

Abstract
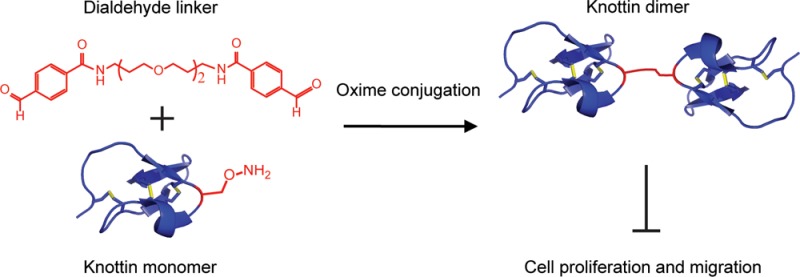
Molecules that target and inhibit αvβ3, αvβ5, and α5β1 integrins have generated great interest because of the role of these receptors in mediating angiogenesis and metastasis. Attempts to increase the binding affinity and hence the efficacy of integrin inhibitors by dimerization have been marginally effective. In the present work, we achieved this goal by using oxime-based chemical conjugation to synthesize dimers of integrin-binding cystine knot (knottin) miniproteins with low-picomolar binding affinity to tumor cells. A non-natural amino acid containing an aminooxy side chain was introduced at different locations within a knottin monomer and reacted with dialdehyde-containing cross-linkers of different lengths to create knottin dimers with varying molecular topologies. Dimers cross-linked through an aminooxy functional group located near the middle of the protein exhibited higher apparent binding affinity to integrin-expressing tumor cells compared with dimers cross-linked through an aminooxy group near the C-terminus. In contrast, the cross-linker length had no effect on the integrin binding affinity. A chemical-based dimerization strategy was critical, as knottin dimers created through genetic fusion to a bivalent antibody domain exhibited only modest improvement (less than 5-fold) in tumor cell binding relative to the knottin monomer. The best oxime-conjugated knottin dimer achieved an unprecedented 150-fold increase in apparent binding affinity over the knottin monomer. Also, this dimer bound 3650-fold stronger and inhibited tumor cell migration and proliferation compared with cilengitide, an integrin-targeting peptidomimetic that performed poorly in recent clinical trials, suggesting promise for further therapeutic development.
Integrins, a family of cell surface adhesion receptors, bind to components of the extracellular matrix (ECM) to provide anchorage necessary for cell division, migration, and invasion.1,2 In particular, αvβ3, αvβ5, and α5β1 integrins are present at high levels on many types of tumor cells or their neovasculature3 and mediate angiogenesis, tumor growth, and metastasis,4−6 generating great interest as targets for therapeutic intervention.7,8 These integrins bind to ECM ligands through an Arg-Gly-Asp (RGD) peptide motif,9 which has been incorporated into a myriad of peptides, peptidomimetics, and protein scaffolds toward the goal of developing cancer diagnostics and therapeutics.10,11 Despite an abundance of integrin-targeting agents, only a limited number have advanced to evaluation in human clinical trials. Cilengitide,12 a cyclic pentapeptide that binds αvβ3 and αvβ5 integrins, was the first peptidomimetic to move to phase-III clinical trials for treatment of glioblastoma multiforme; however, this compound did not show evidence of increased patient survival.10,13
We previously engineered cystine knot (knottin) miniproteins that bind with low-nanomolar affinity to tumor-associated integrin receptors.14,15 Knottins have a compact disulfide-bonded framework16,17 that provides remarkable thermal and proteolytic stability ideal for drug development.18,19 One particular engineered knottin, EETI 2.5F, is a 33 amino acid polypeptide derived from the Ecballium elaterium trypsin inhibitor II (EETI) that binds to αvβ3, αvβ5, and α5β1 integrins.14,20
Here we report an efficient strategy to significantly increase the integrin receptor binding affinity and biological efficacy of knottin-based inhibitors by chemically cross-linking them to form covalent dimers. Dimerization is a well-established approach for creating molecules with increased cell surface receptor binding through avidity effects.21−23 However, previous attempts to create high-affinity integrin-binding small molecules and peptidomimetics through dimerization have resulted in compounds with only marginal affinity improvements,24−31 most likely due to constraints that hinder effective engagement of multiple integrin receptors. In contrast, we show that chemically conjugating EETI 2.5F through flexible polyether linkers generates dimers with apparent integrin binding affinities in the picomolar range and the ability to inhibit tumor cell migration and proliferation.
