Keap1 and Nrf2, whole-cell HNEylation inhibited Nrf2 degradation (Table S2 and Figure S3), consistent with previous reports.6,10b Keap1-specific HNEylation via T-REX similarly raised Nrf2 levels (Figure 3a); however, levels of ribonucleotide reductase small subunit (RRM2), a proteasomally regulated protein with a t1/2 similar to Nrf2 (∼3 h),14 were not altered.
Figure 3.
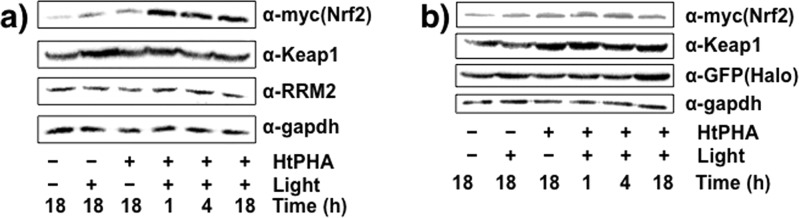
(a) Keap1-alone HNEylation in low stoichiometry is sufficient to stabilize Nrf2, whereas another unrelated, yet proteasomally regulated protein, RRM214 with a half-life similar to Nrf2 is unaffected. Time designates incubation time post-light exposure where applicable. (b) Similar Nrf2 stabilization is not observed in cells subjected to T-REX but expressing Halo and Keap1 separately. In each figure, a representative data set of at least n = 6 (three independent biological replicates × two technical replicates) is shown. Also see Figure S4.
By contrast, treatment with 20 nM Bortezomib, a clinically used proteasome inhibitor, led to time-dependent stabilization of both RRM2 and Nrf2 (Figure S4). These data indicate that T-REX does not impact the degradation of other proteasomally regulated proteins. When T-REX was carried out using cells expressing GFP-Halo as well as non-HaloTagged Keap1, there was no HNEylation of Keap1, and Nrf2 levels were unchanged (Figure 3b). Thus, Nrf2 stabilization likely arises from HaloTag-mediated T-REX-directed HNEylation of Keap1. These results suggest that substoichiometric Keap1-HNEylation (34%, Figure 2b) is alone sufficient to block Nrf2 degradation.
The most common mechanism for ARE stimulation predicts Nrf2 nuclear accumulation upon cell activation by LDEs.9,10,11a,11b We thus measured changes in nuclear and cytosolic Nrf2 upon T-REX-mediated Keap1-specific HNEylation using (1) nuclear/cytosol fractionation after cell lysis and (2) immunostaining (Figures S5 and S6). Both methods suggested that Nrf2 nuclear fraction was not significantly increased relative to the increase in cytosolic Nrf2. Thus, under conditions in which Keap1 alone is HNEylated, Nrf2 did not selectively accumulate in the nucleus; in fact, Nrf2 increased in both nucleus and cytosol. Treatment of the recombinant Keap1–Nrf2 complex with HNE also did not lead to Nrf2 dissociation (Figure S2b), consistent with the cell-based data. Previous data from whole-cell electrophile stimulation implicates Nrf2 nuclear translocation although whether or not Nrf2 dissociates from Keap1 upon electrophile signaling remains controversial.6a,6b,9,10c Since redox-sensitive regulators such as PKC also reportedly regulate Keap1–Nrf2 association,10d,10e the differences observed between T-REX and whole-cell treatment6a,6b,9,10c may be due to synergistic or compensatory effects arising from the global approach. T-REX thus fills a niche to address the effect of HNEylating specific targets in an otherwise largely unperturbed proteome.
