

Abstract
Trisomy 21, the commonest constitutional aneuploidy in humans, causes profound perturbation of stem and progenitor cell growth, which is both cell context dependent and developmental stage specific and mediated by complex genetic mechanisms beyond increased Hsa21 gene dosage. While proliferation of fetal hematopoietic and testicular stem/progenitors is increased and may underlie increased susceptibility to childhood leukemia and testicular cancer, fetal stem/progenitor proliferation in other tissues is markedly impaired leading to the characteristic craniofacial, neurocognitive and cardiac features in individuals with Down syndrome. After birth, trisomy 21-mediated premature aging of stem/progenitor cells may contribute to the progressive multi-system deterioration, including development of Alzheimer's disease.
Keywords: Down syndrome, hematopoietic stem cells, leukemia, neural progenitors, trisomy 21
Introduction
Trisomy 21, trisomy 18 and trisomy 13 are the commonest constitutional trisomies in humans 1. In contrast to trisomy 18 and 13, where fewer than 10% of affected children survive beyond the first year of life 2,3,4, median life expectancy for individuals with trisomy 21 (Down syndrome; DS) is around 60 years 5. Most attention has focused on trisomy 21, not only because it is 20 times and 40 times more frequent than trisomy 18 and 13, respectively, but also because prolonged survival in DS suggests that most cells evolve epigenetic, transcriptional and/or translational regulatory mechanisms which allow them to adapt to the additional copy of chromosome 21 (Hsa21). To some extent, this may reflect the relatively low number of protein-encoding genes on Hsa21 (∼240) (http://www.ensembl.org/biomart/martview/). However, the characteristic phenotypic variability between different individuals with DS points to considerable complexity. Understanding the genomic determinants of this complexity continues to reveal fascinating insights relevant not only to DS, but also to aneuploidy in general.
Here, we review the impact of human trisomies on stem and progenitor cells. We will focus on trisomy 21, and particularly on hematopoiesis, where advances in techniques for characterization of highly purified primary cells and the development of induced pluripotent stem cell (iPSC) and animal models are beginning to answer some of the questions about the mechanisms by which trisomies cause human disease.
Phenotypic variability in constitutional trisomy 21 (DS)
DS is a multisystem disorder caused, in most cases, by meiotic non-disjunction of the maternal Hsa21, resulting in a third copy of the entire Hsa21 in all cells 6,7. The clinical and biological impact of trisomy 21 nevertheless varies widely, not only between individuals with DS, but also in different tissues, the cell types within these tissues and at different ages [reviewed in 8,9,10]. Within this phenotypic variability, certain characteristics, such as the craniofacial abnormalities, hypotonia and cognitive impairment, are common to all individuals with DS (Table1) and may therefore share temporal, biological or genetic mechanisms. Other features, such as cardiac defects or gastrointestinal anomalies, affect only a subset of patients and so may be more strongly influenced by inter-individual differences which interact with trisomy 21-driven changes, in heart and gut development, respectively, during embryogenesis. Many of these phenotypic abnormalities can be modeled using mouse segmental trisomies allowing the consequences of trisomy 21 to be investigated in an appropriate cellular context (Table2).
Table 1.
Phenotypic characteristics of Down syndrome
| Characteristic | Frequency (%) | Reference |
|---|---|---|
| Craniofacial | ∼100 | 10, 114, 194, 195 |
| Epicanthal folds | ||
| Upward slanting palpebral fissures | ||
| Flat nasal bridge | ||
| Small brachycephalic head | ||
| Small ears | ||
| Small mouth | ||
| Other musculoskeletal abnormalities | ∼100 | 10, 196 |
| Hypotonia | ||
| Single transverse palmar crease | ||
| Clinodactyly with wide spacing | ||
| Cognitive impairment | ∼100 | 10, 17, 197 |
| Reduced brain volume | ||
| Learning and memory defects | ||
| Dementia | 40–50, increases with age | 12, 13, 14, 15, 16, 17, 18, 198, 199 |
| Visual | 18–60, increase with age | 10, 18 |
| Hearing | 18–80, increase with age | 10, 18 |
| Thyroid disease | 1–54, increase with age | 10, 18, 196, 200, 201 |
| Cardiac defects | 40–50 | 202, 203, 204 |
| ASD (45%) | ||
| VSD (35%) | ||
| Isolated secundum (8%) | ||
| Isolated PDA (7%) | ||
| Isolated Fallot's (4%) | ||
| Other | ||
| Gastrointestinal defects | 12 | 10 |
| Benign hematological abnormalities | ∼100 | 11 |
| Neonatal thrombocytopenia | ||
| Neonatal polycythemia | ||
| Neonatal neutrophilia, blast cells | ||
| Macrocytosis | ||
| Preleukemia and leukemia | ||
| TAM and silent TAM | 30 | 11, 53 |
| Acute myeloid leukemia | 1 | 53 |
| Acute lymphoblastic leukemia | 1 | 53 |
| Non-hematologic cancers | 50% of risk of individuals without DS | 18, 53, 56, 58, 59 |
Table 2.
Mouse models of Down syndrome
| Ts65Dn | Ts1Cje | Ts1Rhr | Tc1 | Ts1Yeh;Ts2Yeh;Ts3Yeh Dp(16)1Yeh/+; Dp(10)1Yeh/+; Dp(17)1Yeh/+ | Ts1Yah | |
|---|---|---|---|---|---|---|
| Number of trisomic Hsa21 orthologs | ∼100 (also trisomic for ∼60 genes on Mmu17 not syntenic for Hsa21) | ∼80 | 33 | Transchromosomic (trisomic for 200 RefSeq Hsa21 genes*) | ∼175 | ∼12 |
| Craniofacial | Small mandible Brachycephaly | Small mandible | Large mandible | Small mandible | Normal appearance | Not reported |
| Differences in face, palate recapitulate human DS | Differences in face, palate recapitulate human DS | Abnormalities do not recapitulate human DS | ||||
| Learning and memory | Impaired spatial learning and memory Impaired motor coordination | Altered hippocampal dependent learning | Impaired novel object recognition | Defect in short-term memory and motor co-ordination | Recapitulates most of the behavioral features of Ts65Dn | Impaired novel object recognition but improved hippocampal-dependent spatial learning |
| Brain structure | Reduced brain volume | Reduced brain volume during embryogenesis | Reduced brain volume at age 4 months | Hydrocephalus (6.5%) | Not reported | |
| Reduced cerebellar volume | Reduced cerebellar volume | Reduced cerebellar volume | Reduced cerebellar volume | |||
| Impaired neurogenesis: –Impaired neural precursor proliferation and differentiation –Abnormal cell cycle | Impaired neurogenesis: –Impaired neural precursor proliferation and differentiation | |||||
| Cardiac defects | Septal defects similar, but not identical, to human DS | Not reported | None | Mainly VSD; also outflow tract defects and AVSD similar to human DS | Cardiac defects include ASD VSD and AVSD (in several models: Dp(16)1Yeh/+, Dp(16)2Yeh/+ Dp(16)4Yeh/+ and Dp(16)1Yeh/+; Dp (16)2Yeh/+; Dp(16)3Yeh/+ | Not reported |
| Gastro-intestinal defects | Not reported | Not reported | Not reported | Not reported | Not reported | Not reported |
| Thyroid disease | Not reported | Not reported | Not reported | Not reported | Not reported | Not reported |
| Hematopoietic | MPD in adults No leukemia Macrocytosis | No MPD or leukemia Macrocytic anemia | Co-operates with GATA1s and MPL to induce AMKL | No MPD or leukemia even with GATA1s Macrocytosis | Not reported | Not reported |
| Reduced adult HSC Impaired HSC self-renewal in adults | Normal HSC numbers and function in adults Impaired fetal liver HSC and progenitor function | Thrombocytosis, increased MKs and mild anemia in adults | Increased MKs and erythrocytosis in older adults | |||
| Increased GMP in adults | Increased GMP in adults |
AMKL, acute megakaryocytic leukemia; ASD, atrioseptal defect; AVSD, atrioventricular septal defect; DS, Down syndrome; GMP, granulocyte–macrophage progenitor; HSC, hematopoietic stem cell; MPD, myeloproliferative disorder; VSD, ventriculoseptal defect. See text for details.
