

Abstract
Previous studies demonstrated that members of the aminothienopyridazine (ATPZ) class of tau aggregation inhibitors exhibit a promising combination of in vitro activity as well as favorable pharmacokinetic properties (i.e., brain-penetration and oral bioavailability). Here we report the synthesis and evaluation of several new analogues. These studies indicate that the thienopyridazine core is essential for inhibition of tau fibrillization in vitro, while the choice of the appropriate scaffold decoration is critical to impart desirable ADME-PK properties. Among the active, brain-penetrant ATPZ inhibitors evaluated, 5-amino-N-cyclopropyl-3-(4-fluorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (43) was selected to undergo maximum tolerated dose and one-month tolerability testing in mice. The latter studies revealed that this compound is well-tolerated with no notable side-effects at an oral dose of 50 mg/kg/day.
Keywords: Alzheimer’s disease, Tauopathy, Aminothienopyridazine, Tau aggregation inhibitor, K18PL
1. Introduction
Alzheimer’s disease (AD), which is characterized by the presence of intraneuronal aggregates of tau proteins (e.g., neurofibrillary tangles, or NFTs) and extracellular deposits comprised of Aβ peptide (e.g., senile plaques), remains a challenge for biomedical intervention. Although the exact neuropathological mechanisms underlying this disease remain the focus of intense investigation, several lines of evidence indicate that under pathological conditions tau may act as the mediator of neurodegeneration in response to various upstream events.1 As a result, in recent years there has been an increasing interest in the development of therapeutic strategies that could target pathological tau.2 One such strategy calls for the use of small molecules to inhibit the self-assembly of tau into oligomeric/polymeric species, including the well-defined fibrils found within NFTs. Although several classes of tau assembly inhibitors have demonstrated activity in vitro,3,4 aside from selected cases (e.g., methylene blue5), the majority of these inhibitors are unlikely to be considered for in vivo efficacy studies due to potential toxicities and/or limited CNS-uptake.3
Recently, we identified a novel class of tau fibrillization inhibitors, the aminothienopyridazines (ATPZs), which exhibit a favorable combination of efficacy in an in vitro tau fibrillization assay, as well as drug-like properties (i.e., no violations of Lipinski’s ‘rule of five’).6 Furthermore, prototype compounds from this class of inhibitors (e.g., 1, Fig. 1) have been developed that can reach significant concentrations in the brain of mice after intravenous (i.v.) or oral administration, suggesting that the ATPZs may be amenable to efficacy studies in animal models.7 However, ATPZ 1 was found to have limited water solubility (i.e., ~30 μM). Furthermore, evaluation by equilibrium dialysis revealed that 1 is ~99% bound in brain homogenates, indicating that relatively high doses of the compound would be required to achieve efficacious free drug concentration in the brain.
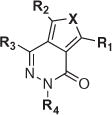
Figure 1.
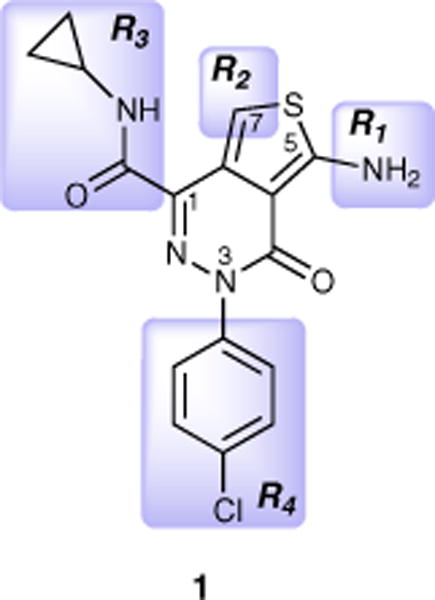
Representative example of a brain-penetrant ATPZ;7 fragments investigated as part of the SAR studies are marked R1–R4.
Thus, to circumvent these shortcomings and to identify preferred candidates for future studies, we designed and synthesized a series of new analogues, including a number of closely related congeners and a series of compounds bearing a range of modifications in the ATPZ heterocyclic core (Schemes 1–3).
Scheme 1.
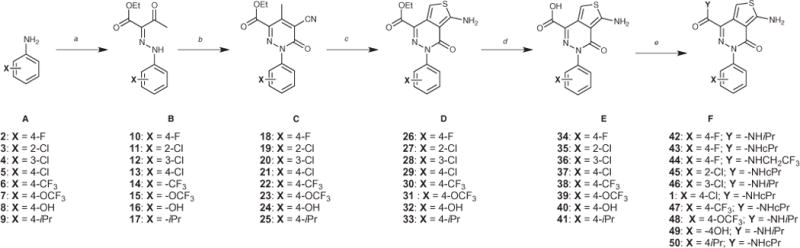
Reagents and conditions: (a) (i) NaNO2, 37% HCl, ethanol, water, 0 °C, 20 min; (ii) ethyl acetoacetate, sodium acetate, ethanol, water, 0 °C, 2 h; (b) ethyl cyanoacetate, ammonium acetate, acetic acid, 170 °C (microwave irradiation), 4 min; (c) S8, morpholine, ethanol, 150 °C (microwave irradiation), 15 min; (d) LiOH•H2O, tetrahydrofuran, water, rt, 16 h; (e) amine, BOP reagent, N,N-diisopropylethylamine, dimethylsulfoxide, rt, 4–16 h.
Scheme 3.
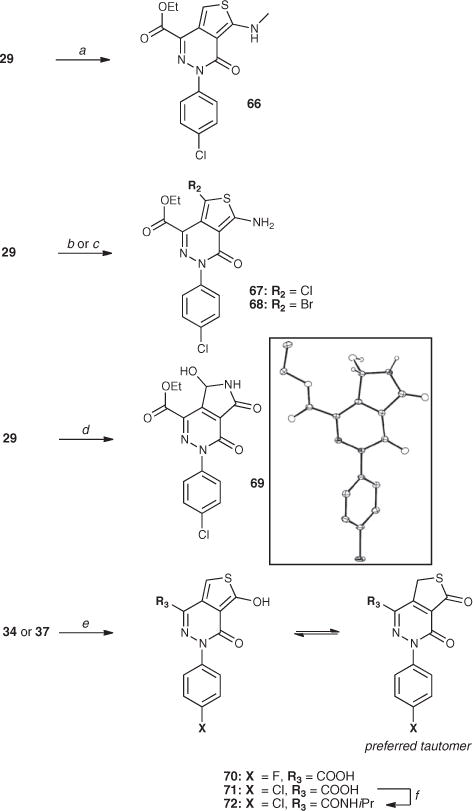
Reagents and conditions: (a) MeI, K2CO3, 65 °C, 10 h.; (b) N-chlorosuccinimmide; (c) N-bromosuccinimmide; (d) DMDO, acetone, rt, 5 min; (e) 1 N HCl, acetonitrile, 120 °C (microwave irradiation), 30 min; (f) isopropyl amine, BOP reagent, N,N-diisopropylethylamine, dimethylsulfoxide, rt, 4 h. The insert shows the X-ray crystal structure of compound 69 (CCDC 853892).
After synthesis, all test ATPZs were evaluated for aqueous solubility and activity in a heparin-induced tau K18PL fibrillization assay.6 Analogues that produced full dose-response curves in the fibrillization assay and whose activity was confirmed in a secondary centrifugation assay6 progressed to an evaluation of pharmacokinetic (PK) properties. Collectively, these studies defined the structure-activity relationship (SAR) of ATPZs and in turn resulted in the identification of selected analogues which exhibit, in addition to excellent brain-penetration and oral bioavailability, more favorable properties compared to 1, such as higher aqueous solubility and higher unbound fraction in brain homogenates. Among these compounds, ATPZ 43 was evaluated for possible dose-limiting toxicities in wild type mice. The results from PK and toxicity studies suggest that nM free drug levels can be achieved in the brains of mice after oral administration of well-tolerated doses of 43.
1.1. Design and synthesis of test aminothienopyridazine (ATPZ) congeners
Previous SAR studies6,7 indicated that the R3 fragment (Fig. 1) is relatively well tolerant of structural variations, while N-acylations (R1) and alkylations at C7 (R2) lead to drastic reduction in the ability of the derived analogues to interfere with tau fibrillization. The SAR of R4, on the other hand, remained incompletely defined. Thus, in addition to a number of congeners of 1 (i.e., compounds of general structure F, Scheme 1), a series of ATPZs bearing modifications at the R3 and R4 fragments (i.e., analogues of general formula G, Scheme 2), as well as congeners modified at the aminothiophene ring (Scheme 3), were synthesized.
Scheme 2.
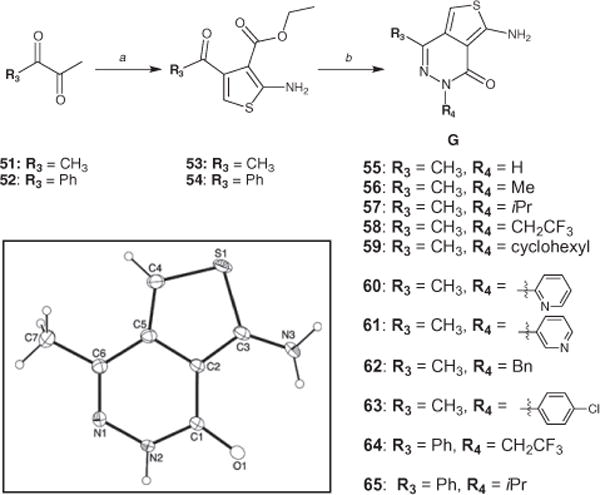
Reagents and conditions: (a) ethyl cyanoacetate, S8, morpholine, ethanol, 150 °C (microwave irradiation), 15 min; (b) appropriate substituted hydrazine, 150 °C (microwave irradiation), 0.5–3 h. The insert shows the X-ray crystal structure of compound 55 (CCDC 853887).
For the synthesis of analogues of general structure F, we employed the reaction sequence highlighted in Scheme 1.7 Central to this strategy is the Gewald reaction protocol8,9 to construct the ATPZ heterocyclic core from pyridazines of general structure C. Next, saponification of esters D to the corresponding acids (E) was followed by coupling reactions with the appropriate amine to provide amides of general structure F (Scheme 1). Such an approach proved both effective and reliable for the synthesis of several closely related analogues of 1. However, as stable diazonium salts can be generally derived from anilines, but not from aliphatic-or heteroaryl-amines, this method could not be exploited to obtain analogues bearing R4 ≠ phenyl/substituted phenyl. Thus, to circumvent this limitation, aminothiophene keto-esters 53 and 54 (Scheme 2) were first prepared by subjecting di-ketones 51 and 52 to the Gewald reaction. Next, condensation of 53 and 54 with a series of alkyl-, aryl-, and (hetero)aryl-hydrazines, under microwave irradiation-promoted conditions, provided the corresponding ATPZs (55–65) of general structure G (Scheme 2).
