

Abstract
Amide bond-containing (ABC) biomolecules are some of the most intriguing and functionally significant natural products with unmatched utility in medicine, agriculture and biotechnology. The enzymatic formation of an amide bond is therefore a particularly interesting platform for engineering the synthesis of structurally diverse natural and unnatural ABC molecules for applications in drug discovery and molecular design. As such, efforts to unravel the mechanisms involved in carboxylate activation and substrate selection has led to the characterization of a number of structurally and functionally distinct protein families involved in amide bond synthesis. Unlike ribosomal synthesis and thio-templated synthesis using nonribosomal peptide synthetases, which couple the hydrolysis of phosphoanhydride bond(s) of ATP and proceed via an acyl-adenylate intermediate, here we discuss two mechanistically alternative strategies: ATP-dependent enzymes that generate acylphosphate intermediates and ATP-independent transacylation strategies. Several examples highlighting the function and synthetic utility of these amide bond-forming strategies are provided.
Keywords: natural products, biosynthesis, ATP-grasp, serine protease superfamily, transacylase, aminolysis, esterase, lipase, α/β hydrolase fold, transpeptidase, β-lactamase, carboxymethyltransferase
Introduction
The formation of an amide bond is one of the major strategies by which biomolecules are assembled from relatively simple precursors in Nature. These amide bond-containing (ABC) biomolecules range from large ribosomally-encoded peptides that are pieced together from the relatively limited pool of proteinogenic amino acids to nonribosomally-encoded peptides as well as essential metabolites and various natural products, which are much smaller in size but are notorious for incorporating a wide array of structurally diverse precursors. Several ABC biomolecules, particularly those considered nonessential to cellular survival (i.e., natural products, also referred to as secondary metabolites), have unparalleled utility in medicine, agriculture, or biotechnology. Due to this significance, there has been considerable effort to unravel the molecular details that guide this thermodynamically challenging event, particularly with respect to how the enzymes select the appropriate precursors for condensation and the catalytic mechanism for carboxylic acid activation that initiates the process. For the latter, coupling with hydrolysis of the phosphoanhydride bond(s) of ATP usually serves as the energy source, and in most cases this occurs through the formation of an acyl-adenylate intermediate as observed during the ribosomally-guided process of peptide biogenesis (Figure 1A). Although adenylating enzymes have received the most attention, the current reality is that alternative, mechanistically unique enzymatic processes have now been established for the biosynthesis of ABC biomolecules, and the enzymes catalyzing these important, convergent reactions, encompass several structural and functional protein families (Figure 2).
Figure 1. Strategies for amide bond formation.

(A) Amide bond formation is typically initiated by enzymes that activate carboxylic acid at the expense of ATP through the formation of an acyl-adenylate or an acylphosphate intermediate. Although some acyl-adenylate forming enzymes are known to directly couple the acyl donor to an amine acceptor as shown, most catalyze thioester or ester formation that serves as an activated intermediate that will be used to ultimately form an amide bond. (B) Several enzymes have been utilized as biocatalysts for amide bond formation in synthesis due to their ability to catalyze transacylation using a mechanism involving an acyl-enzyme intermediate. LG, leaving group. (C) More recently it has been revealed that ABC biomolecules can be assembled using an ATP-independent transacylation strategy starting from a carboxylic acid by the tandem activity of a carboxylmethyltransferase and transacylase.
Figure 2. Conceptual mapping of strategies and enzymes used to generate amide bonds.

Examples are provided in the text for those highlighted in black boxes. ATP-dependent mechanisms include acyl-adenylate forming enzymes that usually, but not always, form esters or thioesters that serve as substrates for distinct condensation catalysts. In contrast ATP-grasp enzymes utilize an acylphosphate intermediate to directly form amide bonds. Transacylation, although technically typically ATP-dependent given that the acyl substrates for these enzymes are derived from an ATP-dependent process, are categorized separately due to the recent discovery of the capuramycin transacylase that takes advantage of an acyl precursor derived from an S-adenosyl-L-methionine-dependent activation of a carboxylic acid.
This review focuses on two alternative, less appreciated mechanisms that do not proceed directly through acyl-adenylate intermediates to achieve amide bond formation: ATP-dependent formation of acylphosphate intermediates and ATP-independent transacylation strategies. As described herein, the enzymes employing these mechanisms are now routinely being reported as central catalysts for assembling natural product scaffolds, and bioinformatic analysis suggests this is just the tip of the iceberg. Importantly, there is a great potential for exploiting these comparatively simple enzyme systems for structural diversification of these scaffolds and generating new ABC biomolecules. The reader is directed to several other excellent articles and reviews for information regarding acyl adenylate-forming enzymes and condensation catalysts involved in the more traditional, templated approach for amide bond formation.1–5
ATP-dependent amide bond formation
ATP-grasp enzymes: mechanism and structure
Enzymatic transformations involving amide bond formation between a carboxylic acid and an amine require the activation of acyl groups to make the overall reaction thermodynamically feasible. More often than not, this energy source is provided by ATP either through hydrolytic coupling of ATP to AMP and PPi or to ADP and Pi (Figure 1a). While aminoacyl-tRNA synthetases and nonribosomal protein synthetases (NRPSs) employ the former, several ABC biomolecules are assembled using the latter route in a reaction catalyzed by enzymes belonging to the so-called ATP-grasp family. Most members of the ATP-grasp family are not only united by the reaction mechanism, wherein a carboxylic acid substrate is activated as an acylphosphate intermediate prior to condensation with a co-substrate nucleophile (two kinase-type enzymes of the family, pyruvate phosphate dikinase and inositol 1,3,4-triphosphate 5/6-kinase, are exceptions to this mechanism), but not surprisingly by similar structures containing a nonclassical fold for ATP binding that is comprised of two α + β domains responsible for “grasping” or holding a molecule of ATP in the active site.6
Despite the conservation in carboxylic acid activation and overall structure, a remarkable feature of the ATP-grasp family is the structurally diverse range of substrates that are utilized by each enzyme. The carboxylic acid substrate can be simple, for example formic acid and bicarbonic acid, to quite complex, such as various organic acids including large proteins serving as substrates. Likewise, the nucleophile substrate, which is predominantly an amine or thiol (D-Ala-D-lactate ligase is a notable exception), ranges from simple ammonium ion to a biotin prosthetic group of a carrier protein. This realization makes it difficult to predict, a priori, the function solely through sequence analysis. However examination of the genomic context of several predicted ATP-grasp enzymes clearly indicates a wide-spread and under-appreciated role for these enzymes.
The reaction mechanism of ATP-grasp enzymes is composed of two half-reactions: the first half-reaction involves activation of the carboxylic acid-containing substrate by a molecule of ATP to form a high energy acylphosphate intermediate (Figure 3).7,8 Evidence for this intermediate has been gleaned in part from isotopic exchange studies involving the transfer of a labeled oxygen atom from the substrate to inorganic phosphate,9 entrapment studies performed utilizing diazomethane trapping10 and inhibition studies with D-Ala:D-Ala ligase.11 The second half-reaction occurs by nucleophilic attack that leads to the formation of a tetrahedral intermediate, which is likewise supported by kinetic evidence as well as the use of transition state analogues (Figure 3).12 Although the formation of a tetrahedral intermediate is likely the case for most organic carboxylic acids, the decomposition of carboxyphosphate for reactions utilizing bicarbonate as the carboxylic acid substrate have been proposed to proceed through decomposition to phosphate and carbon dioxide prior to nucleophilic attack (Figure 3).13 Additionally, an active site base has been implicated for the deprotonation of either thiol- or amine- containing nucleophilic substrate. However, a number of mutational and structural studies have failed to identify the general base.14
Figure 3. Mechanism for ATP-grasp enzymes.
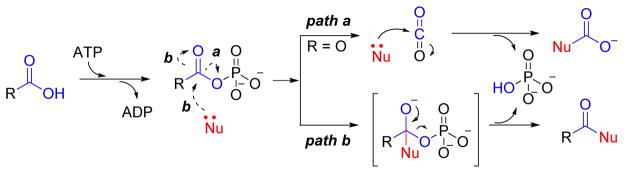
ATP-grasp enzymes form an acylphosphate intermediate followed by decomposition to CO2 prior to nucleophilic (Nu) attack (path a, Nu = biotin) or formation of a tetrahedral intermediate prior to release of phosphate (path b).
