

Abstract
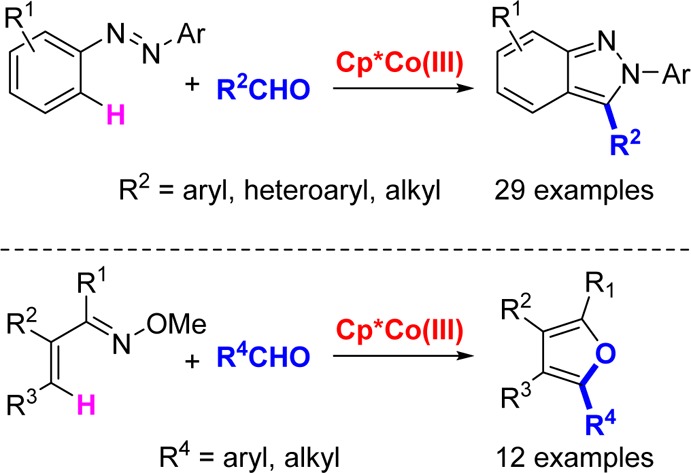
The development of operationally straightforward and cost-effective routes for the assembly of heterocycles from simple inputs is important for many scientific endeavors, including pharmaceutical, agrochemical, and materials research. In this article we describe the development of a new air-stable cationic Co(III) catalyst for convergent, one-step benchtop syntheses of N-aryl-2H-indazoles and furans by C–H bond additions to aldehydes followed by in situ cyclization and aromatization. Only a substoichiometric amount of AcOH is required as an additive that is both low-cost and convenient to handle. The syntheses of these heterocycles are the first examples of Co(III)-catalyzed additions to aldehydes, and reactions are demonstrated for a variety of aromatic, heteroaromatic, and aliphatic derivatives. The syntheses of both N-aryl-2H-indazoles and furans have been performed on 20 mmol scales and should be readily applicable to larger scales. The reported heterocycle syntheses also demonstrate the use of directing groups that have not previously been applied to Co(III)-catalyzed C–H bond functionalizations. Additionally, the synthesis of furans demonstrates the first example of Co(III)-catalyzed functionalization of alkenyl C–H bonds.
Introduction
Remarkable progress in transition-metal-catalyzed C–H bond functionalization has enabled the direct generation of nucleophilic partners without requiring prefunctionalization.1,2 Strategies for the addition of C–H bonds to polarized π bonds are particularly efficient and atom-economical for the assembly of heteroatom substituted products.3 Due to the vast number of diverse aldehydes that are commercially available, aldehydes potentially represent the most versatile of the polarized π-bond inputs. However, C–H bond additions to aldehydes are reversible, with the alcohol products only favored when destabilized, highly electron-deficient aldehydes are used.4 For typical aldehydes, good conversion to the alcohol product can only be achieved by trapping the alcohol to prevent reversibility, for example, through protection5 or oxidation.6 Recently, we have reported a general strategy to access important classes of heterocycles by cyclative capture of the initially formed alcohol intermediate via reaction with the directing group for C–H functionalization (Figure 1).7 In this approach the alcohol can either react as a leaving group with the directing group serving as a nucleophile (as represented by indazole synthesis) or act as a nucleophile with the directing group serving as an electrophile (as depicted for furan synthesis).
Figure 1.
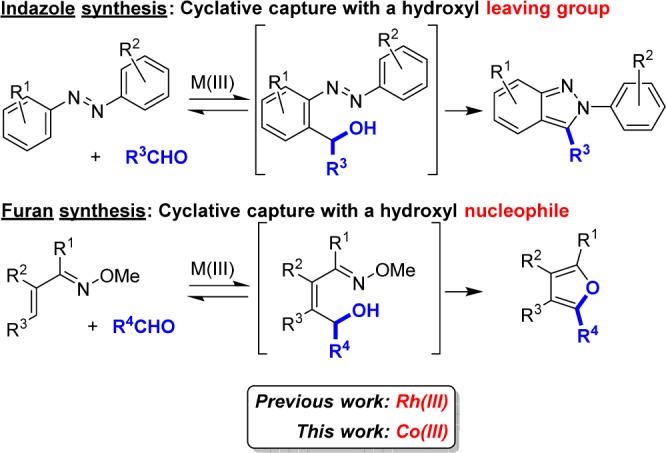
Rhodium- and cobalt-catalyzed heterocycle synthesis via aldehyde addition followed by cyclative capture.
Although our cyclative capture approaches provide efficient and convergent access to heterocycles, this chemistry is limited by the expensive Rh(III) C–H functionalization catalysts that we have so far employed. Due to the high cost of the more extensively developed catalyst systems based upon the precious metals Rh, Pd, Ag, Ir, Pt, and Au, it is not surprising that C–H bond functionalization using the earth-abundant first-row metals such as Mn,8 Fe,9 Co,10−12 Ni,13 and Cu14 has increasingly been investigated as an attractive alternative due to availability and cost.15
In this article we report the development of a new Co catalyst for the synthesis of 2-aryl indazoles and furans (Figure 1). These structural motifs are prevalent in drugs and natural products16,17 and 2-aryl indazoles exhibit interesting spectrophotometric properties.7a The transformations reported herein are the first examples of Co-catalyzed additions of C–H bonds to carbonyl compounds and represent the first use of azo18,19 and oxime directing groups with Co(III) catalysis (Figure 1). Moreover, the C–H bond functionalization of α,β-unsaturated oximes to give furans provides the first example of Co(III)-catalyzed functionalization of alkenyl C–H bonds.20 Both heterocycle syntheses utilize the same air-stable cationic Co(III) catalyst, and reactions can be performed on the benchtop and on a large scale.
Results and Discussion
Seminal reports by Kanai, Matsunaga, and co-workers have established that the high-valent preformed cobalt complex [Cp*Co(C6H6)][PF6]2 (4) catalyzes the addition of 2-phenylpyridyl11g and indole11f C–H bonds to sulfonyl protected imines. We therefore envisioned that a cationic Cp*Co(III) system could also be designed for additions to aldehydes with cyclative capture to obtain heterocycles. To evaluate the potential of high-valent Co catalysts for these transformations, we chose to investigate the addition of azobenzene (1a) to benzaldehyde (2a) (Table 1). The initial attempt utilizing Kanai’s and Matsunaga’s preformed catalyst [Cp*Co(C6H6)][PF6]2 (4) resulted in a poor yield of the desired product (entry 1).
Table 1. Optimization of Reaction Conditions for N-Aryl-2H-indazole Synthesis with [Cp*CoCl2]2a.
| entry | Co(III) source (mol %) | Ag salt (mol %) | acetate additive (mol %) | yield (%)b |
|---|---|---|---|---|
| 1 | 4 (10) | 2 | ||
| 2 | [Cp*CoCl2]2 (5) | AgPF6 (20) | 3 | |
| 3 | [Cp*CoCl2]2 (5) | AgSbF6 (20) | 1 | |
| 4 | [Cp*CoCl2]2 (5) | AgOAc (20) | ||
| 5 | [Cp*CoCl2]2 (5) | AgB(C6F5)4 (20) | 51 | |
| 6 | [Cp*CoCl2]2 (5) | AgB(C6F5)4 (25) | 61 | |
| 7 | [Cp*CoCl2]2 (5) | AgB(C6F5)4 (25) | AgOAc (20) | 97 (91)c |
| 8 | [Cp*CoCl2]2 (2.5) | AgB(C6F5)4 (12.5) | AgOAc (10) | 63 |
| 9 | [Cp*CoCl2]2 (1) | AgB(C6F5)4 (5) | AgOAc (4) | 44 |
| 10 | [Cp*CoCl2]2 (2.5) | AgB(C6F5)4 (12.5) | KOAc (10) | 54 |
| 11d | [Cp*CoCl2]2 (2.5) | AgB(C6F5)4 (12.5) | AgOAc (10) | 45 |
| 12 | AgB(C6F5)4 (25) | AgOAc (20) |
Conditions: 2a (1.0 equiv), 1a (2.0 equiv) in 1,4-dioxane (2.0 M) for 24 h.
Determined by GC analysis relative to tetradecane as an external standard.
Isolated yield at 0.20 mmol scale.
Reverse stoichiometry: 2a (2.0 equiv), 1a (1.0 equiv).
We next evaluated a series of silver halide abstractors using [Cp*CoCl2]2 as the precatalyst to generate cationic Co(III) in situ. AgPF6 provided results similar to those observed with the preformed catalyst (entry 2), and with AgSbF6 or AgOAc little or no product was obtained (entries 3 and 4, respectively). However, switching to the noncoordinating AgB(C6F5)4 significantly increased the yield (entry 5), and the addition of a slight excess of AgB(C6F5)4 resulted in further improvement (entry 6). In previous reports on Co(III) catalysis, the addition of catalytic amounts of acetate base was beneficial.11d−11f During our optimization we found that the addition of 20 mol % of AgOAc also proved to be advantageous to the reaction, resulting in a near-quantitative yield (entry 7).21 Furthermore, although lower concentrations were tolerated, high concentration (2.0 M) was selected for added convenience and efficiency.
