

Abstract
Pseudomonas aeruginosa is an opportunistic Gram-negative bacterial pathogen. A primary contributor to its ability to resist β-lactam antibiotics is the expression, following detection of the β-lactam, of the AmpC β-lactamase. As AmpC expression is directly linked to the recycling of the peptidoglycan of the bacterial cell wall, an important question is the identity of the signaling molecule(s) in this relationship. One mechanism used by clinical strains to elevate AmpC expression is loss of function of penicillin-binding protein 4 (PBP4). As the mechanism of the β-lactams is PBP inactivation, this result implies that the loss of the catalytic function of PBP4 ultimately leads to induction of antibiotic resistance. PBP4 is a bifunctional enzyme having both dd-carboxypeptidase and endopeptidase activities. Substrates for both the dd-carboxypeptidase and the 4,3-endopeptidase activities were prepared by multistep synthesis, and their turnover competence with respect to PBP4 was evaluated. The endopeptidase activity is specific to hydrolysis of 4,3-cross-linked peptidoglycan. PBP4 catalyzes both reactions equally well. When P. aeruginosa is grown in the presence of a strong inducer of AmpC, the quantities of both the stem pentapeptide (the substrate for the dd-carboxypeptidase activity) and the 4,3-cross-linked peptidoglycan (the substrate for the 4,3-endopeptidase activity) increase. In the presence of β-lactam antibiotics these altered cell-wall segments enter into the muropeptide recycling pathway, the conduit connecting the sensing event in the periplasm and the unleashing of resistance mechanisms in the cytoplasm.
Introduction
The Gram-negative members of the Enterobacteriaceae and Pseudomonas aeruginosa bacteria are among the most serious human pathogens and have evolved an inducible resistance mechanism against β-lactam antibiotics.1−3 This induction leads inter alia to the expression of class C β-lactamases, which deactivate the β-lactam antibiotic by hydrolysis of its β-lactam bond. This same bond of these antibiotics is used in their mechanism of action by penicillin-binding protein (PBP) inactivation.4−6 For an inducible system to work, the presence of the antibiotic in the milieu has to be detected by the bacterium. How precisely this process takes place in these Gram-negative rods is not presently known and remains as an important gap in knowledge.
If not intercepted by β-lactamases, β-lactam antibiotics inhibit the PBP enzymes, resulting in cell lysis and death. A typical β-lactam antibiotic often inhibits more than one PBP, of which each bacterium has several. In a series of seminal epidemiological observations using clinical isolates of P. aeruginosa, it was shown that the mutational inactivation of PBP4 leads to high-level expression of the AmpC β-lactamase.7,8 This observation implies that the loss of activity of PBP4 as a result of inhibition by a β-lactam antibiotic would give the same outcome. Hence, knowledge of the identity of the specific substrates and products of PBP4 catalysis is critical to shed first light on this process.
Herein we provide this knowledge. The reactions catalyzed by PBP4 of P. aeruginosa have not been studied previously, but the homologous enzyme in Escherichia coli is a bifunctional dd-carboxypeptidase and endopeptidase.9−11 However, the E. coli PBP4 is not involved in antibiotic resistance. Rather, its role appears to be in alterations of the cell wall for unrelated physiological roles.10,12 We also investigated the consequence of the alteration of cell wall when P. aeruginosa was grown in the presence of a sublethal concentration of cefoxitin, a high inducer of AmpC β-lactamase production. Cefoxitin inhibits PBPs, including PBP4. These results bring into sharper focus, at the molecular level, the signaling events used by P. aeruginosa to detect and to respond to a β-lactam antibiotic challenge.
Results
Expression of P. aeruginosa PBP4
The PA3047 gene corresponding to amino acids 23 to 476 for PBP4 from P. aeruginosa PAO1, with a His-tag in the N-terminal position, was synthesized for optimal expression in E. coli. This synthetic gene was cloned into plasmid pET-28a(+) to give the recombinant plasmid pET28-PBP4. This construct lacks the signal peptide for delivery to the periplasm but possesses the membrane-anchor segment. E. coli Lemo21(DE3) was transformed with this plasmid. Subsequently recombinant PBP4 was purified to homogeneity using the HiTrap Chelating column (Supporting Information).
Catalytic Competence of P. aeruginosa PBP4
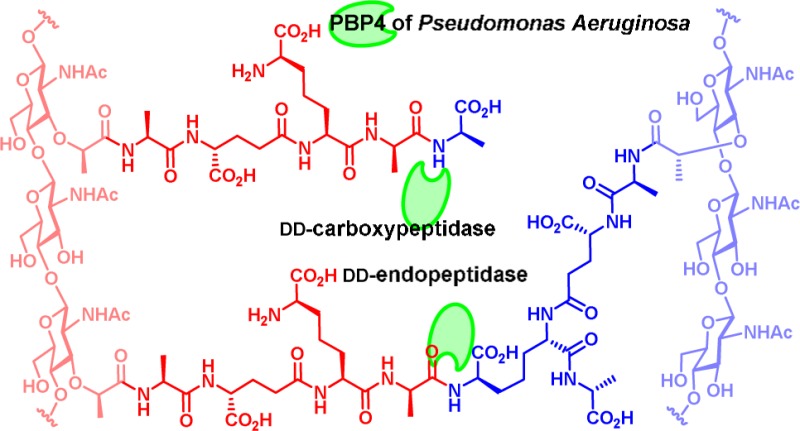
We tested the competence of PBP4 in turnover of the cell wall, using the peptidoglycan sacculus isolated from P. aeruginosa PAO1 as the substrate.13 The sacculus is the polymeric cell wall of an individual bacterium, stripped of all of the proteins and membranes associated with the intact bacterium. At the molecular level, the sacculus is a peptidoglycan polymer, which consists of glycan strands having a repeating disaccharide N-acetylglucosamine (NAG)-N-acetylmuramic acid (NAM) motif. A peptide stem of up to five amino acids is attached to the NAM saccharide. The structure of the pentapeptide stem of P. aeruginosa is l-Ala1-d-γ-Glu2-meso-DAP3-d-Ala4-d-Ala5, where DAP is the amino acid meso-diaminopimelic acid (Figure 1).
Figure 1.

Chemical structure of cross-linked bacterial peptidoglycan. The donor strand and the acceptor strand of cross-links are in red and in blue, respectively. The sites of potential hydrolytic reactions by PBP4 are shown by green arrows.
Cross-linking of neighboring glycan strands is accomplished using the pentapeptide stem by the transpeptidase domain found in certain PBPs. Cross-linking may occur between d-Ala4 of a donor strand (in red in Figure 1) and m-DAP3 of an acceptor strand (in blue in Figure 1) to give a 4,3-cross-link, the common cross-link of many Gram-negative bacteria. A 3,3-cross-link, formed between two m-DAP3 of two neighboring strands, is a minor component. The pentapeptide stems are often trimmed to give tetrapeptide, tripeptide, or dipeptide (referred to in this report as Tetra, Tri, Di: Figure 1) stem structures. Absence of the peptide stem is also possible. In addition, minor stem structures having glycine and lysine extensions to the core tripeptide or tetrapeptide stems (defined as TriGly, TriLys, and TetraGly structures) are also found. This same nomenclature has been used both by us14 and by others.15,16 The amide bonds of these stems having the potential for recognition for hydrolytic cleavage catalyzed by PBP4 of P. aeruginosa are indicated with green arrows in Figure 1, based on the proposed catalytic ability of the homologous PBP4 of E. coli.
Products from PBP4 Hydrolysis of the Sacculus
Incubation of PBP4 with the sacculus generated water-soluble oligomeric muropeptides, having the general formula [NAG-NAM(peptide)]n-[NAG-anhNAM(peptide)] as deduced from analysis of the LC/MS extracted-ion chromatogram (EIC) (Figure 2). This chromatogram shows products with a tetrapeptide stem with different glycan lengths (in blue in Figure 2) and at lower abundance a mix of glycans having tripeptide and tetrapeptide (in red in Figure 2) stems. The stem structures found (Table 1) were mostly tetrapeptide (89%), tripeptide (10%), and TriLys (1.2%). No product with a pentapeptide stem was found. Furthermore, no cross-linked peptide-containing products were found in the soluble fraction. These findings can be interpreted as the result of both endopeptidase and dd-carboxypeptidase activities for PBP4.
Figure 2.