Our approach to knottin dimerization involves the formation of an oxime bond between aldehyde and aminooxy functional groups, allowing cross-linking to occur in a site-selective manner.30,32−35 For this purpose, we introduced a non-natural amino acid with an aminooxy side chain into a knottin monomer using solid-phase peptide synthesis (SPPS) and synthesized polyether cross-linkers containing two terminal aldehyde groups. Knottin dimers have previously been formed by conjugating standard amine-reactive homofunctional cross-linkers with recombinant knottins produced in a bacterial expression system;36 however, this method presumably produces heterogeneous molecules cross-linked through both N-terminal amino groups and lysine side chains. In contrast, our approach produces chemically defined knottin dimers that are site-specifically conjugated through a non-natural amino acid.
Nα-Fmoc-Nβ-(N-Boc-aminooxyacetyl)-l-2,3-diaminopropionic acid was introduced at two separate locations within EETI 2.5F during SPPS: (i) in place of Lys15, which was demonstrated to be tolerant to mutation,14,33 or (ii) at the C-terminus (Figure S1 and Table S1 in the Supporting Information (SI)). These locations were chosen to determine whether EETI 2.5F could tolerate introduction of a non-natural amino acid in the middle or at the end of the protein and to determine the effects of dimers cross-linked through different positions. The resulting knottins are termed 2.5F_AO_1 (3) and 2.5F_AO_2 (5), respectively. Sequences of the knottins used in this study and methods for their synthesis, folding, and purification are described in the SI.
A dialdehyde-containing cross-linker was prepared by conjugating 4-formylbenzoic acid (4FB) to both ends of 4,9-dioxa-1,12-dodecanediamine (1), a diamine polyether chain (Scheme 1). The cross-linker produced from this reaction positions two covalently conjugated knottin monomers at distances of up to 22 Å apart, with the goal of promoting multimeric integrin binding interactions. 4FB was coupled to (1) by addition of dicyclohexylcarbodiimide and HOBt in CH2Cl2 at 0 °C for 2 h. The product (2) was purified by reversed-phase HPLC (RP-HPLC) and used to cross-link (3) in phosphate buffer at 25 °C for 1.5 h. The resulting dimeric knottin (4) was purified using RP-HPLC. A similar strategy was applied to O,O′-bis(2-aminoethyl)octadecaethylene glycol (6) to synthesize a cross-linker (7) used to create dimers with up to 76 Å between the two knottin monomers. The process was repeated with (5) to create knottin dimers coupled through the protein C-termini.
Scheme 1. Site-Specific Knottin Dimerization Strategy.
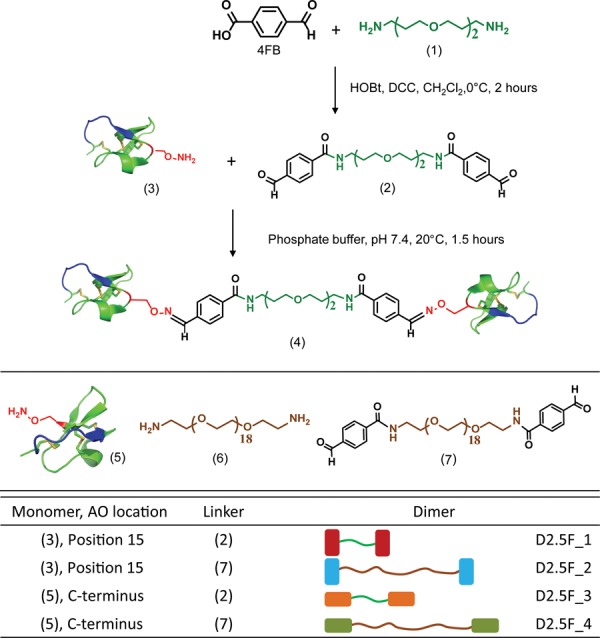
Coupling of 4FB to (1) affords dialdehyde-containing cross-linker (2), which was reacted with an aminooxy functional group present at position 15 in knottin 2.5F_AO_1 (3) to produce knottin dimer (4). The same strategy was repeated with the longer cross-linker (6). Knottin dimers were also created by cross-linking through the C-terminal aminooxy group in knottin 2.5F_AO_2 (5) using (2) or (7). A summary of the four dimeric knottins synthesized and tested in this study is shown in the table.