We then investigated whether T-REX-mediated single-target HNEylation is sufficient to elicit ARE induction. Ectopic expression of ARE-inducible firefly and constitutive Renilla luciferases was achieved in HEK-293 cells alongside either Halo-Keap1 or non-HaloTagged-Keap1 (nontargetable control, exemplified with GFP-Keap1) and myc-Nrf2. T-REX-assisted Keap1-specific HNEylation stimulated ARE with an absolute increase in the luciferase signal comparable to those of global HNE (15 μM) treatment (Figure 4a). The fold increase in the normalized ARE luciferase signal from global electrophile treatment is consistent with previous reports.11b,15 Controls showed that ARE signaling was not caused by HNE transfer from either Halo or HNEylated Keap1 to other proteins within the proteome. When GFP-Keap1 replaced Halo-Keap1, no ARE activation was observed. In-gel fluorescence analyses also revealed that the HNE signal on Keap1 persisted over the time course of the experiment (Figure S7). Quantitative real-time (qRT)-PCR analysis confirmed that Keap1-specific HNEylation positively regulates Nrf2 transcriptional activity (Figure 4b). NQO1, HO-1, Trx, and GCLM1,6b established ARE-driven genes, were upregulated relative to GAPDH (Table S2). We also showed upregulation of NQO1 by western blot (Figures 4c and S8). These data collectively provide direct experimental evidence that substoichiometric HNEylation on a single target is sufficient to stimulate physiologic responses in the same way enzyme-assisted modifications such as phosphorylation16 do.
Figure 4.

(a) ARE induction by Keap1-specific HNEylation phenocopies whole-cell HNEylation. Normalized luciferase activity derived from ratio of ARE-inducible firefly to constitutive Renilla luciferase in HEK-293 cells alongside myc-Nrf2 and either Halo-Keap1 or GFP-Keap1 (nontargetable control). (b) mRNA expression of ARE-driven genes induced subsequent to T-REX HNEylation in HEK-293 cells analyzed by qRT-PCR. (c) The expression level of NQO1 assessed by western blot. Also see Figure S9. Error bars in panels a and b designate SD with n = 3 and ≥9, respectively).
We were interested to test whether a group of Cys residues within the 624-amino-acid Keap1 serves as gatekeeper sensor of HNE. Various models have been proposed for which residues within Keap1 are target(s) of specific electrophiles6,9,10b,17 and each of the 27 Cys residues has been reportedly modified in vitro by at least one electrophile.6a,17 However, obtaining direct LC–MS/MS evidence for HNEylated Keap1 from cells is challenging. The sole report of Keap1 HNEylation in COS-1 cells overexpressing mouse Keap1 treated with excess HNE detected “reduced HNE” at C151.10b Using optimized conditions, we identified HNEylation of four Cys’s, C23, C226, C273, and C368, when HEK-293 cells overexpressing Halo-Keap1 were treated with 100 μM HNE. Only C23 and C368 were hit when 25 μM HNE was used (Tables S4 and S5 and Figures S9 and S10). Keap1-targeted HNEylation using T-REX in cells identified C513 and C518 (Tables S6 and S7). Treatment of recombinant Halo-Keap1 with 1.1 equiv of HNE in vitro modified six Cys’s, C77, C151, C226, C273, C319, and C368 (Table S8), whereas with T-REX only C226 and C368 were HNEylated in vitro (Table S9). C151, the residue with the second-highest ion score in the globally HNEylated sample, was not found to be modified with T-REX in vitro. Differences in Keap1 conformation and contributions from cellular partners likely account for the differences in Cys’s HNEylated using T-REX in cells (Tables S6 and S7) and in vitro (Table S9).
We individually mutagenized each residue HNEylated under T-REX conditions in cell and in vitro to serine, C513S/C518S and C226S/C368S, respectively. T-REX-assisted HNEylation to the respective single and double mutants in each case was as efficient as wild type, and an increase in Nrf2 levels was observed (Table S1 and Figures S11 and S12). ARE upregulation upon targeted HNEylation of C513S/C518S-Keap1 mutants in cells is also in agreement with the observed increase in Nrf2 levels (Figure S13). We also mutagenized C151 and C288, the two residues known to be functionally important for Nrf2 regulation.9e,17 Consistent with previous reports, C151S/C288S affected the Nrf2 levels in unstimulated cells. The mutant proteins were still HNEylated upon T-REX although Nrf2 stabilization was minimal (Figure S14). These data likely imply that Keap1 is a promiscuous sensor with multiple Cys’s able to respond to HNE, and such residue flexibility in sensing HNE may enable robust HNE-induced ARE signaling.