Scheme 3 highlights the synthesis of analogues modified at the aminothiophene ring, which include examples of N-alkylated derivatives, such as 66, obtained by treating 29 with methyl iodide in the presence of potassium carbonate, as well as C7-halogenated derivatives (67 and 68), obtained from 29 upon treatment with N-chloro- and N-bromo-succinimmide, respectively. Interestingly, treatment of 29 with dimethyldioxirane resulted in rapid (~15 min) consumption of the starting material with the formation of the 5,7-dihydroxy-pyrrolopyridazine 69, whose structure was confirmed by single crystal X-ray analysis. Treatment of acids 34 and 37 with hydrochloric acid under microwave-promoted conditions10 then produced, respectively, thienopyridazines 70 and 71, in which the amine is replaced by a hydroxyl. Compound 71 was then converted to the corresponding cyclopropyl amide 72. In agreement with previous reports,10 the NMR spectra of 70–72 are consistent with the thiolactone tautomeric forms shown in Scheme 3.
2. Results
2.1. Evaluation of aminothienopyridazine (ATPZ) analogues in the tau fibrillization assay
Initially, each test compound was evaluated for aqueous solubility in the sodium acetate buffer employed in the fibrillization assay.7 Next, ATPZ analogues were evaluated in vitro for their ability to disrupt the heparin-induced fibrillization of the K18 tau fragment (amino acids 244–369)11 bearing the P301L12 mutation found in inherited frontotemporal lobar degeneration (K18PL). The activity of the ATPZs was determined by a thioflavin-T (ThT) fluorescence assay, which monitors the formation of ThT-positive cross-beta fibrils in the fibrillization reaction.13 In addition, all compounds were evaluated in a secondary assay, which consisted of SDS-PAGE analysis of the soluble and insoluble fractions obtained after centrifugation of the ATPZ-treated fibrillizing mixtures (centrifugation assay).6,13 The results of these experiments, summarized in Table 1, in general reveal a good correlation between the primary and secondary assays, although higher % inhibitions are typically observed in the ThT assay compared to the centrifugation assay. The different % inhibitions seen in the two assays may not be necessarily indicative of some degree of interference of the ATPZ analogues with ThT binding and/or fluorescence, but could be explained considering the differences between the ThT- and centrifugation assay. While the former is dependent on protein-conformation, as it is based on the characteristic red-shift in excitation spectrum of ThT that occurs upon ThT binding to pleated β-sheets found in amyloid fibrils,14 the latter is based on the total amount of pelletable material, regardless of protein-conformation. Consistent with previous results, the majority of active ATPZs were found to exhibit IC50 values in the 1–10 μM range. Given that the tau concentration in the in vitro assay is kept at 20 μM, these results suggest that ATPZ inhibitors may be most effective when present in a ~1:1 ratio with tau protein. Interestingly, the activity data illustrated in Table 1 indicate that all compounds lacking the thienopyridazine heterocycle, such as pyridazine 21, aminothiophene 54, and pyrrolopyridazine 69, were essentially devoid of any activity against K18PL fibrillization as determined by centrifugation assay. Conversely, minimally substituted ATPZs, such as 55 and 56, as well as variously decorated derivatives, including hydroxythienopyridazines (70 and 72), and ATPZs halogenated at C7 (67 and 68), all produced near full inhibition in the K18PL fibrillization assay. These results strongly suggest that the thienopyridazine heterocycle is essential for inhibition of tau fibrillization, while the nature of the R1–R4 substituents may play a role in modulating the potency of the active thienopyridazine, possibly by affecting ATPZ-tau interactions and/or the physicochemical properties of the ATPZs. For example, the relatively lower inhibition observed in the centrifugation assay, which is conducted at 50 μM compound concentration, with ATPZ esters of structure D relative to the corresponding acids (E) and amides (F) may be attributed in part to the generally lower water solubility of these esters.
Table 1.
Activity and Solubility of Test Compounds
|
Solubility in assay buffer | K18PL ThT assay | K18PL centrifugation assay | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Compd number | R1 | R2 | R3 | R4 | X | Upper solubility limit (μM) | IC50 (μM) | Max % inhib. | % Soluble |
| 26 | −NH2 | −H | −COOEt | 4-F-Ph | S | >200 | 1.0 ± 1.11 | 77 | 63 |
| a29 | −NH | −H | −COOEt | 4-Cl-Ph | S | 13 | 4.0 ± 2.99b | 71 | 47 |
| 27 | −NH2 | −H | −COOEt | 2-Cl-Ph | S | 12.8 | 2.0 ± 1.42b | 75 | 71 |
| 28 | −NH2 | −H | −COOEt | 3-Cl-Ph | S | 32 | 3.2 ± 2.02b | 82 | 73 |
| 30 | −NH2 | −H | −COOEt | 4-CF3-Ph | S | 32 | DNC | 48 | 27 |
| 31 | −NH2 | −H | −COOEt | 4-OCF3-Ph | S | 12.8 | DNCb | 41 | 29 |
| 32 | −NH2 | −H | −COOEt | 4-OH-Ph | S | 12.8 | 5.0 ± 1.29 | 81 | 69 |
| 33 | −NH2 | −H | −COOEt | 4-iPr-Ph | S | 32 | DNCb | 8 | 17 |
| 66 | −NHCH3 | −H | −COOEt | 4-Cl-Ph | S | 80 | DNC | 39 | 51 |
| 67 | −NH2 | −Cl | −COOEt | 4-Cl-Ph | S | 32 | 2.0 ± 1.53 | 88 | 65 |
| 68 | −NH2 | −Br | −COOEt | 4-Cl-Ph | S | 12.8 | 4.0 ± 1.10 | 92 | 62 |
| 69 | −OH | −OH | −COOEt | 4-Cl-Ph | NH | >200 | DNC | 57 | 15 |
| 34 | −NH2 | −H | −COOH | 4-F-Ph | S | >200 | 2.0 ± 1.27 | 84 | 83 |
| a37 | −NH | −H | −COOH | 4-Cl-Ph | S | >200 | 5.0 ± 2.15b | 84 | 74 |
| 35 | −NH2 | −H | −COOH | 2-Cl-Ph | S | >200 | 3.2 ± 1.58 | 86 | 64 |
| 36 | −NH2 | −H | −COOH | 3-Cl-Ph | S | 32 | 1.6 ± 1.22 | 92 | 73 |
| 38 | −NH2 | −H | −COOH | 4-CF3-Ph | S | >200 | 19.9 ± 1.10 | 88 | 83 |
| 39 | −NH2 | −H | −COOH | 4-OCF3-Ph | S | >200 | 1.6 ± 1.53 | 92 | 71 |
| 40 | −NH2 | −H | −COOH | 4-OH-Ph | S | >200 | 4.0 ± 1.58b | 84 | 70 |
| 70 | −OH | −H | −COOH | 4-F-Ph | S | >200 | 2.5 ± 1.26b | 92 | 87 |
| 42 | −NH2 | −H | −CONH-iPr | 4-F-Ph | S | >200 | 1.6 ± 1.39 | 89 | 77 |
| 43 | −NH2 | −H | −CONH-cPr | 4-F-Ph | S | >200 | 2.5 ± 2.93b | 88 | 78 |
| 44 | −NH2 | −H | −CONH–CH2CF3 | 4-F-Ph | S | >200 | 8.0 ± 1.28 | 83 | 75 |
| a1 | −NH2 | −H | −CONH-cPr | 4-Cl-Ph | S | 32 | 8.0 ± 2.48b | 83 | 69 |
| 45 | −NH2 | −H | −CONH-cPr | 2-Cl-Ph | S | 80 | DNC | 68 | 44 |
| 46 | −NH2 | −H | −CONH-iPr | 3-Cl-Ph | S | 32 | 3.2 ± 2.02b | 82 | 73 |
| 47 | −NH2 | −H | −CONH-cPr | 4-CF3-Ph | S | 80 | 6.3 ± 2.28 | 64 | 83 |
| 48 | −NH2 | −H | −CONH-iPr | 4-OCF3-Ph | S | 80 | 15.8 ± 1.95 | 67 | 42 |
| 49 | −NH2 | −H | −CONH-iPr | 4-OH-Ph | S | >200 | 15.8 ± 1.38 | 86 | 84 |
| 50 | −NH2 | −H | −CONH-cPr | 4-iPr-Ph | S | 80 | 4.0 ± 2.88 | 82 | 86 |
| 72 | −OH | −H | −CONH-iPr | 4-Cl-Ph | S | ND | 10.0 ± 1.25 | 96 | 85 |
| 55 | −NH2 | −H | −CH3 | −H | S | >200 | 8.0 ± 1.87b | 87 | 81 |
| 56 | −NH2 | −H | −CH3 | −CH3 | S | >200 | 2.5 ± 1.19 | 82 | 76 |
| 57 | −NH2 | −H | −CH3 | iPr | S | 80 | 19.9 ± 1.16 | 95 | 17 |
| 58 | −NH2 | −H | −CH3 | CH2CF3 | S | >200 | 10± 1.18b | 81 | 78 |
| 59 | −NH2 | −H | −CH3 | Cyclohexyl | S | >200 | 31.6 ± 1.12 | 65 | 86 |
| 60 | −NH2 | −H | −CH3 | 2-Py | S | >200 | 2.0 ± 1.27 | 85 | 78 |
| 61 | −NH2 | −H | −CH3 | 3-Py | S | >200 | 4.0 ± 1.41 | 74 | 84 |
| 62 | −NH2 | −H | −CH3 | −Bn | S | 80 | 1.6 ± 1.24 | 89 | 73 |
| 63 | −NH2 | −H | −CH3 | 4-Cl-Ph | S | 80 | 2.0 ± 1.23 | 85 | 70 |
| 64 | −NH2 | −H | −Ph | CH2CF3 | S | >200 | 6.3 ± 1.63b | 103 | 82 |
| 65 | −NH2 | −H | −Ph | iPr | S | >200 | 19.9 ± 1.63b | 89 | 92 |
| 21 |
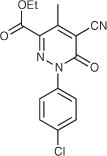
|
12.8 | DNCb | 4.4 | 21 | ||||
| 54 |
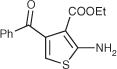
|
>200 | DNC | 29 | 6.5 | ||||
Previously reported data;7 DNC = did not calculate due to absence of asymptote; ND = not determined; all IC50 values were determined through sigmoidal curve fitting of compound concentration-response analyses, with each concentration tested in triplicate.
All results are from a single curve fit, except where indicated by, where the mean of 2 or more separate analyses are reported.
