

Abstract
Neuronal insulin signaling abnormalities have been associated with Alzheimer's disease (AD). However, the specificity of this association and its underlying mechanisms have been unclear. This study investigated the expression of abnormal serine phosphorylation of insulin receptor substrate 1 (IRS1) in 157 human brain autopsy cases that included AD, tauopathies, α-synucleinopathies, TDP-43 proteinopathies, and normal aging. IRS1-pS616, IRS1-pS312 and downstream target Akt-pS473 measures were most elevated in AD but were also significantly increased in the tauopathies: Pick's disease, corticobasal degeneration and progressive supranuclear palsy. Double immunofluorescence labeling showed frequent co-expression of IRS1-pS616 with pathologic tau in neurons and dystrophic neurites. To further investigate an association between tau and abnormal serine phosphorylation of IRS1, we examined the presence of abnormal IRS1-pS616 expression in pathological tau-expressing transgenic mice and demonstrated that abnormal IRS1-pS616 frequently co-localizes in tangle-bearing neurons. Conversely, we observed increased levels of hyperphosphorylated tau in the high-fat diet-fed mouse, a model of insulin resistance. These results provide confirmation and specificity that abnormal phosphorylation of IRS1 is a pathological feature of AD and other tauopathies, and provide support for an association between insulin resistance and abnormal tau as well as amyloid-β.
Keywords: Alzheimer's disease, Tau, Synuclein, TDP-43, Insulin resistance, Insulin receptor substrate 1
Introduction
Accumulating evidence suggests that type 2 diabetes (T2D) and Alzheimer's disease (AD) share certain common pathophysiological mechanisms. Peripheral insulin resistance, which is defined by the failure to respond to normal circulating concentrations of insulin, is one of the earliest and most important metabolic defects in impaired glucose tolerance and T2D [38]. An analogous defect in the response of brain tissue to insulin is now thought to occur in AD. Compared to healthy aging controls, neuro-pathological studies of AD brains have described reduced insulin receptor (IR) expression and alterations of insulin signaling consistent with insulin resistance [39, 43, 44, 48]. Although T2D and AD are both prevalent diseases of aging and frequently overlap, the described abnormalities in brain insulin signaling in AD can occur independently of clinical diabetes [44]. Further supporting a critical role for insulin signaling abnormalities in AD pathology, a recent study has demonstrated that administration of insulin to patients with AD improves cognitive function and memory [8], and insulin and other antidiabetic drugs are currently undergoing clinical investigation as novel therapeutic agents in AD [50].
Insulin receptor substrate 1 (IRS1) is a critical switch in the insulin signaling pathway, and also interacts with other receptor tyrosine kinases including IGF1/2, TrkB and ErbB. While tyrosine phosphorylation of IRS1 leads to the downstream activation of Akt, mTOR, GSK3, among other pathways, the phosphorylation of IRS1 on multiple serine residues can inhibit IRS1 activity, leading to insulin resistance [20]. Abnormal serine phosphorylation of IRS1 is an established marker of peripheral insulin resistance [20, 36], and we have similarly shown that serine-phosphorylated IRS1 in brain is consistently associated with insulin resistance as well as IGF1 resistance in both mouse and human ex vivo insulin stimulation experiments [44]. Using antibodies specifically targeting phosphoserine residues, we and others have previously demonstrated basal elevations in IRS1 serine phosphorylation in the hippocampus in AD [6, 44].
Insulin signaling abnormalities have been proposed to play a role in other neurodegenerative conditions in addition to AD [2, 9], but to our knowledge no studies have provided direct evidence of such in brain. Therefore, it remains unclear whether brain insulin resistance is a general phenomenon of neurodegeneration present in other or all neurodegenerative diseases, or whether it is only associated with particular diseases or disease pathologies. Furthermore, we and others have previously described IRS1 signaling pathway abnormalities in AD in hippocampus only [28, 44], and the degree to which these occur in other brain regions of relevance to neurodegenerative dementias is not known.
To address these questions, we first used immunohistochemistry for IRS1-pS616 and quantitative microscopic image analysis to measure abnormal serine-phosphorylated IRS1 expression in three brain regions in AD and other neurodegenerative diseases including tauopathies [Pick's disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD)], α-synucleinopathies [Parkinson's disease (PD) and PD with dementia (PDD), dementia with Lewy bodies (DLB), multiple system atrophy (MSA)], and TDP-43 proteinopathies [frontotemporal lobar degeneration associated with TDP-43 (FTLD-TDP) and amyotrophic lateral sclerosis (ALS)] as compared to normal-aged controls. We found markedly abnormal expression of IRS1-pS616 in AD and to lesser degrees in all three primary tauopathies, but not in α-synuclein or TDP-43 diseases. We validated the increased IRS1-pS finding with immunohistochemistry for IRS1-pS312 and IRS1's downstream target Akt and, with ELISA for IRS1-pS312. To further demonstrate an association of tau and IRS1-pS616, we performed double immunofluorescence labeling of neuropathological samples. We also separately examined the effect of tau pathology on IRS1-pS616 expression in a tau-overexpressing transgenic mice model and conversely, we examined the effects of insulin resistance on tau phosphorylation in a mouse with high-fat diet-induced insulin resistance.