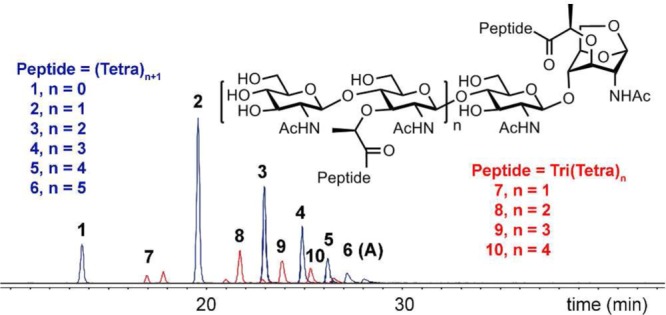
LC/MS EICs of tetrapeptide- (in blue) and mixed tripeptide- and tetrapeptide-containing muropeptides (in red) after the reaction of PBP4 with the sacculus.
Table 1. Product Analysis of Reactions of the Sacculus with the Given Enzymesa,b.
| sacculus/PBP4 | sacculus/PBP4-sol/MltAc | sacculus/PBP4-Insol/MltAc | sacculus/PBP4/MltAd | sacculus/MltA/PBP4e | sacculus/MltA | induced sacculus/MltAf | |
|---|---|---|---|---|---|---|---|
| cross-link/non-cross-linked | |||||||
| noncross-linked | 100 | 87 ± 8 | 68 ± 10 | 70 ± 10 | 97 ± 1 | 53 ± 2 | 50 ± 6 |
| 3,3 cross-link | NDg | 1.5 ± 0.2 | 5 ± 1 | 5 ± 0.3 | 3 ± 1 | 4 ± 1 | 1.4 ± 0.3 |
| 4,3-cross-link | NDg | 11 ± 7 | 26 ± 9 | 25 ± 10 | NDg | 43 ± 3 | 49 ± 5 |
| peptideh | |||||||
| penta | NDg | NDg | 0.06 ± 0.02 | 0.06 ± 0.02 | NDg | 0.05 ± 0.01 | 1.7 ± 0.4 |
| tetra | 89 ± 1 | 81 ± 1 | 64 ± 1 | 66 ± 1 | 79 ± 2 | 75 ± 4 | 82 ± 5 |
| tri | 10 ± 1 | 11 ± 1 | 22 ± 1 | 21 ± 0.2 | 13 ± 3 | 18 ± 3 | 10 ± 3 |
| othersi | 1.2 ± 0.3 | 8 ± 1 | 13 ± 1 | 13 ± 2 | 7 ± 1 | 8 ± 1 | 6 ± 2 |
Amounts are expressed as a percentage of the total EIC peak area.
Average values of two runs with errors.
Sacculus was digested with PBP4, and the soluble and insoluble portions were separated by centrifugation. Each portion was reacted separately with MltA.
Combined results of the soluble and insoluble portions.
Sacculus was reacted with MltA, followed by reaction with PBP4.
Sacculus was isolated from bacteria grown in the presence of sub-MIC concentrations of cefoxitin and reacted with MltA.
ND, not detected.
Cross-linked and non-cross-linked muropeptide containing the corresponding peptide.
Less abundant peptides, TetraGly, TriGly, TriLys, Di, and no peptide.
The products shown in the chromatogram of Figure 2 account only for the water-soluble products released from the polymeric sacculus. Accordingly, an equally important imprint of the reaction might remain within the insoluble fraction of the sacculus after the complete release by PBP4 of water-soluble products. Figure 3 depicts this dilemma and how we overcame it. A simplified structure of the sacculus is given in panel I of Figure 3. Figure 3 shows a schematic structure of three cross-linked glycan strands, both by 4,3- and 3,3-linkages, and the presence of an non-cross-linked pentapeptide (at 7 o’clock in panel I) stem. The cross-linked peptides and the pentapeptide stem are potential sites of reaction by PBP4. We show the three conceivable outcomes for product formation due to PBP4 in panels II, III, and IV. For hydrolysis of the 4,3-cross-linked substrate, PBP4 reaction would give products A and B with all 4,3-cross-links hydrolyzed but not the 3,3-cross-links (panel II, on product B). In the case of hydrolysis of the 3,3-cross-linked species exclusively, the turnover products would be C and D, where all the 4,3-cross-links remain intact (on product C) and the 3,3-cross-link hydrolyzed (panel III). If PBP4 hydrolyzes all cross-linked bonds, regardless of their nature, the reaction would give soluble products as exemplified by A, D, and E (as shown in panel IV). The soluble products shown in Figure 2 encompass some of these possibilities. For example, A corresponds to product 6 in Figure 2. Hence, we know that the endopeptidase activity exists, but the specific nature of the reaction is not revealed. This information exists in the insoluble fraction, which is represented schematically as products B and C in Figure 3. For analysis of the products of PBP4 reactions in the insoluble portion of the sacculus, we used a second sacculus-degrading enzyme, the lytic transglycosylase MltA of E. coli. MltA cleaves between NAM and NAG with formation of 1,6-anhydromuramic acid.14,17 Its reaction sites are indicated with purple arrows in Figure 3 (panel I). This enzyme produces products F, G, H, I, J, and K (in panel V). The reaction of MltA reduces the complexity of the polymer by fragmentation of the glycan strand and so to further reveal the segments comprising the entire sacculus. For example, incubation of B with MltA would give products H, J, and K (in panel VI). This outcome may be compared to products (F–K in panel V) of the control reaction (sacculus degradation by MltA). Similarly, MltA incubation of product C would give products F and G along with J and K. Hence, the collection of experiments shown in Table 1 in a quantitative manner elucidates the scope of the PBP4 reactions with the sacculus. The same experiments with a sacculus synthesized in the presence of a β-lactam antibiotic (here we use cefoxitin, an inhibitor of PBP4 and an excellent inducer of AmpC expression) will also reveal the structural changes that occur to the sacculus in response to the presence of a PBP4-inhibiting β-lactam antibiotic.
Figure 3.

Strategy for analysis of the reactions of PBP4 on the insoluble fraction of the sacculus. The panels are identified by the Roman numeral (I–VIII) in the corners. The schematic abbreviation of the structures of the constituents is given in the panel above.
The soluble and insoluble fractions of the sacculus were separated by centrifugation after PBP4 reaction. The two portions were subjected separately to reaction with MltA. The results are summarized in Table 1. We appreciate that Figure 3 is an idealized representation of the cell-wall structure and that in reality the sacculus is a more complex three-dimensional structure. Accordingly, the reactions of PBP4 (and of MltA) with the sacculus are anticipated to give complex mixtures that reflect additional variations with respect to the stem structure and the degree of cross-linking.
We detect PBP4 reaction products both in the soluble and insoluble portions after treatment with MltA. After the reaction of PBP4 with the sacculus, we observed an increase in the degree of non-cross-linked products (87% for soluble and 68% for insoluble fractions), compared to the control (MltA reaction) value of 53%. Furthermore, pentapeptide-containing muropeptides were no longer detected in the soluble fraction, but they were detected still in the insoluble fraction in levels similar to that of the control (MltA reaction). This result might reflect lack of access to the sites in the insoluble fractions. We can say that the scenario of panel IV is not operative, as we recover both soluble and insoluble fractions. Nonetheless, these observations establish that PBP4 has both endopeptidase and dd-carboxypeptidase activities. Is there selectivity (or preference) for 4,3- versus 3,3-cross-links as substrates for PBP4? The data of Table 1 unequivocally show that the 4,3-cross-link is turned over but does not rule out the 3,3-cross-links as substrates. An additional experiment settled this matter. We processed the sacculus first by MltA (panel V) and then by PBP4. This sequence of reactions would produce products as given in panels VI, VII, and VIII (Figure 3). The reactions of MltA with the sacculus generated compounds 11–17 as major products (Figure 4A and 4B).
Figure 4.