As the name implies, the defining feature of the ATP-grasp family is the ATP-binding site that differs from the more commonly known Walker A motif. The unusual orientation and binding mode of the ATP was initially described as the “palmate (β-sheet) fold” upon structural elucidation of Escherichia coli glutathione synthetase,15 and the ATP-grasp fold was subsequently identified in the E. coli enzymes succinyl-CoA synthetase,16 biotin carboxylase,17 and D-Ala:D-Ala ligase.18 The tertiary structure of all ATP-grasp enzymes consists of three conserved domains termed the N-terminal, central, or C-terminal domains, or alternatively known as A, B, and C domains, respectively.19 The ATP, which coordinates with Mg2+ or Ca2+ ions, is sandwiched between the B- and C-domains, and the B-domain acts as a flexible ‘lid’ in the absence of ATP that undergoes a conformational change and clamps down over the active site upon nucleotide binding.20 The largest structural variability among the family lies in the C domain, which along with the A domain, facilities co-substrate binding and proper orientation of the nucleophile and acylphosphate intermediate. Over 130 different structures of enzymes containing the ATP-grasp fold are now available in the Protein Data Bank.21
ATP-grasp enzymes in primary metabolism
The ATP-grasp family as it relates to primary metabolism includes minimally 25 functionally distinct proteins: 21 that have been previously recognized,8 β-D-Asp:UDP-N-acetylmuramic acid-pentapeptide ligase (Asl) from Enterococcus faecalis that is utilized in peptidoglycan biosynthesis,22 Nε-L-Lys:3R-methyl-D-ornithine ligase (PylC) that catalyzes the second step in pyrrolysine biosynthesis,23 tyramine-glutamate ligase (MfnD) involved in the biosynthesis of the cofactor methanofuran,24 and UDP-N-acetyl-D-fucosamine-4N-α-ketoglutarate synthetase (Pyl) involved polysaccharide biosynthesis of Bacillus cereus and potentially other biofilm-forming organisms.25 Overall, members of this family of enzymes are involved in diverse metabolic pathways including peptidoglycan biosynthesis,26 de novo purine biosynthesis,27 and gluconeogenesis,28 to name a few. One of the most prominent and earliest members of this group is biotin carboxylase (BC),29 a component of the much larger acetyl-CoA carboxylase complex that includes a carboxyltransferase and a biotin carboxyl carrier protein that work in unison to generate malonyl-CoA for fatty acid biosynthesis. BC catalyzes phosphorylation of the “simple” carboxylate bicarbonate, which is subsequently transferred by the enzyme to the biotin prosthetic group to yield carboxybiotin (Figure 4A).13,30 Three other ATP-grasp members, pyruvate carboxylase,30 urea amidolyase,31 and propionyl-CoA carboxylase,32 mechanistically parallel BC by activation and transfer of bicarbonate to biotin. More pertinent to this review, however, are the 10 out of the 25 members that catalyze condensation of an organic carboxylic acid substrate (R3C-COOH) with an organic amine (R3C-NH2): D-Ala:D-Ala ligase, glutathione synthetase, glycinamide ribonucleotide synthetase (PurD), carnosine synthetase, Asl, PylC, MfnD, Pyl, ribosomal protein S6 modification protein (RimK), tubulin-tyrosine ligase (Figure 4B). The first 8 enzymes all catalyze amide bond formation between two relatively small metabolic precursors. Contrastingly, the last two differentiate themselves by utilizing large proteins as substrates, activating the C-terminal carboxylic acid as the acylphosphate to essentially extend the peptide chain by one amino acid. RimK, however, has more recently been shown to catalyze the formation of poly-α-glutamic acid polymers of varying lengths, although the biological function of this reaction, if any, is still unclear.33
Figure 4. Representative ATP-grasp enzymes involved in primary metabolism.

(A) The reaction catalyzed by biotin carboxylase. (B) Substrates used by ATP-grasp enzymes that catalyze amide bond formation including D-Ala:D-Ala ligase (i), glutathione synthetase (ii), glycinamide ribonucleotide synthetase (iii), carnosine synthetase (iv), β-D-Asp:UDP-N-acetylmuramic acid-pentapeptide ligase (v), Nε-L-Lys:3R-methyl-D-ornithine ligase (vi), ribosomal protein S6 modification protein RimK (vii), tubulin-tyrosine ligase (viii), and tyramine-glutamate ligase (ix), and UDP-N-acetyl-D-fucosamine-4N-α-ketoglutarate synthetase (x).
ATP-grasp enzymes in secondary metabolism
The involvement of ATP-grasp enzymes in secondary metabolism was realized shortly after the advent of this family. Genetic evidence has now linked genes encoding ATP-grasp enzymes to the biosynthesis of several peptide-like natural products including those listed in Table 1, and several of these enzymes have been biochemically assigned. The potential for the involvement of ATP-grasp enzymes in natural product biosynthesis has also been recognized by analyzing the genomic context within several organisms, which has revealed that these enzymes potentially participate in the biosynthesis of a number of as-of-yet unknown metabolites.34
Table 1.
ATP-grasp enzymes involved in natural product biosynthesis.
| Enzyme | Function | Biosynthetic pathway | Reference |
|---|---|---|---|
| CphA | Cyanophycin synthetase | Cyanophycin | 35–38 |
| NikS | L-amino acid ligasea | Nikkomycin | 129 |
| PgsBCA | Poly-γ-glutamate synthetase | Poly-γ-glutamate polymers | 130 |
| BacD/YwfE | L-Ala: L-anticapsin ligase | Bacilysin | 43–45 |
| TblF | D-Ala-D-Ala ligasea | Tabtoxin | 131 |
| Ptx18 | biotin carboxylasea | Phaseolotoxin | 132 |
| Ptx21 | L-amino acid ligasea | Phaseolotoxin | 132 |
| RizA | L-Arg:Xaa ligaseb | Rhizocticin | 133 |
| DdaF | Nβ-Fumaramoyl-2,3-diaminopropionic acid ligase | Dapdiamides | 39 |
| MvdD and MvdC | Ligase | Microviridin | 134 |
| FtyB | L-3-formyl-Tyr:L-Thr ligase | Formyl-Tyr dipeptide | 135 |
| RizA | Dipeptide synthetase | Rhizocticin | 133, 136 |
| RizB | Oligopeptide synthetase | Rhizocticin | 137, 138 |
| Ava_3856/MysC | 4-deoxygadusol:Gly ligase | Shinorine | 41 |
| MysD | Shinorine synthetase | Shinorine | 42 |
| MboC | D-Ala-D-Ala ligasea | Mangotoxin | 139 |
| MboD | biotin carboxylasea | Mangotoxin | 140 |
| DcsG | O-ureido-D-Ser cyclase | D-cycloserine | 36 |
Exact function has not been determined; assignment is primarily based on annotation of closest homologs from BLAST analysis.
Nonspecific for C-terminal amino acid (Xaa)
Similarly to the enzymes involved in primary metabolism, the ATP-grasp enzymes involved in secondary metabolism have diverse function and substrate specificities. One of the first enzymes of the family to be functionally assigned was cyanophycin synthetase.35,36 Cyanophycin is an Asp peptide polymer with each β-carboxylate linked to the α-amine of an Arg residue (Figure 5A). This polymer, which putatively functions as a nitrogen reserve, can accumulate to a molecular weight of 100 kD. Heterologous expression of the gene encoding cyanophycin synthetase from Synechocystis sp. PCC 6803 in Escherichia coli revealed this single gene encoding tandem ATP-grasp domains was sufficient for the de novo synthesis of cyanophycin.35,36 In vitro characterization of the recombinant protein from Anabaena 29413 revealed enzyme activity in the presence of Asp, Arg, ATP, Mg2+, and a synthetic cyanophycin primer, and polymer formation occurred in a stepwise fashion with C-terminal addition of L-Asp to form the α-peptide followed by attachment of L-Arg to form the isopeptide bond.37 Although the enzyme is specific for L-Asp, LArg could be replaced by L-Lys. More recently, the enzyme from Thermosynechococcus elongatus BP-1 was shown to not require a cyanophycin primer to initiate polymer assembly.38 This enzyme system highlights several intriguing features including the ability of ATP-grasp enzymes to catalyze classical and nonclassical peptide linkages and the relaxed substrate specificity not only with respect to L-Arg but also in recognizing the growing polymer that is elongated at the C-terminus.
Figure 5. Representative ATP-grasp enzymes in natural product biosynthesis.

(A) Cyanophycin synthetase catalyzes iterative, sequential addition of two different amino acids as acyl acceptors. (B) Dapdiamide amide bonds are formed using two distinct ATP-dependent mechanisms catalyzed by a nonribosomal peptide synthetase (i) and an ATP-grasp enzyme (ii). (C) The final steps during the biosynthesis of shinorine occur through sequential reactions catalyzed by an ATP-grasp enzyme followed by an NRPS (i) or a distinct ATP-grasp enzyme (ii). (D) The final step in bacilysin biosynthesis using an ATP-grasp enzyme that catalyzes amide bond formation between the L-Ala and the unusual acyl acceptor anticapsin.
A thorough bioinformatic analysis of whole genomes has revealed several instances where a gene encoding a putative ATP-grasp enzyme is clustered with genes encoding NRPS systems.34 The biosynthesis of the dapdiamides is one such example that was shown to be assembled using two different mechanisms for amide bond formation (Figure 5B).39,40 DdaG, a member of the adenylate-forming ligases, initiates the biosynthesis by regiospecific condensation of fumarate and 2,3-diaminopropionate (DAP) to form Nβ-fumaroyl-DAP. After modification of the free carboxylic acid to the carboxamide by the amidotransferase DdaH, the ATP-grasp enzyme DdaF catalyzes the formation of the second amide bond. Similarly to cyanophycin synthetase, DdaF utilizes a proteinogenic amino acid as an acyl acceptor, in this case condensing Val, Ile, or Leu to form dapdiamides A-C, respectively. Like dapdiamide, shinorine biosynthesis in Anabaena variabilis features both an ATP-grasp enzyme and an NRPS; in contrast, however, shinorine does not contain any amide bonds (Figure 5C). Characterization of the ATP-grasp enzyme, Ava_3856, revealed this enzyme activated a conjugated vinylogous acid instead of a carboxylate to generate an electrophilic center for attack by Gly, demonstrating that ATP-grasp enzymes not only have the potential to employ various nucleophiles but also alternative acid substrates.41 The NRPS, Ava_3855, completes the pathway by activating L-Ser as the acyl-adenylate, which is condensed with the Ava-3856 product, mycosporine glycine, to yield an ester that undergoes an O- to N-rearrangement to form the imine. Rather interestingly, genetic analysis of several other cyanobacteria known to produce shinorine did not reveal a homologous gene for Ava-3855, but instead a gene encoding a protein with similarity to D-Ala-D-Ala ligase.42 The Nostoc punctiforme mysD gene encoding this distinct ATP-grasp enzyme was indeed shown to be sufficient for heterologous production of shinorine when combined with the ava-3856 homologue mysC and the other structural genes necessary for shinorine biosynthesis (Figure 5C). It was proposed that MysD utilizes a mechanism analogous to that proposed for Ava-3856/MysC by activating the vinylogous acid tautomer of mycosporine-glycine to form an acylphosphate intermediate that serves as the electrophilic center for attack of L-Ser to directly yield shinorine without an O- to N-rearrangement.