2.2. Determination of brain-penetration
Among all active tau aggregation inhibitors shown in Table 1, a representative subset having aqueous buffer solubility >50 μM were selected for studies consisting of determination of brain and plasma levels 1 h after i.p. administration of 5 mg/Kg (Table 2). For each of the ATPZs found to be brain-penetrant, equilibrium dialysis experiments were also performed to determine the unbound fraction in plasma and brain homogenates (Table 2). These values provide for an estimate of the percentage of drug that is freely available in these tissues. The results from the PK studies suggest that substitution of the amino group with an hydroxyl results in derivatives that are not brain-penetrant (cf., 1 and 72). Furthermore, the observation that smaller derivatives lacking the R4 fragment (e.g., 55) exhibit low plasma levels 1 h after dosing suggests that a phenyl group in R4, although not strictly required for biological activity (cf., Table 1), may be an important determinant of metabolic stability and possibly brain-penetration. Notably, among all brain-penetrant ATPZs, 42, 43 and 44 were found to be able to partition freely between plasma and brain with B/P ratio near unity (Table 2). Equally important, evaluation of non-specific plasma/brain protein binding revealed that the unbound fractions in brain homogenates are ~4.3% (43) and 5.0% (44), respectively.
Table 2.
Brain and plasma level of test compounds
| Compd | Brain (ng/g) | Plasma (ng/mL) | B/Pa (total) | Fub (%) brain | Fub (%) plasma | B/P (fu)c |
|---|---|---|---|---|---|---|
| 1e | 1300 ±110 | 820 ± 150 | 1.61 ± 0.28 | 1.1 ± 0.6 | 1.9 ± 0.2 | 0.92 ±0.16 |
| 43 | 670±100 | 610 ± 27 | 1.09 ± 0.15 | 4.3 ± 0.9 | 6.9 ± 0.4 | 0.68 ± 0.09 |
| 42 | 280 ±410 | 110 ± 160 | 2.68 ± 0.47 | 2.9 ± 0.4 | 5.7 ± 0.5 | 1.30 ±0.23 |
| 72 | 16.3 ± 8.8 | 4700 ± 1000 | 0.0035 ± 0.0015 | DNC | DNC | – |
| 49 | 14.1 ± 9.4 | 150 ± 100 | 0.098 ± 0.007 | DNC | DNC | – |
| 44 | 637 ± 75 | 401 ± 14 | 1.59 ± 0.21 | 3.2 ± 0.7 | 2.2 ± 0.2 | 2.31 ± 0.31 |
| 55 | BLDd | BLDd | – | DNC | DNC | – |
| 56 | BLDd | 10± 18 | – | DNC | DNC | – |
| 60 | 7.5 ± 5.3 | 61 ±20 | 0.116 ± 0.039 | DNC | DNC | – |
| 62 | 48 ± 18 | 78 ± 16 | 0.60 ±0.12 | 5.1 ± 1.8 | 4.0 ±1.0 | 0.77 ±0.15 |
Brain-to-plasma ratio.
Fraction unbound.
Ratio of free drug in brain and plasma.
Below limit of detection.
Previously reported data7; DNC = did not calculate due to low B/P.
Finally, a full PK evaluation of 43 confirms that there is good brain exposure to the compound (Fig. 2). Moreover, the oral bioavailability of 43 was found to be ~60% (see Supplementary data).
Figure 2.
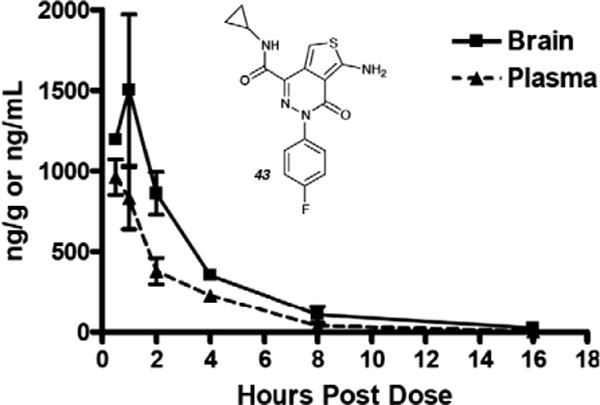
Brain and plasma level of 43 after i.p. administration of 5 mg/kg.
2.3. In vivo tolerability evaluation of ATPZ 43
To evaluate potential dose-limiting toxicities of ATPZ 43 in mice, we determined the maximum tolerated dose (MTD) after a single oral administration of the compound. Notably, no overt signs of toxicity were seen until a dose of 43 of 100 mg/kg, at which point only mild and transient lethargy was observed. Subsequently, 43 was administered in drinking water (containing 3% PEG400 and 0.5% methylcellulose) to normal mice for a 1-month period at a dose equivalent of 50 mg/kg/day (assuming 5 ml water consumption/day/mouse). The amount of water consumed by the compound-treated mice was normal, and no visible adverse events were noted. Importantly, no differences in body weight gain were observed between the compound- and vehicle-treated (PEG400/methylcellulose) groups (n = 5/group; 9.7% and 8.2% increases of body weight, respectively). Moreover, there were no differences in organ weights, including heart, liver, kidney, spleen and brain, between the mice that received compound 43 or vehicle only (see Supplementary data). Similarly, there were no alterations in any blood cell counts in the mice that were administered the ATPZ (see Supplementary data). An evaluation of brain levels of 43 after 3 days of dosing via drinking water at 50 mg/kg/day revealed total brain levels of 139 ± 8 nM, with a calculated free compound level of 6.0 ± 0.4 nM. Notably, the plasma and brain levels of 43 in mice that received compound in drinking water for 30 days was comparable to the levels in mice that received the compound in water for 3 days. This reveals that repeated dosing of the compound over a prolonged interval did not result in the induction of liver enzymes that might alter the metabolism of the compound.
3. Discussion
That ATPZ inhibitors of tau fibrillization hold promise as potential candidates for in vivo testing was suggested by earlier studies from our laboratory.6 The purpose of this study was to investigate further the SAR of this class of compounds and to identify selected candidates that have solubility, brain-penetration, oral bioavailability and safety that might be suitable for longer-term in vivo studies. Based on the observation that the IC50 values of all active ATPZs in our in vitro assay are typically in the range of stoichiometric equivalence with tau, it seems plausible that this chemotype may be interfering with the fibrillization reaction by interacting with tau monomers. If this is the case, similar stoichiometric requirement of tau:ATPZ may be expected in vivo for effective inhibition of tau assembly. The intraneuronal tau concentration is estimated to be ~2 μM. However, under normal circumstances, tau is believed to be almost completely (>99%) bound to microtubules (MTs).15 Thus, the free intraneuronal tau fraction available to enter the fibrillization cascade may be <20 nM, even under pathological conditions.4 Although the values of free tau are only estimates, they suggest that candidate ATPZs should preferably achieve free brain levels in this concentration range to be effective if there is indeed a requirement for stoichiometric amounts of the drug. From the results displayed in Table 1, the pharmacophore of ATPZs would appear to be the thienopyridazine heterocycle, while the R1–R4 substituents may be important determinants of ADME-PK properties. In addition, our results suggest that the presence of a para-fluorophenyl residue at the N3, a primary amino group at the C5 position, and a carboxylamide moiety at C1 of the thienopyridazine heterocycle, comprise the optimal substitution pattern to achieve the desired combination of activity and ADME-PK properties. Indeed, based on the results from our PK studies, administration of a dose of 50 mg/kg/day of compound 43 to mice in drinking water resulted in total brain compound levels of ~140 nM and a predicted brain free drug concentration of 6 nM. Furthermore, since the MTD for compound 43 was found to be >50 mg/kg, it is likely that even higher doses could be utilized during long-term in vivo testing. Taken together, these results indicate that 43 and possibly other closely related congeners, such as 42 and 44, are suitable candidates for further investigating the in vivo properties and potential biological activity of the ATPZ class of tau fibrillization inhibitors.
4. Conclusions
To investigate the SAR of ATPZs and to identify viable candidates for in vivo studies, a series of new analogues were synthesized and evaluated. Among the active, brain-penetrant ATPZ analogues, 43 exhibited good oral bioavailability, as well as acceptable water solubility and non-specific brain tissue binding, such that oral administration of doses lower than the MTD are expected to achieve potentially efficacious free drug concentrations in the mouse brain after oral administration. These results indicate that 43 may be suitable for long-term in vivo testing.
5. Experimental
5.1. Materials and methods
All solvents were reagent grade. All reagents were purchased from Aldrich or Acros and used as received. Thin layer chromatography (TLC) was performed with 0.25 mm E. Merck pre-coated silica gel plates. Flash chromatography was performed with silica gel 60 (particle size 0.040–0.062 mm) supplied by Silicycle and Sorbent Technologies. Spots were detected by viewing under a UV light. Yields refer to chromatographically and spectroscopically pure compounds. Infrared spectra were recorded on a Jasco Model FT/IR-480 Plus spectrometer. All melting points were obtained on a Thomas-Hoover apparatus. Proton (1H) and carbon (13C) NMR spectra were recorded on a Bruker AMX-500 spectrometer. Chemical shifts were reported relative to solvents. High-resolution mass spectra were measured at the University of Pennsylvania, Mass Spectrometry Service on a Waters LCT Premier XE LC-MS system. Single-crystal X-ray structure determinations were performed at the University of Pennsylvania. X-ray intensity data were collected on a Bruker APEXII CCD area detector employing graphite-monochromated Mo-Ka radiation (l = 0.71073 Å) at a temperature of 143(1) K. Analytical reversed-phased (Sunfire™ C18; 4.6 × 50 mm, 5 mL) high-performance liquid chromatography (HPLC) was performed with a Water binary gradient module 2525 equipped with Waters 2996 PDA and Water micromass ZQ. All samples were analyzed employing a linear gradient from 10% to 90% of acetonitrile in water over 8 min and flow rate of 1 mL/min, and unless otherwise stated, the purity level was >95%. Preparative reverse phase HPLC purification was performed employing Waters SunFire™ prep C18 OBD™ columns (5 μm 19 × 50 or 19 × 100 mm). All samples were purified employing a linear gradient from 10% to 90% of acetonitrile in water over 15 min and flow rate of 20 mL/min. The preparative HPLC system was equipped with Gilson 333 pumps, a 215 Liquid Handler, 845Z injection module, and UV detector. Unless otherwise stated, all final compounds were found to be >95% as determined by HPLC/MS and NMR.
5.1.1. General procedure for the synthesis of hydrazones B
To a solution of the appropriate aniline (39.2 mmol) and 37% hydrochloridric acid (9.93 mL) in water (4.96 mL) and ethanol (4.96 mL), a solution of sodium nitrite (2.93 g, 42.5 mmol) in water (13 mL) was added dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 30 min and then a solution of sodium acetate (12.65 g, 154 mmol) in water (20 mL) and a solution of ethyl acetoacetate (5.10 g, 4.96 mL, 39.2 mmol) in ethanol (10 mL) were added maintaining the temperature below 5 °C. The reaction mixture was stirred at 0 °C for 4 h. Water was then added and the resultant mixture was extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. Finally, crystallization from ethanol provided the desired hydrazone B, which was directly used without further purification.