Materials and methods
Case material
Brain tissues were investigated from 132 patients with neurodegenerative diseases belonging to 10 different diagnostic categories and 25 age-matched controls (Table 1). These case materials were obtained from the brain collection of the Center for Neurodegenerative Disease Research (CNDR) at the University of Pennsylvania. Gross inspections, tissue processing and diagnostic microscopic examinations were conducted as previously described [14, 22, 33, 41, 51]. Briefly, after extraction and weighing, visual inspection documented any gross abnormalities of the whole brain, meninges, and extracerebral blood vessels. The hindbrain and cerebral hemispheres were separated and coronally sectioned into 1–2 cm slabs for further inspection of any infarctions, tumors or other lesions. Fresh tissues from multiple CNS areas were fixed in 10 % neutral buffered formalin or 70 % ethanol with 150 mmol sodium chloride, and paraffin embedded. One hundred twenty-eight of the cases had been referred or followed clinically by research clinicians in the University of Pennsylvania Health System. Twenty-nine brains were from patients with frontotemporal dementias who were followed at the University of California San Francisco. These brains were extracted in San Francisco, hemisected, immersion fixed and shipped to the CNDR for processing, microscopic diagnosis and research studies. Sections from all cases were stained with hematoxylin–eosin, thioflavin S and special stains as indicated for establishing diagnosis. These diagnostic examinations also included immunohistochemistry conducted with monoclonal antibodies directed at paired helical filament tau (PHF1, 1:1000, a gift of Peter Davies, PhD), α-synuclein (“303”, 1:4000, generated in the CNDR) and TDP-43 (1:10 000; Novus Biologicals, Littleton, CO, USA) and developed using (1) the avidin–biotin complex detection method (Vectastain ABC kit; Vector Laboratories, Burlingame, CA, USA) or BioGenex Super Sensitive MultiLink IHC Detection System Kit (BioGenex Laboratories, San Ramon, CA, USA) with 3,3-diaminobenzidine as the chromogen.
Table 1.
Subject characteristics and stratification by study group and subgroups
| N | Age | Female sex (%) | PMI | |
|---|---|---|---|---|
| AD | 25 | 73.6 (11.8) | 48 | 10.8 (5.5) |
| Tauopathy | 38 | 69.0 (10.6) | 50 | 14.2 (10.9) |
| CBD | 13 | 69.2 (9.9) | 62 | 11.6 (7.0) |
| PiD | 10 | 64.5 (14.8) | 50 | 20.3 (16.8) |
| PSP | 15 | 71.8 (7.2) | 40 | 11.8 (6.2) |
| α-Synucleinopathy | 41 | 75.8 (9.3) | 34 | 11.8 (6.1) |
| DLB | 9 | 79.6 (8.3) | 22 | 10.8 (6.9) |
| PD | 7 | 74.9 (7.5) | 43 | 11.9 (6.7) |
| PDD | 11 | 73.9 (8.8) | 18 | 10.8 (5.6) |
| MSA | 14 | 71.1 (10.6) | 50 | 13.5 (6.0) |
| TDP-43 proteinopathy | 28 | 68.4 (7.5) | 46 | 13.0 (9.1) |
| ALS | 14 | 68.4 (8.1) | 50 | 13.4 (9.3) |
| FTLD | 14 | 68.3 (7.3) | 43 | 12.6 (9.1) |
| Normal | 25 | 72.4 (13.5) | 52 | 14.3 (7.7) |
Values given are means (standard deviation) or percentages
The cases used in these studies were selected from the entire CNDR brain collection in a stepwise process a priori. Cases were first selected based on the presence of a sole clinical neuropathological diagnosis of a neurode-generative disease within one of four major neuropatho-logical categories: (1) AD diagnosed by the presence of both amyloid-β plaques and tau neurofibrillary tangles and dystrophic neurites according to established criteria [46]; (2) the tauopathies PiD [1], PSP [18], and CBD [10]; (3) the α-synucleinopathies PD and PDD [11], DLB [31], and multiple system atrophy MSA [47]; and (4) the TDP-43 proteinopathies FTLD-TDP-43 [7] and sporadic amyotrophic lateral sclerosis ALS [29]. Mixed neurodegenerative disease pathology cases, i.e., cases with other, secondary neuropathological diagnoses were excluded. Cases with incidental small lesions (e.g., small lacunar infarcts, meningiomas, etc.) that were deemed non-contributory to the patient's neurological status were allowed. In addition, a normal-aged comparison group was selected based on the absence of any history of neurological or psychiatric disorder and no remarkable pathology on examination. Cases were then further screened for group compatibility on the basis of age, sex and postmortem interval to the extent possible among the available cases in the collection.
Histological processing and immunohistochemistry
Paraffin tissue blocks of midfrontal gyrus, angular gyrus and the body of the hippocampus were sectioned at 6 μm and mounted on APES-coated slides. In select diseases in which subcortical pathology may predominate, we also investigated striatum, midbrain, pons, and/ or cervical spinal cord to search for abnormal IRS1-pS616 expression. After dewaxing and rehydration, the sections were prepared for immunohistochemistry. To delineate cytoarchitectural limits of the CA1 and subiculum subfield of interest in this study, adjacent sections were stained in 0.1 % cresyl violet acetate (Acros Organics 229630050, Fisher Scientific) at pH 4.3, differentiated in 95 % ethanol, dehydrated in 100 % ethanol, cleared in xylenes, and coverslipped under Cytoseal 60 (Fisher Scientific). For immunohistochemistry, tissue sections were first dewaxed in xylenes, rehydrated in descending alcohols, and quenched of endogenous peroxidase activity in 5 % H2O2 dissolved in methanol for 30 min. They were then rinsed in distilled water and treated with an epitope retrieval method. For amyloid-β and phosphorylated tau, sections were immersed for 10 min in concentrated formic acid (Fisher Scientific BP1215-500) similar to the method of Kitamoto et al. [25] For other antigens, sections were boiled for 10 min in 1 mM EDTA (pH 8.0).
After antigen retrieval treatment, the tissues were rinsed in water, transferred to 0.1 M Tris buffer with 0.01 % Triton X-100 (TTB), blocked in 10 % normal horse serum, and incubated in primary antibody overnight at 4 °C. Primary antibodies were directed against IRS1-pS616 (Invitrogen 44-550G; Rabbit 1:500), IRS1-pS312 (Invitrogen 44-814G; Rabbit 1:100), Akt-pS473 (Cell Signaling 4051, Mouse, 1:300), Tau pS202/pT205 (Thermo Scientific, AT8 Mouse 1:800), amyloid-β (Santa Cruz 3227, NAB228, Mouse 1:500), α-synuclein and TDP-43 (both antibodies from the laboratory of VMYL and JQT) [15]. For double immunofluorescence labeling, sections were co-incubated in primary antibodies against IRS1-pS616 and against Map-2 (NeoMarkers, AP20, Mouse 1:400), GFAP (Chemicon, MAB360, Mouse 1:200), CD68 (DaKo, M 0814, Mouse 1:50), amyloid-β, tau, α-synuclein, or TDP-43. For secondary antibodies we used Alexa Fluor 488 donkey anti-mouse (A21202, 1:500, Invitrogen) and Alexa Fluor 594 donkey anti-rabbit IgG (A21207, 1:500; Invitrogen).