The application of lower catalyst loadings resulted in a reduced yield, providing 63% yield with 2.5 mol % of [Cp*CoCl2]2 (entry 8) and 44% yield when only 1 mol % of [Cp*CoCl2]2 was employed (entry 9). We decided to continue evaluation at 2.5 mol % of [Cp*CoCl2]2 due to the difficulty in evaluating minor differences at the near-quantitative yield observed at higher catalyst loading. The use of KOAc as an alternative to AgOAc resulted in a slightly lower yield (entries 8 vs 10), as did the reversal of stoichiometry (entry 11). Finally, no reaction occurred when only Ag salts and no [Cp*CoCl2]2 was added (entry 12), indicating that Cp*Co(III) is essential for C–H bond functionalization.
Although a high yield of indazole 3a was obtained under the optimized Co-catalyzed conditions (entry 7, Table 1), our goal of identifying a cost-effective earth-abundant catalyst system was only partially realized because a significant mol % of the precious metal silver was still required. In particular, 25 mol % of AgB(C6F5)4 was necessary to abstract the chlorides to generate the cationic Co(III) catalyst, with 20 mol % more silver contributed by the AgOAc additive needed to achieve high yields.
As a first step to eliminating any use of Ag salts, we developed an independent synthesis of the optimal catalyst [Cp*Co(C6H6)][B(C6F5)4]2 (5) that avoids the use of costly metals. We selected [Cp*Co(C6H6)](PF6)2 (4) as a viable starting point because Kanai and Matsunaga had prepared this complex without the use of Ag salts or other expensive reagents.11g Anion metathesis of complex 4 and commercially available potassium tetrakis(pentafluorophenyl)borate can be conveniently performed on a multigram scale to provide the desired catalyst 5 (Scheme 1). A biphasic mixture of benzene/H2O was selected as the solvent of choice for this reaction to partition KPF6 into the aqueous phase while avoiding any potential exchange of the benzene ligand on the parent [Cp*CoIII(C6H6)] complex.
Scheme 1. Synthesis of [Cp*Co(C6H6)][B(C6F5)4]2 (5).

Preliminary evaluation of 5 in the C–H functionalization cascade to 3a established a large increase in reactivity over 4 (Table 2, entries 1 vs 2). In comparison to the reaction yield for [Cp*CoCl2]2/AgB(C6F5)4 in Table 1, the addition of silver acetate resulted in a dramatic improvement in the reaction yield (entries 3 and 4). We therefore sought to replace the precious metal additive AgOAc with earth-abundant first-row metal acetate salts.
Table 2. Optimization of Reaction Conditions for Indazole Synthesis with Catalyst 5a.
| entry | preformed Co(III) catalyst (mol %) | additive | temp (°C) | yield (%)b |
|---|---|---|---|---|
| 1 | 4 (10) | 130 | 2 | |
| 2 | 5 (10) | 130 | 31 | |
| 3 | 5 (10) | AgOAc (10 mol %) | 130 | 82 |
| 4 | 5 (10) | AgOAc (20 mol %) | 130 | 72 |
| 5 | 5 (10) | Cu(OAc)2 (10 mol %) | 130 | 71 |
| 6 | 5 (10) | Cu(OAc)2 (20 mol %) | 130 | 25 |
| 7 | 5 (10) | Cu(OAc)2(H2O)4 (10 mol %) | 130 | 75 |
| 8 | 5 (10) | Cu(OAc)2(H2O)4 (20 mol %) | 130 | 50 |
| 9 | 5 (10) | H2O (50 mol %) | 130 | 45 |
| 10 | 5 (10) | H2O (1.0 equiv) | 130 | 53 |
| 11 | 5 (10) | H2O (2.0 equiv) | 130 | 27 |
| 12 | 5 (10) | AcOH (10 mol %) | 130 | 85 |
| 13 | 5 (10) | AcOH (10 mol %) | 100 | 87 |
| 14c | 5 (10) | AcOH (10 mol %) | 100 | 90 (83)d |
| 15 | 5 (10) | AcOH (50 mol %) | 130 | 71 |
| 16 | 5 (10) | PivOH (10 mol %) | 130 | 80 |
| 17 | 5 (5) | AcOH (5 mol %) | 130 | 67 |
| 18 | 5 (5) | PivOH (5 mol %) | 130 | 68 |
Conditions:2a (1.0 equiv), 1a (2.0 equiv) in 1,4-dioxane (2.0 M) for 24 h.
Determined by GC analysis relative to tetradecane as an external standard.
Reaction performed on the benchtop under nitrogen.
Isolated yield at 0.20 mmol scale.
We first investigated Cu(OAc)2 because this additive had previously been observed to result in higher yields for Rh(III)-catalyzed transformations, including in transformations where Cu(II) did not serve as an oxidant.22 Promising results were observed at 10 mol % (71%, entry 5), but a significant reduction in yield was obtained at a higher loading (entry 6). Interestingly, although 10 mol % of Cu(OAc)2(H2O)4 did not significantly influence reaction outcome (entry 7), at 20 mol % the tetrahydrate performed substantially better than anhydrous copper(II) acetate (entries 6 versus 8). Water was evaluated as an additive on the basis of this observation, demonstrating that this transformation was not sensitive to moderate amounts of moisture (entries 9–11). In fact, water was slightly beneficial when 1 equiv was employed, providing 3a in 53% yield. Turning our attention to other protic additives we found that AcOH proved highly effective (entry 12), and with these conditions achieved our goal of completely eliminating all precious metal additives from the reaction system.
Additional reaction parameters were investigated to enhance the practicality of the transformation. First, the reaction temperature was explored, and a comparable yield was observed at a temperature below the solvent boiling point (entries 13 and 14). Furthermore, rigorous glovebox conditions were not required, which allowed us to set up reactions conveniently on the benchtop (entry 14).
In previous reports of C–H functionalization, pivalic acid has proven to be superior to other organic acids.23 However, when PivOH was used in place of AcOH (entries 12 vs 16), similar yields were obtained. This trend was also observed at lower acid and catalyst loading (entries 17 vs 18). Ultimately, acetic acid was selected as the additive of choice due to lower cost.
Having identified practical reaction conditions that were free of precious metals for the synthesis of 3a, we next evaluated a series of unsymmetrical azobenzenes to determine the influence of steric and electronic parameters on the regioselectivity of this transformation (Table 3). In addition, the original conditions developed to generate cationic Cp*Co(III) in situ (Table 1) were evaluated for each substrate to provide a comparative analysis of these two catalyst systems.
Table 3. Azobenzene Substrate Scopea,b.
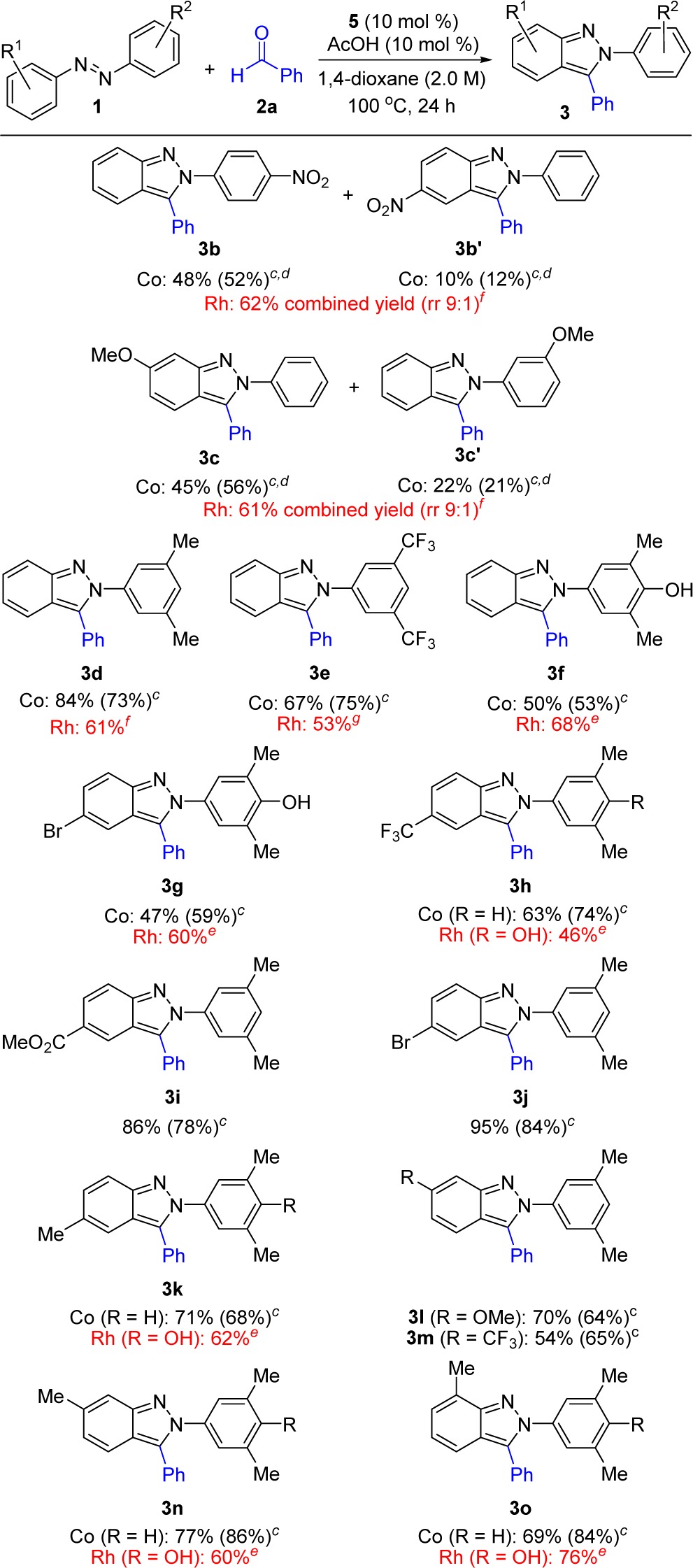
Co(III) conditions: 2a (0.20 mmol), azobenzene 1 (0.40 mmol), 5 (10 mol %), and AcOH (10 mol %) in 1,4-dioxane (2.0 M) at 100 °C for 24 h.