The present study led us to discover the importance of substoichiometric nonenzyme-dependent direct chemical modifications in physiologic cell signaling. This knowledge is unattainable from global bathing with reactive LDEs (Figure 1b). Unlike phosphosignaling in which less than 100% phosphorylation stoichiometry on a target is common,16c the level at which nonenzyme-regulated LDE modifications can induce physiologic response was uncertain1b,5 until this work. T-REX allowed us to establish that HNEylation of Keap1 is alone sufficient to activate pharmaceutically beneficial ARE induction. Such molecular information sheds light on the design of target-specific electrophilic pharmacophores that can regulate ARE. This work is an exciting initial step toward ultimately exploiting the T-REX approach to gain biological tractability within individual redox signaling trajectories.
Acknowledgments
We thank Dr. Mike Fenwick of Prof. Ealick’s laboratory for the generation of HtPHA-bound Halo model (Figure 1c), and the laboratories of Profs. Sylvia Lee and Hening Lin for the use of a real-time PCR instrument and a LCMS instrument, respectively. We acknowledge Dr. Sheng Zhang, Cornell Proteomics Facility, for LC–MS/MS technical support, and the Cornell Biotech Resource Center Cell Imaging Facility. D.K.L thanks the Cornell Pre Professional Program for an undergraduate research fellowship. This research was supported by an NSF CAREER Award (CHE-1351400) and the Beckman Young Investigator Award (to Y.A). Instrumentation used in this research was supported by an NIH Director’s New Innovator Award (1DP2GM114850) (to Y.A).
Supporting Information Available
Experimental details and additional biochemical and cell-based data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ S.P and Y.F contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Rudolph T. K.; Freeman B. A. Sci. Signal. 2009, 2, re7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jacobs A. T.; Marnett L. J. Acc. Chem. Res. 2010, 43, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Schopfer F. J.; Cipollina C.; Freeman B. A. Chem. Rev. 2011, 111, 5997. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Fritz K. Z.; Petersen D. R. Free Radic. Biol. Med. 2013, 59, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wall S. B.; Smith M. R.; Ricart K.; Zhou F.; Vayalil P. K.; Oh J.-Y.; Landar A. Biochim. Biophys. Acta 2014, 1840, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Butterfield D. A.; Stadtman E. R. Adv. Cell Aging Gerontol. 1997, 2, 161. [Google Scholar]; c Guéraud F.; Atalay M.; Bresgen N.; Cipak A.; Eckl P. M.; Huc L.; Jouanin I.; Siems W.; Uchida K. Free Radic. Res. 2010, 44, 1098. [DOI] [PubMed] [Google Scholar]
- Jacob C., Winyard P. G., Eds. Redox Signaling and Regulation in Biology and Medicine; Wiley: Weinheim, Germany, 2009. [Google Scholar]
- a Codreanu S. G.; Zhang B.; Sobecki S. M.; Billheimer D. D.; Liebler D. C. Mol. Cell. Proteomics 2009, 8, 670. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weerapana E.; Wang C.; Simon G. M.; Richter F.; Khare S.; Dillon M. B.; Bachovchin D. A.; Mowen K.; Baker D.; Cravatt B. F. Nature 2010, 468, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Leonard S. E.; Carroll K. S. Curr. Opin. Chem. Biol. 2011, 15, 88. [DOI] [PubMed] [Google Scholar]
- Wang C.; Weerapana E.; Blewett M. M.; Cravatt B. F. Nat. Methods 2014, 11, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bryan H. K.; Olayanju A.; Goldring C. E.; Park B. K. Biochem. Pharmacol. 2013, 85, 705. [DOI] [PubMed] [Google Scholar]; b Hayes J. D.; Dinkova-Kostova A. T. Trends Biochem. Sci. 2014, 39, 199. [DOI] [PubMed] [Google Scholar]; c Holland R.; Fishbein J. C. Antioxid. Redox. Signal. 2010, 13, 1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Liby K. T.; Sporn M. B. Pharmacol. Rev. 2012, 64, 972. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Crunkhorn S. Nat. Rev. Drug Discovery 2012, 11, 96. [DOI] [PubMed] [Google Scholar]