5.1.1.1. Ethyl 2-(2-(4-fluorophenyl)hydrazono)-3-oxobutanoate (10)
Yield: 98%, mp: 81–83 °C. 1H NMR (CDCl3): δ 1.40 (t, J = 7.0 Hz, 3H), 2.59 (s, 3H), 4.34 (q, J = 7.2 Hz, 2H), 7.09 (t, J = 8.7 Hz, 2H), 7.39 (dd, J = 9.0 and 5.0 Hz, 2H) ppm. 13C NMR (CDCl3): δ 14.4, 30.8, 61.1, 116.5 (d, JCF2 = 22.9 Hz), 118.0 (d, JCF3 = 8.1 Hz), 126.1, 138.1, 161.3 (d, JCF1 = 243.9 Hz), 165.0, 197.2 ppm. IR (film): v 3125, 1701, 1620 cm−1. MS [ESI]+: Calcd for C12H14FN2O3+ 253.10. Found 253.25.
5.1.1.2. Ethyl 2-(2-(2-chlorophenyl)hydrazono)-3-oxobutanoate (11)
See Ref. 16.
5.1.13. Ethyl 2-(2-(3-chlorophenyl)hydrazono)-3-oxobutanoate (12)
Yield: 70%. 1H NMR (CDCl3): δ 1.42 (t, J = 7.5 Hz, 3H), 2.60 (s, 3H), 4.36 (q, J =7.2 Hz, 2H), 7.14 (d, J = 8.0 Hz, 1H), 7.24 (d, J = 9.0 Hz, 2H), 7.30 (t, J = 8.2 Hz, 1H), 7.48 (d, J = 3.2 Hz, 1H) ppm. 13C NMR (CDCl3): δ 14.4, 30.8, 61.1, 114.6, 116.4, 125.6, 126.8, 130.6, 135.7, 143.0, 164.8, 197.4 ppm. IR: v 3345, 1725, 1686 cm−1. MS [ESI]+: Calcd for C12H14ClN2O3+ 269.07. Found 269.16.
5.1.1.4. Ethyl 2-(2-(4-chlorophenyl)hydrazono)-3-oxobutanoate (13)
See Ref. 7.
5.1.1.5. Ethyl 3-oxo-2-(2-(4-(trifluoromethyl)phenyl)hydrazono)butanoate (14)
Yield: 94%; 1H NMR (CDCl3): δ 1.39 (t, J = 7.1 Hz, 3H), 2.50 (s, 3H), 4.38 (q, J =7.1 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H), 7.62 (t, J = 6.6 Hz, 2H) ppm.
5.1.1.6. Ethyl 3-oxo-2-(2-(4-(trifluoromethoxy)phenyl)hydrazono)butanoate (15)
Yield: 96%; 1H NMR (CDCl3): δ 1.41 (m, 3H), 2.50 (s, 2H), 2.61 (s, 1H), 4.37 (m, 2H), 7.24–7.26 (m, 2H), 7.24–7.26 (m, 1H), 7.34–7.37 (m, 1H), 7.42–7.45 (m, 1H) ppm. 13C NMR (CDCl3): δ 14.2, 27.0, 61.7, 116.6, 117.5, 122.49, 122.65, 128.0, 140.5, 163.8, 197.4 ppm.
5.1.1.7. Ethyl 2-(2-(4-hydroxyphenyl)hydrazono)-3-oxobutanoate (16)
Yield: 72%, mp: 106–108 °C. 1H NMR (CD3OD): δ 1.36–1.41 (m, 3H), 2.46 (s, 2H), 2.53 (s, 1H), 4.30–438 (m, 2H), 6.84–6.87 (m, 2H), 7.31–7.34 (m, 1H) ppm. MS [ESI]+: Calcd for C12H15N2O4+ 251.10. Found 251.23.
5.1.1.8. (E/Z) Ethyl 2-(2-(4-isopropylphenyl)hydrazono)-3-oxobutanoate (17)
Yield: 94%; 1H NMR (CDCl3): δ 1.25–1.27 (m, 12H, 2 × CH(CH3)2), 1.41 (m, 6H, 2 × CH2CH3), 2.49, 2.59 (s, 6H, 2 × CH3), 2.92 (m, 2H, 2 × CH), 4.32–4.40 (m 4H, 2 × CH2CH3), 7.24–7.30 (m, 6H), 7.36 (d, J = 8.5 Hz, 2H) ppm.
5.1.1.9. General procedure for the synthesis of pyridazines C
Method A: A mixture of the appropriate hydrazone B (1.0 mmol), ethyl cyanoacetate (0.113 g, 1.0 mmol), and sodium acetate (0.17 g, 2.4 mmol) in acetic acid (3.3 mL) was heated to 170 °C using microwave irradiation for 4 min. After cooling, the precipitate was collected and crystallized in ethanol to give the desired pyridazine C. Method B: A mixture of the appropriate hydrazone B (6.29 mmol), ethyl cyanoacetate (1.06 g, 1.0 mL, 9.44 mmol), and 4-aminobutyric acid was heated to 160 °C for 2.5 h. After cooling, the residue was purified by silica gel column chromatography using ethyl acetate-hexanes 1:3 as eluant to provide the desired pyridazine C.
5.1.1.10. Ethyl 5-cyano-1-(4-fluorophenyl)-4-methyl-6-oxo-1,6-dihydropyridazine-3-carboxylate (18)
Method A. Yield: 55%, mp: 146–147 °C. 1H NMR (CDCl3): δ 1.40 (t, J = 7.0 Hz, 3H), 2.76 (s, 3H), 4.43 (q, J = 7.2 Hz, 2H), 7.19 (t, J =8.7 Hz, 2H), 7.63 (dd, J = 9.0 and 5.0 Hz, 2H) ppm. 13C NMR (CDCl3): δ 14.2, 19.3, 62.9, 112.4, 115.9, 116.1 (d, JCF2 = 23.0 Hz), 127.1 (d, JCF3 = 8.6 Hz), 136.1, 137.4, 150.8, 155.9, 162.0, 162.7 (d, JCF1 = 243.9 Hz). IR (film): v 3123, 1727, 1683, 1602 cm−1.
5.1.1.11. Ethyl 1-(2-chlorophenyl)-5-cyano-4-methyl-6-oxo-1,6-dihydropyridazine-3-carboxylate (19)
Method B. Yield: 67%, mp: 93–95 °C (from ethanol). 1H NMR (CDCl3): δ 1.38 (t, J = 7.1 Hz, 3H), 2.78 (s, 3H), 4.41 (q, J = 7.1 Hz, 2H), 7.40–7.49 (m, 3H), 7.57 (dd, J = 7.9 and 1.4 Hz, 1H) ppm. 13C NMR (CDCl3): δ 14.3, 19.6, 63.0, 112.3, 115.9, 128.1, 128.8130.8, 131.5, 131.6, 137.6, 137.8, 151.5, 155.4, 162.0 ppm. IR: v 2235, 1731, 1690 cm−1.
5.1.1.12. Ethyl 1-(3-chlorophenyl)-5-cyano-4-methyl-6-oxo-1,6-dihy-dropyridazine-3-carboxylate (20)
Method B. Yield: 90%. 1H NMR (CDCl3): δ 1.41 (d, J = 7.0 Hz, 6H), 2.76 (s, 3H), 4.45 (q, J = 7.1 Hz, 2H), 7.44–7.45 (m, 2H), 7.57–7.59 (m, 1H), 7.67 (s, 1H) ppm. 13C NMR (CDCl3): δ 14.2, 19.3, 63.0, 112.3, 116.1 123.3, 125.4, 129.7, 130.1, 134.9, 137.6, 140.8, 150.9, 155.7, 161.9 ppm. IR: v 3429, 3221, 1690, 1634 cm−1. MS [ESI]+: Calcd for C15H13ClN3O3+ 318.06. Found 318.08.
5.1.1.13. Ethyl 5-amino-3-(2-chlorophenyl)-4-oxo-3,4-dihydrothieno-[3,4-d]pyridazine-1-carboxylate (21)
See Ref. 7.
5.1.1.14. Ethyl 5-cyano-4-methyl-6-oxo-1-(4-(trifluoromethyl)phenyl)-1,6-dihydropyridazine-3-carboxylate (22)
Method A. Yield: 35%; 1H NMR (CDCl3): δ 1.41 (t, J = 7.1 Hz, 3H), 2.77 (s, 3H), 4.44 (q, J = 7.1 Hz, 2H), 7.77 (d, J = 8.6 Hz, 2H), 7.83 (d, J = 8.6 Hz, 2H) ppm. 13C NMR (CDCl3): δ 14.2, 19.4, 63.0, 112.3, 116.3, 125.5, 123.7 (q, JCF1 = 272.5 Hz), 126.36 (q, JCF2 = 3.7 Hz), 131.38 (q, JCF2 = 37.8 Hz), 137.9, 142.7, 151.1, 155.7, 161.9 ppm.
5.1.1.15. Ethyl 5-cyano-4-methyl-6-oxo-1-(4-(trifluoromethoxy)phenyl)-1,6-dihydropyridazine-3-carboxylate (23)
Method B. Yield: 43%; 1H NMR (CDCl3): δ 1.41 (t, J = 7.1 Hz, 3H), 2.77 (s, 3H), 4.44 (q, J = 7.1 Hz, 2H), 7.36 (d, J = 8.3 Hz, 2H), 7.72 (d, J = 9.1 Hz, 2H) ppm. 13C NMR (CDCl3): δ 14.3, 19.4, 63.0, 112.3, 116.2, 121.5, 126.7, 137.7, 138.3, 150.9, 155.8, 162.0 ppm.
5.1.1.16. Ethyl 5-cyano-1-(4-hydroxyphenyl)-4-methyl-6-oxo-1,6-dihydropyridazine-3-carboxylate (24)
See Ref. 17.
5.1.1.17. Ethyl 5-cyano-1-(4-isopropylphenyl)-4-methyl-6-oxo-1,6-dihydropyridazine-3-carboxylate (25)
Method B. Yield: 85%; 1H NMR (CDCl3): δ 1.28 (d, J = 7.0 Hz, 6H), 1.40 (t, J = 7.0 Hz, 3H), 2.76 (s, 3H), 2.98 (sept, J = 6.8 Hz, 1H), 4.42 (q, J = 7.1 Hz, 2H), 7.35 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.5 Hz, 2H) ppm. 13C NMR (CDCl3): δ 14.3, 19.3, 24.0, 34.1, 62.8, 112.6, 115.8, 124.9, 127.2, 137.1, 137.9, 150.5, 150.6, 156.0, 162.2 ppm.
5.1.2. General procedure for synthesis of aminothienopyridazines D
A mixture of the appropriate pyridazine C (0.31 mmol), sulfur (15 mg, 0.46 mmol), and morpholine (54 mg, 54 μL, 0.62 mmol) in ethanol (1.5 mL) was heated to 150 °C using microwave irradiation for 15 min. After cooling, the precipitate was collected and purified by silica gel column chromatography using ethyl acetate and hexane 1:3 as eluant to obtain the desired aminothienopyridazine D.