Sections used for pathology density measurements and ratings were incubated in species-appropriate biotinylated secondary antibody for 1 h at room temperature, transferred to an avidin–biotin-peroxidase complex for 1 h, and finally reacted with a 0.05 % diaminobenzidine (DAB)—0.03 % hydrogen peroxide solution for 10 min. Immunoreaction signal was enhanced by adding NiSO4 (0.25 % final dilution) to the DAB solution. Sections were then rinsed in water, dehydrated in ascending concentrations of alcohols, cleared in xylenes, and coverslipped under Cytoseal 60.
Neuropathology lesion assessment
For IRS1-pS616, IRS1-pS312, Akt-pS473, tau and amyloid-β pathology quantitation, we used high-throughput computer-assisted percent area determinations, as previously described [4, 5, 32, 40, 42]. Briefly, under uniform lighting conditions, gray-scale photomicrographs at ×100 covering the cortical region of interest (ROI) were taken on a Leitz DMRB microscope (Leica Microsystems) equipped with a Retiga Exi/QEi digital camera (QImaging) distinguishing 4,095 shades of gray and with a MAC 2000 motorized stage (Ludl Electronic Products) driven by the Turboscan feature of Image-Pro Plus software (Media Cybernetics). This software was used to create composite images of the entire ROI in each section. After manually delineating the gray matter to be measured within the section, an image filtering algorithm was applied to differentiate contiguous immunostained pixels from background unstained tissue and then calculate the percent area covered by IRS1-pS616, IRS1-pS312, Akt-pS473, tau neurofibrillary tangles and neuropil threads, and amyloid-β plaques. For α-synuclein and TDP-43, we used semi-quantitative ratings conducted by a single-skilled operator (HK) masked to any clinical information. Lesion densities were scored on a 0 (none), 1 (occasional), 2 (moderate) or 3 (severe) scale for the presence of pathological cytoplasmic and neuritic labeling, and ratings from three separate fields were then averaged into a final expression score for each section.
IRS1-pS312 enzyme-linked immunosorbent assay
A commercial enzyme-linked immunosorbent assay (ELISA) kit (Millipore, Billerica, MA, USA) was used to confirm differences in expression of IRS1-pS312 in brain. One half gram of midfrontal cortex fresh frozen tissue from eight normal, eight AD, and eight tauopathy brains (4 PiD and 4 CBD) was homogenized in RIPA buffer with protease inhibitors. The resultant homogenate was diluted 20-fold, and IRS1-pS312 expression was determined following standard kit procedures. The kit has a detection limit of 1.5 Units/mL, and a range of detection of 1.6–100 Units/mL. We used non-parametric Wilcoxon tests to compare tissue expression of IRS1-pS312 between pairs.
Mouse case material and brain tissue processing
Tau transgenic mice
Brain sections were obtained from old PS19 tau transgenic mice on a B6C3 background that were generated in the laboratory of VMYL as well as young PS19 mice that had been stereotactically inoculated with synthetic preformed fibrils (pffs) assembled from recombinant full length tau or truncated tau containing four microtubule binding repeats, as previously described [23, 52]. PS19 mice were originally generated using a cDNA encoding the human 1N4R tau isoform with the P301S tau gene (MAPT) mutation and typically begin to develop pathological tau aggregates at 8 or 9 months of age. After synthetic tau pff inoculation in hippocampus of young mice (3 months), there is rapid induction of NFT-like inclusions that propagate from injected sites to connected brain regions in a time-dependent manner. Six micron sections from formalin-fixed, paraffinized brains were double labeled for IRS1-pS616 and Tau pS202/pT205 for fluorescence microscopy, as described above in Histological Processing and Immunohistochemistry. The Ser616 phosphorylation sequence is highly conserved across species and the Invitrogen antibody that we used is well-known to recognize this sequence in humans and in mice.
High-fat diet mice
C57BL/6J male mice (Jackson Laboratories) were purchased at 8 weeks of age and housed 5/cage under standard conditions in a temperature- and humidity-controlled facility with a 12-h light–dark cycle (lights on at 07:00) at the University of Pennsylvania Translational Research Laboratories. After 1 week acclimation, mice were fed a very-high-fat diet [“HFD”, 60 % kcal from fat, (D12492, Research Diets, Inc, New Brunswick, NJ, USA) or the normal control diet (“NCD”), 10 % kcal fat diet, D12450B], for 17 days (n = 5/ group). This diet produced less than 20 % weight gain, but mice did have robust hyperglycemia and hyperinsulinemia. Brains were rapidly removed and fresh frontal polar cortex tissue was dissected, lysed in lysis buffer, subjected to SDS-PAGE and immunoblotted for phospho-tau (T231, Invitrogen clone PHF-6, 1:3000) and total tau (phospho-independent, T49 [30], generated in laboratory of V.M.Y.L.) according to previously described methods Bang et al. [3].
All experimental animal procedures were conducted in accordance with the guidelines published in the NIH Guide for Care and Use of Laboratory Animals and approved by the of Pennsylvania Institutional Animal Care and Use Committee.