In all, four distinct knottin dimers were synthesized using the two 2.5F_AO variants and two different cross-linkers (Scheme 1). The yields of dialdehyde cross-linkers and dimeric knottins were typically ∼75% and ∼95%, respectively, as confirmed by RP-HPLC. Chromatograms and mass spectrometry data are provided in Figure S2 and Table S2.
Competition binding assays were performed to compare the relative binding affinities of knottin monomers, dimers, and cilengitide to U87MG human glioblastoma cells, which express αvβ3, αvβ5, and α5β1 integrins,37 and MDA-MB-231 human breast cancer cells, which express αvβ5 integrins38 (Figure S3). Cells (2 × 104) were incubated with varying concentrations of knottins or cilengitide (BOC Sciences) and a constant concentration of competitor for 10 h at 4 °C (see the SI). Fluorescent binding signals were measured using flow cytometry (Guava EasyCyte 8HT, Millipore), and half-maximal inhibitory concentration (IC50) values were determined by nonlinear regression analysis (KaleidaGraph, Synergy Software) (Table 1). Importantly, incorporation of an aminooxy group within 2.5F at either position 15 or the C-terminus did not affect the integrin binding affinity (Figure S4 and Table S3).
Table 1. Tumor Cell Binding Data.
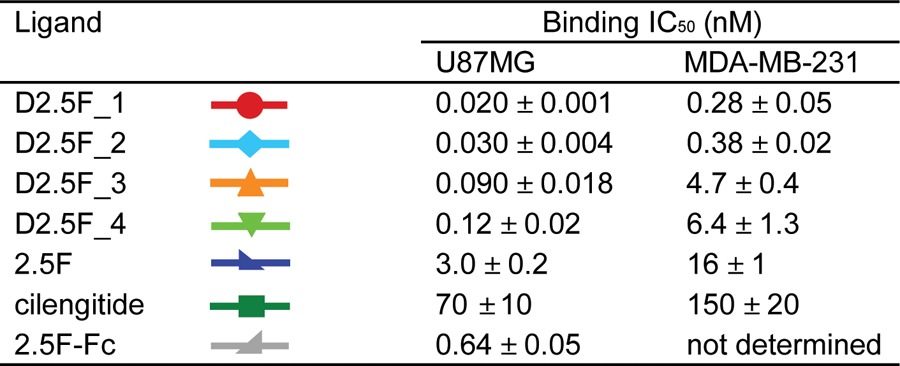
All four knottin dimers showed significantly higher apparent binding affinities compared with the knottin monomer (Figure 1 and Table 1), demonstrating their ability to engage multiple cell surface integrin receptors through avidity effects. Knottin dimers created through the introduction of the aminooxy group at position 15 showed stronger binding affinities than dimers created with the aminooxy group located at the C-terminus, indicating that the orientation of the monomers within a C-terminally cross-linked dimer is less optimal for multivalent binding interactions. In contrast, the cross-linker length did not impact binding, as knottin dimers conjugated with short and long cross-linkers showed similar relative binding affinities. These trends were consistently observed on both cell lines; however, tighter overall relative binding affinities were observed on U87MG cells, likely reflecting their higher integrin receptor density compared with MDA-MB-231 cells (Figure S3).
Figure 1.