The impact of age on the phenotypic expression of DS is increasingly recognized and, for many cells and tissues, is essential to consider in selecting the best experimental model to investigate the role of trisomy 21. Abnormalities of hematopoiesis begin in fetal life and have their maximal expression in the neonatal period when nearly all DS neonates have multiple hematologic defects, including 30% who develop a unique preleukemic syndrome confined to the first few months of life 11. By contrast, the effects of trisomy 21 on visual and hearing impairment, thyroid function and cognitive function increase with age with progressive pathological changes in the brain in almost all DS individuals and clinical evidence of dementia in ∼50% 12,13,14,15,16,17,18,19. These age-related differences in phenotypic expression in DS suggest that trisomy 21, through patterns of gene expression which may be established early in development, causes premature, or accelerated, aging of a range of cell types. Evidence in support of this is now emerging 20, as discussed below.
Although several Hsa21 genes have been linked to the phenotypic expression of specific aspects of DS, such as leukemia and dementia, the mechanism(s) by which trisomy of individual genes or groups of genes contributes to the disorder remains unclear [reviewed in 8,9,21,22,23,24]. Investigators have used three broad approaches to investigate this question: mouse models trisomic for one or more of the genes on Hsa21, genomic association studies and comparative studies between human cells trisomic or disomic for Hsa21.
Mouse models of DS
The phenotypic characteristics of the most well-established mouse models of DS, and the extent to which they recapitulate the human phenotype, are summarized in Table2. These include the only transchromosomic mouse model (Tc1) in which most of Hsa21 is present 25 albeit with several deleted or rearranged genes 26. A number of more recent mouse mutants carrying genomic rearrangements of Hsa21 syntenic regions (on Mmu10, Mmu16 and Mmu17) that are trisomic for some, or most, of the ∼250 mouse genes orthologous to Hsa21 genes have been described 27,28,29,30,31. These interesting models, which may better mimic some aspects of human DS, have so far been used mainly to model the neurocognitive and cardiac defects in DS 27,28,29,30,31. Details of these, and of elegant refinements to narrow down the Hsa21 regions linked to defined phenotypes, are described in several reviews 32,33,34,35,36,37,38,39,40,41,42 and are only briefly discussed here in relation to their insight into the effects of trisomy 21 on stem/progenitor cells.
The impact of trisomy 21 on stem cell function
There is increasing recognition that trisomy 21 impacts on stem cell function in a number of ways (Fig1). In hematopoiesis, for example, trisomy 21 affects the self-renewal, proliferation and differentiation of hematopoietic stem and progenitor cells (HSPC) either directly or via the hematopoietic microenvironment 43,44,45,46,47,48,49,50,51,52. Studies in other tissue types, where stem and progenitor cells are often more difficult to identify and isolate, suggest that trisomy 21 also causes many of the defects in craniofacial, brain and cardiac development through perturbations of stem/progenitor cell growth and differentiation and altered interactions with microenvironmental and temporal cues. These alterations in stem/progenitor proliferation may underlie the increased susceptibility of some cell types, such as HSPC and primordial germ cells to malignant transformation 53,54,55,56,57,58,59,60 and of HSPC to premature aging in DS 20, as discussed in detail below.
Figure 1. Impact of trisomy 21 on stem and progenitor cell function.

Studies in human cells and in animal models of Down syndrome (DS) show that trisomy 21 can affect the self-renewal, proliferation and differentiation of stem and progenitor cells either directly or via the supportive microenvironment. In fetal life in DS, proliferation of hematopoietic and testicular stem/progenitor cells is increased and susceptibility to malignant transformation (leukemia and testicular cancer) is increased in childhood. By contrast, proliferation of stem/progenitor cells of other lineages is impaired and is responsible for many of the developmental defects affecting the brain, craniofacial structures and heart in DS. After birth, trisomy 21 has been shown to cause premature aging of stem and progenitor cells both of hematopoietic and non-hematopoietic lineages, an effect which is likely to contribute to the phenotypic abnormalities in adults with DS, including Alzheimer's disease, bone marrow failure and impaired immunity.
Hematopoiesis and leukemia
The link between childhood leukemia and DS provides strong evidence for a particular susceptibility of hematopoietic cells early in life to perturbation of the normal mechanisms which control their growth and differentiation. Leukemias in DS have several unique features which hint at the ways in which trisomy 21 alters the behavior of HSPC [reviewed in 21,61,62]. First, the frequency of both myeloid leukemias and lymphoid leukemias is increased, by 150-fold and ∼30-fold, respectively 53,59, indicating that trisomy 21 affects both myeloid and lymphoid progenitors. Second, these leukemias have a distinct temporal pattern of onset. Myeloid leukemia of DS (ML-DS) originates in fetal liver HSPC and presents either as a neonatal preleukemic syndrome known as transient abnormal myelopoiesis (TAM) or as full-blown ML-DS in children under the age of 5 years 21,61,62. The peak age at presentation for acute lymphoblastic leukemia in DS (DS-ALL) is 1–4 years and, in contrast to ALL in individuals without DS, never presents in neonates or infants 53. Third, leukemias in DS have distinct biologic and molecular features. Leukemic cells in ML-DS and TAM harbor N-terminal truncating mutations in the key hematopoietic transcription factor GATA1, which result in exclusive production of a short GATA1 protein (Gata1s) with altered functional properties together with loss of expression of full-length Gata1 since the GATA1 gene is on the X chromosome 63,64,65,66,67. Such mutations are not leukemogenic in the absence of trisomy 21 68. In DS-ALL, which in contrast to ALL in children without DS is always a B-precursor disease 69, ∼60% of cases have aberrant expression of the CRLF2 receptor and around half of these have RAS mutations or mutations activating JAK-STAT growth-promoting signaling pathways 70,71,72,73,74,75,76.
Impact of trisomy 21 on fetal, neonatal and adult human hematopoiesis
In contrast to most other tissues, hematopoietic tissues contain a well-characterized hierarchy of stem and progenitor cells, which can be readily isolated for molecular and functional studies. Characterization of the hematologic abnormalities in human DS therefore offers one of the best ways to understand how trisomy 21 perturbs cell biology and how cells adapt to aneuploidy. Recent studies in primary human fetal liver and neonatal cells 11,45, supported by data from human iPSC and hESC 46,47, demonstrate that trisomy 21 causes major disturbance throughout the entire hematopoietic hierarchy from HSC through to progenitors and mature cells (Fig2). In particular, in fetal liver, trisomy 21 alters the balance of HSPC differentiation, promoting expansion and proliferation of megakaryocyte–erythroid progenitors (MEP) and megakaryocytes during the second trimester at the expense of both granulocyte–monocyte progenitors (GMP) and B-cell progenitors (BCP) 45. There is also a 3.5-fold expansion in HSC numbers, and in vitro purified trisomy 21 fetal liver HSCs have erythroid–megakaryocyte-biased gene expression together with reduced expression of lymphoid genes. Consistent with this, fetal liver HSC function is also markedly abnormal in DS. In particular, fetal liver HSCs generate more megakaryocyte and erythroid cells while their B-cell potential is severely impaired 45. Since GATA1 mutations were not detectable in these cells, these data indicate that trisomy 21 itself perturbs fetal liver hematopoiesis.
Figure 2. Perturbation of human fetal hematopoiesis by trisomy 21.
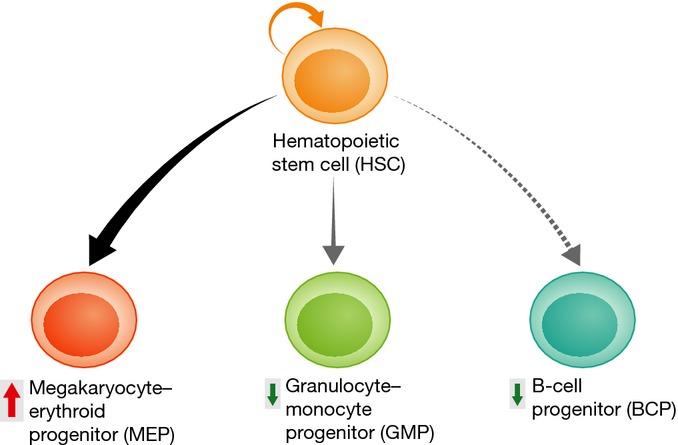
Comparison of the frequency and function of disomic and trisomy 21 second-trimester human fetal hematopoietic stem cells and progenitor cells (HSPC) has shown a consistent pattern of abnormalities in the trisomic populations. Trisomy 21 alters the balance of HSPC differentiation, promoting expansion and proliferation of megakaryocyte–erythroid progenitors (MEP) and megakaryocytes during the second trimester at the expense of both granulocyte–monocyte progenitors (GMP) and B-cell progenitors (BCP), which are both significantly reduced.