Isolated yields after silica gel chromatography.
Co(III) in situ conditions:2a (0.20 mmol), azobenzene 1 (0.40 mmol), [Cp*CoCl2]2 (5 mol % of Co dimer), AgB(C6F5)4 (25 mol %), and AgOAc (20 mol %) in 1,4-dioxane (2.0 M) at 130 °C for 24 h.
Ratio of fully separable regioisomers.
Rh(III) conditions: azobenzene 1 (0.20 mmol), 2a (0.40 mmol), [Cp*RhCl2]2 (5 mol % of Rh dimer), AgSbF6 (20 mol %), and MgSO4 (100 mg) in tetrahydrofuran (0.2 M) at 110 °C for 24 h.
Reaction was conducted in 1,4-dioxane (0.2 M).
Reaction was conducted in 1,4-dioxane (1.0 M).
The major regioisomer obtained from the reaction of the para-nitro-substituted azobenzene, (E)-1-(4-nitrophenyl)-2-phenyldiazene, resulted from C–H functionalization on the more electron-rich aromatic ring, providing fully separable regioisomers 3b and 3b′ in a 4.6:1 ratio when the preformed catalyst 5 was used. Comparable results were seen when cationic Cp*Co(III) was generated in situ and the AgOAc additive was included, providing a 4.4:1 ratio of regioisomers 3b and 3b′. In a similar fashion, the meta-methoxy-substituted azobenzene, (E)-1-(3-methoxyphenyl)-2-phenyldiazene, also underwent C–H functionalization on the more electron-rich aryl ring, furnishing products 3c and 3c′ in 2.1:1 ratio of separable regioisomers with preformed catalyst 5/AcOH and 2.7:1 ratio with [Cp*CoCl2]2/AgB(C6F5)4/AgOAc. Notably, for both catalyst systems reaction on the meta-methoxy-substituted aromatic ring only occurred at the less hindered of the two ortho sites to give regioisomer 3c. These results are consistent with the yields and selectivity that we previously observed with Rh(III), although higher regioselectivities were observed with this precious metal catalyst.7a
Several 3,5-disubstituted azobenzenes containing both electron-withdrawing and electron-donating functionalities were also evaluated. Indazoles 3d and 3e were obtained as single regioisomers in good yield, establishing that steric effects dominate the regioselective outcome of this Cp*Co(III)-catalyzed reaction. With Rh(III) catalysis, high regioselectivities were previously observed for these substrates albeit with slightly lower yields. In addition, a cleavable phenolic derivative was employed to access indazoles 3f and 3g in a reasonable yield,7a though lower than the yield for the corresponding azobenzene lacking the hydroxyl substituent (see 3d). For 3f and 3g, Rh(III) catalysts had previously provided somewhat higher yields.
A series of 3,5-dimethyl-substituted azobenzenes were then applied to the selective introduction of functionality to the indazole core (3h–3o, Table 3). Indazoles containing electron-poor (3h, 3i, 3m), electron-neutral (3g, 3j, 3k, 3n, 3o), and electron-rich (3l) substituents were all obtained in good yields. Moreover, functionality could be placed regiospecifically at the 5- (3g–3k), 6- (3l–3n), and 7-positions (3o) of the indazole ring. In cases where the same or closely related indazole products had previously been prepared using Rh(III) catalysis, comparable yields were observed (3g, 3h, 3k, 3n, 3o).
A variety of aldehydes were next evaluated (Table 4). Both electron-poor (3p–t) and electron-rich (3v–x) aldehydes coupled efficiently. Indazoles bearing diverse functionality such as trifluoromethyl (3p), methyl ester (3q), nitro (3r), fluoro (3s), chloro (3t), bromo (3u), methyl (3v–3w), and methoxy (3x) substituents were obtained in good to excellent yields. Moreover, ortho (3s), meta (3t, 3w), and para (3p–r, 3u, 3v, 3x) substitution patterns were well tolerated. Heterocycles such as an indole (3y), a thiophene (3z), and a furan (3aa) could be introduced in excellent to moderate yields. Even additions to the alkyl aldehydes cyclohexanecarboxaldehyde and isovaleraldehyde proceeded to give indazoles 3ab and 3ac, respectively, in reasonable yields. Notably, in the case of substrate 3aa bearing a furanyl substituent, the Ag-free conditions developed for preformed catalyst 5 provided a noticeably higher yield. For substrates 3ab and 3ac derived from alkyl aldehydes, however, in situ preparation of the Co catalyst with use of the AgOAc additive gave better results, thus demonstrating the complementary nature of these two catalyst systems. We had previously evaluated many of these aldehydes for Rh(III)-catalyzed indazole synthesis (3a, 3p–3s, 3v, 3x, 3ab). Although a direct comparison cannot be made because different azobenzenes were employed in most cases, it is readily apparent that both Co(III) and Rh(III) catalysts show a comparably high level of functional group compatibility.
Table 4. Aldehyde Substrate Scope for N-Aryl-2H-indazole Synthesisa,b.
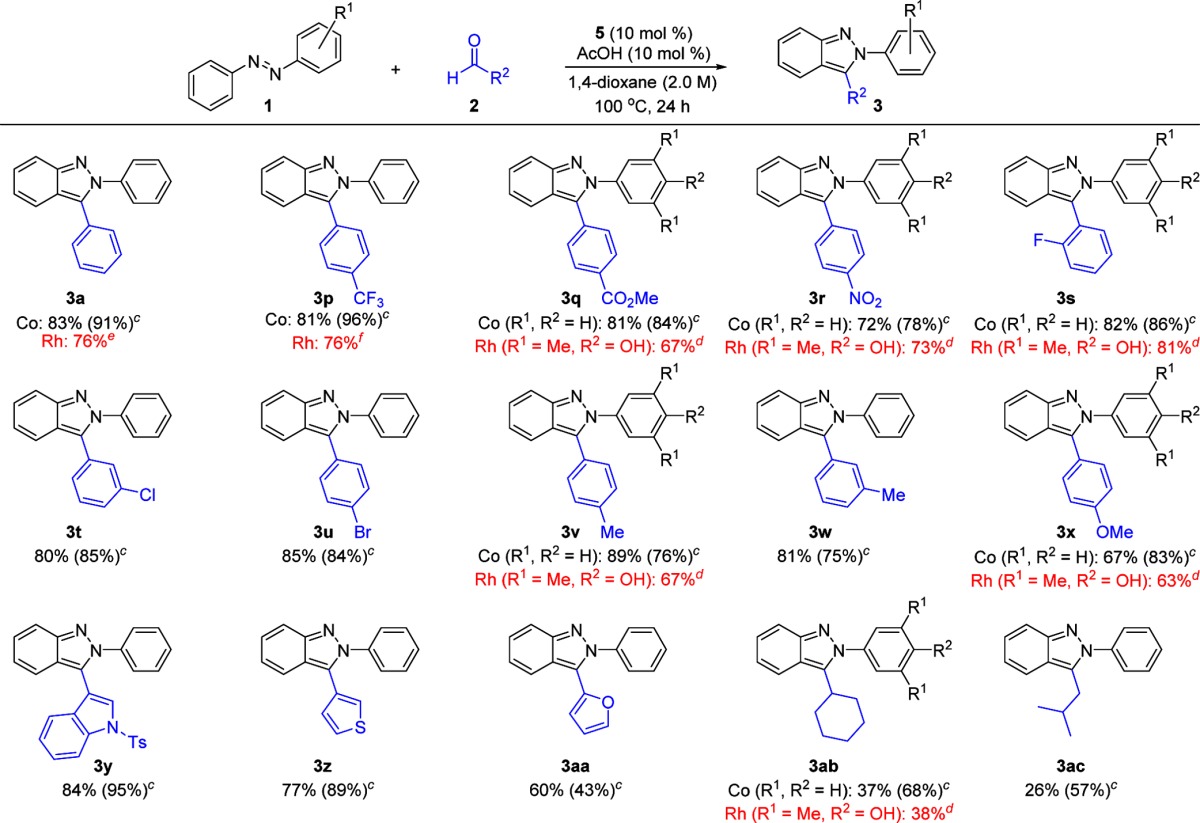
Co(III) conditions: aldehyde 2 (0.20 mmol), 1a (0.40 mmol), 5 (10 mol %), and AcOH (10 mol %) in 1,4-dioxane (2.0 M) at 100 °C for 24 h.
