

Abstract
Food consumption is an important behavior that is regulated by an intricate array of neuropeptides (NPs). Although many feeding-related NPs have been identified in mammals, precise mechanisms are unclear and difficult to study in mammals, as current methods are not highly multiplexed and require extensive a priori knowledge about analytes. New advances in data-independent acquisition (DIA) MS/MS and the open-source quantification software Skyline have opened up the possibility to identify hundreds of compounds and quantify them from a single DIA MS/MS run. An untargeted DIA MSE quantification method using Skyline software for multiplexed, discovery-driven quantification was developed and found to produce linear calibration curves for peptides at physiologically relevant concentrations using a protein digest as internal standard. By using this method, preliminary relative quantification of the crab Cancer borealis neuropeptidome (<2 kDa, 137 peptides from 18 families) was possible in microdialysates from 8 replicate feeding experiments. Of these NPs, 55 were detected with an average mass error below 10 ppm. The time-resolved profiles of relative concentration changes for 6 are shown, and there is great potential for the use of this method in future experiments to aid in correlation of NP changes with behavior. This work presents an unbiased approach to winnowing candidate NPs related to a behavior of interest in a functionally relevant manner, and demonstrates the success of such a UPLC-MSE quantification method using the open source software Skyline.
Keywords: Data-independent acquisition, mass spectrometry, neuropeptide, microdialysis, feeding, quantification
Introduction
Neuropeptides in Feeding
Neuropeptides (NPs) are known to play an important role in regulation of feeding behavior in a variety of organisms. Several NPs have been identified as modulating food consumption on a number of different levels in mammalian systems, including galanin, ghrelin, neuropeptide Y, melanin-concentrating hormone (MCH), alpha-melanocyte stimulating hormone (α-MSH), orexins, oxytocin, insulin, leptin, cholecystokinin (CCK), agouti-related protein (AgRP), neurotensin (NT), neuropeptide W, neuropeptide YY (PYY), and cocaine and amphetamine regulated transcript (CART).1−3 These NPs can act locally as neurotransmitters or at a distance in an endocrine manner.4,5
Although much progress has been made in understanding the effects of NPs on consumption behavior, the mechanisms by which a chemical signal acts to produce a given behavior are not well understood. One challenge lies in understanding behavior generation in the complex brains of animal models such as rodents or primates; another lies in difficulties to accurately characterize NP changes on a physiologically relevant time scale. A number of diverse signals converge on feeding behavior, including reward circuitry, stress signaling, metabolic state signals, somatosensation, and multiple hypothalamic subnuclei.2 Thus, the decision to eat is influenced not only by how “hungry” one is (stomach fullness and/or circulating glucose levels), but also how much one derives pleasure from food, environmental effects, the type of food, whether other rewarding stimuli are present, and so forth. A major concern in this area of research is dissecting the pathways of feeding for energy maintenance and feeding for pleasure (homeostatic vs hedonic), and it appears NPs are involved in both pathways in mammals.3 Second, NPs can be difficult to quantify accurately due to the presence of alternative splice forms and antibody cross-reactivity, and assessing them on a physiologically relevant time scale can also be a challenge.6
With a simple nervous system and more limited suite of behaviors, decapod crustaceans are an attractive model for ingestive behavior. In the past, crabs have been conditioned to press a lever to receive a food reward.7 The crustacean stomatogastric ganglion (STG) and other ganglia form an essential part of well-defined neural circuits responsible mainly for rhythmic movements of the gut in the crab. This well-established model for understanding rhythmic neuron firing and the effects of neuromodulators on activity has been extensively studied and can be modeled using computational neuroscience tools.8,9 Thus, putative NPs can be applied in this system experimentally, and probed for modulation of this critical neural circuit. Certain firing patterns in this circuit are associated with chewing or filtering of food, and thus can be correlated directly to feeding behavior. In addition to providing well-defined circuitry to study NP action, the decapod crustacean also contains major neuroendocrine organs that are rich in NPs, making them amenable for analysis by mass spectrometry (MS)-based techniques.6
MS-Based Techniques for NP Analysis