The effects of trisomy 21 on primary human fetal liver HSPC raise many questions. First, since these studies were confined to second-trimester fetal liver, it is not clear whether the effects are confined to this gestation. Interestingly, Chou et al 47 found that trisomy 21 iPSC differentiated under conditions designed to model yolk sac hematopoiesis showed enhanced erythroid, but not megakaryocyte, differentiation in vitro, suggesting the effects of trisomy 21 may be developmental stage specific. More recently, our group studied hematopoiesis in neonates with DS. In the presence of GATA1 mutations, DS neonates developed the preleukemic condition, TAM. However, even in the absence of GATA1 mutations, DS neonates had trilineage perturbation of hematopoiesis with increased erythroid and myeloid cells and abnormal platelet development consistent with the effects of trisomy 21 on HSPC function persisting after birth 11. In contrast, the few studies in adults with DS suggest that trisomy 21 causes a different profile of hematologic abnormalities later in life. Adults with DS have a high prevalence of red cell macrocytosis and quantitative and qualitative B- and T-lymphocyte abnormalities, while some have unexplained thrombocytopenia and neutropenia 77,78,79, myelodysplasia or bone marrow failure 80. This suggests that in adults, trisomy 21 may induce HSC aging, consistent with recent studies in Ts65Dn mice implicating increased expression USP16 as a possible mechanism for these effects 20.
Second, the mechanisms linking trisomy 21-mediated perturbation of fetal liver hematopoiesis and the high frequency of GATA1 mutations in DS neonates are still unclear. Trisomy 21-mediated proliferation of fetal liver megakaryocyte/erythroid-biased HSPC may simply provide a permissive cellular environment for expansion of preleukemic mutant GATA1 clones. Alternatively, changes to pathways regulating fetal HSPC growth and differentiation in DS may be directly responsible for the increased frequency of GATA1 mutations. Similarly, the link between impaired B-cell development in DS fetal liver HSPC and the increase in B-ALL 69,81,82 and of immune deficiency in children with DS 83 is unclear, although delayed expression of the normal fetal B-cell development program might increase the likelihood of acquiring leukemogenic mutations in lymphoid genes in early childhood.
Third, given that alterations in the microenvironment can promote myeloproliferative disorders and leukemia in mouse models 84,85,86,87, the DS fetal liver microenvironment may support, or even drive, the abnormal growth and differentiation of DS fetal liver HSPC. The natural history of TAM, which resolves within a few weeks of life in most cases and is characterized by infiltration of the liver by mutant GATA1 blast cells 88,89, also suggests that factors produced in the fetal liver microenvironment may be necessary to maintain these cells. In support of this, in vitro survival of TAM blast cells and in vivo survival of leukemia cells in a mouse model of DS-like acute myeloid leukemia has been shown to be dependent on insulin-like growth factors 90.
Finally, the molecular basis for perturbation of fetal liver HSPC growth and differentiation by trisomy 21 remains to be explained. Even using highly purified fetal liver HSPC, we found no significant increase in expression of selected trisomic genes on Hsa21 (RUNX1, ERG, DYRK1A) known to influence HSPC behavior and development of leukemia through gene dosage 51,91. This does not exclude a role for trisomy 21 dose-related changes in these genes given limitations in the sensitivity of the methodology 92 and the confounding influence of interindividual variation 93 as discussed below, especially since even small changes in expression of the Hsa21 genes are associated with DS-like defects in mouse models 94 and multiple genes may be involved 95.
Animal models of leukemia and abnormal hematopoiesis in DS
Although ML-DS and TAM provide a natural human model to interrogate the impact of trisomy 21 on HSPC and the mechanisms which contribute to the development of leukemia in DS, mechanistic experiments to identify the exact role of specific genes are often difficult in human cells. Initial attempts to model ML-DS and TAM in mouse models were disappointing as no spontaneous leukemias developed (Table2). However, this is consistent with human DS where trisomy 21 dysregulates HSPC proliferation and differentiation but is insufficient to promote leukemia without additional, acquired mutations. All of the DS mouse models have abnormal hematopoiesis, typically affecting erythroid and megakaryocyte development 48,49,50, although the defects do not accurately recapitulate those seen in human fetal liver 45. Nevertheless, by co-expressing additional oncogenes, DS mouse models provide potential insight into genes and pathways, which may contribute to perturbation of HSPC development by trisomy 21, including ERG, DYRK1A, HMGN1 and miR125b 51,91,92,96,97,98.
The myeloproliferative disorder in adult Ts65Dn mice 48, for example, is clearly linked to gene dosage of ERG since reducing the number of copies of ERG from 3 to 2 in this model corrects the hematologic abnormalities 91. Since neonatal TsDn mice are not affected, Birger et al 97 used a different approach to modeling TAM. Building on data showing potent effects of ERG overexpression on megakaryocyte proliferation and leukemia in disomic mice 100, they recently created a double transgenic mouse model of TAM/ML-DS on a non-trisomic background in which overexpression of ERG promoted fetal liver MEP expansion similar to that seen in human fetal liver, and this synergized in vivo with expression of GATA1s to cause a TAM-like disease and subsequent progression to megakaryocyte–erythroid leukemia 97. Nevertheless, ERG has not yet been shown to be significantly overexpressed in trisomy 21-containing human hematopoietic cells, including leukemias 45,100 and hESC/iPSC 46,47.
Malinge et al 51 recently used Ts1Rhr mice, trisomic for 33 Hsa21 orthologs (Table2), to create a trisomy 21-dependent ML-DS model by crossing them with GATA1s knock in mice and over-expressing a transforming MPL allele (MPLW515L), which has been reported in ML-DS 101,102. In this model, they showed that DYRK1A was able to act as a megakaryoblastic tumor-promoting gene and they found increased expression of DYRK1A in human ML-DS samples 51. Although this identifies a possible role for increased expression of DYRK1A in the transformation of TAM to ML-DS, this model does not fully recapitulate the human disease. For reasons that are still not clear, this model can only be produced in adult, and not fetal, hematopoietic cells, and indeed, DYRK1A expression does not appear to be significantly increased in human fetal HSPC 45, perhaps indicating altered mechanisms of DYRK1A regulation in fetal cells compared to postnatal or leukemic cells. Furthermore, MPLW515L is able to induce a fatal, rapid onset myeloproliferative disorder even in the absence of Gata1s and a trisomic background 103 highlighting the importance of the cellular context in understanding the contribution of individual genes.
The Ts1Rhr mouse model has also proved useful for investigating the role of Hsa21 orthologs in B-cell development and B-ALL. Lane et al 98 recently showed that, as in human fetal liver 45, B progenitors were reduced in bone marrow from young Ts1Rhr mice but were more clonogenic than wild-type progenitors and could be replated indefinitely in vitro. Furthermore, Ts1Rhr B progenitors were transformed into B-ALL in vivo by CRLF2 with activated JAK2, a known oncogenic stimulus in DS-ALL. Lane et al then identified differential expression of PRC2 targets and sites of H3 K27 trimethylation as a specific ‘signature’ common to DS-ALL and Ts1Rhr B cells and, through a series of elegant experiments, showed that overexpression of HMGN1, an Hsa21 ortholog trisomic in Ts1Rhr mice which encodes a nucleosome remodeling protein, is responsible both for this gene expression signature and for the proliferative and leukemia-promoting effects on Ts1Rhr B cells. These data provide compelling evidence in support of a role for HMGN1 in the perturbation of B-cell development by trisomy 21 and the increased susceptibility of children with DS to B-ALL.
USP16 and defects in HSC self-renewal and stem cell aging
By com-paring hematopoiesis in Ts65Dn, Ts1Cje and wild-type mice, Adorno et al 20 identified a role for the mouse homolog of the Hsa21 gene USP16 in HSC self-renewal. HSC frequency was reduced by greater than threefold in Ts65Dn mice, which are trisomic for USP16, compared to Ts1Cje mice and wild-type mice, which have only 2 copies of USP16. HSC function was also impaired in the USP16 trisomic mice with reduced clonogenicity and multilineage engraftment following secondary transplantation. These features were associated with a 1.5-fold increase in USP16 gene expression and were reversed by short interfering RNAs. Interestingly, similar defects were seen in Ts65Dn neural progenitors and fibroblasts consistent with previously reported defective proliferation of primary human DS fibroblasts 104,105. They also went on to demonstrate a link between trisomy for USP16 and reduced activity of the PRC1 complex and its target CDK2NA, which regulate senescence and self-renewal of several somatic stem cell types [106,107; reviewed in 108]. The reduction in HSC frequency and clonogenicity contrasts with the increase in HSC frequency and clonogenicity in human DS fetal liver 45. However, although this may reflect species-specific differences in hematopoiesis and/or the role played by other genes/pathways in the senescence of Ts65Dn mouse HSC, another important issue is age. The impaired HSC self-renewal reported by Adorno et al in adult Ts65Dn mice 20 is compatible with the increasing recognition of the occurrence of hematologic abnormalities, including myelodysplasia and bone marrow failure, in older adults with DS 80.