Data analysis
Preliminary statistical analyses examined associations of IRS1-pS616, IRS1-pS312, Akt-pS473, amyloid-β, and tau percent area measurements and α-synuclein and TDP-43 ratings with age, sex and postmortem interval. For primary analyses, summary pathological burden scores for IRS1-pS616, IRS1-pS312, Akt-pS473, and other disease lesions (amyloid-β, tau, α-synuclein, TDP-43) in each case were created by averaging the percent area or ratings for the three cortical regions of interest into a global cortical value for that lesion. Secondary analyses examined regions separately within each disease category, within each specific disease, and for associations with ratings of each disease lesion. Analyses of differences of IRS1-pS616 expression among disease categories used ANOVA for global cortical and region-specific values followed by Tukey HSD tests [37] to compare differences between individual diagnostic categories. Associations of IRS1-pS616 with amyloid-β, tau, α-synuclein and TDP-43 were assessed in each disease category with linear regression models adjusting for age and sex. We used one-way ANOVAs to compare differences in IRS1-pS312 and Akt-pS473 expression between normal aging cases and individual diagnostic categories. All statistical analyses were performed using JMP Pro 10.0.0 (SAS Institute Inc., Cary, NC). P values <0.05 were considered significant and all statistical tests were two sided.
Results
Twenty-five cases with a neuropathological diagnosis of AD, 38 with various tauopathies, 48 with α-synucleinopathies, 28 with TDP-43 proteinopathies, and 25 with normal aging brain were included in our analysis of abnormal IRS1-pS616 expression across multiple brain regions. Characteristics of the study groups are displayed in Table 1. We assessed possible cofounders but found no statistically significant relationships between IRS1-pS616 expression and age (p = 0.37), sex (p = 0.42) or postmortem interval (p = 0.16) across the entire cohort or within any diagnostic category.
Serine-phosphorylated IRS1 measures are elevated in AD and other tauopathies
IRS1-pS616 measurements in the three regions for each group are graphically depicted in Fig. 1. IRS-pS616 expression measures were significantly increased in AD as compared to the normal aging group in all three regions (all p < 0.0001). While IRS-pS616 measures were extremely high in the AD group, we also found elevated abnormal IRS1-pS616 in the non-amyloid tauopathy group: PiD, CBD and PSP. In region-specific analyses, we found significant increases in IRS1-pS616 in the hippocampus (p = 0.0005) and frontal cortex (p = 0.0014), but not parietal cortex (p = 0.06). Regression plots of percent area of tau-immunoreactive pixels by IRS1-pS616 immunoreactive pixels for AD and other tauopathies in hippocampus, midfrontal gyrus cortex, and angular gyrus in the parietal cortex are shown in Figure 2a–c. In contrast, the α-synucleinopathies and TDP-43 proteinopathies exhibited no differences in IRS1-pS616 measures compared to the normal group globally or in any region (all p > 0.28).
Fig. 1.

Serine-phosphorylated IRS-1 (IRS1-pS616) percent area measures in neurodegenerative diseases with: AD, tauopathies (including Pick's disease, corticobasal degeneration and progressive supranu-clear palsy), α-synucleinopathies (dementia with Lewy bodies, Parkinson disease, Parkinson dementia and multiple system atrophy) and TDP-43 proteinopathies (frontotemporal dementia, amyotrophic lateral sclerosis), and normal aging. As compared to the normal aging group, IRS1-pS616 measures are most severely elevated in AD but were also significantly elevated in other tauopathies. Error bars represent standard error of the mean
Fig. 2.

Regression plots of IRS1-pS616 and PHF-Tau percent area of immunolabeled cells and neurites in AD and other tauopathies in (a) mid-frontal gyrus cortex, (b) hippocampal CA1/subiculum and (c) angular gyrus cortex in AD and other tauopathies. All p values were <0.0001
The relationships between IRS1-pS616 and the densities of specific pathologic lesions were assessed within each diagnostic category. In correlation analyses, we found highly significant associations between IRS1-pS616 and tau percent area measurements both globally and regionally in all three regions across the entire sample and within the subset of diseases with tau pathology (AD, PiD, CBD and PSP), (all p < 0.0001). In contrast, IRS1-pS616 was not significantly associated with amyloid-β plaque percent area in AD (all p > 0.18) and there were similarly no associations between IRS1-pS616 and α-synuclein or TDP-43 ratings in their respective disease categories.
To determine if the IRS1 abnormality in the tauopathies is specific for IRS1 phosphorylated at S616, or if other serine residues might be affected as they are in AD [44], we performed additional staining and a confirma-tory ELISA on midfrontal gyrus cortex brain tissue. Using immunochemistry for IRS1-pS312, we stained 38 representative cases (14 AD, 13 tauopathy, and 11 normal aging). Results are shown in Supplemental Figure 1a. As with IRS1-pS616, immunohistochemistry and quantitative image analysis showed abnormal IRS1-pS312 was most robustly elevated in the AD group (p < 0.001), but was also significantly elevated in the tauopathy group (p = 0.021). ELISA further confirmed increased expression abnormalities of IRS1-pS312 in midfrontal gyrus cortex tissue lysates from a representative subset of AD, tauopathy, and normal aging samples (each N = 8) where AD and tau cases were both significantly elevated as compared to the normal aging cases (Tau, p = 0.003; AD p = 0.0009, Supplemental Figure 1b). Finally, to understand if the observed differences in serine phosphorylation are associated with downstream abnormalities in the insulin signaling cascade, we additionally investigated phosphorylation of the IRS-1 target Akt. Midfrontal gyrus cortex slices from the same cases used for IRS1-pS312 were immunostained for serine-phosphorylated Akt (pS473). Similar to IRS1, we found that AKT-pS473 was most elevated in the AD group (p = 0.0035), but was also significantly elevated in the tauopathy group (p = 0.043) as compared to normal aging (Supplemental Figure 1c).