Tandem mass spectrometry (MS/MS) is a highly useful analytical tool for both identification of many molecules, especially neuropeptides, and their quantification. However, it typically cannot conduct both identification and quantification simultaneously, because different parameters are typically required for each type of experiment. There are several reasons different parameters would be required for identification and quantification, but the unifying feature is that mass filters (use of the internal electronics of a mass spectrometer to permit only certain ions to pass through for subsequent analysis) are typically used for both approaches, albeit in different ways. These mass filters prevent monitoring of multiple fragments in multiple-reaction monitoring (MRM)-type experiments, and thus preclude identification, or cause too few data points to be obtained for each peak, and thus preclude quantitation. Notable exceptions exist, such as spectral counting and tandem mass tags, but those methods have disadvantages for this study. For MS/MS experiments with high scan rates, the number of times an ion is selected for MS/MS analysis can be used as a measure of its abundance, called its spectral count.10 This method is most robust for quantification at the protein level (as opposed to the peptide level) based on quantification of multiple peptides identified from the same protein, thus is less applicable for the study of endogenous neuropeptides (NPs), which are often present at very low levels and thus can be less reproducible in the frequency with which they trigger a MS/MS scan. Although spectral count has been employed for the quantitative study of NPs across different conditions,11 it was conducted with relatively high sample concentrations: the equivalent of eight rat brain punches pooled into one sample. This improves the reproducibility of MS/MS scan switching by greatly increasing the NP concentration. A NP-compatible approach incorporates isobaric labeling tags at the MS1 level which could produce specific reporter ions upon MS2 fragmentation to offer quantitative information. One example of this approach is DiLeu labeling tags developed by the Li laboratory.12 These tags use isotopes to generate characteristic fragment ions of given masses, and the relative intensities of these ions can be used for quantification. However, high intensity precursor ions must be generated, and this is often not possible with the study of NPs due to their extremely low endogenous concentrations. Finally, several transitions can be monitored with full fragment scans in some high-speed instruments, allowing for simultaneous identification and quantification of those predefined transitions with Skyline SRM software.13 This method will be expanded upon in the current study using data-independent acquisition (DIA) MS analysis, with truly unbiased collection of MSE (MS collected with a different collision Energy) data. A priori knowledge about the analytes of interest will not be used until postacquisition data processing, in order to maximize the potential future utility of acquired data. As novel NPs are discovered with the advent of new analytical technologies, this approach is preferred for analysis in this emerging area of chemical neuroscience.
DIA MS analysis employs no mass filter but produces both precursor and fragment ions. This increases the complexity of data interpretation, but it is possible to quantitate the product ions of specific precursors in a pseudomultiple reaction monitoring (pMRM), or all-reactions monitoring (ARM), manner, as well as to determine identity in a single run. By eliminating mass filtering at the MS1 level (or using large isolation windows), multiple precursors are fragmented simultaneously and their resulting fragments enter the second mass analyzer simultaneously. This results in a highly complex MS/MS spectrum as the fragments cannot initially be assigned to precursors for sequence identification. However, precursors and fragments can be aligned by software using their retention times and mass to charge ratios (m/z). By eliminating filtering at the MS2 level, the intensities of all of these fragments over time can be determined for quantification. By matching precursors and fragments, then quantifying the fragments of a given precursor, all fragmentation reactions are monitored (all reactions monitoring, ARM). Every peptide with several transitions observed can thus be quantified. Several instrument vendors have incorporated this technique into new instruments with slight variations, and the one described and used here is Waters MSE.
This powerful acquisition technique has been employed for simultaneous identification and quantification in a variety of samples at the protein level,14−18 with each protein having multiple peptides detected. However, quantification has not previously been done with analytes at the peptide level. In addition, the SRM software Skyline19,20 has not previously been demonstrated in quantitation of DIA MS/MS data. In this work, the MSE strategy for DIA MS/MS is employed to collect data for quantitation and identification of (neuro)peptides, using a protein digest as internal standard and Skyline software for data analysis. This technique is found to be linear for quantification of several model NP standards, and proof-of-principle experiments were conducted to analyze NP concentration dynamics during feeding in microdialysates obtained from Jonah crab Cancer borealis. Using this method, the entire C. borealis < 2 kDa neuropeptidome (137 NPs from 18 families) was subjected to relative quantification in each of eight replicate feeding experiments. It would be impractical or impossible to obtain information about this many NPs using antibody-based techniques, and some of the slight changes in NP sequence that were able to be differentiated by MS-based techniques would not be detectable by nonmolecular means. This method represents an early stage function-driven discovery method to winnow bioactive peptides related to an experimental perturbation (here, feeding) from a large list (here, the <2 kDa neuropeptidome of C. borealis). This novel approach shows great potential in the application of function-driven discovery neuropeptidomics experiments.