Non-hematologic cancers
It is likely that several mechanisms contribute to the 50% reduction in the frequency of solid tumors with DS, including the effects of trisomy 21 on stem and progenitor proliferation, tumor-associated angiogenesis and tumor suppression [reviewed in 22]. It is notable that the only malignancy, apart from leukemia, to be increased in DS is testicular germ cell tumors. These tumors are derived from primordial germ cells and, in DS, are believed to arise in utero through a pre-invasive stage known as intratubular germ cell neoplasia unclassified (IGCNU), which has been documented in the second trimester 109,110 and which is preceded by activation of signaling pathways leading to increased proliferation and impaired differentiation 60. These cellular abnormalities are similar to those in fetal HSPC, and, interestingly, testicular germ cell tumors also share several key signaling pathways, such as KIT/SCF, K-RAS and PZLF with normal and leukemic HSPC 60 and suggesting that increased susceptibility to both these malignancies in DS may derive from the trisomy 21-mediated proliferative drive to stem cells within fetal hematopoietic and testicular tissues.
Investigations into the mechanisms of reduced tumor susceptibility in DS, largely through studies in mouse models, suggest that 3 Hsa21 genes, ETS2, RCAN1 and DYRK1A, may play a role. Through crossing Ts1Rhr mice with ApcMin mice, which are heterozygous for the adenomatosis polyposis coli gene, and Ets2+/− mice, Sussan et al 111 showed that protection against colonic tumors in this model was in part dependent on the presence of three copies of ETS2. This suggests that ETS2 can act as a tumor suppressor, through as yet unclear mechanisms, but that other genes may contribute to this effect. Subsequently, Baek et al 112 used the Ts65Dn mouse model and a transgenic disomic mouse over-expressing RCAN1 to show that increased expression of RCAN1 was sufficient to suppress growth of lung cancer and melanoma cell lines in vivo through inhibition of VEGF-mediated tumor angiogenesis by suppressing the calcineurin pathway in co-operation with DYRK1A. However, more recent experiments in a more aggressive tumor model (NPcis) found that trisomy improved survival rather than preventing cancer and that neither ETS2 nor tumor angiogenesis was responsible for this protective effect 113. Taken together, these studies indicate that the mechanisms underlying tumorigenesis in DS result from a complex interplay between changes in expression of Hsa21 and other genes, inter-individual differences in genetic susceptibility and acquired changes in the microenvironment.
Craniofacial defects
In contrast to the hematopoietic system, there is limited information about the effects of trisomy 21 on the stem and progenitor cells involved in craniofacial development during fetal life, especially in humans. Most of the insight into the mechanisms by which trisomy 21 causes the craniofacial defects in DS has relied on animal models. Using detailed imaging, Richtsmeier et al 114 have shown that the characteristic craniofacial defects in individuals with DS are closely mimicked early in development in Ts65Dn mice and, to a slightly lesser extent, in Ts1Cje mice 115. In particular, there is hypoplasia of the mandible and mid-facial skeleton 114, structures which in normal mouse development are known to be derived from cells which migrate from the cranial neural crest to populate the craniofacial precursors of the mid and lower face [reviewed in 116]. Using Ts65Dn mice crossed to mice expressing lacZ under the control of the Wnt1 promoter, Roper et al have made a number of important observations about mechanism of these DS-associated craniofacial abnormalities. First, they demonstrated that the number of neural crest cells was significantly reduced in trisomic compared to control (euploid) embryos 117. Second, they showed that this was due both to reduced generation of neural crest cells and to impaired migration into the first pharyngeal arch (PA1), which goes on to form the maxilla and lower jaw. They then found that in vitro proliferation of these trisomic PA1-derived cells in short-term culture was reduced compared to euploid controls. The defect in proliferation of the PA1 cells was partially rescued by addition of the mitogen Sonic Hedgehog (Shh), suggesting that the defects in neural crest generation and proliferation might be due, at least in part, to impaired Shh responsiveness 117. This is interesting because, as discussed below, abnormal Shh signaling is also implicated in the reduced proliferation of cerebellar granule precursors in the Ts65Dn mouse, although whether this is directly or indirectly linked to a specific trisomic gene(s) is not yet clear 118,119.
To identify genes and pathways which underlie the defects in PA1 neural crest cells, Billingsley et al 120 isolated mandibular precursor cells from embryonic day 13.5 (E13.5) Ts65Dn mice and compared their gene expression with the same cell population isolated from euploid controls using microarray. Of the relatively small number of differentially expressed genes, 20 contained homeobox DNA-binding domains, including increased expression of at least two genes (EN2 and OTX2) reported to have a role in mandibular development 121,122, reduced expression of all 12 of the differentially expressed HOX genes and a modest increase (1.2-fold) in expression of SOX9, known to be important for normal skeletal development 120. The extent to which these changes in gene expression are linked to the craniofacial defects in Ts65Dn mice and how they are linked to trisomy is an intriguing puzzle which remains to be investigated particularly given that expression of Ts65Dn trisomic genes in the mandibular precursor cells was not increased compared to euploid controls.
The most studied candidate genes on Hsa21 linked to the craniofacial abnormalities in DS are DYRK1A, RCAN1 (DSCR1) and ETS2 94,123,124,125. Arron et al noted the similarity between the craniofacial defects in calcineurin-deficient and Nfatc-deficient mice and those seen in DS and went on to show that DYRK1A and RCAN1 can act synergistically to prevent activation of NFATc-target genes and would therefore be plausible mediators of the craniofacial defects in DS. However, the role of DYRK1A and RCAN1 in craniofacial development was not directly addressed in this study, and therefore, the extent to which perturbed NFATc-signaling due to increased DYRK1A/RCAN1 expression contributes to craniofacial defects in DS remains unclear 94. Studies in DS mouse models have more directly addressed the role of DYRK1A, RCAN1 and of ETS2 in the craniofacial defects 112,123,126,127,128. Taken together, these studies suggest that trisomy of each of these genes individually is insufficient to cause the characteristic DS-associated craniofacial phenotype.
It is clear that interpreting the impact on craniofacial development of differences in expression of individual genes in DS mouse models is extremely difficult and needs to take into account differing mouse genetic backgrounds, as well as developmental stage, cellular context and interactions between other trisomic and non-trisomic genes 23. As in primary human fetal hematopoietic cells, perturbation of craniofacial development by trisomy 21 may be largely mediated via non-trisomic genes and/or by small changes in the level of expression of multiple trisomic genes which are difficult to detect using standard methods. Nevertheless, the close match between the Ts65Dn mouse and human phenotype, the ability to alter copy number of individual genes or groups of genes in this model and the identification of the relevant stem/progenitor cells now provide crucial tools to investigate candidate genes in DS craniofacial defects and the mechanisms by which they are linked to trisomy 21.
Abnormalities of brain structure and function
Studies in individuals with DS and in DS mouse models indicate that intellectual disability in DS is directly related to impaired development of many areas of the brain, including the cerebellum, the visual, auditory and somatosensory cortex, the motor cortex and the superior temporal gyrus 129. Many of the available DS mouse models recapitulate the structural and functional brain abnormalities of human DS very closely [reviewed in 24,32,39,130]. Here, we briefly discuss recent studies which have investigated the cellular and genomic basis for these defects.
The most consistent finding, both in DS animal models and in primary human samples, is of reduced cell numbers in several specific areas of the brain 119,126,129,130,131,132,133,134,135,136. In second-trimester human DS fetal brain, a number of studies have shown that total cell numbers are reduced in the hippocampus, dentate gyrus, parahippocampal gyrus 131,132 and cerebellum 133. Importantly, there is a particular reduction in neuronal precursor cells while astrocytic cells are preserved 132. Assessment of the proliferative status of these cells using immunohistochemical staining for the cell cycle-associated marker Ki-67 suggests that there are fewer proliferating cells in these regions of the brain in DS samples 131,132,133 compared to controls together with a higher frequency of apoptotic cell death in some areas 132. Detailed functional studies and characterization of the stem and progenitor cells populations have not yet proved possible in these tissues. However, the findings suggest that trisomy 21 causes impaired neurogenesis in DS from early in fetal development and may also affect cell fate specification (from neurones to astrocytes).