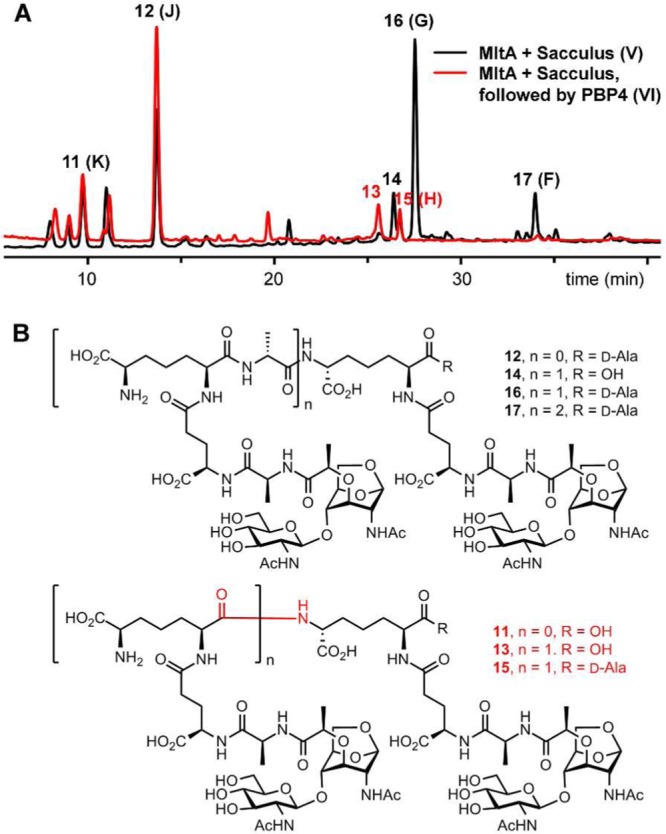
(A) Overlay of LC/MS total-ion chromatograms (TICs) of MltA reactions with the sacculus (in black), followed by that of PBP4 (in red). (B) Chemical structures of the abundant muropeptides of MltA degradation of the sacculus.
Two are non-cross-linked products (11 and 12), and the rest are cross-linked, with both the 4,3- and 3,3-cross-links represented. The reaction of PBP4 on the product mixture of MltA showed that 14, 16, and 17 (all containing the 4,3-cross-link) were consumed, whereas the 3,3-cross-linked (13 and 15) were unreactive (Figure 4A and 4B). The dd-carboxypeptidase activity of PBP4 turns over the pentapeptide stem to give the tetrapeptide derivative. PBP4 also has 4,3-endopeptidase activity but not 3,3-endopeptidase activity.
As indicated earlier, inactivation of PBP4 either by mutagenesis or by inhibition by β-lactam antibiotics results in signaling that culminates in induction of resistance to β-lactam antibiotics in P. aeruginosa.7,8 We grew P. aeruginosa PAO1 in the presence of a sublethal concentration of cefoxitin (1/2 MIC), a strong β-lactamase inducer. The sacculus purified from this bacterial growth was digested with MltA. The products were compared to the products obtained from the “noninduced” sacculus (Table 1). This analysis revealed an increased abundance of pentapeptide stems in the sacculus, the substrates for the dd-carboxypeptidase activity. The quantities of muropeptides containing the full-length peptide stem increased from 0.05% to 1.7%, a 34-fold difference. A smaller increase of the 4,3-cross-linked muropeptide (from 43% to 49%) indicates accumulation of the endopeptidase substrates as well.
PBP4 Has Dual Catalytic Activities
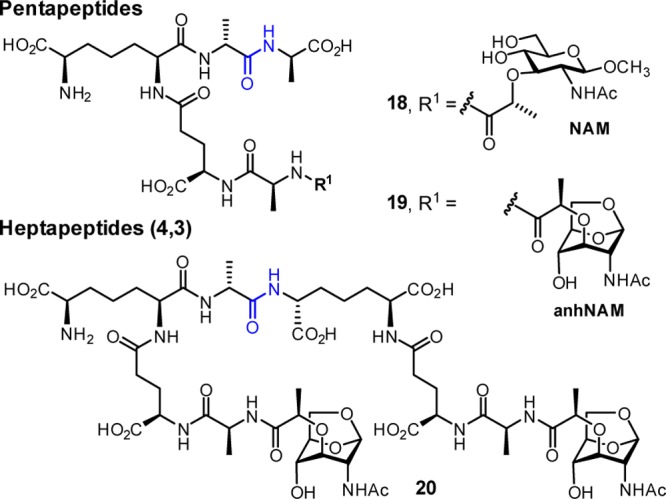
These data demonstrate that PBP4 has dual dd-carboxypeptidase and 4,3-endopeptidase activities with respect to the stem peptide of the sacculus. This conclusion is further supported by additional experiments using synthetic muropeptides as potential substrates. Two peptidoglycan-mimetic compounds containing a pentapeptide stem, synthesized previously by our lab,18 were chosen (Chart 1: 18 and 19) to quantify the dd-carboxypeptidase activity. Both contain a DAP residue in the stem, as is found in the P. aeruginosa sacculus, with two variations on the saccharide structure. One has N-acetyl muramic acid (NAM), as is found in the typical polymeric backbone of peptidoglycan, and the other has N-acetyl 1,6-anhydromuramic acid (anhNAM), as is found in the products of turnover of cell wall by the lytic transglycosylases involved in recycling of cell wall.14,17 To allow quantification of the 4,3-endopeptidase activity, we synthesized a substrate with 1,6-anhNAM as the saccharide (20). To our knowledge, this is the first synthesis of a DAP-containing cross-linked muropeptide (the synthesis of l-Lys-containing cross-linked muropeptides was accomplished by Fujimoto et al.19). The hydrolyzable bond in these compounds is shown in blue in Chart 1.
Chart 1. Synthetic Substrates for PBP4 Used in This Study.
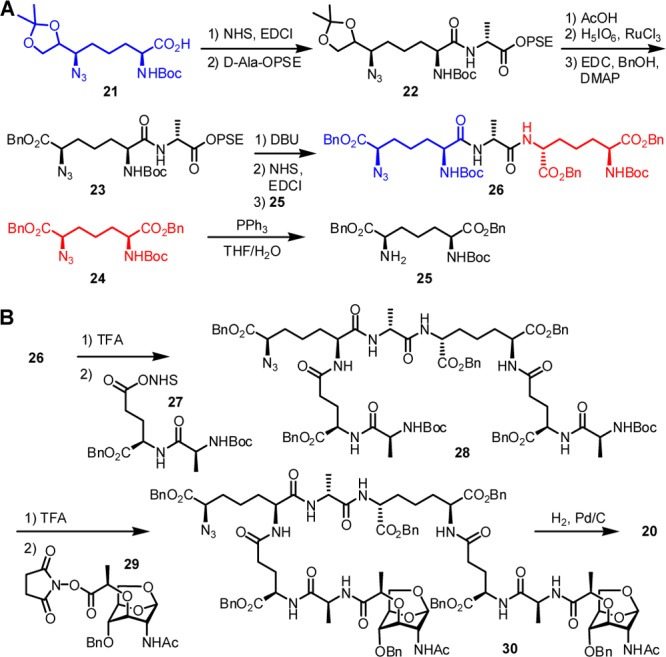
Synthesis of the Discrete PBP4 Substrates
Scheme 1 shows the synthesis of the 4,3-cross-linked substrate (20). The core tripeptide (m-DAP-d-Ala-m-DAP, 26) is prepared and reacted with 2 equiv of dipeptide 27 to arrive at heptapeptide 28. Our orthogonal protection strategy uses groups that can be removed by hydrogenolysis for all peripheral functionalities, and the Boc group for the amine used in the subsequent ligation steps. Additional orthogonal protective groups for carboxylic acids and amines were required for the formation of the cross-linked peptide. We chose 2-(phenylsulfonyl)ethyl (PSE) and azido moieties for this purpose, respectively. The synthesis uses separate DAP precursor units (21 and 24), which were individually prepared from l-Asp in 13 and 14 steps, respectively, according to literature methods.18,20,21 Compound 21 was coupled with d-alanine PSE ester. The acetonide of 22 was deprotected, and the resulting diol was oxidized to the carboxylic acid, subsequently converted to the benzyl ester 23. The base-sensitive PSE group in dipeptide 23 was readily removed by treatment with DBU to give the free carboxylate, which was further transformed to the activated ester and subsequently coupled with the protected DAP derivative 25 to give the key tripeptide 26. The two Boc groups in 26 were removed by TFA treatment, and the resulting amines reacted with the active ester of dipeptide 27 to give heptapeptide 28. The Boc groups in 28 were deprotected, and the resultant amines coupled with saccharide 29. The final compound 20 was obtained by global deprotection using catalytic hydrogenolysis.
Scheme 1.
Kinetic Evaluation of the PBP4 Substrates
These synthetic samples were evaluated as substrates for PBP4. PBP catalysis is a two-step process involving acylation of an active-site serine, followed by acyl transfer in the second step to either a water molecule (carboxypeptidase activity) or to an amine of another peptidoglycan strand (transpeptidase activity). For assessment of the dd-carboxypeptidase reaction of PBP4, the two synthetic pentapeptide substrates (18 and 19) were evaluated for the hydrolytic removal of the terminal d-Ala to give tetrapeptide products (loss of 71 Da in mass). Figure 5 shows the progress of the reaction of compound 18 (NAM-l-Ala-d-γ-Glu-meso-DAP-d-Ala-d-Ala, tR = 5 min) as a substrate for PBP4. A new peak at tR =4 min forms (Figure 5B). This new peak was proven to be compound 31, both by retention time comparison to an authentic synthetic sample of 31 and by MS analysis (both the reaction product and the synthetic standard showed an m/z = 751.3 corresponding to the tetrapeptide) and 71 Da less in mass compared to the pentapeptide 18 (m/z = 822.4) as shown in Figure 5.