Results and Discussion
Linearity for Quantification of Neuropeptide Standards
Plotting the peak areas for the NP standards, normalized to the sum of the areas for the myoglobin tryptic peptides versus the concentrations and performing linear regressions yielded fits with a high degree of linearity over the range of 0.25–10 nM (Figure 1, Table 1). Values for the goodness of fit for these regressions range from 0.9144 to 0.9870. These results indicate that accurate measurement of individual peptide concentrations is possible by making enough measurements of its intensity through analyzing multiple transitions for each of two charge states per peptide (2+, 7 transitions; 3+, 13 transitions). The inclusion of an internal standard (ITSD) with multiple peptides that can be monitored allows for even more measurements of its intensity to be made, and thus, this value is more reproducible. For instance, if interference, signal loss, or artificial enhancement occurs with one of the multiple peptides in some samples, the impact of one irregular measurement is reduced by having other unchanged measurements also possible with the same ITSD. It could also be possible to use several peptide standards spiked into the sample as an ITSD in a similar manner. The method is very sensitive, generated over the range 0.25–10 nM (0.5–20 fmol on column), enabling analysis of NPs in microdialysates obtained from crab hemolymph. This approach therefore permits rapid relative quantification of NPs.
Figure 1.

Linear calibration curves for (A) α-melanocyte stimulating hormone (α-MSH), (B) bradykinin, (C) crustacean cardioactive peptide (CCAP), (D) Homarus americanus FMRFamide-like peptide I (FLP I), (E) H. americanus FMRFamide-like peptide II (FLP II), and (F) substance P over the concentration range 0.25–10 nM (0.5–20 fmol on column). Derived from UPLC-MSE data processed with Skyline.
Table 1. Parameters for Linear Fits of Calibration Curves for α-Melanocyte Stimulating Hormone (α-MSH), Bradykinin (BK), Crustacean Cardioactive Peptide (CCAP), Homarus americanus FMRFamide-Like Peptide I (FLP I), H. americanus FMRFamide-Like Peptide II (FLP II) and Substance P (SP).
| peptide | slope value | SE | intercept value | SE | R2 |
|---|---|---|---|---|---|
| α-MSH | 0.0086 | 0.0004 | 0.0024 | 0.0200 | 0.9870 |
| BK | 0.0121 | 0.0010 | 0.0076 | 0.0045 | 0.9679 |
| CCAP | 0.0081 | 0.0008 | 0.0178 | 0.0037 | 0.9533 |
| FLP I | 0.0049 | 0.0004 | 0.0072 | 0.0020 | 0.9626 |
| FLP II | 0.0168 | 0.0012 | 0.0056 | 0.0057 | 0.9733 |
| SP | 0.0066 | 0.0009 | 0.0309 | 0.0041 | 0.9144 |
Microdialysis Feeding Experiment
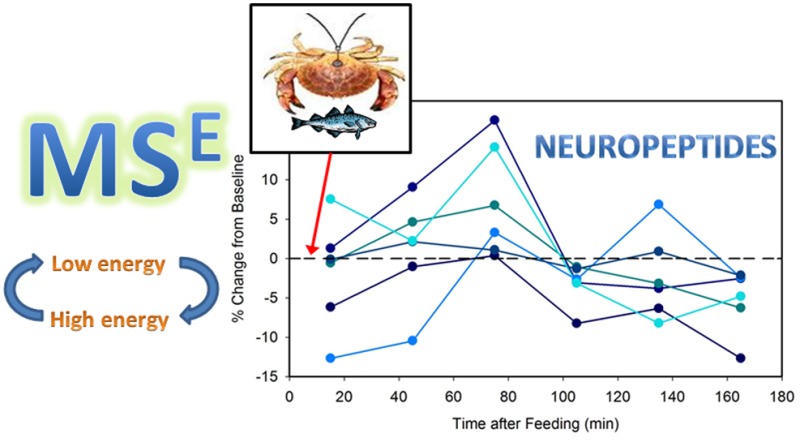
The method was successful at generating relative concentration measurements of NPs in multiple samples from eight experimental replicates from a total of four crabs. A list of the 137 known C. borealis neuropeptides of mass <2 kDa was input into the Skyline software package using DIA quantitation mode. Transitions for the 2+ and 3+ precursors to the parent, bn–3, bn–2, bn–1, yn–3, yn–2, and yn–1 ions were considered. An automatic peak scoring function of Skyline that employs mProphet was used to pick peaks for integration.21 In every sample corresponding to a single feeding experiment time point, the peak areas for each peptide were summed and divided by the total myoglobin internal standard peak area to correct for run-to-run variability and permit relative quantification. The Skyline file containing all data is available for download from Panorama28 at the following URL: https://panoramaweb.org/labkey/project/Li%20Lab%2C%20U.%20of%20Wisconsin-Madison/Cancer%20borealis%20feeding%20MSE%20paper/begin.view. Fifty-five NPs were quantified with less than 10 ppm error across all samples. Although absolute concentrations cannot be determined without generating calibration equations like those above for every NP, which would require the use of authentic standards of known concentration for all 137 appropriate compounds, the general concentration ranges for these NPs are estimated to be 1–100 nM. Relative concentration changes across time can be analyzed using the resulting data. Although this method was successful at providing information on the NP content of the microdialysates, variability