Studies in DS mouse models support the observations in human brain. Several groups have shown that neurogenesis is impaired in several areas of the brain including the hippocampus, neocortex, dentate gyrus and cerebellum and that many of these changes begin during fetal or early postnatal development 118,119,129,134,135,136. Histological studies in Ts65Dn mice indicate that there are reduced numbers of mitotic cells compared to euploid mice 119,135, and more recently, administration of BrdU confirms reduced proliferation of cells in the same areas of the brain 137. There is good evidence, both from in vivo studies and in vitro culture of neural precursor cells 118,129, of altered cell fate specification as a result of which, as in human fetal brain, the reduction in neurogenesis is accompanied by an increase in astrogliogenesis 127,131,132,133,136,137,138.
Insight into mechanisms of impaired neurogenesis has come mainly from investigation of pathways known to be important for normal neurogenesis, such as Shh, and from specific investigation of candidate genes on Hsa21, including DYRK1A, RCAN1, GRIK2 and APP 118,139,140. In particular, recent studies report dramatic improvements in neurogenesis in response to pharmacological agents, thereby implicating defects in the pathways they target in the pathogenesis of the cognitive defects in DS 141,142,143.
Several lines of evidence link abnormalities in the Shh pathway to the defects in neurogenesis in DS. First, cerebellar granule cell precursors isolated from the Ts65Dn DS mouse model have reduced in vitro responsiveness to Shh 119, which is known to be a potent mitogen for normal granule cell precursors 144. Second, administration of a Shh agonist (SAG-1) to neonatal Ts65Dn mice restores cerebellar development to normal in adult mice and improves learning and memory 141, supporting a significant role for the Shh pathway in the pathogenesis of DS-associated cognitive defects. These data are particularly interesting given the putative role of Shh in the craniofacial defects in DS 117,118. Although no direct link between Shh and trisomy 21 was established in those studies, clues to the role of trisomy 21 may lie with studies into the role of the Hsa21 gene APP. Triplication of APP in Ts65Dn impairs neuron precursor proliferation, differentiation and maturation 118,129. These effects are dependent upon the APP intracellular domain (AICD), and increased levels of AICD lead to increased transcription of the Ptch gene, leading to dysregulation of the Shh pathway 118. AICD may also be involved in another pathway important in the impaired neurogenesis in Ts65Dn mice by directly interacting with, and increasing the activity of, glycogen synthase kinase 3b (GSK3β), a key negative regulator of neuron proliferation, differentiation, maturation and migration 145. Trazzi et al 118 recently showed that treatment of Ts65Dn mice with lithium, a GSK3β inhibitor, normalized neural precursor proliferation, cell fate specification and maturation, suggesting that dysregulation of the GSK3β signaling pathway, potentially by the AICD of APP, also plays a significant role in the impaired neurogenesis typical of DS. Interestingly, fluoxetine, a 5-HT1A receptor agonist, has also recently been shown to improve neurogenesis in the Ts65Dn mouse model. Fluoxetine increased total and proliferating neural progenitor cells, corrected defective 5-HT1A receptor expression and rescued defects in contextual memory and behavior typical of DS both in fetal 142,143 and adult Ts65Dn mice 137,143,146. These responses may be due to direct effects of fluoxetine on the serotoninergic system. However, as Trazzi et al 143 showed that activation of 5-HT1A receptors by fluoxetine inhibits GSK3β, this would provide a mechanistic link to Hsa21 (inhibition of the APP-driven increase in GSK3β activity) for the beneficial effects of fluoxetine on neurogenesis, behavior and memory in DS.
Comparison of the defects in Ts65Dn and Ts1Rhr mice (Table2), which are trisomic for only 33 Hsa21 orthologs, with a mouse model monosomic for these genes (Ms1Rhr) identified DYRK1A, GIRK2 and SIM2 as necessary, but not sufficient, for hippocampal-based learning deficits in Ts65Dn 140. Several lines of evidence support a role for increased expression of DYRK1A in DS-associated cognitive defects 125,139,147,148,149,150,151. In disomic transgenic mice, overexpressing DYRK1A by 1.5–2-fold in cortical neurons leads to impaired neural progenitor differentiation and motor and cognitive defects 125 which are ameliorated by selective DYRK1A knockdown 151. More recently, Altafaj et al 139 showed that in vivo knockdown of DYRK1a to normal levels in trisomic mice (Ts65Dn) by shRNA also rescues functional and behavioral defects in these mice consistent with a role for increased DYRK1A in their pathogenesis. These data are supported by Hibaoui et al 152 who reported that impaired neural differentiation of trisomy 21 iPSC was rescued by a selective DYRK1A inhibitor. The exact mechanism(s) by which DYRK1A affects neurogenesis and neuronal differentiation is not yet clear but includes DYRK1A-mediated de-regulation of the master regulator of neuronal differentiation NRSF/REST 153 and inhibition of choline acetyltransferase induction 154.
Cardiac abnormalities
Both Ts65Dn and Tc1 mice (Table2) exhibit heart defects similar to those observed in DS, suggesting that trisomy of one or more of the ∼100 genes common to these models may be responsible for the cardiac defects in DS 30,155,156. Indeed, a recent study has reported that the smallest critical region in a mouse model (Dp(16)4Yeh/+) associated with cardiac defects, including atrial and ventricular septal defects, can be reduced to a 3.7-Mb region containing 35 genes 157. Although elegant experiments in transgenic mice provide a guide to the regions of Hsa21 likely to be critical for cardiac defects in individuals with DS, the frequency of these defects in DS mouse models 30,156 is considerably lower (5–15%) than in humans (40–50%; Table1), suggesting a very complex etiology involving multiple Hsa21 and non-Hsa21 genes.
In comparison with studies in hematopoietic, craniofacial and brain tissue, little is known about the cellular and molecular basis for these defects in cardiac development. Using BrdU labeling, Fuchs et al 158 found that neonatal Ts65Dn mice had fewer proliferating cells in the left and right heart walls and septum compared to euploid mice. Interestingly, in the same experiments, they also found reduced numbers of BrdU-positive cells in the intestine, liver and skin in Ts65Dn mice compared to euploid mice, supporting the contention that trisomy 21 impairs the proliferation of progenitor cells of a wide variety of non-hematopoietic tissues during development. The mechanism(s) is not yet clear. Limited gene expression studies of whole human fetal cardiac tissue suggest that dose-dependent upregulation of Hsa21 genes might alter the expression of mitochondrial function genes although this does not explain why cardiac defects affect only half of DS individuals 159. However, progress in understanding the genetic basis of congenital heart defects, such as AVSD, in the absence of trisomy 21, provides some clues to the potential pathways in DS, including HOX genes 160,161, the Shh pathway 162,163, the VEGF pathway 164 and a number of chromatin remodeling genes, including MLL2 and CHD7 [reviewed in 165]. Interestingly, Ackerman et al 166 recently used a candidate gene resequencing approach to identify potentially damaging variants in six genes at approximately sevenfold higher frequency in DS individuals with AVSD, including 2 Hsa21 genes (COL6A1, COL6A2) and one gene involved in Wnt signaling (FRZB); all six genes identified in this study are implicated in VEGF-A signaling known to be important for normal heart septation 164. The recent refinements in techniques to generate cardiomyocytes and cardiac progenitors in vitro from ESC and iPSC 167 have provided insight into developmental heart defects 168 and may also prove a useful approach for further investigating the mechanisms of defects in cardiac development in DS at the cellular and molecular level 169.
Genomic basis for phenotypic variation in DS
Current estimates identify 243 protein-coding genes on Hsa21 as well as 259 long non-coding RNAs and 138 short non-coding RNAs 5. There is general consensus that changes in the pattern and level of expression of one or more Hsa21 genes are responsible, directly or indirectly, for the abnormalities in stem/progenitor cell function and, ultimately, for the clinical features of DS. The conventional view is that most of these features occur due to imbalanced dosage of Hsa21 genes. However, increasing evidence indicates that the genetic landscape is far more complex than can be accounted for by a simple dosage effect and that trisomy 21 exerts its effects in stem and progenitor cells in several ways.
First, for some Hsa21 genes (probably a minority), mouse models show that in some tissues, increased expression of a single gene is sufficient to produce changes in stem/progenitor cell behavior which correlate with clinical phenotypic read outs, such as leukemia (ERG, DYRK1A, HMGN1 or miR125b) or cognitive defects (APP) 51,91,96,97,98,118,129. Conclusive evidence for a simple dosage effect of individual Hsa21 genes in human stem/progenitor cells is lacking at present. A more likely scenario is that several Hsa21 genes, either acting in a common pathway (e.g., NFAT or Wnt signaling) or independently, cause the phenotypic effects in DS 94,170,95. Interestingly, Emmrich et al 95 demonstrated one mechanism by which increased expression of a group of Hsa21 genes might affect several target genes and alter HSPC behavior. They showed that coordinated expression of 3 Hsa21 miRs as a miR99a/let7c/miR125b tricistron by lentiviral transduction of cord blood CD34+ HSPC caused expansion of megakaryocyte progenitors and modulation of target genes in the TGFβ and Wnt signaling pathways.