Figure 5.
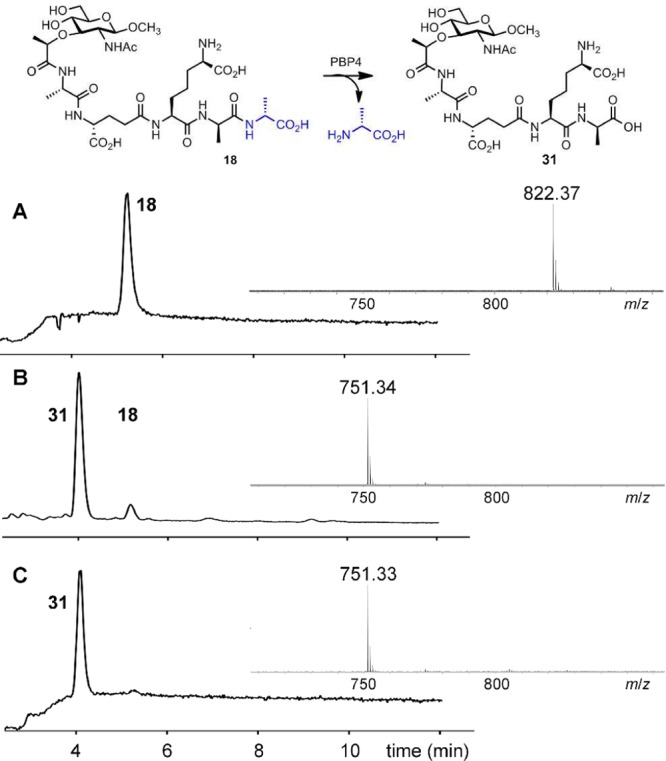
Turnover of compound 18 catalyzed by PBP4. LC/MS TICs of compound 18 with PBP4 at (A) t = 0, and (B) t = 30 min. (C) TIC of the authentic synthetic tetrapeptide standard 31. The mass spectra of compound 18, of product 31, and of authentic synthetic standard 31 are given as inserts in each panel.
The rates of these transformations were evaluated by quantitative HPLC analyses. The steady-state kinetics parameters for turnover of these substrates by PBP4 are given in Table 2. The Km values are in the millimolar range, as these samples are minimalist for the polymeric substrate for the enzyme. The kcat value for 18 is slightly higher than for 19, but the respective kcat/Km values are similar (given the error limits of the analysis). The enzyme turns over equally well muropeptides that contain the muramyl hexopyranose moiety as the variant with the 1,6-anhydromuramyl moiety.
Table 2. Steady-State Kinetic Parameters for Turnover of the Synthetic Substrates by PBP4a.
| kcat (s–1) | Km (mM) | kcat/Km (M–1 s–1) | |
|---|---|---|---|
| 18 | 24.0 ± 1.9 | 3.1 ± 0.5 | 7700 ± 1400 |
| 19 | 14.1 ± 1.5 | 1.3 ± 0.3 | 11000 ± 2800 |
| 20 | 47.0 ± 6.9 | 2.4 ± 0.5 | 20000 ± 5100 |
Reactions were carried out in 50 mM Tris buffer, 100 mM NaCl, pH 7.6 at 25 °C, and they were quenched after 30 min. At pH 8.5, 50 mM Tris buffer, 100 mM NaCl, rate values for substrates were higher by 1.3-fold.
Turnover of the 4,3-cross-linked compound 20 by PBP4 produced 32 and 33 as products (Figure 6A and 6B), whose masses matched the corresponding 1,6-anhNAM tripeptide (m/z = 648.3) and 1,6-anhNAM tetrapeptide (m/z = 719.3).
Figure 6.
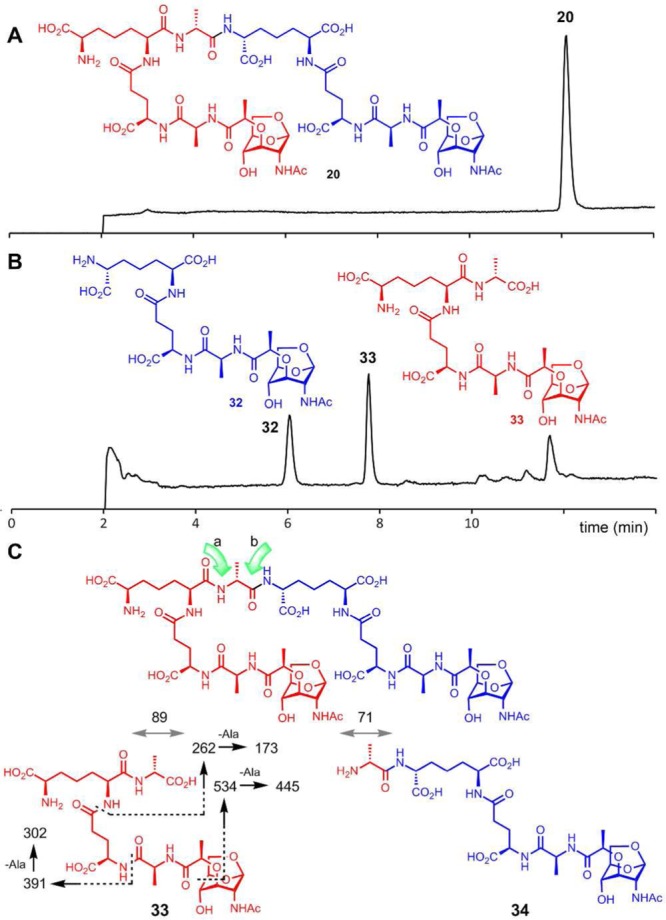
LC/MS TICs of the reaction of cross-linked muropeptide 20 with PBP4 at (A) t = 0 and (B) t = 30 min. (C) The structural assignment from LC/MS/MS of the tetrapeptide product 33.
One could argue that there are potentially two hydrolyzable bonds in compound 20, as indicated by the green arrows a and b in Figure 6C, and hydrolysis of either could give the tripeptide and tetrapeptide products. It is reasonable to assume that PBP4 should cleave the bond indicated by arrow b, based on the correspondence to the scissile bond in the case of the dd-carboxypeptidase activity. Hydrolysis of the bond indicated by arrow a would give tetrapeptide 34 and hydrolysis of the bond of arrow b would give tetrapeptide 33. The difference between 33 and 34 is the position of d-Ala attachment, either at the C-terminus of 33 or attached to the side chain of DAP in 34. One can differentiate between these two possibilities by MS/MS analysis. We obtained tandem mass spectra of tetrapeptide product of PBP4 reaction and compared it to that of synthetic standard 33 (Supporting Information). Their tandem mass spectra were identical. The fragment ions supported the loss of d-Ala at the C-terminus. Therefore, the bond of arrow b in 20 was hydrolyzed by PBP4, and the tetrapeptide product has the chemical structure of 33, as we had suspected to be the case in comparison to the dd-carboxypeptidase reaction. The structure of the tripeptide product was also confirmed by comparison to the authentic synthetic sample for 32 (Supporting Information).
The steady-state kinetic parameters for the endopeptidase reaction of 20 gave values (Table 2) that are very similar to those for the dd-carboxypeptidase activities of 19 and 20. Hence, PBP4 turns over these substrates comparably, regardless of whether it is performing the dd-carboxypeptidase or 4,3-endopeptidase activity.
It is of interest that the variant of compound 20 in which the two diaminopimelate moieties were replaced by lysines was a much poorer substrate, exhibiting a dramatically lower kcat/Km value (by 4200-fold), mostly due to a significantly smaller turnover number (Supporting Information). This observation demonstrates the importance of the side-chain carboxylate of the DAP for interaction with PBP4.