Isolated yields after silica gel chromatography.
Co(III) in situ conditions: aldehyde 2 (0.20 mmol), 1a (0.40 mmol), [Cp*CoCl2]2 (5 mol % of Co dimer), AgB(C6F5)4 (25 mol %), and AgOAc (20 mol %) in 1,4-dioxane (2.0 M) at 130 °C for 24 h.
Rh(III) conditions: azobenzene 1 (0.20 mmol), aldehyde 2 (0.40 mmol), [Cp*RhCl2]2 (5 mol % of Rh dimer), AgSbF6 (20 mol %), and MgSO4 (100 mg) in tetrahydrofuran (0.2 M) at 110 °C for 24 h.
Reaction was conducted in 1,4-dioxane (0.2 M).
Reaction was conducted in 1,4-dioxane (1.0 M).
We next explored the possibility of extending this Co(III)-catalyzed aldehyde addition and cyclative capture approach to include the synthesis of highly functionalized furans 7 from α,β-unsaturated oximes 6 (Table 5). For the optimization studies we employed the oxime as the limiting reagent because aldehydes are generally readily available and inexpensive and the oxime must be prepared from the corresponding α,β-unsaturated ketone. For convenience and practicality, we also focused on preformed catalyst 5 and Ag free conditions. The use of 10 mol % of AcOH with 1,4-dioxane as the solvent was first employed because these conditions were optimal for indazole synthesis (entry 1). Although a good yield was obtained, considerable improvement in yield to 83% was observed upon replacing 1,4-dioxane with THF (entry 2). A significant decrease in yield resulted when the reaction was conducted below the solvent boiling point (entries 3 and 4). However, use of DCE provided a comparable yield at both 100 and 80 °C (entries 5 and 6, respectively). The lower temperature was selected for further experiments to allow for large scale reactions to be conducted under reflux instead of in a sealed tube (vide infra). Finally, removing the AcOH catalyst resulted in much lower conversion (entry 7).
Table 5. Optimization of Reaction Conditions for Furan Synthesis using Catalyst 5a.
| entry | solvent | AcOH loading (mol %) | temp (°C) | yield (%)b |
|---|---|---|---|---|
| 1 | dioxane | 10 | 100 | 64 |
| 2 | THF | 10 | 100 | 83 |
| 3 | THF | 10 | 80 | 74 |
| 4 | THF | 10 | 65 | 56 |
| 5 | DCE | 10 | 100 | 79 |
| 6 | DCE | 10 | 80 | 80 (67)c |
| 7 | DCE | 80 | 25 |
Conditions: 6a (1.0 or 2.0 equiv) and 2a (1.0 or 2.0 equiv) in solvent (2.0 M) for 24 h.
Determined by GC analysis relative to tetradecane as an external standard.
Isolated yield at 0.20 mmol scale.
The substrate scope was explored with a range of α,β-unsaturated oximes 6 and aldehydes 2 (Table 6). Oximes lacking R3 substitution resulted in trisubstituted furans 7a–7k in good to excellent yields. Tetrasubstituted furan 7l with a fused bicyclic framework was also prepared, albeit in lower yield. With respect to aromatic aldehydes, this transformation exhibited a broad scope and was compatible with fluoro (7b), chloro (7c, 7d), bromo (7e), methyl ester (7f), methyl (7g), and trifluoromethyl (7j–l) functionality. Moreover, a good yield was obtained for ortho (7b) and meta (7c, 7g) functionalities. Unactivated alkyl aldehydes were also tolerated, affording furans 7h and 7i in moderate yield.
Table 6. Furan Substrate Scopea,b.
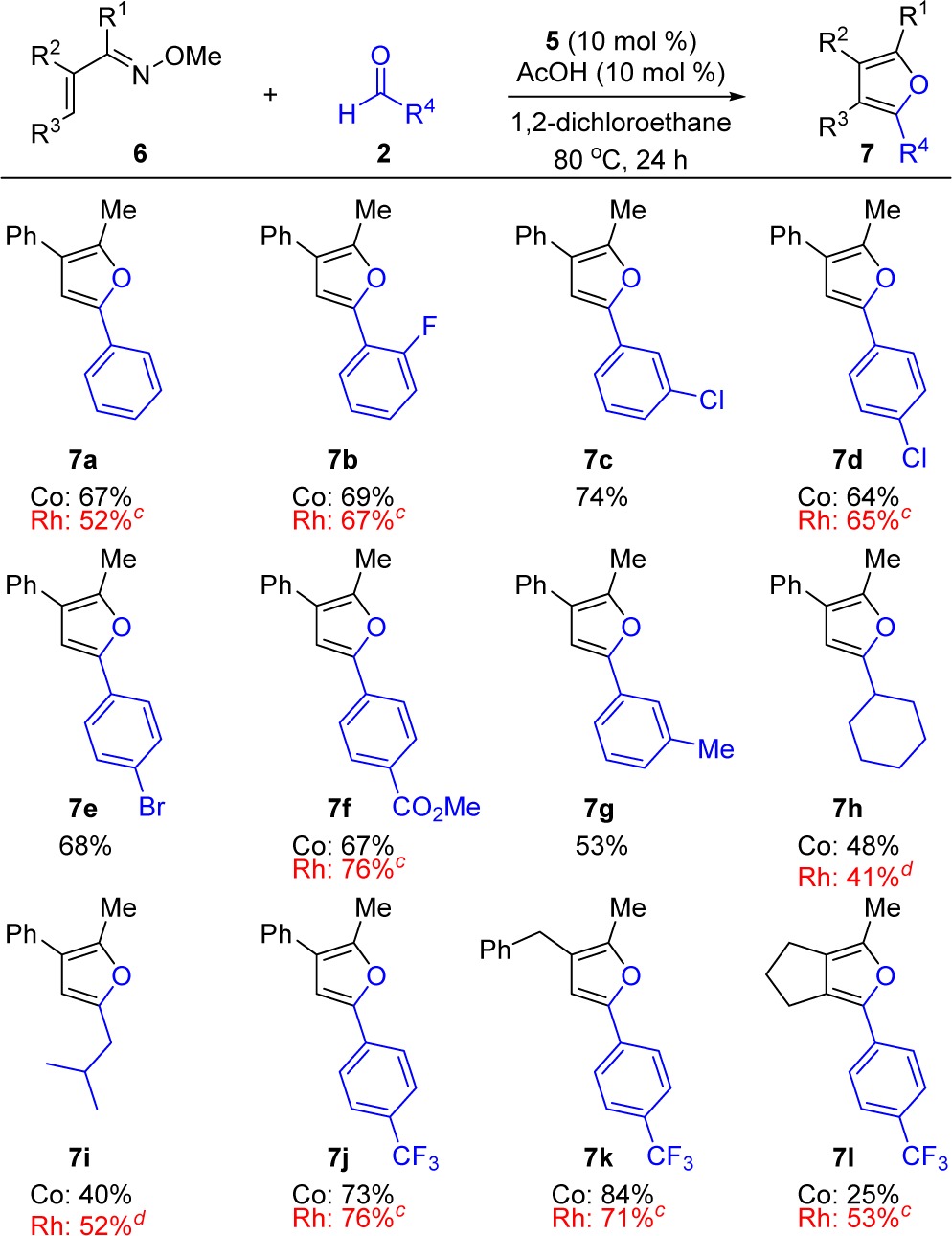
Co(III) conditions: O-methyl oxime 6 (0.20 mmol), aldehyde 2 (0.40 mmol), 5 (10 mol %), and AcOH (10 mol %) in 1,4-dichloroethane (2.0 M) for 24 h.
Isolated yields after silica gel chromatography.
Rh(III) conditions: O-methyl oxime 6 (0.20 mmol), aldehyde (0.40 mmol), [Cp*RhCl2]2 (5 mol % of Rh dimer), and AgSbF6 (20 mol %) in tetrahydrofuran (0.3 M) at 90 °C for 24 h.