was high between animals. This could be due to high background from the protein digest ITSD, or slight differences in the feeding experiment itself. For instance, crabs did not reliably eat during the same time periods of the test. A crab would be given food after having none for at least 24 h, and it had to start eating (as evidenced by mouth part movement) for the experiment to be initiated. However, the duration of eating, number of eating bouts, and timing of those bouts were not consistent across animals. Typically, the initial feeding bout would last 30 min, followed by a 15 min period without eating. The crab then usually started another feeding bout, of a slightly shorter duration. Rarely, the crab would eat a third time. This heterogeneity of behavioral patterns is likely to also result in heterogeneous neurochemical changes. A better understanding of crab feeding behavioral patterns, better time resolution for microdialysis, and/or concurrent video recording of feeding followed by scoring of feeding versus quiescent behavior (to be later correlated with MD sample contents) would improve the quality of information that can be obtained from such a study. The NP content (represented as % change from baseline) versus time graphs for several selected peptides are shown in Figure 2. It should be noted that although several of these NPs share sequence motifs and thus some y-ions, their profiles appear different. It would be highly unlikely that non-MS based techniques could differentiate between these compounds. Although statistical tests did not reveal clear associations between NP content and feeding (data not shown), this might be observed following further refinement of the behavioral protocol (perhaps by adding video analysis) and analysis methods (especially increasing NP detection sensitivity and decreasing background signal by using a less complex peptide mixture as ITSD).
Figure 2.

Changes in the concentrations of 6 selected NPs during the feeding experiment. Samples were collected via microdialysis on a total of four crabs during eight separate experiments. Values shown are means ± SEMs.
Conclusions and Future Work
In this work, a method for quantitation of DIA MS/MS data in a pseudo-MRM manner was developed. It used an open-source, vendor-neutral software for accurate identification of precursors and fragments and integration of the resulting peaks. This quantitation method shows linearity for mixtures of peptide standards over the range 0.25–10 nM using a myoglobin digest as an internal standard. A proof-of-principle experiment was also conducted to determine which NPs are associated with feeding via an unbiased approach, looking at all C. borealis NPs below 2 kDa. Samples were collected using microdialysis from C. borealis during feeding. For each of 7 time points in 8 experimental replicates (4 biological replicates), all 137 known C. borealis NPs and the ITSD peptides were quantified. Fifty-five NPs were quantified with an average mass error of less than 10 ppm. This data set can further be reassessed for quantity changes of other NPs of interest (for instance, if new NPs are discovered), due to the nature of MSE acquisition and Skyline quantification methods. Time-resolved profiles of concentration changes in the 3 h after feeding can also be generated from this data. Additional biological replicates may further illuminate interesting roles for other NPs in feeding behavior. Further refinement of the experiment to better define feeding behavior may permit a better correlation between NP concentration changes and important aspects of feeding behavior. We also suggest additional technical improvements, such as the use of a mixture of reference peptides instead of a messy protein digest as the ITSD and the addition of a DDA-experimental step (run on pooled MD samples following their analysis by DIA) to improve identification confidence. However, in this paper we demonstrate that it is possible to take samples from a species for which a database of the proteins or peptides of interest exists and quantify compounds at the peptide level using this method. This method will be of great use in targeted and untargeted functional analysis studies of NPs.