5.1.2.1. Ethyl 5-amino-3-(4-fluorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylate (26)
Yield: 70%, mp: 178–180 °C 1H NMR (CDCl3): δ 1.42 (t, J = 6.9 Hz, 3H), 4.43 (q, J = 7.1 Hz, 2H), 6.06 (br s, 2H), 7.13 (t, J = 8.3 Hz, 2H), 7.22 (s, 1H), 7.54–7.57 (m, 2H) ppm. 13C NMR (CDCl3): δ 14.4, 62.1, 104.7, 106.2, 115.7 (d, JCF2 = 22.8 Hz), 127.9, (d, JCF3 = 8.6 Hz), 133.9, 136.7, 159.5, 161.7 (d, JCF1 = 269.8 Hz), 163.1 ppm. IR (film): v 3423, 3302, 3128, 1718, 1641 cm−1. MS [ESI]+: Calcd for C15H13FN3O3S+ 334.07. Found 334.11.
5.1.2.2. Ethyl 5-amino-3-(2-chlorophenyl)-4-oxo-3,4-dihydrothieno-[3,4-d]pyridazine-1-carboxylate (27)
Yield: 87%. 1H NMR (CDCl3): δ 1.28 (t, J =7.0 Hz, 3H), 4.31 (q, J = 7.0 Hz, 2H), 7.15 (s, 1H), 7.47 (m, 2H), 7.53–7.60 (m, 3H), 7.64 (s, 1H) ppm. 13C NMR (CDCl3): δ 14.0, 54.9, 61.4, 103.5, 104.1, 126.0, 128.0, 129.7, 130.4, 130.6, 131.7, 133.0, 138.3, 157.9, 162.2, 163.1 ppm. IR: v 3425, 3303, 1710, 1648, 1587 cm−1. HRMS [ESI]+: Calcd for C15H12ClNaN3O3S+ 372.0186. Found 372.0190.
5.1.2.3. Ethyl 5-amino-3-(3-chlorophenyl)-4-oxo-3,4-dihydrothieno-[3,4-d]pyridazine-1-carboxylate (28)
Yield: 72%. 1H NMR (CDCl3): δ 1.44 (t, J = 7.2 Hz, 3H), 4.45 (q, J = 7.1 Hz, 2H), 6.20 (br s, 2H), 7.26 (s, 1H), 7.33 (d, J =8.0 Hz, 1H), 7.39 (t, J = 8.1 Hz, 1H), 7.54 (d, J = 8.1 Hz, 1H), 7.66 (s, 1H) ppm. 13C NMR (CDCl3): δ 14.4, 62.2, 105.0, 106.7, 124.1, 126.2, 127.0, 127.8, 129.7, 134.1, 134.3, 141.7, 159.4, 161.9, 163.0 ppm. MS [ESI]+: Calcd for C15H14N3O4S+ 350.04. Found 350.09.
5.1.2.4. Ethyl 5-amino-3-(4-chlorophenyl)-4-oxo-3,4-dihydrothieno-[3,4-d]pyridazine-1-carboxylate (29)
See Ref. 7.
5.1.2.5. Ethyl 5-amino-4-oxo-3-(4-(trifluoromethyl)phenyl)-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylate (30)
Yield: 90%. 1H NMR (CDCl3): δ 1.43 (t, J = 7.1 Hz, 2H), 4.45 (q, J = 7.1 Hz, 1H), 7.27 (s, 1H), 7.72 (d, J = 8.6 Hz, 1H), 7.80 (d, J =8.4 Hz, 1H), 7.72 (d, J = 8.6Hz, 1H), 7.80 (d, J =8.4Hz, 1H) ppm. 13C NMR (CDCl3): δ 14.4, 62.2, 100.1, 105.3, 106.7, 123.0, 125.2, 125.85, 125.89, 125.91, 127.0, 129.2, 129.5, 134.4, 143.6, 159.4, 162.0, 162.9 ppm. IR (film): v 3438, 3333, 1658, 1604 cm−1. MS [ESI]+: Calcd for C16H13F3N3O3S+ 384.06. Found 384.15.
5.1.2.6. Ethyl 5-amino-4-oxo-3-(4-(trifluoromethoxy)phenyl)-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylate (31)
Yield: 55%. 1H NMR (CDCl3): δ 1.43 (t, J =7.1 Hz, 2H), 4.45 (q, J = 7.1 Hz, 2H), 7.26 (d, J = 6.5 Hz, 1H), 7.31 (d, J = 8.5 Hz, 2H), 7.67 (d, J = 2.0 Hz, 2H) ppm. 13C NMR (CDCl3): δ 14.4, 62.2, 105.1, 119.6, 121.3, 127.1, 127.3, 133.8, 134.1, 139.1, 148.2, 159.4, 161.8, 163.0 ppm. MS [ESI]+: Calcd for C16H13F3N3O4S+ 400.06. Found 400.07.
5.1.2.7. Ethyl 5-amino-3-(4-hydroxyphenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylate (32)
Yield: 46%. 1H NMR (DMSO-d6): δ 1.29 (t, J = 7.2 Hz, 3H), 4.31 (q, J = 7.0 Hz, 2H), 6.83 (d, J = 8.5 Hz, 2H), 7.07 (s, 1H), 7.25 (d, J = 9.0 Hz, 2H), 7.57 (br s, 2H), 9.68 (br s, 1H) ppm. 13C NMR (DMSO-d6): δ 14.7, 61.9, 103.5, 104.8, 115.5, 126.8, 128.3, 132.8, 157.3, 159.1, 163.1, 163.5. IR (film): v 3404, 1639 cm−1. MS [ESI]+: Calcd for C15H14N3O4S+ 332.07. Found 332.04.
5.1.2.8. Ethyl 5-amino-3-(4-isopropylphenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylate (33)
Yield: 72%. 1H NMR (CDCl3): δ 1.28 (d, J = 7.0 Hz, 6H), 1.42 (t, J = 7.5 Hz, 3H), 2.96 (sept, J = 6.9 Hz, 1H), 4.43 (q, J = 7.0 Hz, 2H), 6.20 (br s, 2H), 7.23 (s, 1H), 7.31 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 8.0 Hz, 2H) ppm. 13C NMR (CDCl3): δ 14.1, 24.1, 34.0, 62.0, 104.4, 107.2, 126.0, 126.9, 127.4, 133.5, 138.3, 148.6, 159.7, 161.5, 163.2 ppm. IR (film): v 3426, 3322, 3179, 1705, 1658, 1601 cm−1.
5.1.3. General procedure for ester hydrolysis
To a solution of the appropriate aminothienopyridazine of general structure D (0.43 mmol) in tetrahydrofuran (3 mL) and water (2 mL), lithium hydroxide monohydrate (0.063 g, 1.29 mmol) was added. The reaction mixture was stirred at rt for 16 h. The reaction was quenched with 1 N hydrochloric acid (pH ~2) and filtered. Purification by reverse phase preparative HPLC afforded the desired acid of general structure E.
5.1.3.1. 5-Amino-3-(4-fluorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylic acid (34)
Yield: 77%. 1H NMR (DMSO-d6): δ 7.12 (s, 1H), 7.29 (t, J = 8.7 Hz, 2H), 7.55–7.58 (m, 4H) ppm. 13C NMR (DMSO-d6): δ 104.3, 104.5, 115.8 (d, JCF2 = 22.5 Hz), 126.9, 128.9 (d, JCF3 = 8.5 Hz), 134.2, 137.7, 159.0, 161.3 (d, JCF1 = 242.4 Hz), 163.7, 164.6 ppm. MS [ESI]+: Calcd for C13H9FN3O3S+ 306.03. Found 306.07.
5.1.3.2. 5-Amino-3-(2-chlorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylic acid (35)
Yield: 91%. 1H NMR (DMSO-d6): δ 7.17 (s, 1H), 7.50 (m, 2H), 7.55–7.59 (m, 3H), 7.63–7.65 (m, 1H) ppm. 13C NMR (DMSO-d6): δ 103.7, 104.4, 126.4, 128.0, 129.7, 130.4, 130.7, 131.8, 134.0, 138.4, 158.2, 163.0, 164.0 ppm.
5.1.3.3. 5-Amino-3-(3-chlorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylic acid (36)
Yield: 89%. 1H NMR (DMSO-d6): δ 7.13 (s, 1H), 7.44 (d, J =8.4 Hz, 1H), 7.50 (t, J = 8.5 Hz, 1H), 7.55 (d, J = 8.4 Hz, 1H), 7.65–7.68 (m, 3H) ppm. 13C NMR (DMSO-d6): δ 104.3, 104.7, 125.2, 126.4, 126.8, 127.6, 130.6, 133.1, 142.6, 148.9, 157.7, 158.9, 164.0 ppm. IR: v 3439, 3322,3065 (br, acid band), 1712, 1646, 1590 cm−1. MS [ESI]+: Calcd for C13H9ClN3O3S+ 322.00. Found 322.10.
5.1.3.4. 5-Amino-3-(4-chlorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylic acid (37)
See Ref. 7.
5.1.3.5. 5-Amino-4-oxo-3-(4-(trifluoromethyl)phenyl)-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylic acid (38)
Yield: 81%. 1H NMR (CD3OD): δ 7.21 (s, 1H), 7.76 (d, J = 8.3 Hz, 2H), 7.86 (d, J = 8.3 Hz, 2H) ppm.
5.1.3.6. 5-Amino-4-oxo-3-(4-(trifluoromethoxy)phenyl)-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylic acid (39)
Yield: 51%. 1H NMR (CD3OD): δ 7.17 (s, 1H), 7.35 (d, J = 8.5 Hz, 3H), 7.69 (d, J = 8.9 Hz, 3H) ppm. IR (film): v 3428, 3329, 1725, 1637, 1598 cm−1. 13C NMR (CD3OD): δ 102.7, 103.1, 118.1, 119.3, 120.2, 125.0, 126.1, 132.8, 138.1, 146.4, 157.9, 162.4, 162.8 ppm.
5.1.3.7. 5-Amino-3-(4-hydroxyphenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylic acid (40)
Yield: 76%. 1H NMR (DMSO-d6): δ 6.82 (d, J = 8.6 Hz, 2H), 7.09 (s, 1H), 7.26 (d, J = 8.6 Hz, 2H), 7.55 (s, 2H), 9.63 (s, 1H) ppm. IR (film): v 3425 (br, acid band), 1725, 1635 cm−1.
5.1.4. General procedure for the synthesis of compounds of general structure F
To a mixture of the appropriate acid of general structure E (0.15 mmol), the appropriate amine (0.22 mmol), and benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate (0.097 g, 0.22 mmol) in anhydrous dimethylsulfoxide (2 mL), N,N-diisopropyl-ethylamine (0.028 g, 38 μL, 0.23 mmol) was added. The reaction mixture was stirred at room temperature for 4 h. The reaction was quenched with water and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification by preparative reverse phase HPLC furnished the desired amide of general structure F.