Reaction was conducted using [Cp*RhCl2]2 (10 mol % of Rh dimer) and AgSbF6 (40 mol %).
For almost all of the furans that we had previously prepared with Rh(III) catalysis, Co(III) provided comparable yields. The one exception is for tetrasubstituted furans such as 7l, for which Rh(III)-catalysis provided considerably higher yields.24
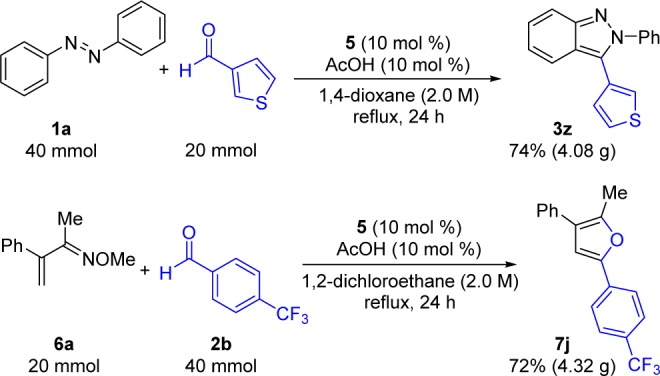
To evaluate the scalability of these C–H functionalization cascades and to further demonstrate the robust nature of catalyst 5, 20 mmol scale reactions were performed on the benchtop under reflux (Scheme 2). Indazole 3z was accessed in 74% yield (4.08 g), comparable to the small scale procedure (Table 3).25 Additionally, furan 7j was synthesized in 72% yield (4.34 g), which was a yield nearly identical to that obtained on smaller scale.
Scheme 2. Gram Scale Indazole and Furan Synthesis Using Catalyst 5.
We performed several experiments to provide insight into the mechanism for aldehyde additions followed by cyclative capture and focused these studies on indazole synthesis. First, several competition experiments were performed at low conversion to determine the effect of electronics upon rate of conversion (Scheme 3).
Scheme 3. Azobenzene and Aldehyde Competition Experiments to Analyze Electronic Effects.
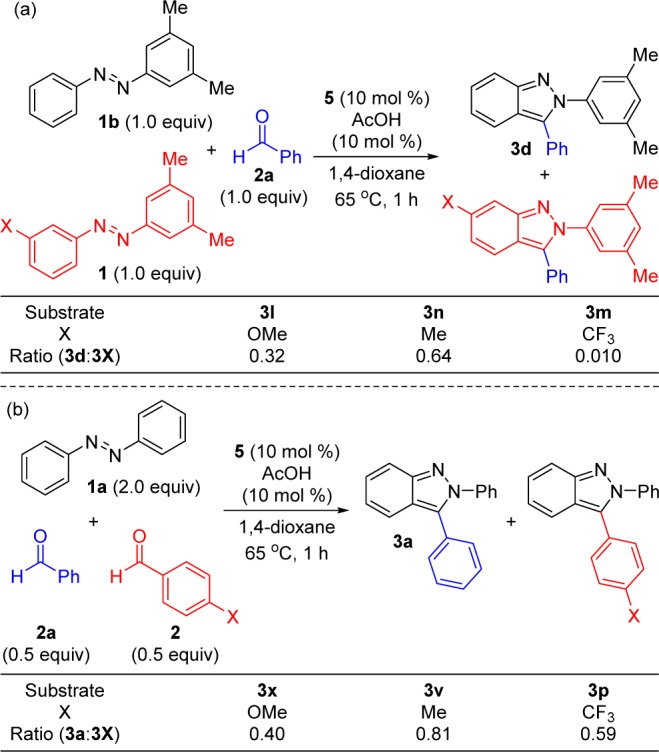
Substituted azobenzenes were first explored (Scheme 3a) because this component is involved in each step of the catalytic cycle (vide infra). For all of the competition experiments, azobenzene 1b provided the highest conversion. Electron-rich methoxy and methyl-substituted azobenzenes (indazoles 3l and 3n, respectively) gave only modestly lower conversion than the unsubstituted derivative. In contrast, the azobenzene bearing a trifluoromethyl group, gave dramatically lower conversion to indazole 3m. We hypothesize that two factors are primarily responsible for the dramatically lower conversion to 3m. First, the electron-deficient trifluoromethyl group may slow the rate of C–H bond activation on the basis of the effect of electronics on concerted metalation–deprotonation for related Cp*Rh(III) complexes.26 Second, the alcohol intermediate resulting from aldehyde addition cyclizes by intramolecular nucleophilic displacement of the hydroxyl group by either an SN1 or SN2 process. This cyclization would be expected to be slower when the electron-deficient trifluoromethyl group is present.
A series of aromatic aldehydes with different electronic properties were also subjected to competition experiments (Scheme 3b). Benzaldehyde (2a) showed higher conversion than both electron-rich (indazoles 3x and 3v) and electron-deficient (indazole 3p) aldehydes. These results suggest that competing effects may lead to a substrate dependent change in the rate-limiting step. The electron-rich aldehyde 4-methoxybenzaldehyde has increased resonance stabilization, and therefore a lower equilibrium for the alcohol product resulting from C–H addition to the aldehyde. However, donating substituents would also be expected to facilitate cyclization by nucleophilic displacement of the hydroxyl group. The electron-deficient 4-trifluoromethylbenzaldehyde should result in a more favorable equilibrium for the alcohol product but might also slow cyclization (vide supra). Regardless of electronic effects, it is important to note that good yields of indazoles 3 are obtained in nearly all cases (see Tables 3 and 4).
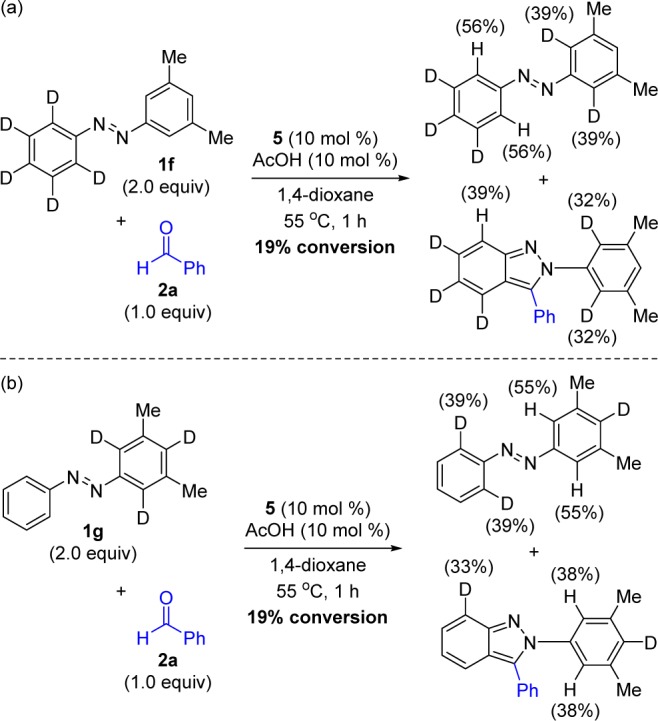
The reversibility of the cyclometalation step was next evaluated by subjecting site-specifically-deuterated azobenzene 1f to the reaction conditions (Scheme 4a). After stopping the reaction at low conversion, we observed that a significant level of protio–deutero scrambling had occurred in both the recovered starting material and product.27 A similar result was obtained for azobenzene 1g that was deuterated solely on the aromatic ring with 3,5-dimethyl substitution (Scheme 4b). These results clearly indicate that although the steric hindrance introduced by 3,5-disubstitution completely prevents indazole formation on this disubstituted ring (see Table 3), cyclometalation readily occurs as evidenced by the extensive protio–deutero scrambling.
Scheme 4. Reversibility of Co(III)-Catalyzed C–H Functionalization Using Deuterated Azobenzenes.
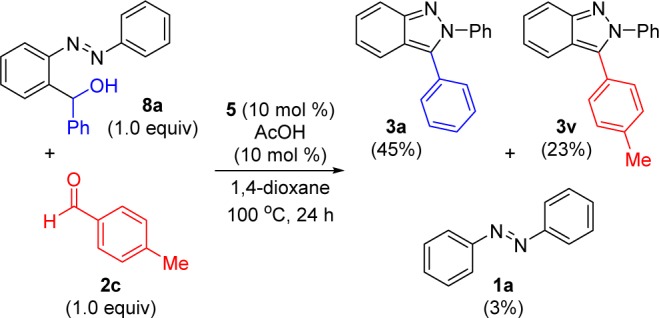
To assess the reversibility of Co(III)-catalyzed aldehyde insertion, the aldehyde addition product 8a was prepared independently and then subjected to the reaction conditions in the presence of 2c (Scheme 5). A mixture of indazoles 3a and 3v was obtained. Even though we had previously shown that 4-methylbenzaldehyde is converted to the product more slowly than benzaldehyde (see Scheme 3), a significant percentage of indazole 3v is still produced. Furthermore, azobenzene (1a) was observed in the final reaction mixture, albeit in only 3% yield. These results conclusively indicate that the Co(III)-catalyzed C–H bond addition to aldehydes is reversible, which is consistent with other metal-catalyzed C–H bond additions to aldehydes.28
Scheme 5. Reversibility of Co(III)-Catalyzed Aldehyde Insertion Using a Synthetic Intermediate.