Methods
Linearity Experiment Sample Preparation
A set of standards to test the linearity of the method was prepared using myoglobin digest and peptide standards. Equine skeletal myoglobin (ERA, Colden, CO) was digested with trypsin (from bovine pancreas, Sigma-Aldrich, St. Louis, MO) following a published procedure.22 Myoglobin was dissolved in 100 mM NH4HCO3 at 0.5 mg/mL. Trypsin was dissolved in the same solution at 1 μg/mL and added to the myoglobin solution at an enzyme/substrate ratio of 1:10. This was then diluted 1:1 with methanol and placed in a water bath at 37 °C for 45 min. The reaction was stopped by adding ice-cold acetic acid to a final concentration of 5%. The sample was then spun at 15 100g for 5 min. The neuropeptide standards, crustacean cardioactive peptide (CCAP), Homarus americanus FMRFamide-like peptide I (FLP I), H. americanus FMRFamide-like peptide II (FLP II), alpha-melanocyte stimulating hormone (α-MSH), bradykinin, and substance P, were obtained from American Peptide Company (Sunnyvale, CA). These were spiked into a solution of 1× diluted crab saline (220 mM NaCl; 5.5 mM KCl; 6.5 mM CaCl2; 13 mM MgCl2; 5 mM HEPES, pH 7.4) with 0.05% formic acid and 1.88 μM myoglobin digest at the following concentrations: 10, 5, 3.75, 2.5, 1.25, 0.63, and 0.25 nM. Samples were mixed in a 96-well plate and kept at 4 °C until analysis.
In Vivo Microdialysis
PAES (20 kDa MWCO, 4 mm membrane length) probes (CMA Microdialysis, Harvard Apparatus, Holliston, MA) were implanted in four separate C. borealis as described in previous work.23,24 Briefly, crabs were anesthetized with ice, the shell in the pericardial region was abraded, a hole was drilled in the shell, and the MD probe was inserted. Several layers of adhesive were employed to obtain a watertight seal. Following implantation, crabs with probes were kept separate from other animals with the aid of plexiglass dividers that the water could flow through and around. The outlet of the probe was connected to an automated refrigerated fraction collector (BASi Automated Refrigerated Fraction Collector, Bioanalytical Systems Inc., West Lafayette, IN). Animals were allowed to recover for a minimum of 24 h prior to collecting samples that were used for analysis. Crabs typically would not eat until a minimum of 48 h had elapsed after surgery, so feeding experiments were conducted beginning the second day after surgery. Crab saline was infused through the system at a flow rate of 0.5 μL/min, and the probe was equilibrated at this rate for a minimum of 30 min before any samples were collected. In many cases, the experiment was started after the perfusate had been running at that rate for several hours and equilibration was already complete. If the rate had to be changed or the syringe refilled, 30 min of equilibration time immediately preceded collection of samples for the feeding experiment. The dead volume of the probe system from the tip of the probe to the collection tube was calculated based on information from the manufacturers of the probe, tubing, and fraction collector, and this was taken into account for timing collections correctly with feeding. Crabs were fed white-fleshed raw fish (cod, tilapia, etc.), thawed and cut into small pieces (1/2″–1″) immediately prior to use. Most crabs took and started eating the food immediately. If it took more than 2–3 min for the crab to start eating, the experiment was abandoned for that day. For crabs that did eat, excess food was always present so that the animal could feed until satiety was reached. Crabs typically stopped eating (as judged by reduced movement of mouth parts) after 45–60 min. At t = 180 min, any remaining food was removed from the tank, even if the crab had to be moved to retrieve it. A second feeding experiment was conducted with most of the crabs a minimum of 24 h after the start of the first one. Crabs were not otherwise disturbed at any point during this process, and the room was maintained on a 12 h on/12 h off (red lights on) light cycle, with feeding experiments taking place during the dark period.