In human tissues, microarray has been used to identify differential expression patterns in trisomic versus euploid cells. Interestingly, in almost all gene expression datasets, only a small minority of the 243 Hsa21 protein-coding genes are significantly differentially expressed (Fig3; Supplementary Tables S1 and S2). The low number of differentially expressed Hsa21 genes may be due to limited sensitivity of microarray to detect the small changes expression (1.2- to 1.8-fold) expected in trisomic cells [discussed in 92]. Such changes might also be masked by inter-individual variation in expression of Hsa21 genes, as seen for non-Hsa21 genes 93. Nevertheless, in principle, small changes in expression of multiple Hsa21 genes, each of which is not identified as statistically significant, may still cause critical dysregulation of stem/progenitor cell function. Figure4 illustrates simple scenarios by which trisomy 21 may lead to a range of possible effects on gene expression and protein production (even without taking into account the impact of epigenetic mechanisms and interindividual variation). Mathematical modeling, supported by experimental evidence 171,172, shows that three dosage effects on gene expression are commonly found in aneuploids: a direct transacting effect, an inverse transacting effect and gene dosage compensation 171,173. While the direct effect would result in a 1.5-fold increase in expression of genes on the trisomic chromosome, inverse transacting effects (where a gene on the trisomic chromosome regulates a gene on another chromosome) account for otherwise unexpected reductions in gene expression below the normal euploid level. On the other hand, dosage compensation arises when direct and inverse effects are counter-balanced, for example, if a gene on the trisomic chromosome regulates another gene on the same chromosome. Thus, dosage compensation (‘buffering’) may explain, at least in part, the small number of significantly differentially expressed genes in mouse and human trisomy 21 datasets.
Figure 3. Differential expression of Hsa21 genes and non-Hsa21 genes in hematopoietic and non-hematopoietic cells.
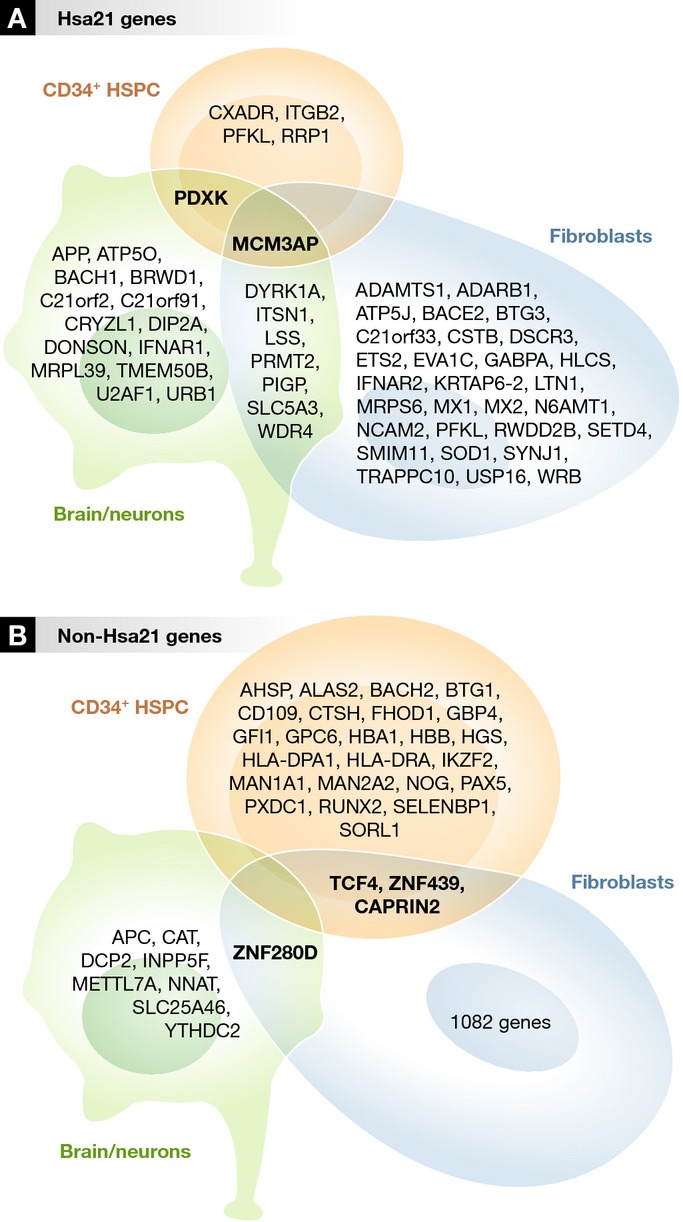
Human Gene Expression Profiling (GEP) datasets from public repositories were extracted, and differential expression analysis was performed using the limma algorithm (see Supplementary Tables S1 and S2). The data analyzed here are from references 187,189,190. In order to compare gene expression by these non-hematopoietic cells with non-malignant hematopoietic cells, we used our own unpublished gene expression data from human trisomy 21 (n = 4) and normal (n = 3) second-trimester fetal liver CD34+ HSPC samples obtained using the Affymetrix HuEX_1-0_st array. An FDR cutoff of 0.15 was used. There was almost no overlap between the differentially expressed genes in different tissue types: Only a single Hsa21 gene, MCM3AP (minichromosome maintenance complex component 3-associated protein) (A), and no non-Hsa21 genes were differentially expressed in both hematopoietic and non-hematopoietic cells (B).
Figure 4. Models predicting the consequence of increased gene dosage due to trisomy in stem and progenitor cells.

This shows various scenarios by which increasing gene dosage to 150% (as in trisomy) may cause a range of effects at the transcriptional and post-transcriptional level which lead to stoichiometric imbalances which may ‘buffer’ the effect of dosage imbalance of trisomic genes. Trisomy 21 is used as an example. For simplicity, these models do not take into account the additional impact of epigenetic mechanisms and interindividual variation. (A) Effects at the transcriptional level. Left: A gene on Hsa21 (red) is regulated by a transcription factor (blue circle) present in a limited and similar amount in normal and trisomy 21 cells. In this case, transcription is the limiting factor: The additional gene will have no impact on the amount of mRNA produced, and the overall level of expression of the gene is the same in both trisomy 21 and normal (euploid) cells. Right: A gene on Hsa21 (red) is co-regulated by a gene on another chromosome (green) by a transcription factor (blue circle) which is present in limited amounts. The total quantity of mRNA produced is still constrained by the limited quantity of transcription factor which must now be shared between expression of five genes rather than four. The expression of the three copies of the (red) Hsa21 gene will now consume 3/5 of the transcription factor (compared to 2/4 in the euploid state); therefore a scaling of (3/5) / (2/4) = 3 × 4 / (2 × 5) = 12/10 = 120%. The expression of the two copies of the (green) non-Hsa21 gene will, instead, consume 2/5 instead of 2/4; therefore a scaling of (2/5) / (2/4) = 4 × 2 / (5 × 2) = 80%. Thus, in this case, the additional copy of Hsa21 causes only a small increase in expression of the Hsa21 gene (to 120%), and this is matched by a decrease in expression (to 80%) of the other, non-Hsa21 gene. (B) Effects at the post-transcriptional level. Buffering effects at the protein level are related to the formation of a complex of proteins. Two examples are illustrated here. Left: A complex AB is formed by a protein A (dark blue circle) and a protein B (light blue circle). If the amount of protein B is increased by a factor of 1.5, but the amount of protein A remains constant, the number of AB complexes will not be increased above the normal level. Right: A complex ABA is formed by a ratio of 2 monomers of protein A and one monomer of protein B through intermediate complexes (AB or BA). When the amount of protein A is exactly twice the amount of protein B, all the proteins are used to form the complexes ABA. However, a 1.5-fold increase in the amount of protein B may lead to a decrease of the amount of ABA complexes since the production of the intermediate AB and BA complexes cannot be completed due to an insufficient amount of A monomers. In addition, as illustrated in the top left of the figure, buffering effects at the protein level may also influence the level of gene expression if the protein complex is itself involved in transcriptional regulation.