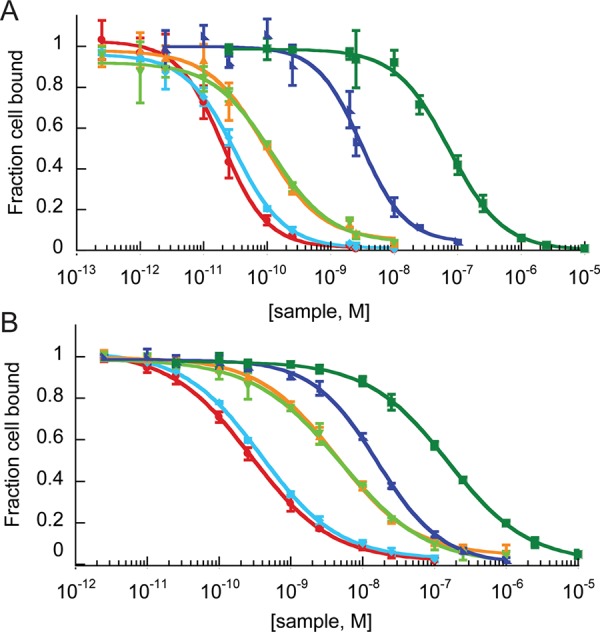
Competitive binding to integrin receptors expressed on (A) U87MG cells and (B) MDA-MB-231 cells by Alexa488–2.5F monomer and knottin monomer, dimers, or cilengitide, as measured by flow cytometry. The fraction of Alexa488–2.5F competitor bound is plotted vs the concentration of unlabeled knottin or cilengitide. Symbols and the corresponding IC50 values are presented in Table 1. Error bars represent standard deviations (SDs) of three replicates.
Our chemical cross-linking strategy was critical for generating high-affinity multimers, as dimeric knottins generated through genetic fusion of EETI 2.5F to an antibody Fc domain39 achieved only a modest increase (less than 5-fold) in apparent integrin binding affinity compared with the knottin monomer (Table 1 and Figure S5). Minimal cell surface binding was observed with a knottin monomer and dimer containing a scrambled integrin binding sequence14 (Figure S5). The highest-affinity knottin dimer, D2.5F_1, exhibited ∼150-fold and 60-fold increases in relative affinity compared with the knottin monomer and ∼3650-fold and 500-fold increases in relative affinity compared with cilengitide on U87MG and MDA-MB-231 cells, respectively (Figure 1 and Table 1). To our knowledge, D2.5F_1 exhibits the strongest apparent binding affinity achieved to date for a peptide-based integrin-targeting agent. D2.5F_1 was subsequently used to study the effects of ligand multimerization on tumor cell migration and proliferation.
We tested the ability of the D2.5F_1 dimer to inhibit cell migration compared with the knottin monomer and cilengitide. Untreated MDA-MB-231 cells did not appear to migrate significantly over a 24 h period, so we used the U87MG cell line for this assay. U87MG cells (4 × 104) were allowed to adhere to wells of a microtiter plate for 12 h. The resulting monolayer was scratched to create a void,40 and the remaining adherent cells were washed gently with medium and incubated with D2.5F_1 dimer, 2.5F monomer, or cilengitide (all at 1 nM) at 37 °C, 5% CO2 (Figure 2). Cell migration was quantified by measuring closure of the scratched area over time. Significant inhibition of migration was observed with U87MG cells treated with D2.5F_1 dimer compared with those treated with knottin monomer 2.5F, cilengitide, or control medium (Figure 2). The ability of knottin dimers to inhibit U87MG cell migration at low-nanomolar concentrations has potential implications for cancer metastasis, in which the ECM facilitates tumor cell migration to other areas of the body. Correlative with these results, we previously showed that EETI 2.5F effectively inhibits interactions between tumor cells and vitronectin and fibronectin,14 two key ECM components important for cell adhesion and migration.41
Figure 2.
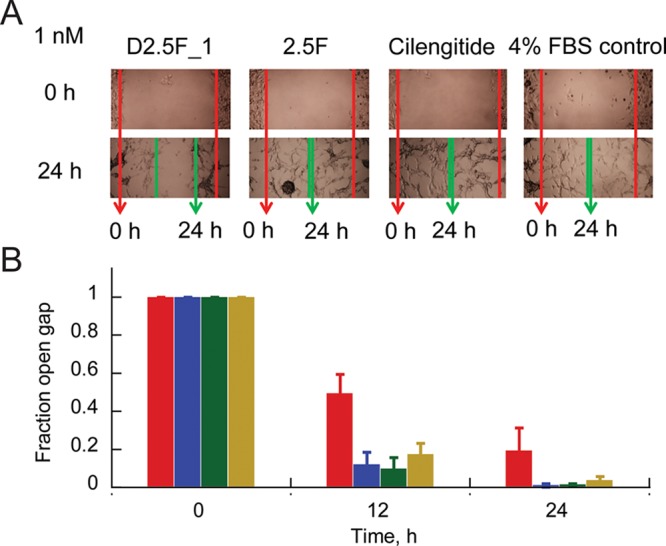
Effects of D2.5F_1 dimer, 2.5F monomer, and cilengitide on U87MG cell migration. (A) Bright-field images showing tumor cell migration into the monolayer defect at 24 h (green arrows) compared with the start of the assay (red arrows). (B) Bar graph quantifying gap closure at different time points for D2.5F_1 (red), 2.5F (blue), and cilengitide (green) relative to untreated control (gold) based on distances measured in (A). Error bars represent SDs of three replicates.