Double immunofluorescence labeling shows frequent co-expression of IRS1-pS616 with pathologic tau
To further examine an association between abnormal serine phosphorylation of IRS1 and pathologic tau, we conducted double labeling experiments for IRS1-pS616 and different types of protein aggregates in multiple samples across the range of neurodegenerative diseases included in our cohort. Representative double-labeled images are shown in Fig. 3. Of particular note, we observed frequent double labeling of IRS1-pS616 and tau in cytoplasmic inclusions and dystrophic neurites in multiple tauopathies in multiple brain regions. In cortical regions, statistically significant differences in abnormal IRS1-pS616 measures were observed in AD, PiD, and CBD, but not PSP. We considered whether the absence of IRS1-pS616 expression abnormalities in PSP might be due to the predominant subcortical localization of tau pathology in this disease, in contrast to PiD and CBD. We therefore examined the pons, a region of high pathology in PSP and frequently saw abnormal tau/IRS1-pS616 co-expression in cytoplasm and neurites (Fig. 3h). In contrast to its association with tau lesions, we did not observe any double labeling or other evident relationship between IRS1-pS616 expression and α-synuclein inclusions or neurites in PD, PDD, DLB or MSA or TDP-43 inclusions or neurites in FTLD-TDP or ALS in cortical regions. To further ensure the specificity of the observed association between IRS1-pS616 and AD and other tauopathies, we also examined sections from pons and substantia nigra in PD and in cervical spinal cord in ALS. Even in these vulnerable regions for these respective diseases, no abnormal IRS-1 pS616 or pathological inclusion double labeling was observed as compared to normal controls.
Fig. 3.

Double immunofluorescence images of IRS1-pS616 (red) and neurodegenerative disease lesion proteins (green) in AD (a–d), tauopathies (e–h), α-synucleinopathies (i, j) and TDP43 proteinopathies (k, l). Yellow indicates proteins’ co-localization. a 72-year-old female with AD double labeled for IRS1-pS616 and amyloid-β shows pathological IRS1-pS616 expression in neuronal cytoplasm and neu-rites interspersed among diffuse amyloid-β plaque deposits in mid-frontal cortex. In this and all images except (d), magnification is 400× and scale bar = 50 μm. b 58-year-old male with AD showing IRS1-pS616 immunoreactive neurites enveloped in neuritic amyloid-β plaque and surrounding IRS1-pS616 immunoreactive neurons in mid-frontal cortex; c Same case as in (a) double labeled for IRS1-pS616 and abnormally phosphorylated tau (p-tau) in midfrontal cortex. Note the frequent co-localization (yellow) of perikaryal and neuritic IRS1-pS616 and tau in neurofibrillary tangle-bearing neurons as well as p-tau-immunoreactive neurites; d High magnification (1,000×) image of midfrontal cortex from 68-year-old male with AD shows neurofibrillary tangle that is intensely double labeled for IRS1-pS616 and p-tau neurofibrillary tangle. e 57-year-old male with PiD midfrontal cortex labeled for IRS1-pS616 and p-tau shows double-labeled fibrillar perikaryon of neuron as well as extensive p-tau-immunoreactive neuritic pathology, with some double labeling for IRS1-pS616. f 52-year-old male with CBD shows IRS1-pS616 and p-tau double-labeled neurons. g 68-year-old female with PSP midfrontal cortex double labeled for pS-IRS1 and p-tau shows co-localization in several cells. Such abnormal IRS-1 pS616 expression and p-tau pathology was relatively rare in cortical regions in our sample of PSP cases, prompting us to evaluate IRS-1 pS616 in subcortical regions known to be especially vulnerable in PSP (see text for details). h Pons in 63-year-old female with PSP exhibits extensive IRS1-pS616 and tau co-localization. i Midfrontal cortex in 70-year-old male with PDD immunolabeled for IRS1-pS616 and α-synuclein. Intracellular Lewy inclusions did not co-localize with abnormal IRS1-pS616. j Pons locus coeruleus in 81-year-old male with PD double labeled for IRS-1 pS616 and α-synuclein shows Lewy pathology and no abnormal IRS1-pS616 expression or double labeling. k Midfrontal cortex in 66-year-old female with FTD-TDP immunolabeled for IRS1-pS616 and TDP-43 shows normal-appearing IRS1-pS616 nuclear labeling amidst frequent cytoplasmic and neuritic TDP-43 lesions without co-localization. l Cervical spinal cord from 82-year-old female with ALS shows motor neuron with cytoplasmic TDP-43 surrounding normal IRS1-pS616 labeled nucleus, without co-localization
To ascertain the cell-type specificity of pathological IRS1-pS616 expression in AD and other tauopathies, we conducted double labeling experiments with antibodies directed against IRS1-pS616 and microtubule-associated protein 2 (MAP2, neurons), glial fibrillary acidic protein (GFAP, astocytes) and CD68 antigen (microglia). IRS1-pS616 immunoreactivity in the brain in both young and normal elderly subjects without CNS disease is mostly confined to the nucleus with very little immunoreactivity evident in cytoplasm, dendrites or axons, whereas IRS1-pS616 is abnormally expressed in cytoplasm and neuritic threads in AD and other tauopathies [44]. In AD cases (n = 3, Supplemental Figure 2a), normal nuclear IRS1-pS616 expression was observed in both neurons and astrocytes, but not in microglia, while pathological perikaryal IRS1-pS616 expression was seen only in neurons. Similarly, in both CBD and PSP (each n = 2), we again observed pathological perikaryal IRS1-pS616 expression in neurons but not in astrocytes (Supplemental Figure 2b). Therefore, pathological IRS1-pS616 is present in the perikarya of cells of neuronal morphology, but not astrocytes or microglia, in AD and other tauopathies.
Mouse models further support an association between IRS1-pS616 and pathologic tau
We further investigated an association between tau pathology and IRS1-pS616 by exploring whether primary tau pathology was associated with abnormal IRS1-pS616 expression in human tau-overexpressing transgenic mice. We examined old PS19 mice (19 months old) and younger adult (5–7 months) mice that were inoculated with synthetic tau preformed fibrils in the hippocampus at 3 months of age. This mouse model is known to produce dramatic and robust tau pathology morphologically very similar to human AD and tauopathies. We found abnormal IRS1-pS616 frequently co-localized in tangle-bearing neurons in these mice (Fig. 4). Conversely, to investigate whether insulin resistance could alter or promote pathologic tau in mice, we compared immunoblot levels of phosphorylated tau (normalized to total tau) in brain frontal cortex homogenates from C57BL6/J mice randomized to either a 60 % high-fat diet (a mouse model of insulin resistance) or control diet conditions. Using an antibody specific for tau phosphorylation at T231, a common site of abnormal hyperphosphorylation in AD, we found that the mice receiving the high-fat diet exhibited approximately 60 % increased phospho-tau levels compared to control diet mice (Fig. 5).