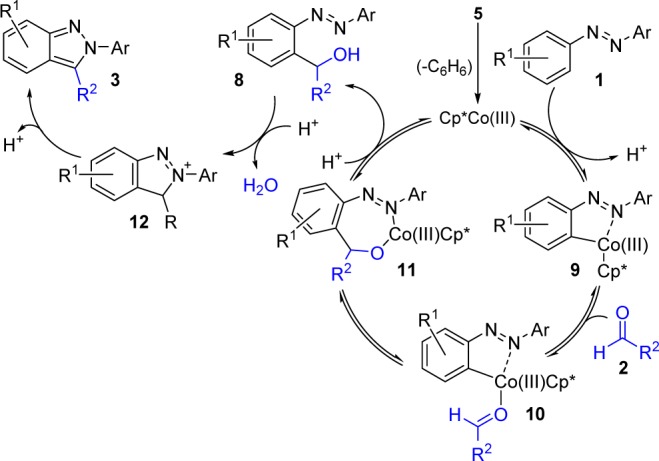
A potential mechanistic pathway for this C–H functionalization/addition/cyclization cascade is depicted in Scheme 6.29 Thermal loss of the benzene from 5 provides the active catalyst. Coordination of azobenzene 1 is followed by reversible formation of cobaltacycle 9.30 Reversible aldehyde coordination and migratory insertion affords cobaltacycle 11. Protonation of this metallacycle releases the alcohol intermediate 8 and regenerates the active catalyst. Cyclative capture of 8 then proceeds via an intramolecular nucleophilic substitution reaction with concomitant release of H2O to furnish 12. This species then rearomatizes to afford the desired indazole 3.
Scheme 6. Proposed Mechanism of Co(III)-Catalyzed C–H Functionalization/Addition/Cyclization Cascade.
Conclusion
In summary, we have developed the first examples of cobalt-catalyzed C–H bond functionalization with additions to aldehydes. Employing azo and oxime directing groups, which have not previously been applied to Cp*Co(III)-catalyzed C–H bond functionalization, led to cyclative capture and aromatization to enable the single-step convergent assembly of N-aryl-2H-indazoles and furans, respectively. A wide range of aryl, hetero, and alkyl aldehydes participate in these reactions to provide efficient access to highly substituted heterocycles. These transformations can be performed on the benchtop, under standard reflux conditions, and on a large scale.
Successful implementation of these practical conditions necessitated that we develop a new air-stable cationic catalyst, [Cp*Co(C6H6)][B(C6F5)4]2 (5). Additionally, although AgOAc was initially identified as a key additive for obtaining acceptable reaction yields, after extensive evaluation we established that this precious metal additive could be replaced with substoichiometric quantities of AcOH as a very low-cost and easy to handle alternative. The air-stable cobalt catalyst and convenient benchtop reaction conditions should hopefully be applicable to other C–H bond functionalization reactions.
Acknowledgments
Support has been provided by the National Institutes of Health (R01-GM069559).
Supporting Information Available
General methods, preparation of starting material and catalysts, catalytic indazole and furan syntheses, gram scale syntheses, mechanistic studies, and spectral characterization data. This content is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For select recent reviews on C–H functionalization, see:; a Wencel-Delord J.; Glorius F. Nat. Chem. 2013, 5, 369. [DOI] [PubMed] [Google Scholar]; b Arockiam P. B.; Bruneau C.; Dixneuf P. H. Chem. Rev. 2012, 112, 5879. [DOI] [PubMed] [Google Scholar]; c Li B.-J.; Shi Z.-J. Chem. Soc. Rev. 2012, 41, 5588. [DOI] [PubMed] [Google Scholar]; d Yamaguchi J.; Yamaguchi A. D.; Itami K. Angew. Chem., Int. Ed. 2012, 51, 8960. [DOI] [PubMed] [Google Scholar]; e Engle K. M.; Mei T.-S.; Wasa M.; Yu J.-Q. Acc. Chem. Res. 2012, 45, 788. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Ackermann L. Chem. Rev. 2011, 111, 1315. [DOI] [PubMed] [Google Scholar]; g Cho S. H.; Kim J. Y.; Kwak J.; Chang S. Chem. Soc. Rev. 2011, 40, 5068. [DOI] [PubMed] [Google Scholar]; h Lyons T. W.; Sanford M. S. Chem. Rev. 2010, 110, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. Chem. Rev. 2010, 110, 890. [DOI] [PubMed] [Google Scholar]; j Colby D. A.; Bergman R. G.; Ellman J. A. Chem. Rev. 2010, 110, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Seregin I. V.; Gevorgyan V. Chem. Soc. Rev. 2007, 36, 1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent reviews on Rh(III)-catalyzed C–H functionalization, see:; a Song G.; Wang F.; Li X. Chem. Soc. Rev. 2012, 41, 3651. [DOI] [PubMed] [Google Scholar]; b Patureau F. W.; Wencel-Delord J.; Glorius F. Aldrichimica Acta 2012, 45, 31. [Google Scholar]; c Satoh T.; Miura M. Chem.—Eur. J. 2010, 16, 11212. [DOI] [PubMed] [Google Scholar]
- For reviews on C–H bond additions to polarized π bonds, see:; a Zhang X.-S.; Chen K.; Shi Z.-J. Chem. Sci. 2014, 5, 2146. [Google Scholar]; b Yan G.; Wu X.; Yang M. Org. Biomol. Chem. 2013, 11, 5558. [DOI] [PubMed] [Google Scholar]
- For examples of Rh(III)-catalyzed C–H bond additions to destabilized, highly electron-deficient aldehydes to provide alcohols, see:; a Li Y.; Zhang X.-S.; Chen K.; He K.-H.; Pan F.; Li B.-J.; Shi Z.-J. Org. Lett. 2012, 14, 636. [DOI] [PubMed] [Google Scholar]; b Li Y.; Zhang X.-S.; Zhu Q.-L.; Shi Z.-J. Org. Lett. 2012, 14, 4498. [DOI] [PubMed] [Google Scholar]; c Yang L.; Correia C. A.; Li C.-J. Adv. Synth. Catal. 2011, 353, 1269. [Google Scholar]
- For examples of Ir-, Re-, and Mn-catalyzed C–H bond addition to aldehydes with capture of alcohol products as silyl ethers, see:; a Li B.-J.; Shi Z.-J. Chem. Sci. 2011, 2, 488. [Google Scholar]; b Kuninobu Y.; Asanoma D.; Takai K. Synlett 2010, 2883. [Google Scholar]; c Kuninobu Y.; Fujii Y.; Matsuki T.; Nishina Y.; Takai K. Org. Lett. 2009, 11, 2711. [DOI] [PubMed] [Google Scholar]; d Kuninobu Y.; Nishina Y.; Takeuchi T.; Takai K. Angew. Chem., Int. Ed. 2007, 46, 6518. [DOI] [PubMed] [Google Scholar]; e Fukumoto Y.; Sawada K.; Hagihara M.; Chatani N.; Murai S. Angew. Chem., Int. Ed. 2002, 41, 2779. [DOI] [PubMed] [Google Scholar]
- For examples of Rh(III)-catalyzed C–H bond addition to aldehydes with alcohol capture via in situ oxidation, see:; a Sharma S.; Park E.; Park J.; Kim I. S. Org. Lett. 2012, 14, 906. [DOI] [PubMed] [Google Scholar]; b Zhou B.; Yang Y.; Li Y. Chem. Commun. 2012, 48, 5163. [DOI] [PubMed] [Google Scholar]; c Park J.; Park E.; Kim A.; Lee Y.; Chi K.-W.; Kwak J. H.; Jung Y. H.; Kim I. S. Org. Lett. 2011, 13, 4390. [DOI] [PubMed] [Google Scholar]; For a Pd-catalyzed example, see: ; d Xiao F.; Shuai Q.; Zhao F.; Basle O.; Deng G.; Li C.-J. Org. Lett. 2011, 13, 1614. [DOI] [PubMed] [Google Scholar]
- For examples of Rh(III)-catalyzed C–H bond addition to aldehydes followed by cyclative capture of alcohol intermediates, see the following.; a For indazole synthesis, see: Lian Y.; Bergman R. G.; Lavis L. D.; Ellman J. A. J. Am. Chem. Soc. 2013, 135, 7122. [DOI] [PMC free article] [PubMed] [Google Scholar]; b For furan synthesis, see: Lian Y.; Huber T.; Hesp K. D.; Bergman R. G.; Ellman J. A. Angew. Chem., Int. Ed. 2013, 52, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]; c For phthalide synthesis, see: Lian Y.; Bergman R. G.; Ellman J. A. Chem. Sci. 2012, 3, 3088. [DOI] [PMC free article] [PubMed] [Google Scholar]; d For a subsequent phthalide synthesis, see:Shi X.; Li C.-J. Adv. Synth. Catal. 2012, 354, 2933. [Google Scholar]; e For an early example of cyclative capture in the Re-catalyzed synthesis of isobenzofurans, see: Kuninobu Y.; Nishina Y.; Nakagawa C.; Takai K. J. Am. Chem. Soc. 2006, 128, 12376. [DOI] [PubMed] [Google Scholar]
- For a review of Mn-catalyzed C–H functionalization, see:; a Wang C. Synlett 2013, 24, 1606. [Google Scholar]; b Also see refs (5b) and (5d).