We next compared the abilities of D2.5F_1, knottin monomer, and cilengitide to inhibit cell proliferation. U87MG and MDA-MB-231 cells were incubated with varying concentrations of knottins or cilengitide for 12–72 h, and proliferation was measured by quantifying hydrogenase activity in living cells (Dojindo Cell Counting Kit-8 assay). In MDA-MB-231 cells, D2.5F_1 was significantly more potent at inhibiting cell proliferation compared with cilengitide, and the knottin monomer 2.5F displayed intermediate levels of inhibition (Figure 3). In contrast, none of the compounds significantly affected U87MG proliferation, despite a dramatic change in cell morphology after treatment with knottin dimer and a lesser change with knottin monomer (Figure S6). These results are consistent with previous studies showing resistance of U87MG cells to detachment-induced apoptosis.42,43
Figure 3.
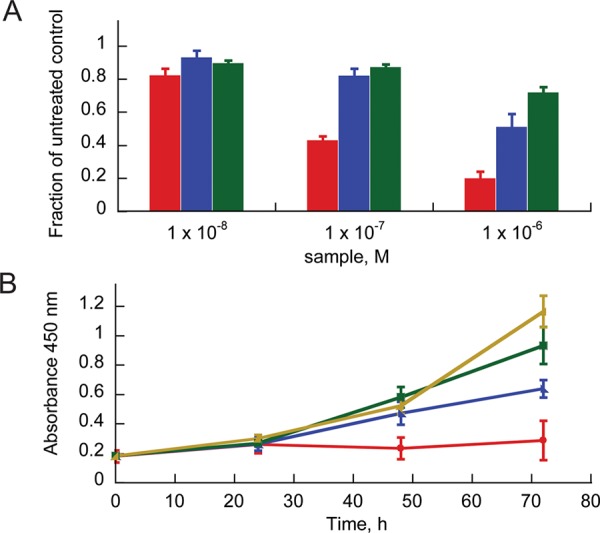
Effects of D2.5F_1 dimer (red), 2.5F monomer (blue), and cilengitide (green) on MDA-MB-231 cell proliferation relative to untreated control (gold). Integrin-binding knottins inhibit cell proliferation in both (A) concentration- and (B) time-dependent manners. Proliferation was measured at 72 h in (A) and using 1 μM protein in (B).
In conclusion, we have reported an effective chemical strategy for multimerization of knottin miniproteins through oxime formation between an introduced aminooxy functional group and a dialdehyde-functionalized cross-linker. Oxime cross-linking of knottin monomers affords homogeneous products in high yield after a simple purification step. This approach was validated using knottins with aminooxy groups incorporated at two different locations within the protein and cross-linkers of different lengths. Using this strategy, we created a dimeric knottin with unprecedented integrin binding affinity. This knottin dimer more strongly inhibited tumor cell migration and proliferation compared with its monomeric counterpart and cilengitide, potentially explaining the poor performance of the latter in recent clinical trials. To our knowledge, D2.5F_1 is the first RGD-containing compound with the ability to inhibit tumor cell migration and proliferation at nanomolar concentrations, suggesting promise for its development as a cancer therapeutic. Furthermore, this work demonstrates that a non-natural amino acid can be incorporated at different locations within a knottin without perturbing protein folding or function. While we have used the introduced aminooxy group to create covalent knottin dimers, this chemical function also enables site-specific orthogonal conjugation of small molecules or imaging probes for targeted drug delivery or diagnostic applications.44
Acknowledgments
This work was supported in part by NIH NCI R21CA143498 and the Stanford Bio-X Program. J.W.K. is grateful for graduate student support from the Stanford Bioengineering Department and the Stanford Bio-X Graduate Fellowship Program. The authors thank Dr. Corey Liu at Stanford Magnetic Resonance Laboratory for assistance with NMR analysis, Dr. Spencer Alford for helpful discussions, the Molecular Structure Facility at UC Davis for assistance with amino acid analysis, and the Stanford Vincent Coates Foundation Mass Spectrometry Laboratory for assistance with peptide analysis.