Discussion
The combined ability of P. aeruginosa to adapt to different ecological niches, and therein to refine an ensemble of resistance mechanisms (notably porin deletion, efflux transporter expression, and resistance-enzyme expression), accounts for its emergence as an opportunistic pathogen.22−24 While the judicious selection of a β-lactam antibiotic remains an effective means for the control of infections by P. aeruginosa,25 the clinical emergence of extensively drug-resistant P. aeruginosa, wherein inter alia high-level expression of the AmpC β-lactamase (and increasingly other β-lactamases as well) limits chemotherapy, is truly problematic.26,27 The genetic mechanisms used to secure high-level AmpC expression include mutational deletion of PBP4 activity, mutational deletion of AmpD activity, and mutation to AmpR so as to abrogate its ability to repress expression of the amp operon.28−30 The commonality of each of these three proteins is the (seemingly paradoxical) functional requirement of this Gram-negative bacterium for concomitant degradation of its peptidoglycan so as to enable peptidoglycan growth. These two processes are directly interconnected by the pathway for the efficient recycling of the muropeptides liberated by the degradation and brought into the cytoplasm for reuse.2,3,31,32
The direct relationship in the Enterobacteriaceae and P. aeruginosa between peptidoglycan recycling and the induction of AmpC β-lactamase expression has been known for some time. Recent studies emphasize the complexity of this relationship. Within this relationship the role of the AmpD enzyme is known with good certainty, the role of the AmpR transcription regulator is known in part with good certainty, and the role of PBP4 is known hardly at all. AmpD is a zinc-dependent amidase, located in the bacterial cytosol, that participates in muropeptide recycling by catalyzing the hydrolytic separation of the stem peptide from the anhNAM saccharide21,33,34 (itself liberated by the combined action of the lytic transglycosylase enzymes, imported from the periplasm into the cytoplasm through the AmpG permease, and cleaved in the cytosol by the NagZ glycosidase) (Figure 7).
Figure 7.

Degradation of cell wall by lytic transglycosylases (LTases) in the periplasm leads to the formation of muropeptides, which are internalized by the permease AmpG. The internalized muropeptides serve as the substrate for NagZ, which removes the NAG moiety from the muropeptide to initiate recycling of the muropeptide and also gene derepression for the onset of antibiotic resistance.
AmpR is a LysR-type transcription regulator that is controlled by ligand binding to an effector domain of the protein. In P. aeruginosa (more than in the Enterobacteriaceae) AmpR controls a myriad of virulence responses.35−42 With respect to AmpR control of AmpC expression, the status of the effector domain of AmpR determines whether AmpR acts as a repressor of its own synthesis, or when derepressed it behaves as a transcriptional activator of AmpC synthesis. The long-standing presumption is that peptidoglycan-derived ligand(s) control the status of the effector domain of AmpR and in particular for enforcing the repressor status for AmpR.43−46 This particular presumption is fully consistent with all experimental observations, although the particular chemical entity or entities involved are not proven. The relationship of AmpD to such a muropeptide structure(s) is the preservation (by the mutational inactivation of AmpD) of an anhNAM saccharide having an intact stem (when AmpD is active, the stem is removed, and the ampC gene is not transcribed; Figure 7) in the muropeptide pool. With respect to the peptide stem structure of the muropeptide, the absence of certainty as to its structure (there are several possibilities, as is evident from panels VI–VIII of Figure 3) follows from the previously uncertain knowledge as to the catalytic ability of PBP4. The recent discovery that PBP4 separately exerts control of AmpR function,7,29,30 presumably as a result of its ability to either create or destroy a particular stem structure, dramatically underscores the importance of understanding the molecular function of PBP4.
The carefully interwoven experiments of this study reveal this molecular function. PBP4 possesses the dual catalytic activities of dd-carboxypeptidase trimming of the peptide stem and endopeptidase cleavage of 4,3-cross-linked muropeptides. Loss of function of PBP4, as a result of the irreversible acylation of its active-site serine by a β-lactam antibiotic, induces AmpC expression. Hence, the loss of one (or both) of these activities must coincide with the preservation of a particular muropeptide structure in the muropeptide pool being recycled from the periplasm to the cytoplasm and so controls AmpR expression.
The identity of the active muropeptide(s) that initiate the antibiotic-resistance response has eluded us for 20 years. During growth Gram-negative bacteria constantly release muropeptides from their cell wall, concurrent with biosynthesis of new peptidoglycan. While the muropeptides released into the recycling pool are exposed in the periplasm to a host of peptidoglycan-transforming enzymes, including no less than ten different lytic transglycosylases,8,47 the role here of PBP4 is special. Exposure of the bacterium to β-lactam antibiotics incapacitates PBP4 and results (for the lack of its activity) in the appearance in the cytoplasm of the muropeptide(s) recognized by AmpR. As there is no understanding whatsoever of the structural dynamics between muropeptide release in cell-wall remodeling, and new peptidoglycan incorporation in cell-wall growth, our study here cannot by itself identify the key muropeptide(s). Yet, our data sharply narrow the possibilities. Table 1 shows a sharp increase in the relative amount of muropeptides having a pentapeptide stem in the sacculus of P. aeruginosa grown under the conditions where partial to complete loss of PBP4 activity has occurred (compare the pentapeptide data in column 7 “induced sacculus/MltA” to the data in column 6). This result is consistent with PBP4 as a dd-carboxypeptidase. Table 1 further shows a decrease in the soluble muropeptides having a preserved 4,3-cross-link (compare the 4,3-cross-link data in column 2 to the data in columns six and seven) liberated by PBP4/MltA compared to MltA alone. This outcome coincides with PBP4 acting as an endopeptidase on the 4,3-cross-link. We confirmed both activities by the use of synthetic samples as well. The seeming subtlety of these changes must not be interpreted as demeaning their significance. During active growth of the peptidoglycan, a pool of muropeptides may be envisioned, and the perturbation of this pool by PBP4 inactivation results in a sharper contrast of structures between the two pools. The altered pool would have the effector muropeptide(s) within it to exert the derepression of the resistance genes. Nonetheless, our analysis clearly focuses onto a small group of possible structures. The reactions of lytic transglycosylases on the cell-wall peptidoglycan would liberate the sugar backbone from the cell wall as NAG-anhNAM disaccharide entities with various peptide stems. Some of these peptide stems will be cross-linked (terminating in additional NAG-anhNAM disaccharide), and others might not (Figure 7). After internalization of the muropeptide pool, they serve as substrates for the glycosidase NagZ, whereby the glucosamine moiety is excised from the muropeptides. The structures of the potential effector ligands within this pool of muropeptides may be surmised as either an anhNAM with a pentapeptide stem (19, Figure 8), a 4,3-cross-linked version with a trailing peptide stem of appropriate length (20, 35, and 36), or a variation on these structural motifs. The identification of the active muropeptide(s) has been the focus of considerable effort in the past 20 years. The enormity of the task in identifying the requisite muropeptide(s) is reflected in the complexity of the cell wall itself, compounded by the diversity of the catalytic outcomes for the reactions of lytic transglycosylases, of which there are 10 in P. areuginosa.8,47 The specific catalytic functions of each lytic transglycosylase in the growing P. aeruginosa bacterium remain enigmatic. Whereas E. coli does not have the β-lactam-sensing function that has been attributed to PBP4 of P. aeruginosa, and is hence different, the reactions of its seven lytic transglycosylases have been studied.14 The E. coli lytic transglycosylases produce 50 muropeptides in the stationary phase and 59 in the logarithmic phase of growth.14 The 10 lytic transglycosylases of P. aeruginosa must produce a comparable number of muropeptides (or possibly more), one or a handful of which would have the signaling function that activates this system.
Figure 8.

Chemical structures of possible effector ligands.
Yet, the static sacculus is an imperfect model for the dynamic sacculus. The former shows us the universe of muropeptide structures (from the side wall to the caps). However, the latter is acted upon by a specific subset of lytic transglycosylases and PBPs. This specific subset of enzymes creates a (matching) specific subset of muropeptides, the identity of which awaits elucidation, and the effect of PBP4 inactivation within this subset, in our opinion, will certainly be larger than what is seen with the entire sacculus. We include exposure to antibiotics at various concentrations as a variable that modulates the nature of the dynamic sacculus.
The elucidation of the nature of the active muropeptide(s) in P. aeruginosa, the inducer of antibiotic resistance, must await full characterization of the reactions of all 10 lytic transglycosylases8,47 and the judicious syntheses of the structural entities among the products that conform to the knowledge of catalysis by PBP4 that has emerged from this study for direct evaluation with the AmpR–DNA complex. These studies also will verify that the changes in the muropeptide pool of the recycling pathway coincide to the changes we report here for the entire sacculus using sub-MIC concentrations of the β-lactam inducer.