Perhaps the most studied ATP-grasp enzyme in secondary metabolism is BacD involved in bacilysin biosynthesis (Figure 5D). BacD, also known as YwfE, was initially shown to function as a dipeptide synthetase that was able to catalyze amide bond formation using multiple combinations of the proteinogenic amino acids.43 Follow up studies demonstrated BacD is an L-Ala:anticapsin ligase catalyzing the ultimate step in bacilysin biosynthesis, revealing an ATP-grasp enzyme that utilizes the unusual, non-proteinogenic amino acid anticapsin as an acyl acceptor.44 The crystal structure of BacD has recently been solved, providing the first opportunity for an in depth analysis of the molecular details driving specificity and catalysis for an ATP-grasp enzyme involved in natural product biosynthesis, thus opening the door for structure-guided enzyme evolution.45
The utility of peptides for many different applications has spawned considerable efforts toward defining the fundamental mechanisms of ATP-dependent amide bond formation outside of the ribosome, which is perhaps best explored for NRPS.46,47 Similar to NRPS systems, the ATP-grasp enzymes utilize a diverse structural range of substrates, which includes various unusual nonproteinogenic amino acids and other acids. This feature of ATP-grasp enzymes, along with the relative simplicity compared to NRPS, gives these enzymes a potentially unequalled value in synthesis and synthetic biology for generating designer ABC biomolecules.
Transacylation
Transacylation is a fundamental reaction catalyzed by the ribosome and condensation domains of NRPS during peptide elongation.4,46–48 The former involves an aminoacyl donor in the form of an ester within a charged transfer RNA, while the latter involves a thioester within a phosphopantetheinyl prosthetic group of an acyl carrier protein (Figure 1A). Although some specific aspects of their mechanisms still remain unclear, it is generally agreed that neither the ribosome nor NRPS utilize covalent catalysis via formation of an acyl-enzyme intermediate. However, it is now established that several enzymes can do just that: catalyze substitution of an acyl acceptor through covalent tethering of the acyl donor during the reaction coordinate (Figure 1B). One of the naturally occurring and well characterized examples of this type of transacylase is bacterial serine-type D-Ala-D-Ala transpeptidase, an enzyme which catalyzes peptide cross-linking that is critical for the integrity of the peptidoglycan cell wall.49 Likewise, many enzymes of the serine protease family and α/β hydrolase fold superfamily have been shown to catalyze transacylation under artificial in vitro conditions despite their primary metabolic role as hydrolases, suggesting the possibility that transacylation using this mechanism may occur in Nature to assemble ABC biomolecules. The following section covers representative enzymes of the serine protease family or α/β hydrolase fold superfamily and their utility as catalysts in synthetic chemistry, which is followed by some examples of enzymes whose biological role as transacylases—either dependent or independent of ATP carboxylate activation—have been established.
Utility and mechanism of hydrolytic enzymes in peptide synthesis
Two challenges of traditional peptide synthesis in medicinal chemistry are the potential racemization that occurs during the activation steps and the laborious purification of the final product from an isomeric mixture of peptides. To circumvent these challenges, a number of proteases and hydrolases have been explored as catalysts that function under artificial in vitro conditions to generate peptides from a variety of synthesized or naturally occurring substrates.50 Initial work with proteases revealed that, in contrast to the normal aqueous conditions where hydrolysis is heavily favored, the reverse reaction can occur in water-restricted conditions resulting in the synthesis of peptide bonds.51 The two principal strategies enabling this chemistry are (i) thermodynamic control to favor the reverse reaction52 (by inclusion of water miscible organic media,53 biphasic systems,54 and water mimetics55) and (ii) the use of activated substrate esters or N-protected amino acids (fragment-based approach).56 The latter strategy has been particularly effective for enzymes that operate via an acyl-enzyme intermediate such as the serine proteases. However, ex vivo peptide synthesis by proteases has several drawbacks including the hydrolysis of the products, the rather strict substrate specificities, and the relative instability of these enzymes in anhydrous environments. These limitations inspired the search for non-protease catalysts, which resulted in the discovery of lipases and esterases that catalyze ester aminolysis.57, 58 Esterases represent a large and diverse group of hydrolytic enzymes (EC 3.1.1.x) that are stable under a variety of conditions and, in some cases, exhibit activity in organic solvents. The two classes of relative interest belong to the α/β hydrolase fold superfamily: the lipases (triacylglycerol hydrolases; E.C. 3.1.1.3) and the ‘true’ esterases (carboxylesterase; E.C. 3.1.1.1).
Serine proteases and esterases/lipases of the α/β hydrolase fold superfamily have similar mechanisms with most members employing a nucleophilic serine at the active site (Figure 6).59 This critical Ser is often found as part of a His, Ser, Asp catalytic triad, although several variations to this structural feature have now been described. The catalytic events involving these enzymes proceed via the following steps: (i) reaction of a substrate amide or ester with the hydroxyl of the active site serine that leads to the formation of a tetrahedral adduct that is stabilized by main chain hydrogen bonding in the oxyanion hole, (ii) subsequent breakdown of the high energy transition state adduct to the acyl-enzyme intermediate and concomitant elimination of the amine or alcohol product, (iii) activation and nucleophilic attack of water to generate a second tetrahedral adduct, and (iv) breakdown to regenerate Ser and the carboxylic acid product. Although the natural reaction occurs by nucleophilic attack with water, in theory, any properly oriented nucleophile could attack the acyl-enzyme intermediate leading to the synthesis of a variety of products (Figure 6). In principle, an enzymatic transformation involving an amine nucleophile is highly exothermic and thereby thermodynamically favorable.60
Figure 6. Mechanism for serine proteases and esterases/lipases.
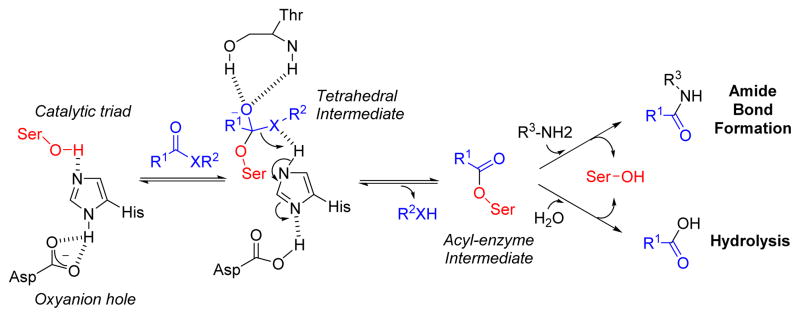
The active site serine in the catalytic triad (Ser-Asp-His) is activated by histidine and aspartate residues, which in turn leads to nucleophilic attack of the substrate generating a tetrahedral intermediate stabilized by an oxyanion hole. Breakdown of this intermediate, elimination of the alcohol (X = O) or amine (X = NH) product, and reformation of the active site serine occur sequentially to set up the stage for further nucleophilic attack with an organic amine or water.
Proteases in peptide synthesis
Proteases are some of the best characterized enzymes and much of our current knowledge of protein structures and functions are associated with those investigations.49 As previously noted, exclusion of water is usually essential for exploiting proteases as amide bond-forming catalysts. Thus, several strategies for improving operational stability of proteases in non-aqueous or biphasic systems have been reported, including the use of a variety of immobilization processes such as covalent attachment on various surfaces to encapsulation in polymers.61 In addition to structural stability, immobilization offers the added advantage of easy recovery and separation as well as economic viability. Protein engineering has also been routinely used to improve catalytic properties—for example, the introduction of a methyl group to the ε-2 N of the active His in chymotrypsin improves aminolysis over unwanted hydrolysis62. Alternatively, the implementation of an exopeptidase like carboxypeptidase Y instead of an endopeptidase such as chymotrypsin has further expanded the utility of N- to C-terminus peptide synthesis while limiting hydrolysis. Another example of protein engineering to improve catalysis is with the protease subtilisin, which has been modified to an acyltransferase by mutating the active site serine to cysteine63 or selenocysteine.64
A number of dipeptides and other small peptides used commercially for human and animal nutrition and as pharmaceutical entities have been synthesized by utilizing proteases in vitro (Table 2). In addition to the utility of preparing relatively small peptides, some proteases have been employed in the modification or synthesis of large polypeptides and proteins. For example, the enzymatic synthesis of Leu-enkephalin, a pentapeptide that binds opioid receptors, was achieved by adopting four different proteases for chain elongation (Figure 7A).65 A similar controlled stepwise, convergent synthesis using an assortment of proteases was adapted for the synthesis of a functionalized octapeptide.66 Other examples wherein proteases have been used to modify or semisynthetically prepare larger polypeptides include the biologically active 493–515 sequence of human thyroid protein kinase A-anchoring protein Ht31 (Figure 7B),67 native and mutant RNaseA,68 bovine ribonuclease,69 staphylococcal nuclease,70 human insulin and [Glyα142]-hemoglobin,71 somatostatin,72 vasopressin, and oxytocin.73
Table 2.