Supporting Information Available
Procedures and additional data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): The authors are listed as inventors on a pending patent application related to technology described in this work.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hynes R. O. Cell 1992, 69, 11. [DOI] [PubMed] [Google Scholar]
- Grassian A. R.; Coloff J. L.; Brugge J. S. Cold Spring Harbor Symp. Quant. Biol. 2011, 76, 313. [DOI] [PubMed] [Google Scholar]
- Mizejewski G. J. P. Soc. Exp. Biol. Med. 1999, 222, 124. [DOI] [PubMed] [Google Scholar]
- Brooks P. C.; Clark R. A. F.; Cheresh D. A. Science 1994, 264, 569. [DOI] [PubMed] [Google Scholar]
- Desgrosellier J. S.; Cheresh D. A. Nat. Rev. Cancer 2010, 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Bell K.; Mousa S. A.; Varner J. A. Am. J. Pathol. 2000, 156, 1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D.; Brennan M.; Moran N. Nat. Rev. Drug Discovery 2010, 9, 804. [DOI] [PubMed] [Google Scholar]
- Goodman S. L.; Picard M. Trends Pharmacol. Sci. 2012, 33, 405. [DOI] [PubMed] [Google Scholar]
- Pierschbacher M. D.; Ruoslahti E. Nature 1984, 309, 30. [DOI] [PubMed] [Google Scholar]
- Marelli U. K.; Rechenmacher F.; Sobahi T. R.; Mas-Moruno C.; Kessler H. Front. Oncol. 2013, 3, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaertner F. C.; Schwaiger M.; Beer A. J. Q. J. Nucl. Med. Mol. Imaging 2010, 54, 309. [PubMed] [Google Scholar]
- Mas-Moruno C.; Rechenmacher F.; Kessler H. Anti-Cancer Agents Med. Chem. 2010, 10, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele G.; Wick A.; Eisele A. C.; Clement P.; Tonn J.; Tabatabai G.; Ochsenbein A.; Schlegel U.; Neyns B.; Krex D.; Simon M.; Nikkhah G.; Picard M.; Stupp R.; Wick W.; Weller M. J. Neuro-Oncol. 2014, 117, 141. [DOI] [PubMed] [Google Scholar]
- Kimura R. H.; Levin A. M.; Cochran F. V.; Cochran J. R. Proteins 2009, 77, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman A. P.; Levin A. M.; Lahti J. L.; Cochran J. R. J. Mol. Biol. 2009, 385, 1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallaghy P. K.; Nielsen K. J.; Craik D. J.; Norton R. S. Protein Sci. 1994, 3, 1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Nguyen D.; Heitz A.; Chiche L.; Castro B.; Boigegrain R. A.; Favel A.; Coletti-Previero M. A. Biochimie 1990, 72, 431. [DOI] [PubMed] [Google Scholar]
- Werle M.; Schmitz T.; Huang H. L.; Wentzel A.; Kolmar H.; Bernkop-Schnurch A. J. Drug Targeting 2006, 14, 137. [DOI] [PubMed] [Google Scholar]
- Colgrave M. L.; Craik D. J. Biochemistry 2004, 43, 5965. [DOI] [PubMed] [Google Scholar]
- Kimura R. H.; Cheng Z.; Gambhir S. S.; Cochran J. R. Cancer Res. 2009, 69, 2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen M.; Choi S.-K.; Whitesides G. M. Angew. Chem., Int. Ed. 1998, 37, 2754. [DOI] [PubMed] [Google Scholar]
- Kiessling L. L.; Gestwicki J. E.; Strong L. E. Curr. Opin. Chem. Biol. 2000, 4, 696. [DOI] [PubMed] [Google Scholar]
- Cheng Z.; Wu Y.; Xiong Z.; Gambhir S. S.; Chen X. Bioconjugate Chem. 2005, 16, 1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Hsieh W. Y.; Jiang Y.; Kim Y. S.; Sreerama S. G.; Chen X.; Jia B.; Wang F. Bioconjugate Chem. 2007, 18, 438. [DOI] [PubMed] [Google Scholar]