Second, gene expression studies in human DS tissues (Supplementary Tables S1 and S2) 61,71,72,90,93,95,100,102,152,159,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191, as well as DS mouse models, show extensive dysregulation of non-trisomic (euploid) genes as well as trisomic genes in DS (Fig3), which are linked to DS-specific phenotypes in DS mouse models 35,93,191,192. In a recent fascinating study in fibroblasts and iPSC from a unique set of monozygotic twins where one twin had trisomy 21 and the other did not (as a result of abnormal chromosome segregation prior to twinning), Letourneau et al 93 reported changes in gene expression across every chromosome. Furthermore, they found a consistent pattern of alternating regions of increased and decreased gene expression across large chromosomal segments which they called ‘gene expression dysregulation domains’ (GEDDs). Remarkably, GEDDs with increased expression in trisomic cells corresponded to areas which were normally repressed, while GEDDs with decreased expression corresponded to areas where transcription would normally be active. The mechanism(s) by which the chromatin environment is altered in this way by trisomy 21 has not yet been identified. No comparable data exist for other human trisomies, and so, it is also possible that these effects are due to the physical presence of an additional chromosome in the nucleus rather than specific to Hsa21. It also remains to be seen whether such altered global gene expression patterns are also seen in stem and progenitor cells from other tissues (e.g., HSPC) in individuals with DS.
Since the effects of trisomy 21 on stem/progenitor cells vary depending on the cellular context and stage of development, it is likely that the gene expression patterns responsible for these effects will also vary in different cell types. To address whether there was any overlap in differentially expressed genes between different tissue types in DS, we performed differential expression analysis of non-hematopoietic microarray datasets (Supplementary Tables S1 and S2) with our own gene expression dataset of primary fetal liver CD34+ HSPC. We found almost no overlap between the differentially expressed genes in different tissue types (Fig3). Indeed, no non-Hsa21 genes and only a single Hsa21 gene, MCM3AP, were differentially expressed in both hematopoietic and non-hematopoietic cells. Although MCM3AP may be of interest since it is essential for initiation of DNA replication and mutations in families with inherited intellectual disability have been reported 193, the impact of increased levels of expression on hematopoietic and non-hematopoietic cells is unknown.
Sidebar A:In need of answers.
The molecular mechanism by which gene dosage is regulated in a developmental stage- and tissue-specific context in trisomic stem and progenitor cells
Reasons for the variable penetrance of abnormal phenotypes seen in individuals with aneuploidy
The role of non-coding RNAs in the abnormalities of hematopoietic and non-hematopoietic stem and progenitor cells in human trisomies
Mechanisms by which epigenetic changes are driven by trisomic genes; either directly or via their effects on other (disomic) genes
Conclusion
There is increasing recognition that trisomy 21 impacts on stem and progenitor cell function in many different ways. These changes in stem/progenitor cell behavior reflect adaptive epigenetic, transcriptional and/or translational regulatory mechanisms which allow cells to survive and function despite the presence of an additional copy of an entire chromosome. The effects of trisomy 21 on stem and progenitor cells are cell context dependent and developmental stage specific (Fig1). During fetal and embryonic life, proliferation of hematopoietic and testicular stem and progenitor cells is increased and coupled with altered differentiation, which may underlie the unique susceptibility of individuals with DS to tumors of these two cell types. By contrast, the effects of trisomy 21 on progenitor cells of other lineages (e.g., cardiac, neural and intestinal) during early development are manifested mainly as impaired, rather than enhanced, cell proliferation which may protect these cells from subsequent malignant transformation and explain the reduced frequency of non-hematopoietic cancers in DS. Finally, trisomy 21-mediated premature aging of stem/progenitor cells may contribute to the phenotypic abnormalities in many tissue types, particularly in adults with DS. Although changes in the pattern and level of expression of one or more of these genes on Hsa21 are likely to be responsible, for the abnormalities in stem/progenitor cell function and, ultimately, for the clinical features of DS, increasing evidence indicates that these effects are mediated by complex genetic and epigenetic mechanisms beyond increased Hsa21 gene dosage. Uncovering the molecular mechanisms underpinning these defects in stem and progenitor cell function remains an exciting challenge and is at last beginning to offer real prospects of translation of these finding into useful therapeutic advances for individuals with DS.
Acknowledgments
The authors would like to thank members of the Roberts and Karadimitris labs (especially Tassos Karadimitris, Centre for Haematology, Imperial College London, UK), Professor Michael Stumpf (Centre for Bioinformatics, Division of Molecular Biosciences, Imperial College London, UK) and Professor Paresh Vyas (University of Oxford, UK) and members of his laboratory (especially Kelly Perkins) for helpful discussions over many years. We would also like to thank Professor Vaskar Saha and his laboratory (University of Manchester, UK) and Dr. Ollie Tunstall for help with the microarray experiments in human fetal CD34+ cells. This work was supported by Leukaemia Lymphoma Research, Children with Cancer and the Medical Research Council.
Glossary
- AICD
APP intracellular domain
- ALL
acute lymphoblastic leukemia
- AVSD
atrioventricular septal defect
- BCP
B-cell progenitor
- DS
Down syndrome
- FDR
false discovery rate
- GEDD
gene expression dysregulation domain
- GEP
gene expression profile
- GMP
granulocyte–monocyte progenitor
- GSK3b
glycogen synthase kinase 3b
- hESC
human embryonic stem cell
- Hsa21
Homo sapiens chromosome 21
- HSC
hematopoietic stem cell
- HSPC
hematopoietic stem and progenitor cell
- IGF
insulin-like growth factor
- iPSC
induced pluripotent stem cell
- MEP
megakaryocyte–erythroid progenitor
- miR
microRNA
- ML-DS
myeloid leukemia of Down syndrome
- Mmu
Mus musculus
- NFAT
nuclear factor of activated T cells
- qPCR
quantitative polymerase chain reaction
- Shh
sonic hedgehog
- T21
trisomy 21
- TAM
transient abnormal myelopoiesis
- VEGF
vascular endothelial growth factor
Author contributions
BL, SF, AR and IR analyzed data and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Table S1
Supplementary Table S2
References
- Morris JK, Springett A. The National Down Syndrome Cytogenetic Register for England and Wales: 2012 Annual Report. London: Public Health England; 2014. [Google Scholar]
- Rasmussen SA, Wong LC, May KM, Friedman JM. Population-based analyses of mortality in trisomy 13 and trisomy 18. Pediatrics. 2003;111:777–784. doi: 10.1542/peds.111.4.777. [DOI] [PubMed] [Google Scholar]
- Nelson KE, Hexem KR, Feudtner C. Inpatient hospital care of children with trisomy 13 and trisomy 18 in the United States. Pediatrics. 2012;129:869–876. doi: 10.1542/peds.2011-2139. [DOI] [PubMed] [Google Scholar]
- Liang CA, Braddock BA, Heithaus JL, Christensen KM, Braddock SR, Carey JC. Reported communication ability of persons with trisomy 18 and trisomy 13. Dev Neurorehabil. 2013 doi: 10.3109/17518423.2013.847980. doi: 10.3109/17518423.2013.847980. [DOI] [PubMed] [Google Scholar]
- Wu J, Morris JK. The population prevalence of Down's syndrome in England and Wales in 2011. Eur J Hum Genet. 2013;21:1016–1019. doi: 10.1038/ejhg.2012.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis SE. 10 years of Genomics, chromosome 21, and Down syndrome. Genomics. 1998;51:1–16. doi: 10.1006/geno.1998.5335. [DOI] [PubMed] [Google Scholar]
- Ross JA, Spector LG, Robison LL, Olshan AF. Epidemiology of leukemia in children with Down syndrome. Pediatr Blood Cancer. 2005;44:8–12. doi: 10.1002/pbc.20165. [DOI] [PubMed] [Google Scholar]
- Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet. 2004;5:725–738. doi: 10.1038/nrg1448. [DOI] [PubMed] [Google Scholar]
- Roper RJ, Reeves RH. Understanding the basis for Down syndrome phenotypes. PLoS Genet. 2006;2:e50. doi: 10.1371/journal.pgen.0020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull MJ the Committee on Genetics. Health supervision for children with Down syndrome. Pediatrics. 2011;128:393–406. doi: 10.1542/peds.2011-1605. [DOI] [PubMed] [Google Scholar]