Protease-catalyzed synthesis of commercially used peptides
| Peptide | Peptide bond formeda | Enzyme | Reference |
|---|---|---|---|
| Kyotorphin | Tyr-Arg | α-Chymotrypsin | 141 |
| Enkephalin (Enkephalinamide) | Tyr-Gly-Gly-Phe-Leu | α-Chymotrypsin, Papain | 142 |
| Dynorphin | Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Ile | α-Chymotrypsin, Papain, Trypsin | 143 |
| Vasopressin | Tyr-Phe-Phe-Gln | α-Chymotrypsin, Thermolysin | 143 |
| Aspartame | Asp-Phe | Thermolysin, Papain | 144, 145 |
| RGD Tripeptide | Boc-Arg-Gly-OEt | Alcalase, Trypsin, Papain, α-Chymotrypsin | 146 |
| Cholecystokinin | Asp-Tyr-Met-Gly-Trp-Met-Asp-Phe | α-Chymotrypsin, Papain, Thermolysin | 147 |
OEt=Ethoxy ester, Boc=tert butyl carbamate
Figure 7. Representative protease enzymes used in in vitro peptide synthesis.

(A) Convergent (4 + 4) synthesis of a highly functionalized octapeptide with an overall yield of ~ 40% and utilization of seven different proteases. (B) Convergent approach towards the synthesis of the human thyroid PKA (protein kinase A)-anchoring protein Ht31. The carboxyl component Boc-Asp-Leu-Ile-Glu-Glu-Ala-Ala-Ser-OGp was synthesized by oxime-resin strategy, and the hexadecapeptide fragment synthesized by standard peptide chemistry. The final ligation step utilized chymotrypsin in vitro, to afford the protected oligopeptide which could be deprotected in the final step to obtain a fully functionalized Ht31(final yield ~ 30%).
Although the aforementioned examples incorporate L-amino acids to generate classical peptide bonds, the utility of proteases in peptide synthesis has been expanded to non-proteinogenic substrates. For example, certain proteases such as chymotrypsin have been shown to directly incorporate D-amino acids.73 As observed for chymotrypsin, the inclusion of a D-amino acid is often advantageous since the product tends to be less prone to the reverse hydrolytic reaction. Peptides with nonclassical peptide bonds have also been prepared; a peptidase from Staphylococcus aureus strain V8 has been shown to catalyze isopeptide bond formation via intermolecular transacylation using a thioesterified side chain of Asp or Glu as the acyl donor.74 Likewise, subtiligase-catalyzed cyclization using a C-terminus glycolate phenylalanylamide ester as the acyl donor has been successful for generating marcrocycles ranging from 12- to 31-amino acids in size.68
Esterases and lipases of the α/β hydrolase fold superfamily
The three-dimensional crystallographic structures of numerous esterases and lipases have revealed a characteristic α/β hydrolase fold composed of an ordered sequence of α-helices and β-strands.75 A distinguishing feature of lipases, in contrast to esterases in the superfamily, is the unique property of interfacial activation.76 Biochemical and structural studies of lipases attribute this feature to a helical ‘lid’ covering the active site, which undergoes a conformational change to increase the nonpolarity of the active site while simultaneously exposing the catalytic triad for substrate binding.77 Moreover, lipases prefer long-chained as opposed to short-chained acylglyceride substrates, while esterases tend to use relatively polar substrates.
In contrast to the large number of commercially available microbial lipases, the availability of true commercial esterases is limited.78 The bulk of esterase-mediated reactions have been conducted with porcine liver esterase (PLE; also called porcine liver carboxyesterase) and its applicability is restricted to reactions performed in aqueous media.79 PLE was first demonstrated to generate dipeptides with N-protected amino acid esters (Table 3),80 and subsequently this enzyme was shown to catalyze intramolecular amide bond formation from γ-amino esters in water to give a mixture of the γ-lactam and the hydrolysis product.81 Similar results were observed using PLE with the ‘degradation’ of racemic ethyl 4-phenyl-4-aminobutanoate, where the stereoselective formation of (S)-5-phenyl-2-pyrrolidine was observed alongside the hydrolysis product (Figure 8).
Table 3.
Esterase-catalyzed amide formation80
| Enzyme used | Acyl Donor | Acyl Acceptora | Yield |
|---|---|---|---|
| PLE | Carbobenzyloxy-L-Tyr-OMe | L-Met-NH2, HCl | 11% |
| PLE | Carbobenzyloxy-L-Tyr-OMe | L-Met-NH2, HCl | 66% |
| CCL | Carbobenzyloxy-L-Phe-OMe | L-Ala-OBus* | 85% |
| CCL | Carbobenzyloxy-L-Phe-OMe | D-Ala-OBus* | 60% |
BuS= sec-butyl.
Figure 8. Esterase mediated amide bond formation.
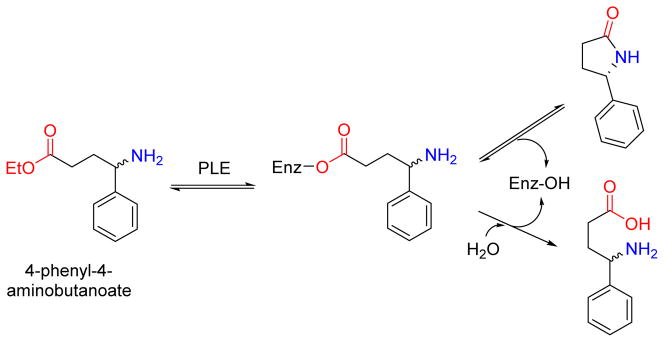
Pig liver esterase (PLE) mediated resolution of racemic ethyl 4-phenyl-4-aminobutanoate into the predominantly (S)-5-phenyl-2-pyrrolidine along with the hydrolysis product. Unlike the hydrolysis, intramolecular aminolysis is stereoselective for one enantiomer.
Another group of esterases exhibiting aminolysis activity are the α-amino acid ester hydrolases (AEH; E.C. 3.1.1.43), which were initially explored for the synthesis of β-lactam antibiotics from D-aminoacyl ester donors and a β-lactam acceptor such as 7-amino-3-deacetoxycephalosporanic acid (Figure 9).82,83 The AEH from Xanthomonas citri IFO 3835, among others characterized, was shown to catalyze this reaction, and importantly the enzyme did not display caseinolytic activity. AEHs from Acetobacter turbidans, Xanthomonas campestris, and X. citri have since been cloned and overexpressed in E. coli, crystallized for structural elucidation, and/or biochemically characterized.84–86 Apart from the signature catalytic triad and the α/β-hydrolase-like fold, their unique specificity towards α-amino group acceptors have been attributed to the presence of an acidic carboxylate cluster in the active site (Asp-208, Glu-309, and Asp-310 with respect to X. citri AEH).86 An α-amino esterase isolated from Bacillus mycoides, proposed to be related to the other AEHs although the sequence is not yet known, was shown to extend the transacylation utility by catalyzing the formation of several dipeptides by incorporating not only L- but D-amino acids in various combinations. Although there is precedent for the synthesis of peptides of DD- or LD- configurations utilizing proteases,87 the enzymatic synthesis of peptides with DL-configuration is rare yet was readily achieved using the B. mycoides AEH (Table 4).
Figure 9. AEH (α-amino ester hydrolases) in the synthesis of β-lactam antibiotics.
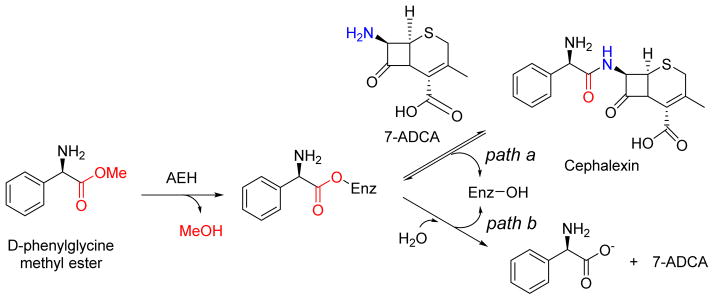
AEH from Acetobacter turbidans ATCC 9325 catalyzes the synthesis of 7-(D-α-amino-α-phenylacetoamido)-3-cephem-3-methyl-4-carboxylic acid (cephalexin) from methyl D-α-aminophenylacetate and 7-ADCA (7-amino-3-deacetoxycephalosporanic acid) (path a). AEHs synthesize β-lactam antibiotics from acyl compounds and β-lactam building blocks obtained from the hydrolysis of natural antibiotics, but without the major disadvantage feedback inhibition by phenylacetic acid that leads to the hydrolysis of the product (path b).
Table 4.