Clarke et al.11 make the insightful observation that within the broad PBP family of enzymes, only the class C low-molecular-mass PBPs (of which the PBP4 of P. aeruginosa is an example) demonstrate recognition of the amino acid substructure of the stem of the peptidoglycan. This is a characteristic expected as required for an enzyme having a pivotal role as a sentinel (here, to detect the presence of β-lactam antibiotics) and where the presence or absence of the products of its catalytic reaction regulates gene transcription. Our study has established this character for PBP4, has identified the reaction products of PBP4, and has brought clarity to the structural conception of the negative effector used by the AmpR system of P. aeruginosa. These new perspectives are essential to the further understanding of the central role of the PBP4–AmpR system as a control point for resistance and virulence in this pathogenic bacterium.
Materials and Methods
Cloning of the PA3047 Gene from P. aeruginosa PAO1
The PA3047 gene was synthesized by Genescript for optimal expression in E. coli from nucleotide position 67 to 1431. The gene was digested with NdeI and XhoI, and the resulting piece was ligated into NdeI and XhoI sites of the T7 expression vector pET-28a-c(+). The recombinant plasmid pET28-PBP4 (from amino acid 23 to 476 and with N-terminal His-Tag) was confirmed by sequencing of both strands.
Purification of P. aeruginosa PBP4
Escherichia coli Lemo21(DE3) (New England Biolabs) cells containing the pET28-PBP4 plasmid were incubated overnight in 12 mL of LB medium supplemented with 50 μg/mL of kanamycin and 34 μg/mL of chloramphenicol. The cells were diluted into 1 L of LB medium (kanamycin/chloramphenicol supplemented) that was grown at 37 °C with agitation (at 170 rpm) until the culture reaches OD600 of 0.8. Isopropyl β-d-thiogalactoside (IPTG) was added at this stage to a concentration of 0.4 mM, and the culture was incubated at 20 °C overnight. Cells were harvested by centrifugation for 35 min at 4 °C at 4000 g and resuspended in 30 mL of 50 mM Tris buffer pH 7.6, 0.5 M NaCl, 10 mM imidazole. The cells were disrupted by sonification (1 min bursts with 2 min of break, for 30 min total). Bacterial debris were removed by centrifugation for 45 min at 4 °C at 18 000 g. The supernatant was loaded onto a 5 mL HiTrap Chelating column (GE Healhcare). The column was washed with 50 mM Tris pH 7.6, 0.5 M NaCl, 10 mM imidazole. Elution was performed using a gradient from 10 to 500 mM imidazole in 50 mM Tris pH 7.6, 0.5 M NaCl. The PBP4 fractions were pooled, and the imidazole concentration was reduced by dilution with 50 mM Tris pH 7.6, 0.5 M NaCl to give a final buffer composition of 100 mM imidazole in 50 mM Tris pH 7.6, 0.5 M NaCl. Then PBP4 was passed a second time onto a 5 mL HiTrap Chelating column (GE Healhcare) and eluted under the same conditions. All purification steps were carried out at 4 °C. The fractions were dialyzed against 50 mM Tris pH 7.6, 0.5 M NaCl. The purified PBP4 was kept in this solution, and it was used within the 5 days. The PBP4 concentration was determined by measuring the absorbance of the solution at 280 nm and using a theoretical extinction coefficient of 62 005 M–1cm–1. The protein was at apparent homogeneity by SDS-PAGE (Figure S1, Supporting Information). Liquid chromatography/mass spectrometry (LC/MS) analysis revealed a molecular mass of 51,617 ± 5 Da, in agreement with the mass calculated from the gene sequence (51 619 Da) (Figure S2, Supporting Information).
Synthesis of Compounds
Compounds 18, 19, 31–33 were prepared by literature procedures.18,20,21,34
Compound 22
EDCI (0.56 g, 2.9 mmol) was added to a chilled suspension of N-hydroxysuccinimide (0.33 g, 2.9 mmol) and compound 21(18,21) (1.0 g, 2.7 mmol) in CH2Cl2 (10 mL). The mixture was stirred for 16 h at room temperature. d-Alanine phenylsulfonylethyl ester (0.78 g, 3.0 mmol) was added to the activated ester solution (prepared above). Stirring was continued for 16 h, and the mixture was washed with water. The organic layer was dried over anhydrous Na2SO4 and filtered, and the filtrate was concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (0.95 g, 58%). 1H NMR (500 MHz, CDCl3) δ 1.22–1.44 (m, 23H), 1.50–1.84 (m, 4H), 3.35–3.53 (m, 3H; CH2SO2Ph and DAP-H6), 3.83 (dd, J = 7.9, 6.5 Hz, 1H; DAP-H8a), 3.98 (dd, J = 8.2, 6.4 Hz, 1H; DAP-H8b), 4.04 (q, J = 6.0 Hz, 1H; DAP-H7), 4.09 (q, J = 7.1 Hz, 1H; DAP-H2), 4.34 (m, 1H; Ala-α-H), 4.46 (t, J = 5.9 Hz, 2H; CO2CH2CH2SO2Ph), 5.18 (d, J = 8.2 Hz, 1H; NHBoc), 6.78 (d, J = 6.6 Hz, 1H; CONH), 7.58 (t, J = 7.7 Hz, 2H), 7.68 (t, J = 7.4 Hz, 1H), 7.91 (d, J = 7.4 Hz, 2H); 13C NMR (126 MHz, CDCl3) d 17.8 (q, Ala-CH3), 22.5 (t), 25.3, 26.3 (2 × q, C(CH3)2), 28.4 (q, C(CH3)3), 30.6, 32.2 (2 × t), 48.0 (d, Ala-α-C), 54.0 (d, DAP-C2), 55.0 (t, CH2SO2Ph), 58.4 (t, CO2CH2CH2SO2Ph), 63.7 (d, DAP-C6), 65.9 (t, DAP-C8), 77.9 (d, DAP-C7), 80.3 (s, C(CH3)3), 109.8 (s, C(CH3)2), 128.2, 129.6, 134.3, 139.2, 139.2, 155.9, 171.7, 172.0; HRMS (ESI), calcd for C27H42N5O9S (M + H+), 612.2698, found 612.2714.
Compound 23
An acetic acid solution (AcOH:water, 4:1, 15 mL) of compound 22 (0.9 g, 1.5 mmol) was stirred for 36 h at room temperature, and the volatiles were removed in vacuo to dryness. The residue was dried further under high vacuum. The resulting diol was dissolved in MeCN:CCl4:water (2:2:3, 10 mL), and the solution was cooled in an ice–water bath. H5IO6 (1.2 g, 5.3 mmol) and RuCl3·xH2O (5.4 mg, 0.03 mmol) were added to this solution. After stirring for 2 h at room temperature, the solution was diluted with CH2Cl2 and water. The organic layer was dried over anhydrous Na2SO4 and filtered, and the filtrate was concentrated under reduced pressure. The crude product was dissolved in CH2Cl2 (10 mL), and benzyl alcohol (0.14 mL, 1.4 mmol) was added. After the solution was cooled in an ice–water bath, EDCI (0.25 g, 1.3 mmol) and DMAP (16 mg, 0.13 mmol) were added. The mixture was stirred for 16 h at room temperature, at which point it was washed with water. The organic layer was dried over anhydrous Na2SO4 and filtered, and the filtrate was concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (0.40 g, 42% over three steps). 1H NMR (500 MHz, CDCl3) δ 1.23 (d, J = 7.2 Hz, 3H; Ala-CH3), 1.41 (s, 9 H), 1.35–1.88 (m, 6H), 3.43 (t, J = 5.8 Hz, 2H; CH2SO2Ph), 3.85 (dd, J = 8.6, 5.0 Hz, 1H; DAP-H6), 4.09 (m, 1H; DAP-H2), 4.30 (m, 1H; Ala-α-H), 4.43 (t, J = 5.9 Hz, 2H; CO2CH2CH2SO2Ph), 5.17 (s, 2H; CH2Ph), 5.20 (d, J = 8.4 Hz, 1H; NHBoc), 6.82 (m, 1H; CONH), 7.24–7.40 (m, 5H), 7.50–7.61 (m, 2H), 7.65 (t, J = 7.4 Hz, 1H), 7.89 (d, J = 7.6 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 17.7 (q, Ala-CH3), 21.9 (t), 28.4 (q, C(CH3)3), 31.0, 31.8 (2 × t), 48.0 (d, Ala-α-C), 54.0 d, DAP-C2), 54.9 (t, CH2SO2Ph), 58.3 (t, CO2CH2CH2SO2Ph), 61.8 (d, DAP-C6), 67.6 (t, CH2Ph), 80.3 (s, C(CH3)3), 127.0, 127.5, 128.1, 128.5, 128.7, 128.8, 129.6, 134.3, 135.1, 139.1, 155.8, 170.3, 171.6, 172.0; HRMS (ESI), calcd for C30H40N5O9S (M + H+), 646.2541, found 646.2523.