For every experiment, one baseline sample was obtained from t = −30 min to t = 0 min. The first baseline sample was collected approximately 2.5 h after the lights turned off. Collection tubes were changed every 30 min during the experiment by the refrigerated fraction collector until seven samples were obtained, for a final time of 180 min postfeeding. Samples were graphed according to the time midway through their collection time. The resulting 15 μL samples were immediately acidified by adding 1.5 μL of pure formic acid (FA) to them, bringing their acid content to approximately 10%, as this has been shown to reduce NP degradation.25 The collected microdialysates were then kept at −20 °C until they could be analyzed. The feeding experiment was conducted once on each of two crabs and twice on each of two crabs, for a total number of replicates of eight. Samples were placed in LC vials, and an internal standard (1 μL of 56.4 μM myoglobin digest solution) was added. Immediately prior to analysis on UPLC-MS/MS, 1 μL of a 0.91 M NH4HCO3 solution was added to increase the sample’s pH so as not to damage the UPLC column.
Instrumental Analysis
Ultrahigh performance liquid chromatography (UPLC)-MSE analysis was conducted on a Waters nanoAcquity UPLC coupled to a Waters Synapt G2 quadrupole-time-of-flight (QTOF) mass spectrometer (Waters, Milford, MA). Samples (2 μL) were trapped on a preconcentration column and desalted online. This column was then put in line with a C18 reversed-phase column (BEH130 C18, 1.7 μm, 75 μm × 100 mm, Waters, Milford, MA). The outlet of this column was connected to a fused silica capillary with a pulled tip of internal diameter ∼5 μm (Sutter Instrument Company, Novato, CA). This was used as the ESI inlet. A 75 min reversed-phase run with solvent A as 0.1% FA in water and B as 0.1% FA in ACN was used. The instrument was operated in data-independent MS/MS mode with the high energy scan having a voltage ramp from 25 to 65 V and Glu-fibrinopeptide was infused for lockspray calibration.
Data Analysis
Linearity Samples Analysis
The sequences of the neuropeptide standards (CCAP, PFC(−H)NAFTGC(−H)amide; FLP I, SDRNFLRFamide; FLP II, TNRNFLRFamide; α-MSH, acetyl-SYSMEHFRWGKPVamide; bradykinin, RPPGFSPFR; and substance P, RPKPQQFGGLMamide, with (−H) indicating a C in a disulfide bond that loses a hydrogen) and nine myoglobin internal standard (ITSD) tryptic peptides (FDKFK, FKHLK, TEAEMK, TEAEM(Ox)K, TEAEMKASEDLK, ALEFLR, ALEFRNDIAAK, NDIAAK, and ELGFQG, with M(O) indicating an oxidized methionine) were input into the Skyline SRM software (version 2.6.0.6851). Modifications were added to peptides manually. The iRT function with retention time predictor was used,26 calibrated off of results from Mascot (Matrix Science, London, U.K.) identifications of equine myoglobin tryptic peptides in a previous LC run of the myoglobin digest alone under the same UPLC conditions. The 2+ and 3+ precursors were monitored to the six most abundant 1+ and 2+ b and y ions. MS/MS filtering parameters were set to DIA with isolation set as “MSe” and 10 000 resolving power. Peak integration was checked manually for a good match to the most abundant transitions and agreement with the predicted retention times. Between runs, which were all conducted sequentially on the same day, variation of less than 0.2 min was observed in retention time. Peak areas were exported to Microsoft Excel. All transitions for each peptide were summed, and the nine myoglobin peptides were summed together. Peptide peak areas were normalized to the total myoglobin internal standard peak area. In SigmaPlot 10 (Systat Software, Inc. San Jose, CA), data was plotted and linear regressions to the equation f = y0 + ax, where y0 is the y-intercept and a is the slope, using a least-squares method were conducted.