5.1.4.1. 5-Amino-3-(4-fluorophenyl)-N-isopropyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (42)
Yield: 74%. 1H NMR (CDCl3): δ 1.24 (d, J = 7.0 Hz, 6H), 4.17–4.24 (m, 1H), 5.83 (br s, 2H), 6.89 (br s, 1H), 7.15–7.18 (m, 2H), 7.50–7.58 (m, 3H) ppm. 13C NMR (CDCl3): δ 22.8, 41.5, 106.6, 106.9, 115.7 (d, JCF2 = 22.6 Hz), 126.8, 128.0 (d, JCF3 = 8.5 Hz), 135.5, 136.8, 159.7, 161.5, 161.7 (d, JCF1 = 245.5 Hz), 162.0 ppm. IR (film): v 3405, 3301, 1651, 1598 cm−1. MS [ESI]+: Calcd for C16H16FN4O2S+ 347.10. Found 347.11.
5.1.4.2. 5-Amino-N-cyclopropyl-3-(4-fluorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (43)
Yield: 71%. 1H NMR (DMSO-d6): δ 0.54–0.72 (m, 4H), 2.74–2.81 (m, 1H), 7.20 (s, 1H), 7.23–7.31 (m, 2H), 7.53–7.71 (m, 4H), 8.25 (br s, 1H) ppm. 13C NMR (DMSO-d6): δ 6.3, 23.2, 104.6, 104.9, 115.5 (d, JCF2 = 22.2 Hz), 126.6, 128.7 (d, JCF3 = 9.2 Hz), 136.7, 137.5, 159.0, 161.2 (d, JCF1 = 246.7 Hz), 163.4, 164.4 ppm. IR (film): v 3298, 3105 (br band), 1660, 1643, 1587 cm−1. MS [ESI]+: Calcd for C16H14FN4O2S+ 345.08. Found 345.14.
5.1.4.3. 5-Amino-3-(4-fluorophenyl)-4-oxo-N-(2,2,2-trifluoroethyl)-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (44)
Yield 54%; 1H NMR (CDCl3): δ 4.04 (qd, J=8.9, 6.8 Hz, 2H), 6.19 (s, 2H), 7.16–7.19 (m, 2H), 7.38 (t, J = 6.4 Hz, 1H), 7.52–7.49 (m, 2H), 7.55 (s, 1H) ppm. 13C NMR (CDCl3): δ 40.4, 40.7, 106.5, 106.8, 115.79, 115.97, 123.0, 126.4, 127.92, 127.99, 134.2, 136.48, 136.51, 159.6, 160.9, 161.6, 162.92, 162.97 ppm. HRMS [ESI]−: Calcd for C15H9F4N4O2S− 385.0382. Found 385.0397.
5.1.4.4. 5-Amino-3-(2-chlorophenyl)-N-cyclopropyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (45)
Yield 62%; 1H NMR (CDCl3): δ 0.59–0.62 (m, 2H), 0.82 (dd, J =6.9, 5.4 Hz, 2H), 2.79 (m, 1H), 6.12 (bs, 2H), 7.12 (d, J = 4.2 Hz, 1H), 7.46–7.40 (m, 3H), 7.56 (dd, J = 5.8, 3.5 Hz, 1H), 7.64 (s, 1H) ppm. 13C NMR (CDCl3): δ 6.6, 22.5, 106.8, 107.1, 126.7, 127.7, 130.06, 130.23, 130.5, 132.9, 135.3, 138.2, 159.5, 161.3, 164.3 ppm. HRMS [ESI]+: Calcd for C16H13N4O2SCl+ 361.0526. Found 361.0537.
5.1.4.5. 5-Amino-3-(3-chlorophenyl)-N-isopropyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (46)
Yield: 53%. 1H NMR (CD3OD): d1.27 (d, J = 6.6 Hz, 6H), 4.12–4.16 (m, 1H), 7.28 (s, 1H), 7.38 (dt, J = 8.0, 0.9 Hz, 1H), 7.46 (t, J = 8.0 Hz, 1H), 7.58 (dt, J = 8.0, 0.9 Hz, 1H), 7.71 (s, 1H) ppm. 13C NMR (CD3OD): δ 19.7, 40.0, 103.13, 103.28, 122.9, 124.6, 124.9, 125.6, 128.1, 132.2, 135.2, 140.6, 157.9, 161.4, 162.2 ppm. IR(film): v 3413, 3305, 2971, 1652, 1591 cm−1. MS [ESI]+: Calcd for C16H16ClN4O2S+ 363.84. Found 363.16.
5.1.4.6. 5-Amino-3-(4-chlorophenyl)-N-cyclopropyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (1)
See Ref. 7.
5.1.4.7. 5-Amino-N-cyclopropyl-4-oxo-3-(4-(trifluoromethyl)-phenyl)-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (47)
Yield: 53%. 1H NMR (DMSO-d6): δ 0.57–0.60 (m, 2H), 0.66–0.70 (m, 2H), 2.79 (m, 1H), 7.21 (s, 1H), 7.64 (s, 2H), 7.80 (d, J = 8.6 Hz, 2H), 7.91 (d, J = 8.5 Hz, 2H), 8.32 (d, J = 4.1 Hz, 1H) ppm. 13C NMR (DMSO-d6): δ 6.4, 23.2, 104.3, 105.5, 125.85, 125.88, 125.91, 126.4, 126.8, 137.4, 144.6, 158.9, 164.0, 164.3 ppm. MS [ESI]+: Calcd for C17H14F3N4O2S+ 395.08. Found 394.98.
5.1.4.8. 5-Amino-N-isopropyl-4-oxo-3-(4-(trifluoromethoxy)-phenyl)-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (48)
Yield 50%; 1H NMR (CDCl3): δ 1.25 (d, J = 6.6 Hz, 6H), 4.18–4.25 (m, 1H), 6.19 (s, 2H), 6.89 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 8.2 Hz, 2H), 7.60–7.62 (m, 3H) ppm. 13C NMR (CDCl3): δ 22.8, 41.6, 106.89, 106.98, 121.4, 126.7, 127.4, 135.8, 139.2, 159.6, 161.5, 161.9 ppm. IR (film): v 3411, 3281, 3176, 1647 cm−1. MS [ESI]+: Calcd for C17H16F3N4O3S+ 413.09. Found 413.20.
5.1.4.9. 5-Amino-3-(4-hydroxyphenyl)-N-isopropyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (49)
Yield: 49%. 1H NMR (DMSO-d6): δ 1.14 (d, J = 6.6 Hz, 6H), 4.02–4.07 (m, 1H), 6.82 (d, J = 8.8 Hz, 2H), 7.16 (s, 1H), 7.34 (d, J = 8.8 Hz, 2H), 7.50 (s, 2H), 7.85 (d, J = 8.1 Hz, 1H), 9.61 (s, 1H) ppm. IR (film): v 3429, 3323, 3269, 2975, 1640, 1507 cm−1. MS [ESI]+: Calcd for C16H17N4O3S+ 345.10. Found 345.23.
5.1.4.10. 5-Amino-N-cyclopropyl-3-(4-isopropylphenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (50)
Yield: 55%. 1H NMR (DMSO-d6): δ 0.57–0.60 (m, 2H), 0.65–0.69 (m, 2H), 2.79 (td, J = 7.3, 4.0 Hz, 1H), 2.94 (dt, J =13.8, 6.9 Hz, 1H), 7.19 (s, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.49 (d, J = 8.4 Hz, 2H), 7.54 (s, 2H), 8.20 (d, J = 4.3 Hz, 1H) ppm. 13C NMR (DMSO-d6): δ 6.3, 23.2, 24.5, 33.8, 104.64, 104.81, 126.58, 126.68, 126.75, 136.5, 139.1, 147.8, 159.1, 163.3, 164.5 ppm. MS [ESI]+: Calcd for C19H19N4O2S+ 369.14. Found 369.13.
5.1.4.11. Ethyl 4-acetyl-2-aminothiophene-3-carboxylate (53)
To a mixture of 2,3-butanedione (20 mg, 20 μL, 0.23 mmol), ethyl cyanoacetate (26 mg, 25 μL, 0.23 mmol), and sulfur (11 mg, 0.35 mmol) in N,N-dimethylformammide (0.5 mL) morpholine (40 mg, 40 μL, 0.46 mmol) was added. The reaction mixture was stirred at rt for 16 h. A saturated solution of sodium chloride was added and the resulting mixture was extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over sodium sulfate, filtered and concentrated in vacuo. Crystallization from acetate-hexanes furnished 53. Yield: 32%, mp: 105–106 °C. 1H NMR (CDCl3): 1.32 (t, J = 7.2 Hz, 3H), 2.42 (s, 3H), 4.28 (q, J = 7.2 Hz, 2H), 6.07 (br s, 2H), 6.42 (s, 1H) ppm. 13C NMR (CDCl3): δ 14.2, 30.6, 60.5, 104.6, 109.3, 143.0, 163.7, 164.7, 198.6 ppm. IR (film): v 3429, 3324, 1677, 1593 cm−1. MS [ESI]+: Calcd for C9H12NO3S+ 214.05. Found 214.22.
5.1.4.12. Ethyl 2-amino-4-benzoylthiophene-3-carboxylate (54)
A mixture of the 1-phenylpropane-1,2-dione (500 mg, 0.45 mL, 3.37 mmol), sulfur (162 mg, 5.05 mmol), ethyl cyanoacetate (382 mg, 0.36 mL, 3.37 mmol), and triethylamine (682 mg, 940 mL, 6.00 mmol) in N,N-dimethylformamide (10 mL) was stirred at rt for 12 h. The reaction was quenched with water and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over sodium sulfate, filtered and concentrated in vacuo. Purification by silica gel column chromatography using ethyl acetate-hexanes 2:3 as eluant provided 54. Yield: 95%, mp: 103–105 °C. 1H NMR (CDCl3): δ 0.78 (t, J = 7.2 Hz, 3H), 3.90 (q, J = 7.2 Hz, 2H), 5.95 (br s, 2H), 6.44 (s, 1H), 7.42 (t, J = 7.7 Hz, 2H), 7.53 (t, J = 7.2 Hz, 1H), 7.84 (d, J = 8.5 Hz, 2H) ppm. 13C NMR (CDCl3): δ 13.5, 60.1, 106.0, 109.3, 128.4, 129.4, 133.0, 138.0, 140.0, 163.5, 164.7, 193.0 ppm. IR (film): v 3431, 3327, 1667, 1595 cm−1.
5.1.5. General procedure for the synthesis of compounds of general structure G
A mixture of the appropriate aminothiophene keto-ester (53 or 54, 0.23 mmol), the appropriate hydrazine (0.23 mmol) in ethanol was stirred for 0.5–3 h at 150 °C (microwave irradiation). After cooling, the solvent was removed and the residue was purified by either preparative reverse phase HPLC, or silica gel column chromatography using gradients of ethyl acetate in hexanes 1:3 as eluent, to give the desired compound of general structure G.