- Dijkgraaf I.; Rijnders A. Y.; Soede A.; Dechesne A. C.; van Esse G. W.; Brouwer A. J.; Corstens F. H.; Boerman O. C.; Rijkers D. T.; Liskamp R. M. Org. Biomol. Chem. 2007, 5, 935. [DOI] [PubMed] [Google Scholar]
- Liu S. Bioconjugate Chem. 2009, 20, 2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garanger E.; Boturyn D.; Coll J. L.; Favrot M. C.; Dumy P. Org. Biomol. Chem. 2006, 4, 1958. [DOI] [PubMed] [Google Scholar]
- Kok R. J.; Schraa A. J.; Bos E. J.; Moorlag H. E.; Asgeirsdottir S. A.; Everts M.; Meijer D. K.; Molema G. Bioconjugate Chem. 2002, 13, 128. [DOI] [PubMed] [Google Scholar]
- Thumshirn G.; Hersel U.; Goodman S. L.; Kessler H. Chem.—Eur. J. 2003, 9, 2717. [DOI] [PubMed] [Google Scholar]
- Janssen M.; Oyen W. J.; Massuger L. F.; Frielink C.; Dijkgraaf I.; Edwards D. S.; Radjopadhye M.; Corstens F. H.; Boerman O. C. Cancer Biother. Radiopharm. 2002, 17, 641. [DOI] [PubMed] [Google Scholar]
- Rose K. J. Am. Chem. Soc. 1994, 116, 30. [Google Scholar]
- Shao J.; Tam J. P. J. Am. Chem. Soc. 1995, 117, 3893. [Google Scholar]
- Canne L. E.; Ferredamare A. R.; Burley S. K.; Kent S. B. H. J. Am. Chem. Soc. 1995, 117, 2998. [Google Scholar]
- Dirksen A.; Dawson P. E. Bioconjugate Chem. 2008, 19, 2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause S.; Schmoldt H. U.; Wentzel A.; Ballmaier M.; Friedrich K.; Kolmar H. FEBS J. 2007, 274, 86. [DOI] [PubMed] [Google Scholar]
- Bruning A.; Runnebaum I. B. Gene Ther. 2003, 10, 198. [DOI] [PubMed] [Google Scholar]
- Taherian A.; Li X.; Liu Y.; Haas T. A. BMC Cancer 2011, 11, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore S. J.; Hayden Gephart M. G.; Bergen J. M.; Su Y. S.; Rayburn H.; Scott M. P.; Cochran J. R. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 14598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitorino P.; Meyer T. Gene Dev. 2008, 22, 3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas T. A.; Plow E. F. Curr. Opin. Cell Biol. 1994, 6, 656. [DOI] [PubMed] [Google Scholar]
- Galli R.; Binda E.; Orfanelli U.; Cipelletti B.; Gritti A.; De Vitis S.; Fiocco R.; Foroni C.; Dimeco F.; Vescovi A. Cancer Res. 2004, 64, 7011. [DOI] [PubMed] [Google Scholar]
- Westhoff M. A.; Zhou S.; Bachem M. G.; Debatin K. M.; Fulda S. Oncogene 2008, 27, 5169. [DOI] [PubMed] [Google Scholar]
- a Axup J. Y.; Bajjuri K. M.; Ritland M.; Hutchins B. M.; Kim C. H.; Kazane S. A.; Halder R.; Forsyth J. S.; Santidrian A. F.; Stafin K.; Lu Y. C.; Tran H.; Seller A. J.; Biroce S. L.; Szydlik A.; Pinkstaff J. K.; Tian F.; Sinha S. C.; Felding-Habermann B.; Smider V. V.; Schultz P. G. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 16101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fleissner M. R.; Brustad E. M.; Kalai T.; Altenbach C.; Cascio D.; Peters F. B.; Hideg K.; Peuker S.; Schultz P. G.; Hubbell W. L. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 21637. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Garbuio L.; Bordignon E.; Brooks E. K.; Hubbell W. L.; Jeschke G.; Yulikov M. J. Phys. Chem. B 2013, 117, 3145. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Li X. G.; Haaparanta M.; Solin O. J. Fluorine Chem. 2012, 143, 49. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.