Microdialysis Feeding Study Data Analysis
A list of all NPs previously identified in C. borealis and the nine myoglobin tryptic peptides mentioned above was selected for data extraction and integration. The iRT function with retention time predictor was used, calibrated off of results from Mascot (Matrix Science) identifications of equine myoglobin tryptic peptides in a previous LC run of the myoglobin digest alone under the same UPLC conditions. Other parameters were identical to those used in the linearity experiment. Peak picking and integration was conducted with the aid of the mProphet integration refinement feature, using second-best peaks to train the model. A Skyline file used for peak picking and integration, with all relevant software settings, integrated NPs, and data scans included, is available for download from Panorama28 at the following URL: https://panoramaweb.org/labkey/project/Li%20Lab%2C%20U.%20of%20Wisconsin-Madison/Cancer%20borealis%20feeding%20MSE%20paper/begin.view. Peak areas were exported to Microsoft Excel. All transitions for each peptide were summed, and the nine myoglobin peptides were summed together. Peptide peak areas were divided by the total myoglobin internal standard peak area. This data was explored using RMANOVA in SPSS 21 (IBM, Armonk, NY) software (data not shown). The percent change from baseline was calculated as % change = (value – baseline)/(baseline). This software also used to calculate the means and standard errors in the percent changes from baseline that are depicted in Figure 2, generated with SigmaPlot 10 (Systat Software, Inc.).
Acknowledgments
Heidi L. Behrens is acknowledged for initially developing the crab microdialysis technique24,27 and training.
Author Contributions
C.M.S. designed and carried out experiments and wrote the paper. Experiments were carried out while C.M.S. was a member of L.L.’s research group at the University of Wisconsin—Madison. Statistical analyses and writing occurred while C.M.S. was employed at Duke University Medical Center. Z.L. aided in experimental procedures and paper revision. L.L. was responsible for the initial ideas and theory, and revised the paper.
C.M.S. acknowledges NIH training Grant 5T32GM08349. This work was supported by NIH grants R01DK071801 and R56DK071801 (to L.L.). L.L. acknowledges an H. I. Romnes Faculty Research Fellowship.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Fulton S. (2010) Appetite and reward. Front. Neuroendocrinol. 31, 85–103. [DOI] [PubMed] [Google Scholar]
- Parker J. A.; Bloom S. R. (2012) Hypothalamic neuropeptides and the regulation of appetite. Neuropharmacology 63, 18–30. [DOI] [PubMed] [Google Scholar]
- Menzies J. W., Skibicka K., Egecioglu E., Leng G., and Dickson S. (2012) Peripheral Signals Modifying Food Reward, In Appetite Control (Joost H.-G., Ed.), pp 131–158, Springer, Berlin/Heidelberg. [DOI] [PubMed] [Google Scholar]
- Skibicka K. P.; Dickson S. L. (2011) Ghrelin and food reward: The story of potential underlying substrates. Peptides 32, 2265–2273. [DOI] [PubMed] [Google Scholar]
- Thompson J. L.; Borgland S. L. (2011) A role for hypocretin/orexin in motivation. Behav. Brain Res. 217, 446–453. [DOI] [PubMed] [Google Scholar]
- Li L.; Sweedler J. V. (2008) Peptides in the brain: Mass spectrometry-based measurement approaches and challenges. Annu. Rev. Anal. Chem. 1, 451–483. [DOI] [PubMed] [Google Scholar]
- Abramson C. I.; Feinman R. D. (1990) Lever-press conditioning in the crab. Physiol. Behav. 48, 267–272. [DOI] [PubMed] [Google Scholar]
- Stein W. (2009) Modulation of stomatogastric rhythms. J. Comp. Physiol. 195, 989–1009. [DOI] [PubMed] [Google Scholar]
- Marder E.; Bucher D. (2007) Understanding Circuit Dynamics Using the Stomatogastric Nervous System of Lobsters and Crabs. Annu. Rev. Physiol. 69, 291–316. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Lemeer S.; Savitski M. M.; Kuster B. (2012) Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal. Bioanal. Chem. 404, 939–965. [DOI] [PubMed] [Google Scholar]