5.1.5.1. 7-Amino-4-methylthieno[3,4-d]pyridazin-1(2H)-one (55)
Yield: 77%. 1H NMR (DMSO-d6): δ 2.18 (s, 3H), 6.62 (s, 1H), 7.26 (br s, 2H), 11.04 (br s, 1H) ppm. 13C NMR (DMSO-d6): δ 31.3, 101.4, 104.9, 31.8, 141.5, 160.9, 162.0 ppm. IR: v 3422, 3260, 1642 cm−1. MS [ESI]+: Calcd for C7H8N3OS+ 182.04. Found 182.20.
5.1.5.2. 7-Amino-2,4-dimethylthieno[3,4-d]pyridazin-1(2H)-one (56)
Yield: 79%, mp: 158–160 °C. 1H NMR (CDCl3): δ 2.29 (s, 3H), 3.57 (s, 3H), 6.13 (br s, 2H), 6.36 (s, 1H) ppm. 13C NMR (CDCl3): δ 18.4, 36.6, 100.9, 107.3, 132.3, 141.5, 159.9, 160.1 ppm. IR: v 3386, 3266, 1604 cm−1. MS [ESI]+: Calcd for C8H10N3OS+ 196.05. Found 196.17.
5.1.5.3. 7-Amino-2-isopropyl-4-methylthieno[3,4-d]pyridazin-1(2H)-one (57)
Yield: 59%. 1H NMR (DMSO-d6): δ 1.20 (d, J = 6.7 Hz, 6H), 2.22 (s, 3H), 5.01 (7, J = 6.7 Hz, 1H), 6.61 (s, 1H), 7.31 (s, 2H) ppm. 13C NMR (DMSO-d6): δ 19.0, 21.5, 45.2, 100.8, 105.0, 131.4, 141.2, 159.0, 162.0 ppm. MS [ESI]+: Calcd for C10H14N3OS+ 224.08. Found 224.00.
5.1.5.4. 7-Amino-4-methyl-2-(2,2,2-trifluoroethyl)thieno[3,4-d]pyridazin-1(2H)-one (58)
Yield: 47%. 1H NMR (DMSO-d6): δ 2.23 (s, 3H), 4.65 (q, J = 9.2 Hz, 2H), 6.75 (s, 1H), 7.46 (s, 2H) ppm. 13C NMR (DMSO-d6): δ 18.5, 48.13 (d, JCF2 =33.1 Hz), 103.0, 103.4, 131.0, 142.9, 159.5, 163.2 ppm. HRMS [ESI]−: Calcd for C9H7N3OF3S− 262.0262. Found 262.0272. X-ray crystal structure shown in Supplementary data.
5.1.5.5. 7-Amino-2-cyclohexyl-4-methylthieno[3,4-d]pyridazin-1(2H)-one (59)
Yield: 64%. 1H NMR (DMSO-d6): δ 1.10–1.18 (m, 1H), 1.34 (qt, J = 13.1, 3.4 Hz, 2H), 1.61–1.73 (m, 6H), 1.79 (d, J = 13.3 Hz, 2H), 2.22 (s, 3H), 4.58 (m, 1H), 6.61 (s, 1H), 7.32 (s, 2H) ppm. 13C NMR (DMSO-d6): δ 18.9, 25.7, 26.0, 31.5, 53.0, 100.7, 105.0, 131.3, 140.9, 159.0, 162.1 ppm. MS [ESI]+: Calcd for C13H18N3OS+ 264.12. Found 264.07.
5.1.5.6. 7-Amino-4-methyl-2-(pyridin-2-yl)thieno[3,4-d]pyridazin-1(2H)-one (60)
Yield: 53%, mp: 227–229 °C. 1H NMR (CDCl3): 2.41 (s, 3H), 6.02 (br s, 2H), 6.46 (s, 1H), 7.29 (t, J = 8.5 Hz, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.83 (t, J = 8.5 Hz, 1H), 8.66 (d, J = 4.0 Hz, 1H) ppm. 13C NMR (CDCl3): δ 18.7, 102.2, 106.5, 121.5, 122.5, 131.7, 138.0, 143.3, 148.9, 153.0, 159.8, 161.8 ppm. IR: v 3415, 3113, 1637, 1594 cm−1. MS [ESI]+: Calcd for C12H11N4OS+ 259.06. Found 259.11.
5.1.5.7. 7-Amino-4-methyl-2-(pyridin-3-yl)thieno[3,4-d]pyridazin-1(2H)-one (61)
Yield: 43%. 1H NMR (DMSO-d6): δ 2.29 (s, 3H), 6.80 (s, 1H), 7.45–7.52 (m, 3H), 7.95 (dd, J = 8.1, 1.3 Hz, 1H), 8.46 (d, J = 0.7 Hz, 1H), 8.78 (d, J = 0.8 Hz, 1H) ppm. 13C NMR (DMSO-d6): δ 18.7, 102.9, 103.9, 123.9, 131.0, 133.2, 143.5, 146.9, 147.4, 149.9, 159.2, 163.7 ppm. MS [ESI]+: Calcd for C12H11N4OS+ 259.06, Found 259.05.
5.1.5.8. 7-Amino-2-benzyl-4-methylthieno[3,4-d]pyridazin-1(2H)-one (62)
Yield: 97%, mp: 123–125 °C. 1H NMR (CDCl3): 2.30 (s, 3H), 5.17 (s, 2H), 5.99 (br s, 2H), 6.35 (s, 1H), 7.25 (t, J = 8.5 Hz, 1H), 7.32 (t, J = 8.2 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H) ppm. 13C NMR (CDCl3): δ 18.5, 52.2, 101.0, 107.3, 127.4, 128.2, 128.6, 132.3, 138.1, 141.9, 159.9, 160.3 ppm. IR (film): v 3397, 3112, 1600 cm−1. MS [ESI]+: Calcd for C14H14N3OS+ 272.09. Found 272.17.
5.1.5.9. 7-Amino-2-(4-chlorophenyl)-4-methylthieno[3,4-d]pyridazin-1(2H)-one (63)
Yield: 73%. 1H NMR (CDCl3): δ 2.38 (s, 3H), 6.09 (br s, 2H), 6.47 (s, 1H), 7.40 (d, J = 9.0 Hz, 2H), 7.56 (d, J = 9.0 Hz, 2H) ppm. 13C NMR (CDCl3): δ 18.6, 101.7, 107.2, 127.1, 128.8, 132.1, 132.5, 139.8, 142.7, 159.6, 162.3 ppm. IR: v 3411, 3329, 1633, 1590 cm−1. MS [ESI]+: Calcd for C13H11ClN3OS+ 292.03. Found 292.12.
5.1.5.10. 7-Amino-4-phenyl-2-(2,2,2-trifluoroethyl)thieno[3,4-d]pyridazin-1(2H)-one (64)
Yield: 46%. 1H NMR (DMSO-d6): δ 4.77 (q, J = 9.2 Hz, 2H), 6.69 (s, 1H), 7.51–7.53 (m, 3H), 7.59 (s, 2H), 7.67–7.69 (m, 2H) ppm. 13C NMR (DMSO-d6): δ 48.2, 48.5, 103.8, 104.3, 128.4, 128.9, 129.3, 130.1, 135.4, 144.5, 159.2, 163.6 ppm. HRMS [ESI]+: Calcd for C14H11N3OF3S+ 326.0575. Found 326.0571.
5.1.5.11. 7-Amino-2-isopropyl-4-phenylthieno[3,4-d]pyridazin-1(2H)-one (65)
Yield: 36%. 1H NMR (DMSO-d6): δ 1.27 (d, J = 6.6 Hz, 6H), 5.11 (dt, J = 13.3, 6.6 Hz, 1H), 6.62 (s, 1H), 7.51–7.45 (m, 5H), 7.71 (dd, J = 7.9, 1.5 Hz, 2H) ppm. 13C NMR (DMSO-d6): δ 21.6, 45.8, 101.9, 105.4, 128.3, 129.13, 129.21, 129.6, 136.5, 142.5, 158.8, 162.5 ppm. MS [ESI]+: Calcd for C15H16N3OS+ 286.10. Found 285.96.
5.1.5.12. Ethyl 3-(4-chlorophenyl)-5-(methylamino)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylate (66)
A mixture of 29, potassium carbonate and iodometane in acetonitrile was stirred at 70 °C for 10 h. The reaction was quenched by addition of water and then extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. Purification by silica gel column chromatography using ethyl acetate/hexanes 1:3 as eluant provided 68. Yield 27%; 1H NMR (CDCl3): δ 1.43 (t, J = 7.0 Hz, 3H), 3.07 (d, J = 5.5 Hz, 3H), 4.44 (q, J = 7.0 Hz, 2H), 7.19 (s, 1H), 7.42 (d, J = 9.0 Hz, 2H), 7.54 (bs, 1H), 7.58 (d, J = 9.0 Hz, 2H) ppm. 13C NMR (CDCl3): δ 14.39, 33.99, 62.05, 102.83, 104.32, 127.07, 127.60, 128.82, 133.07, 134.08, 139.34, 159.39, 163.15, 165.50 ppm. IR (film): v 3370, 2923, 1718, 1653, 1567 cm−1. MS [ESI]+: Calcd for C16H15ClN3O3S+ 364.05. Found 364.07.
5.1.5.13. Ethyl 5-amino-7-chloro-3-(4-chlorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylate (67)
A mixture 30 (30 mg, 0.086 mmol) and N-chlorosuccinimide (11 mg, 0.086 mmol) in anhydrous N,N-dimethylformamide (0.5 mL) was heated to 80 °C for 30 minutes. After cooling, the reaction was quenched with water and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over sodium sulfate, filtered and concentrated in vacuo. Purification by preparative reverse phase HPLC furnished 50. Yield: 79%. 1H NMR(CDCl3): δ 1.42 (t, J = 7.5 Hz, 3H), 4.45 (q, J = 7.2 Hz, 2H), 6.25 (br s, 2H), 7.40 (d, J = 9.0 Hz, 2H), 7.51 ppm(d, J = 9.0 Hz, 2H). 13CNMR(CDCl3): δ 14.1, 62.8, 105.7, 106.7, 122.7, 127.1, 128.9, 133.3, 136.2, 138.7, 158.5, 159.1, 162.9 ppm. IR (film): v 3435, 3324, 1725, 1656, 1594 cm−1. MS [ESI]+: Calcd for C15H12Cl2N3O3S+ 384.00. Found 384.01.
5.1.5.14. Ethyl 5-amino-7-bromo-3-(4-chlorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylate (68)
Prepared as 67 from 29 and N-bromosuccinimmide. Yield: 75%. 1H NMR (CDCl3): δ 1.43 (t, J = 7.1 Hz, 3H), 4.46 (q, J = 7.2 Hz, 2H), 6.34 (br s, 2H), 7.40 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 9.0 Hz, 2H) ppm. IR: v 3425, 3317, 1732, 1656, 1589 cm−1. MS [ESI]+: Calcd for C15H12BrClN3O3S+ 427.95. Found 427.73.