Compound 25
A solution containing compound 24(20,21) (0.30 g, 0.60 mmol) and PPh3 (0.17 g, 0.66 mmol) in THF:water (7 mL, 10:1) was stirred for 16 h at room temperature and was concentrated under reduced pressure. The resulting amine 25 was used for the coupling without further purification.
Compound 26
Compound 23 (0.40 g, 0.62 mmol) was dissolved in CH2Cl2 (6 mL), and DBU (92 μL, 0.62 mmol) was added. After stirring for 2 h at room temperature, the mixture was concentrated to yield the corresponding carboxylate. The transformation of carboxylate to NHS ester and coupling with amine 25 was performed in the same manner as described for compound 22. The crude product was purified by column chromatography on silica gel (0.22 g, 39%). 1H NMR (500 MHz, CDCl3) δ 1.07–1.85 (m, 33H), 3.87 (dd, J = 8.6, 5.0 Hz, 1H), 4.09 (m, 1H), 4.26 (s, 1H), 4.46–4.62 (m, 2H), 5.07–5.22 (m, 7H), 5.46 (m, 1H), 6.90–7.02 (m, 2H), 7.30–7.40 (m, 15H); 13C NMR (126 MHz, CDCl3) d 17.8, 21.2, 21.3, 22.2, 28.4, 31.0, 31.3, 32.2, 48.8, 52.2, 53.4, 54.3, 61.9, 67.2, 67.6, 80.1, 80.3, 128.4, 128.5, 128.6, 128.7, 128.8, 135.1, 135.4, 155.6, 156.0, 170.4, 171.7, 172.0, 172.6; HRMS (ESI), calcd for C48H64N7O12 (M + H+), 930.4607, found 930.4617.
Compound 28
Trifluoroacetic acid (2 mL) was added to a chilled solution of compound 26 (0.20 g, 0.22 mmol) in CH2Cl2 (2 mL), and the solution was stirred for 1 h. After concentration of the solution to dryness under reduced pressure, the resultant residue was suspended in toluene and the mixture was evaporated in vacuo. The crude amine was dissolved in CH2Cl2, was cooled in an ice–water bath, and was neutralized with iPr2EtN (0.45 mL, 2.6 mmol). Boc-Ala-γ-d-Glu-ONHS (27, 0.24 g, 0.47 mmol) was added to the free amine solution (prepared above), and the resultant mixture was stirred for 16 h. After addition of water, the organic layer was separated and concentrated to dryness. The crude compound was subjected to column chromatography on silica gel to yield the desired compound (0.18 g, 55%). 1H NMR (500 MHz, 5% CD3OD in CDCl3) δ 1.10, 1.13, 1.19 (3 × d, J = 7.0 Hz, 9H), 1.24, 1.25 (2 × s, 18H), 1.16–2.15 (m, 20H), 3.69 (dd, J = 8.7, 4.7 Hz, 1H), 3.94 (m, 1H), 4.00 (s, 1H), 4.09 (m, 1H), 4.17 (s, 1 H), 4.21–4.36 (m, 4H), 4.86–5.04 (m, 10H), 5.70 (d, J = 4.8 Hz, 1H), 5.74 (d, J = 4.8 Hz, 1H), 7.07–7.21 (m, 25 H), 7.32 (m, 1H), 7.51 (s, 2H), 7.58 (s, 1H), 7.72 (d, J = 7.6 Hz, 1H), 7.88 (d, J = 7.6 Hz, 1H); 13C NMR (126 MHz, 5% CD3OD in CDCl3) δ 16.8, 17.6, 18.3, 21.6, 22.0, 24.7, 25.5, 26.3, 27.2, 28.1, 30.5, 30.6, 30.9, 31.0, 31.7, 33.7, 48.7, 50.1, 50.5, 50.7, 51.9, 52.1, 54.0, 61.4, 66.4, 66.5, 66.7, 67.1, 77.0, 77.5, 79.3, 79.6, 127.6, 127.7, 127.8, 127.8, 127.8, 127.9, 128.0, 128.0, 128.2, 128.3, 128.4, 129.2, 133.5, 134.8, 135.1, 135.3, 135.3, 138.1, 151.7, 155.2, 155.7, 170.0, 171.1, 171.2, 171.5, 172.1, 172.19, 172.23, 172.5, 173.3, 173.5; HRMS (ESI), calcd for C78H100N11O20 (M + H+), 1510.7141, found 1510.7111.
Compound 20
The deprotection of the Boc groups in compound 28 and its coupling reaction with activated ester 29 was performed in the same manner as described for 26 and 27. Compound 30 (50 mg, 33 μmol) was stirred in acetic acid (2 mL) in the presence of 10% Pd/C (30 mg) under a hydrogen atmosphere for 16 h. The resulting suspension was filtered through a layer of Celite and washed with acetic acid. The combined filtrate was concentrated to dryness under reduced pressure. The crude compound was triturated in acetonitrile, and the desired product was obtained by filtration (20 mg, 59%). 1H NMR (500 MHz, D2O) δ 1.42, 1.44, 1.49 (3 × d, J = 7.2 Hz, 9H), 2.06, (s, 6H), 1.30–2.55 (m, 20H), 3.48 (s, 2H), 3.86 (t, J = 6.8 Hz, 2H), 3.92 (s, 2H), 3.98 (s, 2H), 4.02 (s, 1H), 4.24 (d, J = 6.8 Hz, 2H), 4.27–4.48 (m, 8H), 4.74 (d, J = 5.0 Hz, 2H), 5.54 (s, 2H); 13C NMR (126 MHz, D2O) δ 16.6, 17.0, 17.1, 18.1, 18.2, 20.6, 21.0, 21.8, 22.0, 26.6, 26.8, 29.7, 30.01, 30.03, 30.4, 31.3, 31.6, 49.49, 49.55, 49.63, 49.8, 52.1, 52.3, 52.7, 52.8, 53.3, 54.1, 65.3, 68.28, 68.33, 76.0, 76.1, 78.6, 78.7, 100.1, 172.8, 173.7, 174.0, 174.7, 174.8, 174.9, 175.1, 175.3, 175.4, 175.6, 175.7, 175.8, 176.8; HRMS (ESI), calcd for C55H86N11O28 (M + H+), 1348.5638, 674.7856, found 1348.5627.
Reaction of PBP4 with the Bacterial Sacculus and Reaction Products Quantification
Preparations of the pseudomonal sacculus and MltA of E. coli were published previously.13,14 PBP4 was incubated with 100 μL of the sacculus preparation in 50 mM Tris, pH 7.6 with 0.1 M NaCl at 37 °C. After 24 h, the reaction was stopped by boiling for 3 min. The reaction mixture was centrifuged, and supernatant and pellet were separated. The supernatant was analyzed by LC/MS, and the results are given in the column “sacculus/PBP4” in Table 1. The supernatant and pellet were incubated with MltA separately, for 24 h at 37 °C, and the reaction was stopped by boiling for 3 min. The reaction mixture was concentrated under reduced pressure and dissolved in water, and the resulting solution was centrifuged. LC/MS analysis of supernatant is given in the columns “sacculus/PBP4-sol/MltA” and “sacculus/PBP4-Insol/MltA”. LC/MS and LC/MS/MS conditions were described previously.14 Quantification and characterization of reaction products were performed using the method reported earlier by our laboratory.14 LC/MS/MS spectra of PBP4 reaction products of compound 20 and comparison to synthetic standards 32 and 33 are given in Figure S3, Supporting Information.
Kinetic Studies
The enzymatic reactions with synthetic compounds were carried out in 50 mM Tris buffer, 0.1 M NaCl, pH 7.6 at 25 °C. After 30 min, one volume of 0.2% TFA was added to stop reactions. The substrate concentrations were varied between 0.125 and 5 mM. The reaction products were quantified with an internal standard on Dionex Acclaim PolarAdvantage II C18 column. The kinetic data were fit to the Michaelis–Menten equation by nonlinear regression using GraphPad Prism (GraphPad Software, Inc. La Jolla, CA). The internal standard was compound 32 for the analysis of compounds 18 and 19, and compound 31 was used for the analysis of compound 20.
Acknowledgments
This work was supported by a grant from the NIH (GM61629). The Mass Spectrometry & Proteomics Facility of the University of Notre Dame is supported by grant CHE0741793.