- Southey B. R.; Lee J. E.; Zamdborg L.; Atkins N.; Mitchell J. W.; Li M.; Gillette M. U.; Kelleher N. L.; Sweedler J. V. (2014) Comparing Label-Free Quantitative Peptidomics Approaches to Characterize Diurnal Variation of Peptides in the Rat Suprachiasmatic Nucleus. Anal. Chem. 86, 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang F.; Ye H.; Chen R.; Fu Q.; Li L. (2010) N,N-dimethyl leucines as novel isobaric tandem mass tags for quantitative proteomics and peptidomics. Anal. Chem. 82, 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrod S. D.; Myers M. V.; Li M.; Myers J. S.; Carpenter K. L.; MacLean B.; MacCoss M. J.; Liebler D. C.; Ham A.-J. L. (2012) Label-Free Quantitation of Protein Modifications by Pseudo Selected Reaction Monitoring with Internal Reference Peptides. J. Proteome Res. 11, 3467–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geromanos S. J.; Vissers J. P. C.; Silva J. C.; Dorschel C. A.; Li G.-Z.; Gorenstein M. V.; Bateman R. H.; Langridge J. I. (2009) The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependant LC-MS/MS. Proteomics 9, 1683–1695. [DOI] [PubMed] [Google Scholar]
- Gillet L. C.; Navarro P.; Tate S.; Rost H.; Selevsek N.; Reiter L.; Bonner R.; Aebersold R. (2012) Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin Y.; Hradetzky E.; Bahn S. (2011) Quantification of proteins using data-independent analysis (MSE) in simple andcomplex samples: a systematic evaluation. Proteomics 11, 3273–3287. [DOI] [PubMed] [Google Scholar]
- Martins-de-Souza D.; Guest P. C.; Guest F. L.; Bauder C.; Rahmoune H.; Pietsch S.; Roeber S.; Kretzschmar H.; Mann D.; Baborie A.; Bahn S. (2012) Characterization of the human primary visual cortex and cerebellum proteomes using shotgun mass spectrometry-data-independent analyses. Proteomics 12, 500–504. [DOI] [PubMed] [Google Scholar]
- Mbeunkui F.; Goshe M. B. (2011) Investigation of solubilization and digestion methods for microsomal membrane proteome analysis using data-independent LC-MSE. Proteomics 11, 898–911. [DOI] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J. (2010) Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling B.; Rardin M. J.; MacLean B. X.; Zawadzka A. M.; Frewen B. E.; Cusack M. P.; Sorensen D. J.; Bereman M. S.; Jing E.; Wu C. C.; Verdin E.; Kahn C. R.; Maccoss M. J.; Gibson B. W. (2012) Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: Application to protein acetylation and phosphorylation. Mol. Cell. Proteomics 11, 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter L.; Rinner O.; Picotti P.; Huttenhain R.; Beck M.; Brusniak M. Y.; Hengartner M. O.; Aebersold R. (2011) mProphet: Automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods 8, 430–435. [DOI] [PubMed] [Google Scholar]
- Li F.; Schmerberg C. M.; Ji Q. C. (2009) Accelerated tryptic digestion of proteins in plasma for absolute quantitation using a protein internal standard by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 23, 729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmerberg C. M.; Li L. (2013) Mass spectrometric detection of neuropeptides using affinity-enhanced microdialysis with antibody-coated magnetic nanoparticles. Anal. Chem. 85, 915–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens H. L.; Chen R.; Li L. (2008) Combining microdialysis, NanoLC-MS, and MALDI-TOF/TOF to detect neuropeptides secreted in the crab, Cancer borealis. Anal. Chem. 80, 6949–6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Zubieta J. K.; Kennedy R. T. (2009) Practical aspects of in vivo detection of neuropeptides by microdialysis coupled off-line to capillary LC with multistage MS. Anal. Chem. 81, 2242–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escher C.; Reiter L.; MacLean B.; Ossola R.; Herzog F.; Chilton J.; MacCoss M. J.; Rinner O. (2012) Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 12, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens H. L.; Li L. (2010) Monitoring neuropeptides in vivo via microdialysis and mass spectrometry. Methods Mol. Biol. 615, 57–73. [DOI] [PubMed] [Google Scholar]
- Sharma V.; Eckels J.; Taylor G. K.; Shulman N. J.; Stergachis A. B.; Joyner S. A.; Yan P.; Whiteaker J. R.; Halusa G. N.; Schilling B.; Gibson B. W.; Colangelo C. M.; Paulovich A. G.; Carr S. A.; Jaffe J. D.; MacCoss M. J.; MacLean B. (2014) Panorama: A Targeted Proteomics Knowledge Base. J. Proteome Res. 13, 4205–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]