- For recent examples of C–H functionalization catalyzed by low-valent iron complexes, see:; a Shang R.; Ilies L.; Asako S.; Nakamura E. J. Am. Chem. Soc. 2014, 136, 14349. [DOI] [PubMed] [Google Scholar]; b Fruchey E. R.; Monks B. M.; Cook S. P. J. Am. Chem. Soc. 2014, 136, 13130. [DOI] [PubMed] [Google Scholar]; c Ilies L.; Matsubara T.; Ichikawa S.; Asako S.; Nakamura E. J. Am. Chem. Soc. 2014, 136, 13126. [DOI] [PubMed] [Google Scholar]; d Monks B. M.; Fruchey E. R.; Cook S. P. Angew. Chem., Int. Ed. 2014, 53, 11065. [DOI] [PubMed] [Google Scholar]; e Matsubara T.; Asako S.; Ilies L.; Nakamura E. J. Am. Chem. Soc. 2014, 136, 646. [DOI] [PubMed] [Google Scholar]; f Asako S.; Ilies L.; Nakamura E. J. Am. Chem. Soc. 2013, 135, 17755. [DOI] [PubMed] [Google Scholar]; g Shang R.; Ilies L.; Matsumoto A.; Nakamura E. J. Am. Chem. Soc. 2013, 135, 6030. [DOI] [PubMed] [Google Scholar]; For a review, see:Nakamura E.; Yoshikai N. J. Org. Chem. 2010, 75, 6061. [DOI] [PubMed] [Google Scholar]
- For recent reviews of Co-catalyzed C–H functionalization, see:; a Gao K.; Yoshikai N. Acc. Chem. Res. 2014, 47, 1208. [DOI] [PubMed] [Google Scholar]; b Tilly D.; Dayaker G.; Bachu P. Catal. Sci. Technol. 2014, 4, 2756. [Google Scholar]
- For Co(III)-catalyzed C–H functionalization, see:; a Grigorjeva L.; Daugulis O. Angew. Chem., Int. Ed. 2014, 53, 10209. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Grigorjeva L.; Daugulis O. Org. Lett. 2014, 16, 4684. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Grigorjeva L.; Daugulis O. Org. Lett. 2014, 16, 4688. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Sun B.; Yoshino T.; Matsunaga S.; Kanai M. Adv. Synth. Catal. 2014, 356, 1491. [Google Scholar]; e Ikemoto H.; Yoshino T.; Sakata K.; Matsunaga S.; Kanai M. J. Am. Chem. Soc. 2014, 136, 5424. [DOI] [PubMed] [Google Scholar]; f Yoshino T.; Ikemoto H.; Matsunaga S.; Kanai M. Chem.—Eur. J. 2013, 19, 9142. [DOI] [PubMed] [Google Scholar]; g Yoshino T.; Ikemoto H.; Matsunaga S.; Kanai M. Angew. Chem., Int. Ed. 2013, 52, 2207. [DOI] [PubMed] [Google Scholar]
- For leading references on Co(0) and Co(I)-catalyzed C–H functionalization, see:; a Xu W.; Yoshikai N. Angew. Chem., Int. Ed. 2014, 10.1002/anie.201408028. [DOI] [Google Scholar]; b Chen Q.-A.; Kim D. K.; Dong V. Y. J. Am. Chem. Soc. 2014, 136, 3772. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhao C.; Crimmin M. R.; Toste F. D.; Bergman R. G. Acc. Chem. Res. 2014, 47, 517. [DOI] [PubMed] [Google Scholar]; d Yamamoto S.; Saga Y.; Andou T.; Matsunaga S.; Kanai M. Adv. Synth. Catal. 2014, 356, 401. [Google Scholar]; e Andou T.; Saga Y.; Komai H.; Matsunaga S.; Kanai M. Angew. Chem., Int. Ed. 2013, 52, 3213. [DOI] [PubMed] [Google Scholar]; f Punji B.; Song W.; Shevchenko G. A.; Ackermann L. Chem.—Eur. J. 2013, 19, 10605. [DOI] [PubMed] [Google Scholar]; g Song W.; Ackermann L. Angew. Chem., Int. Ed. 2012, 51, 8251. [DOI] [PubMed] [Google Scholar]; h Ding Z.; Yoshikai N. Angew. Chem., Int. Ed. 2012, 51, 4698. [DOI] [PubMed] [Google Scholar]; i Yao T.; Hirano K.; Satoh T.; Miura M. Angew. Chem., Int. Ed. 2012, 51, 775. [DOI] [PubMed] [Google Scholar]; j Gao K.; Yoshikai N. Chem. Commun. 2012, 48, 4305. [DOI] [PubMed] [Google Scholar]; k Lee P.-S.; Fujita T.; Yoshikai N. J. Am. Chem. Soc. 2011, 133, 17283. [DOI] [PubMed] [Google Scholar]; l Ilies L.; Chen Q.; Zeng X.; Nakamura E. J. Am. Chem. Soc. 2011, 133, 5221. [DOI] [PubMed] [Google Scholar]; m Chen Q.; Ilies L.; Nakamura E. J. Am. Chem. Soc. 2011, 133, 428. [DOI] [PubMed] [Google Scholar]; n Gao K.; Yoshikai N. J. Am. Chem. Soc. 2011, 133, 400. [DOI] [PubMed] [Google Scholar]; o Gao K.; Yoshikai N. Angew. Chem., Int. Ed. 2011, 50, 6888. [DOI] [PubMed] [Google Scholar]; p Li B.; Wu Z.-H.; Gu Y.-F.; Sun C.-L.; Wang B.-Q.; Shi Z.-J. Angew. Chem., Int. Ed. 2011, 50, 1109. [DOI] [PubMed] [Google Scholar]; q Chen Q.; Ilies L.; Yoshikai N.; Nakamura E. Org. Lett. 2011, 13, 3232. [DOI] [PubMed] [Google Scholar]; r Gao K.; Lee P.-S.; Fujita T.; Yoshikai N. J. Am. Chem. Soc. 2010, 132, 12249. [DOI] [PubMed] [Google Scholar]; s Bolig A. D.; Brookhart M. J. Am. Chem. Soc. 2007, 129, 14544. [DOI] [PMC free article] [PubMed] [Google Scholar]; t Lenges C. P.; White P. S.; Brookhart M. J. Am. Chem. Soc. 1998, 120, 6965. [Google Scholar]; u Lenges C. P.; Brookhart M. J. Am. Chem. Soc. 1997, 119, 3165. [Google Scholar]
- For examples of C–H bond functionalization with Ni complexes, see:; a Aihara Y.; Tobisu M.; Fukumoto Y.; Chatani N. J. Am. Chem. Soc. 2014, 136, 15509. [DOI] [PubMed] [Google Scholar]; b Aihara Y.; Chatani N. J. Am. Chem. Soc. 2014, 136, 898. [DOI] [PubMed] [Google Scholar]; c Muto K.; Yamaguchi J.; Lei A.; Itami K. J. Am. Chem. Soc. 2013, 135, 16384. [DOI] [PubMed] [Google Scholar]; d Amaike K.; Muto K.; Yamaguchi J.; Itami K. J. Am. Chem. Soc. 2012, 134, 13573. [DOI] [PubMed] [Google Scholar]; e Muto K.; Yamaguchi J.; Itami K. J. Am. Chem. Soc. 2012, 134, 169. [DOI] [PubMed] [Google Scholar]; f Shiota H.; Ano Y.; Aihara Y.; Fukumoto Y.; Chatani N. J. Am. Chem. Soc. 2011, 133, 14952. [DOI] [PubMed] [Google Scholar]; g Meng L.; Kamada Y.; Muto K.; Yamaguchi J.; Itami K. Angew. Chem., Int. Ed. 2013, 52, 10048. [DOI] [PubMed] [Google Scholar]; h Canivet J.; Yamaguchi J.; Ban I.; Itami K. Org. Lett. 2009, 11, 1733. [DOI] [PubMed] [Google Scholar]; i Mukai T.; Hirano K.; Satoh T.; Miura M. J. Org. Chem. 2009, 74, 6410. [DOI] [PubMed] [Google Scholar]; j Nakao Y.; Kashihara N.; Kanyiva K. S.; Hiyama T. J. Am. Chem. Soc. 2008, 130, 16170. [DOI] [PubMed] [Google Scholar]; k Nakao Y.; Kanyiva K. S.; Hiyama T. J. Am. Chem. Soc. 2008, 130, 2448. [DOI] [PubMed] [Google Scholar]; l Kanyiva K. S.; Nakao Y.; Hiyama T. Angew. Chem., Int. Ed. 2007, 46, 8872. [DOI] [PubMed] [Google Scholar]; m Liang L.-C.; Chien P.-S.; Huang Y.-L. J. Am. Chem. Soc. 2006, 128, 15562. [DOI] [PubMed] [Google Scholar]; n Nakao Y.; Kanyiva K. S.; Oda S.; Hiyama T. J. Am. Chem. Soc. 2006, 128, 8146. [DOI] [PubMed] [Google Scholar]; o Chement N. D.; Cavell K. J. Angew. Chem., Int. Ed. 2004, 43, 3845. [DOI] [PubMed] [Google Scholar]