Dipeptides synthesized by AEH from Bacillus mycoides87
| Producta | Acyl Donor | Acyl Acceptor | Yield |
|---|---|---|---|
| Ac-D-Phe-D-Phe-NH2 | Ac-D-Phe-OMe | D-Phe-NH2 | 4.7% |
| Ac-D-Phe-L-Phe-NH2 | Ac-D-Phe-OMe | L-Phe-NH2 | 21.3% |
| Ac-D-Phe-D-Leu-NH2 | Ac-D-Phe-OMe | D-Leu-NH2 | 2.1% |
| Ac-D-Phe-L-Leu-NH2 | Ac-D-Phe-OMe | L-Leu-NH2 | 4.8% |
Ac=acyl
Lipases normally function at an oil-water interface catalyzing the hydrolysis of lipids to fatty acids and glycerols.76 Perhaps not surprisingly, in the first demonstration of lipase-catalyzed aminolysis, it was reported that representative lipases retain their activity even when the bulk of water in the reacting media is replaced by certain organic solvents.88 The successful application of organic solvents in lipase-catalyzed aminolysis has lead to the search for other versatile lipases that function well in a hydrophobic environment and accept a variety of acyl donors and acceptors. The lipases from porcine pancreas (PPL)89 and Candida cylindracea (CCL)90,91 from earlier studies have gradually been replaced by recombinant microbial lipases of increased purity and substrate promiscuity. Currently, the Candida antarctica lipase B (CalB) is used extensively owing to its catalytic versatility.77,92
Numerous synthetic applications of lipases have been reported, and the following examples highlight the utility regarding their broad range of substrates and the unique chemistry associated with these transformations. Using CalB, aminolysis of β-keto methyl esters with aliphatic, allyl- and benzyl-amines gave the desired products in good yield (Figure 10A).93 Other studies have revealed that the ionization state of the amine determines the outcome of CalB-catalyzed aminolysis.94–96 CalB has also been used to convert triolein (olive oil) to its corresponding oleamide by using ammonia as the acyl acceptor (Figure 10B).60 The scope of acyl donor variability of CalB has been extensively examined, revealing the enzyme can use a diverse range of esters97–101 as well as dialkyl and dibenzyl carbonates.102 Intramolecular aminolysis is also possible with lipases, as PPL has been shown to catalyze the synthesis of both small lactam rings from 4- and 5- amino-alkanoic esters (Figure 10C) and macrocyclic bislactams via condensation of diamines and diesters (Figure 10D).103 In addition to the previous lipase examples using esters as acyl donors, CCL was shown to catalyze transacylation using an activated N-2,2,2-trifluoroethyl-2-chloropropionamide donor, thus extending the potential utility of these enzymes (Figure 10E).104 Further extension of the utility has been realized upon replacement of the ester with a carboxylic acid, wherein direct amidation of oleic acid using taurine as the acyl acceptor was shown to be catalyzed by Rml (a lipase from Rhizomucor miehei)102 as well as CalB in hexane and other organic solvents (Figure 10F).105
Figure 10. Representative lipase enzymes involved in amide bond synthesis.

(A) Aminolysis of the methyl ester of acetoacetate using allyl- and benzyl-amines and hydroxylamine acceptors resulting in high product yields. (B) CalB-catalyzed transformation of triolein (olive oil) to its corresponding oleamide via ammonialysis, i.e., direct utilization of ammonia as the nucleophile. (C) The synthesis of lactams by intramolecular aminolysis with amino-alkanoic esters. (D) The synthesis of macrocyclic bislactams by inter/intramolecular aminolysis of diamines and diesters. (E) Lipase-mediated transamidation reaction with a highly activated N-2,2,2-trifluoroethyl-2-chloropropionamide donor. (F) Amidation of oleic acid using taurine as the acyl acceptor.
Transacylation in bacterial metabolism
The previous sections highlight the utility of several unique enzymes employing an acyl-enzyme intermediate for assembling peptides and ABC biomolecules ex vivo. Although not very common to date, the following two examples provide direct evidence for the existence and utility of this enzymatic strategy in bacterial metabolism.
Penicillin-binding proteins (PBPs) catalyze nucleophilic displacement of the terminal D-Ala in the peptidoglycan backbone with either water (D-Ala-D-Ala carboxypeptidase) or the amine of an adjacent peptidoglycan polymer (D-Ala-D-Ala transpeptidase), the latter affording a cross-linked cell wall. These enzymes are members of the serine protease superfamily and hence utilize covalent catalysis for hydrolysis or amine exchange (Figure 11A). Penicillins and β-lactams function as a blend of transition state analogs and mechanism based inhibitors that form covalent adducts with the active site serine of these enzymes, and given the essential role of crosslinking in survival, bacteria have developed several resistance strategies to these antibiotics. Of note organisms often harbor multiple PBP-encoding genes with mutations in some isozymes rendering them less prone to inhibition or resulting in the evolutionary transformation of these enzymes into families of serine-dependent hydrolytic enzymes (Class A, C, and D β-lactamases) that inactivate β-lactams.106–108
Figure 11. Amide bond formation in peptidoglycan biosynthesis.
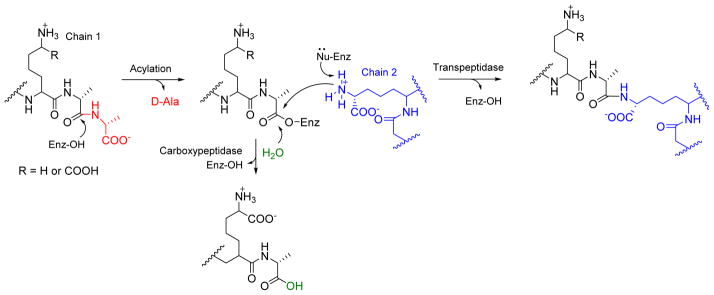
Nucleophilic transpeptidation or hydrolysis of the terminal D-Ala in the peptidoglycan backbone with the amine group of an adjacent peptidoglycan polymer strand (cross-linking) by D-Ala-D-Ala transpeptidase or water by D-Ala-D-Ala carboxypeptidase.
Recently it has been shown that an E. coli D-Ala-D-Ala transpeptidase (PBP1A*) is able to exchange the terminal D-Ala of the pentapeptide of peptidoglycan with D-amino acids of every classification.109 Subsequently, Bacillus subtilis PBP1 was shown to exchange D-Ala with D-Phe or D-Phe carboxamide, and the relaxed specificity for the amino acid side chain was exploited to incorporate a fluorescent probe.110 A similar intermolecular transacylation with select D-amino acids and the fluorescent probe has been reported for one (PBP4) of the four PBPs found within Staphylococcus aureus.111 These results provide evidence to suggest serine-dependent transacylation is a feasible mechanism to establish a new amide bond. Interestingly, cysteine-dependent transpeptidase variants catalyzing unique reactions compared to the aforementioned PBPs also can be exploited to incorporate D-amino acids through an exchange mechanism.112
The peptidoglycan transpeptidase reaction employs an “activated” acyl donor in the form of D-Ala-D-Ala that is initially synthesized by the ATP-grasp enzyme D-Ala-D-Ala ligase. A variation of the serine-dependent transacylase mechanism has been reported that employs an overall ATP-independent strategy for amide bond formation starting from the carboxylic acid as the penultimate acyl donor. The enzyme CapW, identified as a putative Class C β-lactamase encoded within the biosynthetic gene cluster for the capuramycin family of nucleoside antibiotics,113 was found to catalyze a transacylation resulting in the addition of an L-aminocaprolactam at the expense of the methyl ester (Figure 12A).114 The methyl ester was shown to be produced by CapS, an S-adenosyl-L-methionine-dependent carboxylmethyltransferase that activates the carboxylic acid component of the capuramycin precursor to the methyl ester, thereby providing a kinetically competent substrate for the transacylase. The putative active site Ser of CapW was mutated to Ala resulting in loss of enzyme activity, lending support to the hypothesis that the reaction proceeds by a serine-dependent acylation/deacylation mechanism typical of Class C β-lactamases.
Figure 12. Cryptic carboxylmethylation in bacterial metabolism.
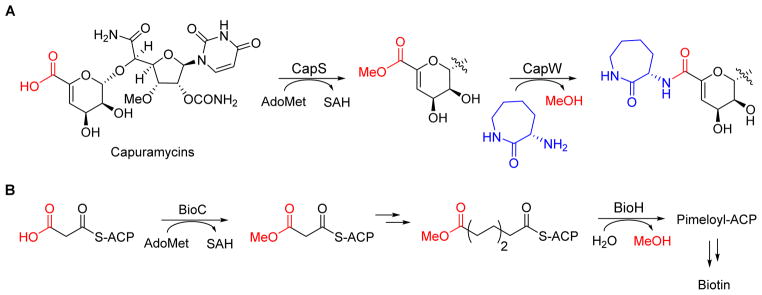
(A) The enzyme CapW catalyzes an unconventional transacylation resulting in incorporation of L-aminocaprolactam at the expense of methanol. The enzyme works in association with a carboxylmethyltransferase (CapS) that activates the carboxylic acid component to the methyl ester. (B) Carboxylmethylation in biotin biosynthesis functions to divert malonyl-ACP from fatty acid biosynthesis to cofactor production. AdoMet, S-adenosyl-L-methionine.