Fig. 4.

Abnormal IRS1-pS616 expression in hippocampus tangle-bearing neurons of PS19 tau transgenic mice. Double immunofluorescence for tau (a–e) and IRS-1 pS616 (a′–e′) and merged images (a″–e″) in 19-month-old PS19 human tau Tg mouse without pff inoculation (a), 3 PS19 human tau Tg mice (b–d) injected with syn thetic tau pffs at 3 months of age and surviving until 5 months (b, c) or 7 months (d) or WT control (e). Note frequent abnormal IRS1-pS616 expression in many, but not all tangle-bearing neurons of the transgenic animals
Fig. 5.
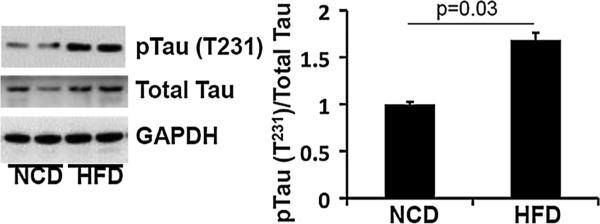
Phosphorylated tau (T231) in high-fat diet mice. Immunob-lots of prefrontal cortex from mice fed a normal control diet (NCD) or high-fat diet (HFD, 60 % kcal by fat) for 17 days. Total tau was slightly increased in the HFD mice. Average phosphorylated tau (normalized to total tau) was increased approximately 60 % in the HFD group compared to NCD mice
Discussion
Abnormal insulin signaling is increasingly recognized for its association with neurodegenerative dementia. Here, we confirm previous reports that abnormal serine phosphorylation of IRS1 is common in the hippocampus in AD and we newly demonstrate that this occurs in other cortical regions in this disease as well. While abnormal IRS1-pS616 and IRS1-pS312 expression was extremely elevated in AD, we also unexpectedly found increased expression of serinephosphorylated IRS-1 in other tauopathies, including PiD, CBD and PSP, albeit to a lesser degree than in AD. These other tauopathies share a common biochemical hallmark with AD of hyperphosphorylated tau, but are pathologically distinct from AD in that the distributions of pathological tau pathology differ and AD also features extracellular amyloid-β plaques. We also found increased expression of Akt-pS473 associated with IRS1 abnormalities in AD and other tauopathies, indicating disruption of insulin signaling downstream to IRS1 as well. We found no increase in serine phosphorylation of IRS1 levels in the other neurodegenerative diseases, i.e., α-synuclein or TDP-43 diseases in our sample. The association of IRS1 abnormalities and tau was further supported by double immunofluorescence experiments demonstrating frequent co-expression of IRS1-pS616 with tau lesions in neurons and dystrophic neurites, but not with lesions of other diseases. IRS1-pS616 partial co-localization with PHF tau-immunoreactive neurofibrillary lesions was shown previously in the 3 × Tg-AD mouse model [28] in which amyloid is present, we newly show partial co-localization in pure tauopathies in which there is absence of amyloid-β. We further demonstrate that IRS1-pS616 and tau pathology are both present in mouse models of primary tau pathology and increased tau phosphorylation is present in high-fat diet-induced insulin resistance in the mouse. These results demonstrate that IRS-1 pS616 and abnormal tau frequently coexist and we surmise they may have reciprocal relationships even in the absence of amyloid-β.
Determining the specific mechanisms underlying the association between serine-phosphorylated IRS1 and abnormal tau requires investigation. We speculate that they may be linked by common regulatory kinases. For example, glycogen synthase kinase-3 (GSK-3) is a multifunctional serine/threonine kinase [24] that has been implicated both in the phosphorylation and aggregation of tau [17, 21], and in the serine phosphorylation of IRS1 leading to insulin resistance [12, 19, 44]. We previously demonstrated abnormal GSK-3 phosphorylation in association with increased serine-phosphorylated IRS1 and other features of insulin resistance in MCI and AD [44]. Activated GSK-3 has also been found to accumulate in the cytoplasm of pre-tangle neurons in brains staged for neurofibrillary changes [34]. Unlike many other kinases, GSK-3 is normally constitutively active and is regulated primarily through inhibition of its activity. Since one of the biological responses of insulin action is downstream activation of the IRS1–PI3k–Akt pathway and inhibition of GSK-3 (for review, [24]), we hypothesize that brain insulin resistance disinhibits GSK-3 activity, leading to increased abnormally phosphorylated tau, tau aggregation, microtubule dysfunction and ultimately neurodegeneration. Another mechanism through which IRS1–PI3k–Akt pathway dysfunction may directly affect tau is via mTOR. In AD, abnormal levels and activation of mTOR and its downstream targets have been found in association with tau pathology [26, 45] and with IRS1-pS616 and other markers of insulin resistance [44]. On the other hand, it is also possible that IRS1 serine phosphorylation and abnormal tau phosphorylation occur as independent processes, perhaps downstream of a common kinase. One candidate kinase that is known to phosphorylate both IRS1 and tau is the Jun N-terminal kinase (JNK) [6, 35].
The AD group in our study had significantly higher overall IRS1-pS616 and IRS1-pS312 measures than the other tauopathies. This may be due to additive or synergistic effects of amyloid-β on IRS1 serine phosphorylation acting through a JNK signaling pathway as previously demonstrated by Ma et al. [28]. Amyloid-β may contribute to abnormal IRS-1 signaling through other mechanisms as well. For example, pathologic amyloid-β has been shown to trigger pro-inflammatory signaling including increased tumor necrosis factor-α (TNF-α) expression, leading to IRS-1 inhibition [6, 27]. Additionally, amyloid-β may activate GSK-3 [16], promoting downstream IRS-1 inhibition [13]. Since amyloid-β shares a consensus sequence with insulin, it may also potentiate insulin resistance by binding to the insulin receptor directly [49]. Thus, insulin resistance, hyperphosphorylated tau, and amyloid-β may have reciprocal relationships in AD that together drive neuronal dysfunction.