- For leading references on Cu-catalyzed C–H functionalization, see:; a Wang Z.; Kuninobu Y.; Kanai M. Org. Lett. 2014, 16, 4790. [DOI] [PubMed] [Google Scholar]; b Truong T.; Klimovica K.; Daugulis O. J. Am. Chem. Soc. 2013, 135, 9342. [DOI] [PMC free article] [PubMed] [Google Scholar]; c For a related review, see: Zhang M. Appl. Organomet. Chem. 2010, 24, 269. [Google Scholar]; d Do H.-Q.; Kashif Khan R. M.; Daugulis O. J. Am. Chem. Soc. 2008, 130, 15185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on several first row transition metals, see:Kulkarni A. A.; Daugulis O. Synthesis 2009, 4087. [Google Scholar]
- a Nasonex (Mometasone furoate), Zantac (ranitidine HCl), lapatinib, and furadantin monohydrate are approved drugs containing a furan moiety. Detailed information on these drug candidates including compound structure, bioactivity, published studies, and information regarding ongoing clinical trials, applications, and usage can be obtained at PubChem ; (http://pubchem.ncbi.nlm.nih.gov/).; Also see:; b Katritzky A. R.Comprehensive Heterocyclic Chemistry III, 1st ed.; Elsevier: Amsterdam, 2008. [Google Scholar]
- a Niraparib and pazopanib are 2-substituted 2H-indazoles currently in clinical trials. Detailed information on these drug candidates including compound structure, bioactivity, published studies, and information regarding ongoing clinical trials, applications, and usage can be obtained at PubChem; (http://pubchem.ncbi.nlm.nih.gov/).; b For leading references on pharmaceutical applications and syntheses of 2-substituted 2H-indazoles, see: Genung N. E.; Wei L.; Aspnes G. E. Org. Lett. 2014, 16, 3114. [DOI] [PubMed] [Google Scholar]
- For azobenzene C–H functionalization with Co(0) and Co(I) complexes, see:; a Halbritter G.; Knoch F.; Wolski A.; Kisch H. Angew. Chem., Int. Ed. Engl. 1994, 33, 1603. [Google Scholar]; b Klein H.-F.; Helwig M.; Koch U.; Flörke U.; Haupt H.-J. Z. Naturforsch., B: J. Chem. Sci. 1993, 48, 778. [Google Scholar]; c Heck R. F. J. Am. Chem. Soc. 1968, 90, 313. [Google Scholar]; d Murahashi S.; Horiie S. J. Am. Chem. Soc. 1956, 78, 4816. [Google Scholar]
- For selected recent examples of azobenzene C–H functionalization with Cp*Rh(III), see:; a Wangweerawong A.; Bergman R. G.; Ellman J. A. J. Am. Chem. Soc. 2014, 136, 8520. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ryu T.; Min J.; Choi W.; Jeon W. H.; Lee P. H. Org. Lett. 2014, 16, 2810. [DOI] [PubMed] [Google Scholar]; c Wang H.; Yu Y.; Hong X.; Tan Q.; Xu B. J. Org. Chem. 2014, 79, 3279. [DOI] [PubMed] [Google Scholar]; d Lian Y.; Hummel J. R.; Bergman R. G.; Ellman J. A. J. Am. Chem. Soc. 2013, 135, 12548. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Zhao D.; Wu Q.; Huang X.; Song F.; Lv T.; You J. Chem.—Eur. J. 2013, 19, 6239. [DOI] [PubMed] [Google Scholar]; f Maralirajan K.; Cheng C.-H. Chem.—Eur. J. 2013, 19, 6198. [DOI] [PubMed] [Google Scholar]; g Also see ref (7a).
- For Co(0)-catalyzed alkenyl C–H functionalization, see:Yamakawa T.; Yoshikai N. Org. Lett. 2013, 15, 196. [DOI] [PubMed] [Google Scholar]
- Cationic Cp*Co with the noncoordinating B(C6H5)4 counterion in combination with AgOAc is presumably successful because Ag+ and Cp*Co2+ bind with comparable strength to acetate such that only partial exchange occurs.
- For examples where Cu(OAc)2 was beneficial for redox neutral Rh(III)-catalyzed C–H functionalization reactions, see:; a Liu B.; Zhou T.; Li B.; Xu S.; Song H.; Wang B. Angew. Chem., Int. Ed. 2014, 53, 4191. [DOI] [PubMed] [Google Scholar]; b Li B.-J.; Wang H.-Y.; Zhu Q.-L.; Shi Z.-J. Angew. Chem., Int. Ed. 2012, 51, 3948. [DOI] [PubMed] [Google Scholar]; c Tsai A. S.; Brasse M.; Bergman R. G.; Ellman J. A. Org. Lett. 2011, 13, 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples employing pivalic acid as an additive in Pd catalysis, see:; a Lafrance M.; Fagnou K. J. Am. Chem. Soc. 2006, 128, 16496. [DOI] [PubMed] [Google Scholar]; In Cp*Rh(III)-catalysis, see: ; b Zhang X.; Li Y.; Shi H.; Zhang L.; Zhang S.; Xu X.; Liu Q. Chem. Commun. 2014, 50, 7306. [DOI] [PubMed] [Google Scholar]; c Schipper D. J.; Hutchinson M.; Fagnou K. J. Am. Chem. Soc. 2010, 132, 6910. [DOI] [PubMed] [Google Scholar]
- Acetophenone O-methyl oxime was evaluated under the optimized conditions but gave a mixture of products. For the Re-catalyzed synthesis of isobenzofurans, see ref (7e).
- Indazole 3a was also prepared on 20 mmol scale, providing a yield of 75% (4.04 g). See the Supporting Information for details.
- For a study demonstrating the effect of electronics on concerted metalation–deprotonation for related Cp*Rh(III) complexes, see:Li L.; Brennessel W. W.; Jones W. D. Organometallics 2009, 28, 3492. [Google Scholar]
- To demonstrate that Co(III) is required for protio–deutero scrambling to occur, a control reaction was run in which azobenzene 1f was subjected to the reaction conditions in the absence of catalyst 5. Under these conditions, no protio–deutero exchange occurred. See the Supporting Information for details.
- The reversibility of Rh(III)-catalyzed C–H bond addition to aldehydes has been previously demonstrated. See:; a Chen K.; Li H.; Lei Z.-Q.; Li Y.; Ye W.-H.; Zhang L.-S.; Sun J.; Shi Z.-J. Angew. Chem., Int. Ed. 2012, 51, 9851. [DOI] [PubMed] [Google Scholar]; b Zhang X.-S.; Li Y.; Li H.; Chen K.; Lei Z.-Q.; Shi Z.-J. Chem.—Eur. J. 2012, 18, 16214. [DOI] [PubMed] [Google Scholar]; c Li H.; Li Y.; Zhang X.-S.; Chen K.; Wang X.; Shi Z.-J. J. Am. Chem. Soc. 2011, 133, 15244. [DOI] [PubMed] [Google Scholar]
- For relevant mechanistic studies demonstrated for Rh(III) catalysis, see:; a Reference (26); b Tauchert M. E.; Incarvito C. D.; Rheingold A. L.; Bergman R. G.; Ellman J. A. J. Am. Chem. Soc. 2012, 134, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Li Y.; Zhang X.-S.; Li H.; Wang W.-H.; Chen K.; Li B.-J.; Shi Z.-J. Chem. Sci. 2012, 3, 1634. [Google Scholar]; d Li L.; Brennessel W. W.; Jones W. D. J. Am. Chem. Soc. 2008, 130, 12414. [DOI] [PubMed] [Google Scholar]
- For an independent synthesis of a cobaltacycle, see ref (11e).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.