Methyl esters are common in natural products of plant origin and are particularly prevalent in alkaloids exemplified by cocaine and vinblastine and small molecules employed in defense or regulatory processes such as jasmonate and franesoic acid (Figure 13A). This conceptually simple modification has a profound effect on the properties, and is generally considered as a strategy for plants to prepare volatile metabolites.115–118 Methyl esters are less commonly encountered in bacterial metabolites but nonetheless are known, for example within the porphyrin pheophytin that are produced by several photosynthetic bacteria and certain anthracycline polyketides from Streptomyces sp such as nogalamycin and aclacinomycin A (Figure 13B).119,120 Methyl esterification in these two bacterial metabolites significantly increases the acidity of the αposition, thus enabling downstream chemistry. While the methyl ester is directly observed in the final product for these examples, cryptic carboxylmethylation has been reported during the biosynthesis of biotin,121 streptonigrin,122 and certain thiopeptide antibiotics.123 For the former methyl esterification is used to divert the fatty acid building block malonly-S-acyl carrier protein into the biotin biosynthetic pathway (Figure 12B).121 In contrast to biotin and thiopeptide biosynthesis, however, cryptic carboxylmethylation in capuramycin biosynthesis by CapS serves a distinct purpose: to activate the carboxylic acid for intermolecular amide bond formation, thus effectively replacing ATP as the thermodynamic driving force (Figure 1C). A similar carboxylmethylation of isoaspartic acid side chains within eukaryotic proteins that promotes intramolecular peptide bond formation has been described.124 Perhaps the most important aspect of the discovery of the tandem reactions catalyzed by CapS/CapW is the fact that many of the aforementioned serine-dependent proteases and enzymes of the α/β hydrolase superfamily readily convert methyl esters to a variety of amides in vitro, suggesting this chemistry is feasible within an organism and carboxylmethylation/transacylation catalyzed by CapS/CapW may not be an unique example.
Figure 13. Natural products with methyl esters.

(A) Representative plant secondary metabolites and (B) bacterial metabolites.
Conclusion
We have highlighted mechanisms of amide bond formation that are distinct from the templated systems utilized for ribosomally encoded peptides and peptides produced using modular NRPSs. An important feature of the enzyme catalysts described here, along with other examples mediating amide bond formation by transacylation that were not covered such as translgutaminases,125,126 sortases,127 and cysteine protease family enzymes,128 among others, is that the specificity for the acyl acceptor and donor is directly dictated by the enzyme, a property that has been exploited to generate structurally diverse, unnatural ABC biomolecules. Protein engineering via directed evolution or structure-guided mutagenesis, and the co ntinued discovery and characterization of natural versions of these enzymes with unique substrate specificity profiles and catalytic properties—which bioinformatic analysis of whole genomes clearly indicates is highly probable—will undoubtedly expand upon the scope of substrates that are utilized by these enzyme catalysts and provide unmatched tools for applications in drug discovery and molecular design.
Acknowledgments
This work is supported by National Institute of Health National Institute of Allergy and Infectious Diseases (AI087849) and National Center for Advancing Translational Sciences (UL1TR000117).
References
- 1.Leong KW, Pavlov MY, Kwiatkowski M, Forster AC, Ehrenberg M. J Am Chem Soc. 2012;134:17955–17962. doi: 10.1021/ja3063524. [DOI] [PubMed] [Google Scholar]
- 2.Gulick AM. ACS Chem Biol. 2009;4:811–827. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strieker M, Tanović A, Marahiel MA. Curr Opin Struct Biol. 2010;20:234–240. doi: 10.1016/j.sbi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Hur GH, Vickery CR, Burkart MD. Nat Prod Rep. 2012;29:1074–1098. doi: 10.1039/c2np20025b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams GJ. Curr Opin Struct Biol. 2013;23:603–612. doi: 10.1016/j.sbi.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galperin MY, Koonin EV. Prot Sci. 1997;6:2639–2643. doi: 10.1002/pro.5560061218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Attwood PV. Int J Biochem Cell Biol. 1995;27:231–249. doi: 10.1016/1357-2725(94)00087-r. [DOI] [PubMed] [Google Scholar]
- 8.Fawaz MV, Topper ME, Firestine SM. Bioorg Chem. 2011;39:185–191. doi: 10.1016/j.bioorg.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang HC, Ciskanik L, Dunaway-Mariano D, von der Saal W, Villafranca JJ. Biochemistry. 1988;27:625–633. doi: 10.1021/bi00402a020. [DOI] [PubMed] [Google Scholar]
- 10.Powers SG, Meister A. Proc Natl Acad Sci U S A. 1976;73:3020–3024. doi: 10.1073/pnas.73.9.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDermott AE, Creuzet F, Griffin RG, Zawadzke LE, Ye QZ, Walsh CT. Biochemistry. 1990;29:5767–5775. doi: 10.1021/bi00476a018. [DOI] [PubMed] [Google Scholar]
- 12.Thoden JB, Holden HM, Firestine SM. Biochemistry. 2008;47:13346–13353. doi: 10.1021/bi801734z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeczycki TN, Menefee AL, Adina-Zada A, Jitrapakdee S, Surinya KH, Wallace JC, Attwood PV, StMaurice M, Cleland WW. Biochemistry. 2011;50:9724–9737. doi: 10.1021/bi2012788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chou CY, Linda P, Tong L. J Biol Chem. 2009;284:11690–11697. doi: 10.1074/jbc.M805783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaguchi H, Kato H, Hata Y, Nishioka T, Kimura A, Oda Ji, Katsube Y. J Mol Biol. 1993;229:1083–1100. doi: 10.1006/jmbi.1993.1106. [DOI] [PubMed] [Google Scholar]
- 16.Wolodko WT, Fraser ME, James MN, Bridger WA. J Biol Chem. 1994;269:10883–10890. doi: 10.2210/pdb1scu/pdb. [DOI] [PubMed] [Google Scholar]
- 17.Waldrop GL, Rayment I, Holden HM. Biochemistry. 1994;33:10249–10256. doi: 10.1021/bi00200a004. [DOI] [PubMed] [Google Scholar]
- 18.Fan C, Moews P, Walsh C, Knox Science. 1994;266:439–443. doi: 10.1126/science.7939684. [DOI] [PubMed] [Google Scholar]
- 19.Wang W, Kappock TJ, Stubbe J, Ealick SE. Biochemistry. 1998;37:15647–15662. doi: 10.1021/bi981405n. [DOI] [PubMed] [Google Scholar]
- 20.Thoden JB, Blanchard CZ, Holden HM, Waldrop GL. J Biol Chem. 2000;275:16183–16190. doi: 10.1074/jbc.275.21.16183. [DOI] [PubMed] [Google Scholar]
- 21.http://www.rcsb.org/pdb/home/home.do.
- 22.Bellais S, Arthur M, Dubost L, Hugonnet JE, Gutmann L, van Heijenoort J, Legrand R, Brouard JP, Rice L, Mainardi JL. J Biol Chem. 2006;281:11586–11594. doi: 10.1074/jbc.M600114200. [DOI] [PubMed] [Google Scholar]
- 23.Quitterer F, List A, Beck P, Bacher A, Groll M. J Mol Biol. 2012;424:270–282. doi: 10.1016/j.jmb.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Xu H, Harich KC, White RH. Biochemistry. 2014;53:6220–6230. doi: 10.1021/bi500879h. [DOI] [PubMed] [Google Scholar]
- 25.Hwang S, Li Z, Bar-Peled Y, Aronov A, Ericson J, Bar-Peled M. J Biol Chem. 2014 doi: 10.1074/jbc.M114.614917.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan C, Moews PC, Shi Y, Walsh CT, Knox JR. Proc Natl Acad Sci U S A. 1995;92:1172–1176. doi: 10.1073/pnas.92.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Fast W, Benkovic SJ. Protein Sci. 2009;18:881–892. doi: 10.1002/pro.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimroth P, Guchhait RB, Stoll E, Lane MD. Proc Natl Acad Sci U S A. 1970;67:1353–1360. doi: 10.1073/pnas.67.3.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guchhait RB, Polakis SE, Dimroth P, Stoll E, Moss J, Lane MD. J Biol Chem. 1974;249:6633–6645. [PubMed] [Google Scholar]