Notable strengths of this investigation include the large sample of diverse neurodegenerative disease dementias, including fairly rare diseases and relatively “pure” diseases without mixed pathologies that might confound comparisons, the use of highly sensitive and specific antibodies for the molecular lesions of interest, and the use of both double labeling and quantitative analyses to investigate relationships between IRS1 abnormalities and signature neurode-generative disease lesions. Relative weaknesses include selection biases inherent in our cohort such as a tertiary care practice referral bias and the fact that most cases died in advanced stages of their neurodegenerative diseases. These may limit generalizability of findings. Although we confirmed our finding of a specific association between abnormal IRS1 phosphorylation and tau pathology by examining several sites of phosphorylation, we cannot exclude the possibility that abnormal IRS1 signaling is a feature of other neurodegenerative diseases but is manifest in through different epitopes in the IRS-1 signaling pathway. In conclusion, this study demonstrates that abnormal serine phosphorylation of IRS1 is widespread in brain in AD and other tauopathies but not other neurodegenerative diseases. Our results have implications for the potential treatment or prevention of AD and other tauopathies, as they raise interest in a role for antidiabetic compounds that restore IRS-1 signaling in the brain.
Supplementary Material
Acknowledgments
We acknowledge the special contributions to case recruitment and evaluation of Christopher. M. Clark, Stephen J. DeArmond, Mark S. Forman, Murray Grossman, Howard I. Hurtig, Jason H. Karlawish and Leo F. McCluskey, Bruce L. Miller, and William Seeley, as well as neuropathology fellows and staff at the University of Pennsylvania. This work was supported by grants from the NIH P30 AG010124, P01 AG017586, AG039478, NS084965 and a gift from the Allen H. and Selma W. Berkman Charitable Trust.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00401-014-1328-5) contains supplementary material, which is available to authorized users.
Contributor Information
Mark Yarchoan, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA mark.yarchoan@uphs.upenn.edu.
Jon B. Toledo, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
Edward B. Lee, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
Zoe Arvanitakis, Rush Alzheimer's Disease Center, Rush University, Chicago, IL 60612, USA.
Hala Kazi, Department of Psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Li-Ying Han, Department of Psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Natalia Louneva, Department of Psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Virginia M.-Y. Lee, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
Sangwon F. Kim, Department of Psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
John Q. Trojanowski, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
Steven E. Arnold, Department of Psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA Departments of Psychiatry and Neurology, Penn Memory Center, Perelman School of Medicine, University of Pennsylvania, 3615 Chestnut Street, Philadelphia, PA 19104, USA.
References
- 1.Armstrong RA, Cairns NJ, Lantos PL. Quantification of pathological lesions in the frontal and temporal lobe of ten patients diagnosed with Pick's disease. Acta Neuropathol. 1999;97:456–462. doi: 10.1007/s004010051014. [DOI] [PubMed] [Google Scholar]
- 2.Aviles-Olmos I, Limousin P, Lees A, Foltynie T. Parkinson's disease, insulin resistance and novel agents of neuroprotection. Brain. 2012 doi: 10.1093/brain/aws009. doi:10.1093/brain/aws009. [DOI] [PubMed] [Google Scholar]
- 3.Bang S, Kim S, Dailey MJ, et al. AMP-activated protein kinase is physiologically regulated by inositol polyphosphate multikinase. Proc Natl Acad Sci. 2012;109:616–620. doi: 10.1073/pnas.1119751109. doi:10.1073/p nas.1119751109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett DA, Schneider JA, Wilson RS, et al. Amyloid mediates the association of apolipoprotein E e4 allele to cognitive function in older people. J Neurol Neurosurg Psychiatry. 2005;76:1194–1199. doi: 10.1136/jnnp.2004.054445. doi:10.1136/jnnp.2004.054445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett DA, Schneider JA, Wilson RS, et al. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61:378–384. doi: 10.1001/archneur.61.3.378. doi:10.1001/archneur.61.3.378. [DOI] [PubMed] [Google Scholar]
- 6.Bomfim TR, Forny-Germano L, Sathler LB, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease-associated Aβ oligomers. J Clin Invest. 2012;122:1339–1353. doi: 10.1172/JCI57256. doi:10.1172/JCI57256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cairns NJ, Bigio EH, Mackenzie IRA, et al. Neuropatho-logic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. doi:10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craft S, Baker LD, Montine TJ, et al. Intranasal insulin therapy for alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2011 doi: 10.1001/archneurol.2011.233. doi:10.1001/archn eurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3:169–178. doi: 10.1016/S1474-4422(04)00681-7. doi:10.1016/S1474-4422(04)00681-7. [DOI] [PubMed] [Google Scholar]
- 10.Dickson DW, Bergeron C, Chin SS, et al. Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61:935–946. doi: 10.1093/jnen/61.11.935. [DOI] [PubMed] [Google Scholar]
- 11.Dickson DW, Braak H, Duda JE, et al. Neuropatho-logical assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol. 2009;8:1150–1157. doi: 10.1016/S1474-4422(09)70238-8. doi:10.1016/ S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 12.Eldar-Finkelman H, Krebs EG. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc Natl Acad Sci. 1997;94:9660–9664. doi: 10.1073/pnas.94.18.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eldar-Finkelman H, Krebs EG. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. PNAS. 1997;94:9660–9664. doi: 10.1073/pnas.94.18.9660. doi:10.1073/pnas.94.18.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geser F, Brandmeir NJ, Kwong LK, et al. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol. 2008;65:636–641. doi: 10.1001/archneur.65.5.636. doi:10.10 01/archneur.65.5.636. [DOI] [PubMed] [Google Scholar]
- 15.Giasson BI, Duda JE, Quinn SM, et al. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 16.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 17.Hanger DP, Hughes K, Woodgett JR, et al. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. doi:10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- 18.Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology. 1994;44:2015–2019. doi: 10.1212/wnl.44.11.2015. [DOI] [PubMed] [Google Scholar]