- Scannevin R. H.; Chollate S.; Jung M. Y.; Shackett M.; Patel H.; Bista P.; Zeng W.; Ryan S.; Yamamoto M.; Lukashev M.; Rhodes K. J. J. Pharmacol. Exp. Ther. 2012, 341, 274. [DOI] [PubMed] [Google Scholar]
- a Motohashi H.; Yamamoto M. Trends. Mol. Med. 2004, 10, 549. [DOI] [PubMed] [Google Scholar]; b Kensler T. W.; Wakabayashi N.; Biswal S. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89. [DOI] [PubMed] [Google Scholar]; c Hur W.; Gray N. S. Curr. Opin. Chem. Biol. 2011, 15, 162. [DOI] [PubMed] [Google Scholar]; d Nguyen T.; Nioi P.; Pickett C. B. J. Biol. Chem. 2009, 284, 13291. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Zhang D. D. Drug Metab. Rev. 2006, 38, 769. [DOI] [PubMed] [Google Scholar]
- a Eggler A. L.; Liu G.; Pezzuto J. M.; Mesecar A. D. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 10070. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McMahona M.; Lamont D. J.; Beattie K. A.; Hayes J. D. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 18838. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Baird L.; Lleres D.; Swift S.; Dinkova-Kostova A. T. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 15259. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Huang H.-C.; Nguyen T.; Pickett C. B. J. Biol. Chem. 2002, 277, 42769. [DOI] [PubMed] [Google Scholar]; e Bloom D. A.; Jaiswal A. K. J. Biol. Chem. 2003, 278, 44675. [DOI] [PubMed] [Google Scholar]; f Zhang D. D.; Hannink M. Mol. Cell. Biol. 2003, 23, 8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a He X.; Ma Q. Mol. Pharmacol. 2009, 76, 1265. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Huang Y.; Li W.; Kong A.-N. Cell Biosci. 2012, 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Leonarduzzi G.; Robbesyn F.; Poli G. Free. Radic. Biol. Med. 2004, 11, 1694. [DOI] [PubMed] [Google Scholar]; d Numazawa S.; Ishikawa M.; Yoshida A.; Tanaka S.; Yoshida T. Am. J. Physiol. Cell Physiol. 2003, 285, C334. [DOI] [PubMed] [Google Scholar]; e Castello L.; Marengo B.; Poli G.; Chiarpotto E. Biochim. Biophys. Acta 2005, 1737, 83. [DOI] [PubMed] [Google Scholar]; f Dozza B.; Smith M. A.; Perry G.; Tabaton M.; Strocchi P. J. Neurochem. 2004, 89, 1224. [DOI] [PubMed] [Google Scholar]; g Shearn C. T.; Smathers R. L.; Backos D. S.; Reigan P.; Orlicky D. J.; Petersen D. R. Free. Radic. Biol. Med. 2013, 65, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X.; Fu Y.; Long M. J. C. L.; Haegele J. H.; Ge E. J.; Parvez S.; Aye Y. J. Am. Chem. Soc. 2013, 135, 14496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los G. V.; Encell L. P.; McDougall M. G.; Hartzell D. D.; Karassina N.; Zimprich C.; Wood M. G.; Learish R.; Ohana R. F.; Urh M.; Simpson D.; Mendez J.; Zimmerman K.; Otto P.; Vidugiris G.; Zhu J.; Darzins A.; Klaubert D. H.; Bulleit R. F.; Wood K. V. ACS Chem. Biol. 2008, 3, 373. [DOI] [PubMed] [Google Scholar]
- Aye Y.; Li M.; Long M. J. C.; Weiss R. S. Oncogene 2014, 10.1038/onc.2014.155. [DOI] [PubMed] [Google Scholar]
- Levonen A.-L.; Landar A.; Ramachandran A.; Ceaser E. K.; Kickinson D. A.; Zanoni G.; Morrow J. D.; Darley-Usmar V. M. Biochem. J. 2004, 378, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Walsh C. T. In Posttranslational Modification of Proteins: Expanding Nature’s Inventory; Roberts & Co.: Greenwood Village, CO, 2005. [Google Scholar]; b Tarrant M. K.; Cole P. A. Annu. Rev. Biochem. 2009, 78, 797. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Prabakaran S.; Lippens G.; Steen H.; Gunawardena J. WIREs Syst. Biol. Med. 2012, 4, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkova-Kostova A. T. Scientifica 2012, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.