- 30.Jitrapakdee S, Stmaurice M, Rayment I, Cleland WW, Wallace JC, Attwood PV. Biochem J. 2008;413:369–387. doi: 10.1042/BJ20080709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanamori T, Kanou N, Atomi H, Imanaka T. J Bacteriol. 2004;186:2532–2539. doi: 10.1128/JB.186.9.2532-2539.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang H, Rao KS, Yee VC, Kraus JP. J Biol Chem. 2005;280:27719–27727. doi: 10.1074/jbc.M413281200. [DOI] [PubMed] [Google Scholar]
- 33.Kino K, Arai T, Arimura Y. Appl Environ Microbiol. 2011;77:2019–2025. doi: 10.1128/AEM.02043-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyer LM, Abhiman S, Maxwell Burroughs A, Aravind L. Mol Biosyst. 2009;5:1636–1660. doi: 10.1039/b917682a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziegler K, Diener A, Herpin C, Richter R, Deutzmann R, Lockau W. Eur J Biochem. 1998;254:154–159. doi: 10.1046/j.1432-1327.1998.2540154.x. [DOI] [PubMed] [Google Scholar]
- 36.Aboulmagd E, Oppermann-Sanio FB, Steinbüchel A. Arch Microbiol. 2000;174:297–306. doi: 10.1007/s002030000206. [DOI] [PubMed] [Google Scholar]
- 37.Berg H, Ziegler K, Piotukh K, Baier K, Lockau W, Volkmer-Engert R. Eur J Biochem. 2000;267:5561–5570. doi: 10.1046/j.1432-1327.2000.01622.x. [DOI] [PubMed] [Google Scholar]
- 38.Arai T, Kino K. Biosci Biotechnol Biochem. 2008;72:3048–3050. doi: 10.1271/bbb.80439. [DOI] [PubMed] [Google Scholar]
- 39.Hollenhorst MA, Clardy J, Walsh CT. Biochemistry. 2009;48:10467–10472. doi: 10.1021/bi9013165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hollenhorst MA, Bumpus SB, Matthews ML, Bollinger JM, Kelleher NL, Walsh CT. J Am Chem Soc. 2010;132:15773–15781. doi: 10.1021/ja1072367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balskus EP, Walsh CT. Science. 2010;329:1653–1656. doi: 10.1126/science.1193637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao Q, Garcia-Pichel F. J Bacteriol. 2011;193:5923–5928. doi: 10.1128/JB.05730-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tabata K, Ikeda H, Hashimoto S-i. J Bacteriol. 2005;187:5195–5202. doi: 10.1128/JB.187.15.5195-5202.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parker JB, Walsh CT. Biochemistry. 2013;52:889–901. doi: 10.1021/bi3016229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsuda T, Asami M, Koguchi Y, Kojima S. Biochemistry. 2014;53:2650–2660. doi: 10.1021/bi500292b. [DOI] [PubMed] [Google Scholar]
- 46.Giessen TW, Marahiel MA. FEBS Lett. 2012;586:2063–2075. doi: 10.1016/j.febslet.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 47.Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 48.Leung EKY, Suslov N, Tuttle N, Sengupta R, Piccirilli JA. In: Annu Rev Biochem. Kornberg RD, Raetz CRH, Rothman JE, Thorner JW, editors. Vol. 80. 2011. pp. 527–555. [DOI] [PubMed] [Google Scholar]
- 49.Pratt RF. Cell Mol Life Sci. 2008;65:2138–2155. doi: 10.1007/s00018-008-7591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woessner AJBDRF, editor. Handbook of Proteolytic Enzymes. 2. Academic Press; London: 2004. p. iv. [Google Scholar]
- 51.Bordusa F. Chem Rev. 2002;102:4817–4868. doi: 10.1021/cr010164d. [DOI] [PubMed] [Google Scholar]
- 52.Jakubke HD, Kuhl P, Könnecke A. Angew Chem Int Ed Engl. 1985;24:85–93. [Google Scholar]
- 53.Homandberg GA, Mattis JA, Laskowski M. Biochemistry. 1978;17:5220–5227. doi: 10.1021/bi00617a023. [DOI] [PubMed] [Google Scholar]
- 54.Martinek K, Semenov AN, Berezin IV. Biochim Biophys Acta – Enzymol. 1981;658:76–89. doi: 10.1016/0005-2744(81)90251-5. [DOI] [PubMed] [Google Scholar]
- 55.Kitaguchi H, Klibanov AM. J Am Chem Soc. 1989;111:9272–9273. [Google Scholar]
- 56.Barbas CF, Matos JR, West JB, Wong CH. J Am Chem Soc. 1988;110:5162–5166. [Google Scholar]
- 57.Kullmann W. Enzymatic Peptide Synthesis. CRC Press; Boca Raton: 1987. [Google Scholar]
- 58.Schellenberger V, Jakubke HD. Angew Chem Int Ed Engl. 1991;30:1437–1449. [Google Scholar]
- 59.Pleiss J, Fischer M, Schmid RD. Chem Phys Lipids. 1998;93:67–80. doi: 10.1016/s0009-3084(98)00030-9. [DOI] [PubMed] [Google Scholar]
- 60.de Zoete MC, Kock-van Dalen AC, van Rantwijk F, Sheldon RA. J Mol Catal B: Enzym. 1996;2:19–25. [Google Scholar]
- 61.Wilson SA, Daniel RM, Peek K. Biotechnol Bioeng. 1994;44:337–346. doi: 10.1002/bit.260440311. [DOI] [PubMed] [Google Scholar]
- 62.West JB, Scholten J, Stolowich NJ, Hogg JL, Scott AI, Wong CH. J Am Chem Soc. 1988;110:3709–3710. [Google Scholar]
- 63.Nakatsuka T, Sasaki T, Kaiser ET. J Am Chem Soc. 1987;109:3808–3810. [Google Scholar]
- 64.Wu ZP, Hilvert D. J Am Chem Soc. 1989;111:4513–4514. [Google Scholar]
- 65.Kleln JU, Cerovský V. Int J Pept Protein Res. 1996;47:348–352. [PubMed] [Google Scholar]
- 66.Gill I, Lopez-Fandino R, Vulfson E. J Am Chem Soc. 1995;117:6175–6181. [Google Scholar]
- 67.Cerovský V, Kockskämper J, Glitsch HG, Bordusa F. ChemBioChem. 2000;1:126–129. doi: 10.1002/1439-7633(20000818)1:2<126::AID-CBIC126>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 68.Jackson DY, Burnier JP, Wells JA. J Am Chem Soc. 1995;117:819–820. [Google Scholar]
- 69.Homandberg GA, Laskowski M. Biochemistry. 1979;18:586–592. doi: 10.1021/bi00571a006. [DOI] [PubMed] [Google Scholar]
- 70.Homandberg GA, Komoriya A, Chaiken IM. Biochemistry. 1982;21:3385–3389. doi: 10.1021/bi00257a021. [DOI] [PubMed] [Google Scholar]
- 71.Looker D, Abbott-Brown D, Cozart P, Durfee S, Hoffman S, Mathews AJ, Miller-Roehrkh J, Shoemaker S, Trimble S, Fermi G, Komiyama NH, Nagai K, Stetler GL. Nature. 1992;356:258–260. doi: 10.1038/356258a0. [DOI] [PubMed] [Google Scholar]
- 72.Bille V, Ripak C, Van Aasche I, Forni I, Degelaen L, Searso A, Giralt E, Andreu D. Pept. 1990. Proc Eur Pept Symp, 21st. 1991:253–254. [Google Scholar]
- 73.Rizo J, Gierasch LM. Annu Rev Biochem. 1992;61:387–416. doi: 10.1146/annurev.bi.61.070192.002131. [DOI] [PubMed] [Google Scholar]
- 74.Wehofsky N, Alisch M, Bordusa F. Chem Commun. 2001:1602–1603. doi: 10.1039/b105752a. [DOI] [PubMed] [Google Scholar]
- 75.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verschueren KHG, Goldman A. Prot Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 76.Desnuelle P. In: The Enzymes. Paul DB, editor. Vol. 7. Academic Press; 1972. pp. 575–616. [Google Scholar]
- 77.Anderson EM, Larsson KM, Kirk O. Biocat Biotransform. 1998;16:181–204. [Google Scholar]
- 78.Jones JB. Pure Appl Chem. 1990;62:1445–1448. [Google Scholar]
- 79.Yamada H, Shimizu S. Angew Chem Int Ed Engl. 1988;27:622–642. [Google Scholar]
- 80.West JB, Wong CH. Tetrahedron Lett. 1987;28:1629–1632. [Google Scholar]
- 81.Vivienne Barker C, Page MI, Korn SR, Monteith M. Chem Commun. 1999:721–722. [Google Scholar]
- 82.Alkema WB, Floris R, Janssen DB. Anal Biochem. 1999;275:47–53. doi: 10.1006/abio.1999.4300. [DOI] [PubMed] [Google Scholar]
- 83.Kato K, Kawahara K, Takahashi T, Kakinuma A. Agric Biol Chem Japan. 1980;44:1069–1074. [Google Scholar]
- 84.Blum JK, Bommarius AS. J Mol Catal B: Enzym. 2010;67:21–28. doi: 10.1016/j.molcatb.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Polderman-Tijmes JJ, Jekel PA, Jeronimus-Stratingh CM, Bruins AP, van der Laan JM, Sonke T, Janssen DB. J Biol Chem. 2002;277:28474–28482. doi: 10.1074/jbc.M204143200. [DOI] [PubMed] [Google Scholar]
- 86.Barends TR, Polderman-Tijmes JJ, Jekel PA, Hensgens CM, de Vries EJ, Janssen DB, Dijkstra BW. J Biol Chem. 2003;278:23076–23084. doi: 10.1074/jbc.M302246200. [DOI] [PubMed] [Google Scholar]
- 87.Petkov D, Stoineva I. Tetrahedron Lett. 1984;25:3751–3754. [Google Scholar]
- 88.Zaks A, Klibanov AM. Proc Natl Acad Sci U S A. 1985;82:3192–3196. doi: 10.1073/pnas.82.10.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Faber K. Biotransformations in organic chemistry: a textbook. Springer; 2011. [Google Scholar]
- 90.Garner CW, Smith LC. J Biol Chem. 1972;247:561–565. [PubMed] [Google Scholar]
- 91.Cygler M, Schrag JD. Biochim Biophys Acta-Mol Cell Biol Lip. 1999;1441:205–214. doi: 10.1016/s1388-1981(99)00152-3. [DOI] [PubMed] [Google Scholar]