Supporting Information Available
Supplementary figures and the NMR spectra of the new compounds are given. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Boudreau M. A.; Fisher J. F.; Mobashery S. Biochemistry 2012, 51, 2974–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. W.; Fisher J. F.; Mobashery S. Ann. N.Y. Acad. Sci. 2013, 1277, 54–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J. F.; Mobashery S. Bioorg. Chem. 2014, 56, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livermore D. M.; Woodford N. Trends Microbiol. 2006, 14, 413–420. [DOI] [PubMed] [Google Scholar]
- Jacoby G. A. Clin. Microbiol. Rev. 2009, 22, 161–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testero S. A.; Fisher J. F.; Mobashery S.. β-Lactam Antibiotics. In Burger’s Medicinal Chemistry, Drug Discovery and Development (Antiinfectives); Abraham D. J., Rotella D. P., Eds.; Wiley and Sons: New York, 2010; Vol. 7, pp 259–404. [Google Scholar]
- Moyá B.; Dotsch A.; Juan C.; Blazquez J.; Zamorano L.; Haussler S.; Oliver A. PLoS Pathog. 2009, 5, e1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallari J. F.; Lamers R. P.; Scheurwater E. M.; Matos A. L.; Burrows L. L. Antimicrob. Agents Chemother. 2013, 57, 3078–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korat B.; Mottl H.; Keck W. Mol. Microbiol. 1991, 5, 675–684. [DOI] [PubMed] [Google Scholar]
- Meberg B. M.; Paulson A. L.; Priyadarshini R.; Young K. D. J. Bacteriol. 2004, 186, 8326–8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke T. B.; Kawai F.; Park S. Y.; Tame J. R. H.; Dowson C. G.; Roper D. I. Biochemistry 2009, 48, 2675–2683. [DOI] [PubMed] [Google Scholar]
- Ghosh A. S.; Chowdhury C.; Nelson D. E. Trends Microbiol. 2008, 16, 309–317. [DOI] [PubMed] [Google Scholar]
- Martínez-Caballero S.; Hesek D.; Lee M.; Artola-Recolons C.; Carrasco-López C.; Hesek D.; Spink E.; Lastochkin E.; Zhang W.; Hellman L. M.; Boggess B.; Mobashery S.; Hermoso J. A. J. Am. Chem. Soc. 2013, 135, 10318–10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Hesek D.; Llarrull L. I.; Lastochkin E.; Pi H.; Boggess B.; Mobashery S. J. Am. Chem. Soc. 2013, 135, 3311–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glauner B. Anal. Biochem. 1988, 172, 451–464. [DOI] [PubMed] [Google Scholar]
- Glauner B.; Höltje J. V.; Schwarz U. J. Biol. Chem. 1988, 263, 10088–10095. [PubMed] [Google Scholar]
- Scheurwater E.; Reid C. W.; Clarke A. J. Int. J. Biochem. Cell Biol. 2008, 40, 586–591. [DOI] [PubMed] [Google Scholar]
- Lee M.; Hesek D.; Shah I. M.; Oliver A. G.; Dworkin J.; Mobashery S. ChemBioChem. 2010, 11, 2525–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto Y.; Konishi Y.; Kubo O.; Hasegawa M.; Inohara N.; Fukase K. Tetrahedron Lett. 2009, 50, 3631–3634. [Google Scholar]
- Hernández N.; Martín V. S. J. Org. Chem. 2001, 66, 4934–4938. [DOI] [PubMed] [Google Scholar]
- Lee M. J.; Zhang W. L.; Hesek D.; Noll B. C.; Boggess B.; Mobashery S. J. Am. Chem. Soc. 2009, 131, 8742–8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister P. D.; Wolter D. J.; Hanson N. D. Clin. Microbiol. Rev. 2009, 22, 582–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenstein E. B. M.; de la Fuente-Núñez C.; Hancock R. E. W. Trends Microbiol. 2011, 19, 419–426. [DOI] [PubMed] [Google Scholar]
- Poole K. Front. Microbiol. 2011, 2, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Ortega C.; Weigand I.; Olivares J.; Hancock R. E. W.; Martinez J. L. Virulence 2011, 2, 144–146. [DOI] [PubMed] [Google Scholar]
- Cabot G.; Ocampo-Sosa A. A.; Domínguez M. A.; Gago J. F.; Juan C.; Tubau F.; Rodríguez C.; Moyà B.; Peña C.; Martínez-Martínez L.; Oliver A. Antimicrob. Agents Chemother. 2012, 56, 6349–6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiorakos A. P.; Srinivasan A.; Carey R. B.; Carmeli Y.; Falagas M. E.; Giske C. G.; Harbarth S.; Hindler J. F.; Kahlmeter G.; Olsson-Liljequist B.; Paterson D. L.; Rice L. B.; Stelling J.; Struelens M. J.; Vatopoulos A.; Weber J. T.; Monnet D. L. Clin. Microbiol. Infect. 2012, 18, 268–281. [DOI] [PubMed] [Google Scholar]
- Schmidtke A. J.; Hanson N. D. Antimicrob. Agents Chemother. 2008, 52, 3922–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamorano L.; Moyá B.; Juan C.; Oliver A. J. Antimicrob. Chemother. 2010, 65, 1540–1542. [DOI] [PubMed] [Google Scholar]
- Moyá B.; Beceiro A.; Cabot G.; Juan C.; Zamorano L.; Alberti S.; Oliver A. Antimicrob. Agents Chemother. 2012, 56, 4771–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark B. L.; Vocadlo D. J.; Oliver A. Future Microbiol. 2011, 6, 1415–1427. [DOI] [PubMed] [Google Scholar]
- Zeng X.; Lin J. Front. Microbiol. 2013, 4, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco-Lopez C.; Rojas-Altuve A.; Zhang W.; Hesek D.; Lee M.; Barbe S.; Andre I.; Ferrer P.; Silva-Martin N.; Castro G. R.; Martinez-Ripoll M.; Mobashery S.; Hermoso J. A. J. Biol. Chem. 2011, 286, 31714–31722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Lee M.; Hesek D.; Lastochkin E.; Boggess B.; Mobashery S. J. Am. Chem. Soc. 2013, 135, 4950–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong K. F.; Jayawardena S. R.; Indulkar S. D.; Del Puerto A.; Koh C. L.; Hoiby N.; Mathee K. Antimicrob. Agents Chemother. 2005, 49, 4567–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D.; Kong K. F.; Jayawardena S. R.; Leal S. M.; Sautter R. T.; Mathee K. J. Med. Microbiol. 2011, 60, 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D.; Schneper L.; Merighi M.; Smith R.; Narasimhan G.; Lory S.; Mathee K. PLoS One 2012, 7, e34067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D.; Kumari H.; Jaric M.; Fernandez M.; Turner K. H.; Dove S. L.; Narasimhan G.; Lory S.; Mathee K. Nucleic Acids Res. 2014, 42, 979–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D.; Kumari H.; Mathee K. Pathog. Dis. 2014, 10.1111/2049-632X.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caille O.; Zincke D.; Merighi M.; Balasubramanian D.; Kumari H.; Kong K.-F.; Silva-Herzog E.; Narasimhan G.; Schneper L.; Lory S.; Mathee K. J. Bacteriol. 2014, 196, 3890–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari H.; Balasubramanian D.; Zincke D.; Mathee K. J. Med. Microbiol. 2014, 63, 544–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari H.; Murugapiran S. K.; Balasubramanian D.; Schneper L.; Merighi M.; Sarracino D.; Lory S.; Mathee K. J. Proteomics 2014, 96, 328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs C.; Huang L. J.; Bartowsky E.; Normark S.; Park J. T. EMBO J. 1994, 13, 4684–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs C.; Joris B.; Jamin M.; Klarsov K.; Van Beeumen J.; Mengin-Lecreulx D.; van Heijenoort J.; Park J. T.; Normark S.; Frère J. M. Mol. Microbiol. 1995, 15, 553–559. [DOI] [PubMed] [Google Scholar]
- Jacobs C.; Frère J. M.; Normark S. Cell 1997, 88, 823–832. [DOI] [PubMed] [Google Scholar]
- Balcewich M. D.; Reeve T. M.; Orlikow E. A.; Donald L. J.; Vocadlo D. J.; Mark B. L. J. Mol. Biol. 2010, 400, 998–1010. [DOI] [PubMed] [Google Scholar]
- Jorgenson M. A.; Chen Y.; Yahashiri A.; Popham D. L.; Weiss D. S. Mol. Microbiol. 2014, 93, 113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.