- 19.Henriksen EJ, Kinnick TR, Teachey MK, et al. Modulation of muscle insulin resistance by selective inhibition of GSK-3 in Zucker diabetic fatty rats. Am J Physiol Endocrinol Metab. 2003;284:E892–E900. doi: 10.1152/ajpendo.00346.2002. doi:10.1152/ajpendo.00346.2002. [DOI] [PubMed] [Google Scholar]
- 20.Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. doi:10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 21.Hong M, Lee VM-Y. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem. 1997;272:19547–19553. doi: 10.1074/jbc.272.31.19547. doi:10.1074/jbc.272.31.19547. [DOI] [PubMed] [Google Scholar]
- 22.Hurtig HI, Trojanowski JQ, Galvin J, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology. 2000;54:1916–1921. doi: 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- 23.Iba M, Guo JL, McBride JD, et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci. 2013;33:1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013. doi:10.1523/JNEUROSCI.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. doi:10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Kitamoto T, Ogomori K, Tateishi J, Prusiner SB. Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest. 1987;57:230–236. [PubMed] [Google Scholar]
- 26.Li X, Alafuzoff I, Soininen H, et al. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J. 2005;272:4211–4220. doi: 10.1111/j.1742-4658.2005.04833.x. doi:10.1111/j.1742-4658.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- 27.Lourenco MV, Clarke JR, Frozza RL, et al. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer's β-amyloid oligomers in mice and monkeys. Cell Metab. 2013;18:831–843. doi: 10.1016/j.cmet.2013.11.002. doi:10.1016/j.cmet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Ma Q-L, Yang F, Rosario ER, et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci. 2009;29:9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009. doi :10.1523/JNEUROSCI.1071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackenzie IRA, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. doi:10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 30.Mawal-Dewan M, Henley J, Van de Voorde A, et al. The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J Biol Chem. 1994;269:30981–30987. [PubMed] [Google Scholar]
- 31.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. doi:10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell TW, Mufson EJ, Schneider JA, et al. Parahippocampal tau pathology in healthy aging, mild cognitive impairment, and early Alzheimer's disease. Ann Neurol. 2002;51:182–189. doi: 10.1002/ana.10086. [DOI] [PubMed] [Google Scholar]
- 33.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. doi:10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 34.Pei JJ, Braak E, Braak H, et al. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- 35.Ploia C, Antoniou X, Sclip A, et al. JNK plays a key role in tau hyperphosphorylation in Alzheimer's disease models. J Alzheimers Dis. 2011;26:315–329. doi: 10.3233/JAD-2011-110320. doi:10.3233/JAD-2011-110320. [DOI] [PubMed] [Google Scholar]
- 36.Qiao LY, Goldberg JL, Russell JC, Sun XJ. Identification of enhanced serine kinase activity in insulin resistance. J Biol Chem. 1999;274:10625–10632. doi: 10.1074/jbc.274.15.10625. [DOI] [PubMed] [Google Scholar]
- 37.Ramsey PH, Ramsey PP. Power of pairwise comparisons in the equal variance and unequal sample size case. Br J Math Stat Psychol. 2008;61:115–131. doi: 10.1348/000711006X153051. doi:10.1348/000711006X153051. [DOI] [PubMed] [Google Scholar]
- 38.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 39.Rivera EJ, Goldin A, Fulmer N, et al. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer' s disease: link to brain reductions in acetylcholine. Insulin. 2005;8:247–268. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]
- 40.Robinson JL, Geser F, Corrada MM, et al. Neocortical and hippocampal amyloid-β and tau measures associate with dementia in the oldest-old. Brain. 2011;134:3708–3715. doi: 10.1093/brain/awr308. doi:10.1093/brain/awr308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt ML, Lee VM, Hurtig H, Trojanowski JQ. Properties of antigenic determinants that distinguish neurofibrillary tangles in progressive supranuclear palsy and Alzheimer's disease. Lab Invest. 1988;59:460–466. [PubMed] [Google Scholar]
- 42.Soetanto A, Wilson RS, Talbot K, et al. Association of anxiety and depression with microtubule-associated protein 2- and synaptopodin-immunolabeled dendrite and spine densities in hippocampal CA3 of older humans. Arch Gen Psychiatry. 2010;67:448–457. doi: 10.1001/archgenpsychiatry.2010.48. doi:10.1001/archgenpsychiatry.2010.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steen E, Terry BM, Rivera EJ, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease—is this type 3 diabetes? Insulin. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- 44.Talbot K, Wang H, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–1338. doi: 10.1172/JCI59903. doi:10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang Z, Bereczki E, Zhang H, et al. mTor mediates tau dyshomeostasis: implication for Alzheimer disease. J Biol Chem. 2013;288:15556–15570. doi: 10.1074/jbc.M112.435123. doi:10.1074/jbc.M112.435123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.The National Institute on Aging, Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 47.Trojanowski JQ, Revesz T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol. 2007;33:615–620. doi: 10.1111/j.1365-2990.2007.00907.x. doi:10.1111/j.1365-2990.2007.00907.x. [DOI] [PubMed] [Google Scholar]
- 48.Wands JR. Alzheimer's disease is type 3 diabetes—evidence reviewed. Society. 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie L, Helmerhorst E, Taddei K, et al. Alzheimer's beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 2002;22:1–5. doi: 10.1523/JNEUROSCI.22-10-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yarchoan M, Arnold SE. Repurposing diabetes drugs for brain insulin resistance in alzheimer disease. Diabetes. 2014;63:2253–2261. doi: 10.2337/db14-0287. doi:10.2337/db14-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yarchoan M, Xie SX, Kling MA, et al. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neuro-degenerative dementias. Brain. 2012;135:3749–3756. doi: 10.1093/brain/aws271. doi:10.1093/brain/aws271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. doi:10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.