- 92.De Goede A, Benckhuijsen W, Van Rantwijk F, Maat L, Van Bekkum H. Recueil des Travaux Chimiques des Pays-Bas. 1993;112:567–572. [Google Scholar]
- 93.García MJ, Rebolledo F, Gotor V. Tetrahedron Lett. 1993;34:6141–6142. [Google Scholar]
- 94.Gotor V, Brieva R, Rebolledo F. J Chem Soc Chem Commun. 1988:957–958. [Google Scholar]
- 95.Asensio G, Andreu C, Marco JA. Tetrahedron Lett. 1991;32:4197–4198. [Google Scholar]
- 96.Maugard T, Remaud-Simeon M, Petre D, Monsan P. Tetrahedron. 1997;53:5185–5194. [Google Scholar]
- 97.Puertas S, Brieva R, Robelledo F, Gotor V. Tetrahedron. 1993;49:4007–4014. [Google Scholar]
- 98.Gotor V. Bioorg Med Chem. 1999;7:2189–2197. doi: 10.1016/s0968-0896(99)00150-9. [DOI] [PubMed] [Google Scholar]
- 99.Chamorro C, González-Muñiz R, Conde S. Tetrahedron: Asymmetry. 1995;6:2343–2352. [Google Scholar]
- 100.Puertas S, Rebolledo F, Gotor V. Tetrahedron. 1995;51:1495–1502. [Google Scholar]
- 101.Conde S, López-Serrano P, Fierros M, Biezma MI, Martínez A, Rodríguez-Franco MI. Tetrahedron. 1997;53:11745–11752. [Google Scholar]
- 102.Hacking M, Van Rantwijk F, Sheldon R. J Mol Catal B: Enzym. 2000;9:201–208. [Google Scholar]
- 103.Gutman AL, Meyer E, Yue X, Abell C. Tetrahedron Lett. 1992;33:3943–3946. [Google Scholar]
- 104.Gotor V, Brieva R, González C, Rebolledo F. Tetrahedron. 1991;47:9207–9214. [Google Scholar]
- 105.Maugard T, Remaud-Simeon M, Petre D, Monsan P. Biotechnol Lett. 1997;19:751–753. [Google Scholar]
- 106.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. FEMS Microbiol Rev. 2008;32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 107.Meroueh SO, Minasov G, Lee W, Shoichet BK, Mobashery S. J Am Chem Soc. 2003;125:9612–9618. doi: 10.1021/ja034861u. [DOI] [PubMed] [Google Scholar]
- 108.Fisher JF, Meroueh SO, Mobashery S. Chem Rev. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 109.Lupoli TJ, Tsukamoto H, Doud EH, Wang TSA, Walker S, Kahne D. J Am Chem Soc. 2011;133:10748–10751. doi: 10.1021/ja2040656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lebar MD, May JM, Meeske AJ, Leiman SA, Lupoli TJ, Tsukamoto H, Losick R, Rudner DZ, Walker S, Kahne D. J Am Chem Soc. 2014;136:10874–10877. doi: 10.1021/ja505668f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Qiao Y, Lebar MD, Schirner K, Schaefer K, Tsukamoto H, Kahne D, Walker S. J Am Chem Soc. 2014 doi: 10.1021/ja508147s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cava F, de Pedro MA, Lam H, Davis BM, Waldor MK. EMBO J. 2011;30:3442–3453. doi: 10.1038/emboj.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Funabashi M, Nonaka K, Yada C, Hosobuchi M, Masuda N, Shibata T, Van Lanen SG. J Antibiot. 2009;62:325–332. doi: 10.1038/ja.2009.38. [DOI] [PubMed] [Google Scholar]
- 114.Funabashi M, Yang Z, Nonaka K, Hosobuchi M, Fujita Y, Shibata T, Chi X, Van Lanen SG. Nat Chem Biol. 2010;6:581–586. doi: 10.1038/nchembio.393. [DOI] [PubMed] [Google Scholar]
- 115.Kim EH, Kim YS, Park S-H, Koo YJ, Do Choi Y, Chung Y-Y, Lee I-J, Kim J-K. Plant Physiol. 2009;149:1751–1760. doi: 10.1104/pp.108.134684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Qin G, Gu H, Zhao Y, Ma Z, Shi G, Yang Y, Pichersky E, Chen H, Liu M, Chen Z. Plant Cell. 2005;17:2693–2704. doi: 10.1105/tpc.105.034959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kwon S, Hamada K, Matsuyama A, Yasuda M, Nakashita H, Yamakawa T. Plant Biotechnol. 2009;26:207–215. [Google Scholar]
- 118.Hippauf F, Michalsky E, Huang R, Preissner R, Barkman TJ, Piechulla B. Plant Mol Biol. 2010;72:311–330. doi: 10.1007/s11103-009-9572-0. [DOI] [PubMed] [Google Scholar]
- 119.Esquenazi E, Jones AC, Byrum T, Dorrestein PC, Gerwick WH. Proc Natl Acad Sci U S A. 2011;108:5226–5231. doi: 10.1073/pnas.1012813108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kallio P, Sultana A, Niemi J. J Mol Biol. 2006;357:210–220. doi: 10.1016/j.jmb.2005.12.064. [DOI] [PubMed] [Google Scholar]
- 121.Lin S, Hanson RE, Cronan JE. Nat Chem Biol. 2010;6:682–688. doi: 10.1038/nchembio.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xu F, Kong D, He X, Zhang Z, Han M, Xie X, Wang P, Cheng H, Tao M, Zhang L, Deng Z, Lin S. J Am Chem Soc. 2013;135:1739–1748. doi: 10.1021/ja3069243. [DOI] [PubMed] [Google Scholar]
- 123.Liao R, Liu W. J Am Chem Soc. 2011;133:2852–2855. doi: 10.1021/ja1111173. [DOI] [PubMed] [Google Scholar]
- 124.Vigneswara V, Lowenson JD, Powell CD, Thakur M, Bailey K, Clarke S, Ray DE, Carter WG. J Biol Chem. 2006;281:32619–32629. doi: 10.1074/jbc.M605421200. [DOI] [PubMed] [Google Scholar]
- 125.Ramos D, Rollin P, Klaffke W. J Org Chem. 2001;66:2948–2956. doi: 10.1021/jo001439c. [DOI] [PubMed] [Google Scholar]
- 126.Fortin PD, Walsh CT, Magarvey NA. Nature. 2007;448:824–827. doi: 10.1038/nature06068. [DOI] [PubMed] [Google Scholar]
- 127.Wu Z, Guo X, Wang Q, Swarts BM, Guo Z. J Am Chem Soc. 2010;132:1567–1571. doi: 10.1021/ja906611x. [DOI] [PubMed] [Google Scholar]
- 128.Llewellyn NM, Li Y, Spencer JB. Chem Biol. 2007;14:379–386. doi: 10.1016/j.chembiol.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 129.Lauer B, Russwurm R, Schwarz W, Kálmánczhelyi A, Bruntner C, Rosemeier A, Bormann C. Mol Gen Genet. 2001;264:662–673. doi: 10.1007/s004380000352. [DOI] [PubMed] [Google Scholar]
- 130.Ashiuchi M, Shimanouchi K, Nakamura H, Kamei T, Soda K, Park C, Sung MH, Misono H. Appl Environ Microbiol. 2004;70:4249–4255. doi: 10.1128/AEM.70.7.4249-4255.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kinscherf TG, Willis DK. J Antibiot. 2005;58:817–821. doi: 10.1038/ja.2005.109. [DOI] [PubMed] [Google Scholar]
- 132.Aguilera S, López-López K, Nieto Y, Garcidueñas-Piña R, Hernández-Guzmán G, Hernández-Flores JL, Murillo J, Alvarez-Morales A. J Bacteriol. 2007;189:2834–2843. doi: 10.1128/JB.01845-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kino K, Kotanaka Y, Arai T, Yagasaki M. Biosci Biotechnol Biochem. 2009;73:901–907. doi: 10.1271/bbb.80842. [DOI] [PubMed] [Google Scholar]
- 134.Philmus B, Guerrette JP, Hemscheidt TK. ACS Chem Biol. 2009;4:429–434. doi: 10.1021/cb900088r. [DOI] [PubMed] [Google Scholar]
- 135.Blasiak LC, Clardy J. J Am Chem Soc. 2009;132:926–927. doi: 10.1021/ja9097862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Borisova SA, Circello BT, Zhang JK, van der Donk WA, Metcalf WW. Chem Biol. 2010;17:28–37. doi: 10.1016/j.chembiol.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kino K, Arai T, Tateiwa D. Biosci Biotechnol Biochem. 2010;74:129–134. doi: 10.1271/bbb.90649. [DOI] [PubMed] [Google Scholar]
- 138.Arai T, Kino K. Biosci Biotechnol Biochem. 2010;74:1572–1577. doi: 10.1271/bbb.100148. [DOI] [PubMed] [Google Scholar]
- 139.Carrión VJ, Arrebola E, Cazorla FM, Murillo J, de Vicente A. PLoS ONE. 2012;7:e36709. doi: 10.1371/journal.pone.0036709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Uda N, Matoba Y, Kumagai T, Oda K, Noda M, Sugiyama M. Antimicrob Agents Chemother. 2013;57:2603–2612. doi: 10.1128/AAC.02291-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Meos H, Tõugu V, Haga M, Aaviksaar A, Schuster M, Jakubke HD. Tetrahedron: Asymmetry. 1993;4:1559–1564. [Google Scholar]
- 142.Kimura Y, Nakanishi K, Matsuno R. Enz Microb Technol. 1990;12:272–280. doi: 10.1016/0141-0229(90)90099-c. [DOI] [PubMed] [Google Scholar]
- 143.Jorba X, Clapés P, Xaus N, Calvet S, Torres J, Valencia G, Mata J. Enz Microb Technol. 1992;14:117–124. [Google Scholar]
- 144.Eichhorn U, Bommarius A, Drauz K, Jakubke HD. J Pept Sci. 1997;3:245–251. doi: 10.1002/(sici)1099-1387(199707)3:4<245::aid-psc98>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 145.Chen ST, Wang KT. J Org Chem. 1988;53:4589–4590. [Google Scholar]
- 146.Chen YX, Zhang XZ, Zheng K, Chen SM, Wang QC, Wu XX. Enz Microb Technol. 1998;23:243–248. [Google Scholar]
- 147.Kullmann W. Proc Natl Acad Sci U S A. 1982;79:2840–2844. doi: 10.1073/pnas.79.9.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]