5.1.5.15. Ethyl 2-(4-chlorophenyl)-5-hydroxy-1,7-dioxo-2,5,6,7-tetrahydro-1H-pyrrolo[3,4-d]pyridazine-4-carboxylate (69)
A solution of 29 in acetone (2 mL) was added at room temperature with a freshly prepared solution of dimethyldioxirane in acetone (0.07–0.09 M solution prepared as described in Organic Syntheses, Coll. Vol 9, p.288 (1998); Vol 74, p.91 (1997)). The reaction was monitored by TLC and after ~20 minutes the starting material was completely consumed. The reaction mixture was then evaporated to dryness and the resulting material purified by preparative HPLC obtaining 71. Yield: 20%. 1H NMR tautomeric mixture (DMSO-d6): δ 1.31 (t, J = 7.1 Hz, 3H), 3.30 (d, J = 11.8 Hz, 1H), 4.33–4.40 (m, 2H), 6.12 (d, J = 7.1 Hz, 1H), 6.65 (d, J = 8.6 Hz, 0.5H), 7.60–7.65 (m, 4H), 9.22 (s, 0.5H) ppm. IR (film): v 3359, 2923, 2852, 1734 cm−1. HRMS [ESI]+: Calcd for C15H12ClN3NaO5+ 372.0363. Found 372.0374. See X-ray crystal structure report in the Supplementary data.
5.1.5.16. 3-(4-Fluorophenyl)-4,5-dioxo-3,4,5,7-tetrahydrothieno[3,4-d]pyridazine-1-carboxylic acid (70)
1H NMR (DMSO-d6): δ 4.93 (s, 2H), 7.39–7.43 (m, 2H), 7.62–7.64 (m, 2H) ppm. MS [ESI]+: Calcd for C13H8FN2O4S+ 307.02. Found 306.97.
5.1.5.17. 3-(4-Chlorophenyl)-4,5-dioxo-3,4,5,7-tetrahydrothieno[3,4-d]pyridazine-1-carboxylic acid (71)
To a solution of 41 (50 mg, 0.15 mmol) in acetonitrile (1 mL) 1 N hydrochloric acid (1.5 mL) was added. The reaction mixture was stirred at 130 °C (microwave irradiation) for 30 minutes. Water was then added and the desired product was filtered and washed with water. Yield: 58%. 1H NMR (DMSO-d6): δ 4.93 (s, 2H), 7.61–.66 (m, 4H) ppm.
5.1.5.18. 3-(4-Chlorophenyl)-N-isopropyl-4,5-dioxo-3,4,5,7-tetrahydrothieno[3,4-d]pyridazine-1-carboxamide (72)
Prepared following the general procedure described for the synthesis of compounds of general structure F. 1H NMR (DMSO-d6): δ 1.17 (d, J = 6.6 Hz, 6H), 4.07–4.13 (m, 1H), 4.96 (s, 2H), 7.62–7.65 (m, 2H), 7.70–7.72 (m, 2H), 8.36 (d, J =8.2 Hz, 1H) ppm. 13C NMR (DMSO-d6): δ 22.6, 35.1, 41.6, 114.4, 128.8, 129.4, 131.1, 133.9, 136.8, 155.3, 157.5, 161.2 ppm.
5.1.6. Pharmacokinetic studies
Test compounds were administered to 2-month old female B6C3F1 mice (20–25 g average body weight). Typical studies involved i.p. injection of test ATPZ at a dose of 5 mg/kg, followed by sacrifice 1 h later. For full PK analysis of compound 43, mice (n = 3/time point) received i.p. injections at a dose of 5 mg/kg, followed by sacrifice at times ranging from 0.5–16 h post-dosing. Perfused whole brain hemispheres, obtained from mice euthanized according to protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committee, were homogenized in 10 mM ammonium acetate, pH 5.7 (50% w/v), using a hand-held sonic homogenizer. Plasma was obtained from blood that was collected into a 1.5 mL tube containing 0.5 M EDTA solution and subjected to centrifugation for 10 min at 4500g at 4 °C. Aliquots (50 μL) of brain homogenate or plasma were mixed with 0.2 mL of acetonitrile and centrifuged at 15000g, and the resulting supernatants were used for subsequent LC-MS/MS analysis. The LC-MS/MS system comprised an Aquity UPLC and a TQ MS that was controlled using MassLynx software (Waters Corporation, Mil-ford, MA). Compounds were detected using multiple reaction monitoring (MRM) of their specific collision-induced ion transitions. Samples (5 μL) were separated on an Aquity BEH C18 column (1.7 μm, 2.1 mm × 50 mm) at 35 °C. Operation was in positive electrospray ionization mode, with mobile phase A of 0.1% (v/v) formic acid and mobile phase B of acetonitrile with 0.1% (v/v) formic acid at a flow rate of 0.6 mL/min using a gradient from 5% to 95% B over 2 min, followed by wash and re-equilibration steps. The mass spectrometer was operated with a desolvation temperature of 450 °C and a source temperature of 150 °C. Desolvation and source nitrogen gas flows were 900 and 50 L/h, respectively. Source and MS/MS voltages were optimized for each compound using the MassLynx autotune utility. Standard curves were generated for each compound from brain homogenate and plasma samples and extracted as above. Peak areas were plotted against concentration and a linear regression curve was used to quantify the unknowns. In all cases, the tissue-derived sample peak areas fell within the linear portion of standard curves that were prepared and analyzed concurrently with the samples.
5.1.7. Determination of unbound fraction in plasma and brain homogenates
The unbound fractions of drug in mouse plasma and brain were determined in vitro using rapid equilibrium dialysis (Pierce Biotechnology, Rockford, IL). Test compounds were mixed at a concentration of 1 μM with mouse plasma or brain homogenate that was diluted threefold with 100 mM sodium phosphate, pH 7.4. Triplicate 100 μL aliquots were placed in sample chambers and dialyzed against 300 μL of 100 mM sodium phosphate, pH 7.4, for 5 h at 37°C with shaking at 100 rpm. After incubation, 20uL aliquots were removed from each side of the dialysis membrane and an equal volume of plasma, brain homogenate, or buffer added in order to create identical matrix compositions. Compounds were subsequently extracted with 120 μL acetonitrile, vortexed and centrifuged at 15,000g for 15 min. Supernatants were analyzed by LC-MS/MS. Unbound fraction (fu) was calculated as the ratio of the buffer chamber response to the sample chamber response. Brain unbound fraction was calculated using the following equation, adjusting for tissue dilution (D).
5.1.8. Mouse tolerability studies
To determine the maximum tolerated dose of ATPZ 43, 2-month old female B6C3F1 mice (n = 5) were administered compound by oral gavage (in 0.5% methylcellulose) starting at 1 mg/kg. The mice were carefully monitored for the next 48 h for signs of ataxia, lethargy, piloerection, salivation or other unusual behaviors. If no signs of intolerance were observed, doing was increased in gradual steps (2–3 fold increases) until a noticeable change occurred in at least 2 mice. For the 1-month tolerability testing of compound 43, groups of 2-month old female B6C3F1 mice (n = 5) were administered compound in drinking water containing 3% PEG400 and 0.5% methylcellulose. Control mice received the vehicle only. Water consumption was monitored, and normal water intake was observed (~5 ml/mouse/day). At the completion of dosing, body weights were determined. The mice then underwent perfusion and euthanization as described above, and organs weights were recorded and blood was collected for complete blood cell counts. Samples of plasma and brain tissue were also kept for LC-MS/MS analysis of the levels of compound 43.
Supplementary Material
Acknowledgments
Financial support for this work was provided in part by the Alzheimer’s Drug Discovery Foundation, Coins for Alzheimer’s Research Trust, Marian S. Ware Drug Discovery Program, and the NSF (Grant CHE-0840438, X-ray facility).
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2012.05.027.
References and notes
- 1.Ballatore C, Lee VMY, Trojanowski JQ. Nat Rev Neurosci. 2007;8:663. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 2.Brunden KR, Trojanowski JQ, Lee VMY. Nat Rev Drug Disc. 2009;8:783. doi: 10.1038/nrd2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballatore C, Brunden KR, Trojanowski JQ, Lee VM, Smith AB, Huryn DM. Curr Top Med Chem. 2011;11:317. doi: 10.2174/156802611794072605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunden KR, Ballatore C, Crowe A, Smith AB, III, Lee VMY, Trojanowski JQ. Exp Neurol. 2010;223:304. doi: 10.1016/j.expneurol.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wischik CM, Edwards PC, Lai RYK, Roth M, Harrington CR. Proc Natl Acad Sci U S A. 1996;93:11213. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crowe A, Huang W, Ballatore C, Johnson RL, Hogan AM, Huang R, Wichterman J, McCoy J, Huryn D, Auld DS, Smith AB, III, Inglese J, Trojanowski JQ, Austin CP, Brunden KR, Lee VM-Y. Biochemistry. 2009;48:7732. doi: 10.1021/bi9006435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballatore C, Brunden K, Piscitelli F, James M, Crowe A, Yao Y, Hyde E, Trojanowski J, Lee V, Smith A., III J Med Chem. 2010;53:3739. doi: 10.1021/jm100138f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gewald K. Chem Ber. 1965;98:3571. [Google Scholar]
- 9.Wang K, Kim D, Do mling A. J Comb Chem. 94 [Google Scholar]
- 10.Al-Saleh B, El-Apasery MA, Hilmy NM, Elnagdi MH. J Heterocycl Chem. 2006;43:1575. [Google Scholar]
- 11.Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Biochemistry. 1994;33:9511. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- 12.Goedert M, Jakes R. Biochim Biophys Acta. 2005;1739:240. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Crowe A, Ballatore C, Hyde E, Trojanowski JQ, Lee VMY. Biochem Biophy Res Commun. 2007;358:1. doi: 10.1016/j.bbrc.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gamblin TC, King ME, Dawson H, Vitek MP, Kuret J, Berry RW, Binder LI. Biochemistry. 2000;39:6136. doi: 10.1021/bi000201f. [DOI] [PubMed] [Google Scholar]
- 15.Congdon EE, Kim S, Bonchak J, Songrug T, Matzavinos A, Kuret J. J Biol Chem. 2008;283:13806. doi: 10.1074/jbc.M800247200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagakura M, Ota T, Shimidzu N, Kawamura K, Eto Y, Wada Y. J Med Chem. 1979;22:48. doi: 10.1021/jm00187a012. [DOI] [PubMed] [Google Scholar]
- 17.Ferguson GN, Valant C, Horne J, Figler H, Flynn BL, Linden J, Chalmers DK, Sexton PM, Christopoulos A, Scammells PJ. J Med Chem. 2008;51:6165. doi: 10.1021/jm800557d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.