- Roberts I, Alford K, Hall G, Juban G, Richmond H, Norton A, Vallance G, Perkins K, Marchi E, McGowan S, et al. GATA1-mutant clones are frequent and often unsuspected in babies with Down syndrome: identification of a population at risk of leukemia. Blood. 2013;122:3908–3917. doi: 10.1182/blood-2013-07-515148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigman WB, Schupf N, Sersen E, Silverman W. Prevalence of dementia in adults with and without Down syndrome. Am J Ment Retard. 1996;100:403–412. [PubMed] [Google Scholar]
- Sekijima Y, Ikeda S, Tokuda T, Satoh S, Hidaka H, Hidaka E, Ishikawa M, Yanagisawa N. Prevalence of dementia of Alzheimer type and apolipoprotein E phenotypes in aged patients with Down's syndrome. Eur Neurol. 1998;39:234–237. doi: 10.1159/000007940. [DOI] [PubMed] [Google Scholar]
- Holland AJ, Hon J, Huppert FA, Stevens F. Incidence and course of dementia in people with Down's syndrome: findings from a population-based study. J Intellect Disabil Res. 2000;44:138–146. doi: 10.1046/j.1365-2788.2000.00263.x. [DOI] [PubMed] [Google Scholar]
- Tyrrell J, Cosgrave M, McCarron M, McPherson J, Calvert J, Kelly A, McLaughlin M, Gill M, Lawlor BA. Dementia in people with Down's syndrome. Int J Geriatr Psychiatry. 2001;16:1168–1174. doi: 10.1002/gps.502. [DOI] [PubMed] [Google Scholar]
- Coppus A, Evenhuis H, Verberne GJ, Visser F, van Gool P, Eikelenboom P, van Duijn C. Dementia and mortality in persons with Down's syndrome. J Intellect Disabil Res. 2006;50:768–777. doi: 10.1111/j.1365-2788.2006.00842.x. [DOI] [PubMed] [Google Scholar]
- Lott IT. Neurological phenotypes for Down syndrome across the life span. Prog Brain Res. 2012;197:101–121. doi: 10.1016/B978-0-444-54299-1.00006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasson EJ, Dye DE, Bittles AH. The triple challenges associated with age-related comorbidities in Down syndrome. J Intellect Disabil Res. 2014;58:393–398. doi: 10.1111/jir.12026. [DOI] [PubMed] [Google Scholar]
- Hartley SL, Handen BL, Devenney DA, Hardison R, Mihaila I, Proce JC, Cohen AD, Klunk WE, Mailick MR, Johnson SC, et al. Cognitive functioning in relation to brain amyloid-β in healthy adults with Down syndrome. Brain. 2014;137:2556–2563. doi: 10.1093/brain/awu173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adorno M, Sikandar S, Mitra SS, Kuo A, Nicolis Di Robilant B, Haro-Acosta V, Ouadah Y, Quarta M, Rodriguez J, Qian D, et al. Usp16 contributes to somatic stem-cell defects in Down's syndrome. Nature. 2013;501:380–384. doi: 10.1038/nature12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts I, Izraeli S. Haematopoietic development and leukaemia in Down syndrome. Br J Haematol. 2014;167:587–599. doi: 10.1111/bjh.13096. [DOI] [PubMed] [Google Scholar]
- Nizetic D, Groet J. Tumorigenesis in Down's syndrome: big lessons from a small chromosome. Nat Rev Cancer. 2012;12:721–732. doi: 10.1038/nrc3355. [DOI] [PubMed] [Google Scholar]
- Deitz SL, Roper RJ. Trisomic and allelic differences influence phenotypic variability during development of Down syndrome mice. Genetics. 2011;189:1487–1495. doi: 10.1534/genetics.111.131391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruparelia A, Pearn ML, Mobley WC. Aging and intellectual disability: insights from mouse models of Down syndrome. Dev Disabil Res Rev. 2013;18:43–50. doi: 10.1002/ddrr.1127. [DOI] [PubMed] [Google Scholar]
- O'Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, Sesay A, Modino S, Vanes L, Hernandez D, et al. An aneuploid mouse strain carrying human chromosome 21 with down syndrome phenotypes. Science. 2005;309:2033–2037. doi: 10.1126/science.1114535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble SM, Wiseman FK, Clayton S, Prigmore E, Langley E, Yang F, Maguire S, Fu B, Rajan D, Sheppard O, et al. Massively parallel sequencing reveals the complex structure of an irradiated human chromosome on a mouse background in the Tc1 model of Down syndrome. PLoS ONE. 2013;8:e60482. doi: 10.1371/journal.pone.0060482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Yu T, Morishima M, Pao A, LaDuca J, Conroy J, Nowak N, Matsui S, Shiraishi I, Yu YE. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum Mol Genet. 2007;16:1359–1366. doi: 10.1093/hmg/ddm086. [DOI] [PubMed] [Google Scholar]
- Yu T, Li Z, Jia Z, Clapcote SJ, Liu C, Li S, Asrar S, Pao A, Chen R, Fan N, et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet. 2010;19:2780–2791. doi: 10.1093/hmg/ddq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Liu C, Belichenko P, Clapcote SJ, Li S, Pao A, Kleschevnikov A, Bechard AR, Asrar S, Chen R, et al. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res. 2012;1366:162–171. doi: 10.1016/j.brainres.2010.09.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveau M, Lignon JM, Naleso V, Duchon A, Groner Y, Sharp AJ, Dembele D, Brault V, Herault Y. The App-Runx1 region is critical for birth defects and electrocardiographic dysfunctions observed in a Down syndrome mouse model. PLoS Genet. 2012;8:e1002724. doi: 10.1371/journal.pgen.1002724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes Pereira P, Magnol L, Sahun I, Brault V, Duchon A, Prandini P, Gruart A, Bizot J-C, Chadefaux-Vekemans B, Deutsch S, et al. A new mouse model for the trisomy of the Abcg1-U2af1 region reveals the complexity of the combinatorial genetic code of down syndrome. Hum Mol Genet. 2009;18:4756–4769. doi: 10.1093/hmg/ddp438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahun I, Marechal D, Pereira PL, Nalesso V, Gruart A, Garcia JMD, Antonarakis SE, Dierssen M, Herault Y. Cognition and hippocampal plasticity in the mouse is altered by monosomy of a genomic region implicated in Down syndrome. Genetics. 2014;197:899–912. doi: 10.1534/genetics.114.165241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS, Roper RJ. The power of comparative and developmental studies for mouse models of Down syndrome. Mamm Genome. 2007;18:431–443. doi: 10.1007/s00335-007-9030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman FK, Alford KA, Tybulewicz VL, Fisher EM. Down syndrome- recent progress and future prospects. Hum Mol Genet. 2009;18:R75–R83. doi: 10.1093/hmg/ddp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana-Elola E, Watson-Scales SD, Fisher EM, Tybulewicz VL. Down syndrome: searching for the genetic culprits. Dis Model Mech. 2011;4:586–595. doi: 10.1242/dmm.008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das I, Reeves RH. The use of mouse models to understand and improve cognitive deficits in Down syndrome. Dis Model Mech. 2011;4:596–606. doi: 10.1242/dmm.007716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchon A, Raveau M, Chevalier C, Nalesso V, Sharp AJ, Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling Down syndrome. Mamm Genome. 2011;22:674–684. doi: 10.1007/s00335-011-9356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinholdt LG, Ding Y, Gilbert GJ, Czechanski A, Solzak JP, Roper RJ, Johnson MT, Donahue LR, Lutz C, Davisson MT. Molecular characterization of the translocation breakpoints in the Down syndrome mouse model Ts65Dn. Mamm Genome. 2011;22:685–691. doi: 10.1007/s00335-011-9357-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgin JO, Mason GM, Spanò G, Fernández A, Nadel L. Human and mouse cognitive phenotypes in Down syndrome: implications for assessment. Prog Brain Res. 2012;197:123–151. doi: 10.1016/B978-0-444-54299-1.00007-8. [DOI] [PubMed] [Google Scholar]
- Zhang L, Fu D, Belichenko PV, Liu C, Kleschevnikov AM, Pao A, Liang P, Clapcote SJ, Mobley WC, Yu YE. Genetic analysis of Down syndrome facilitated by mouse chromosome engineering. Bioeng Bugs. 2012;3:8–12. doi: 10.4161/bbug.3.1.17696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda N, Florez J, Martinez-Cue C. Mouse models of Down syndrome to unravel causes of mental disabilities. Neural Plast. 2012;2012:584071. doi: 10.1155/2012/584071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Morishima M, Jiang X, Yu T, Meng K, Kay D, Pao A, Ye P, Parmacek MS, Yu YE. Engineered chromosome-based genetic mapping establishes a 3.7 Mb critical genomic region for Down syndrome-associated heart defects in mice. Hum Genet. 2014;133:743–753. doi: 10.1007/s00439-013-1407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunstall-Pedoe O, Roy A, Karadimitris A, de la Fuente J, Fisk NM, Bennett P, Norton A, Vyas P, Roberts I. Abnormalities in the myeloid progenitor compartment in Down syndrome fetal liver precede acquisition of GATA1 mutations. Blood. 2008;112:4507–4511. doi: 10.1182/blood-2008-04-152967. [DOI] [PubMed] [Google Scholar]
- Chou ST, Opalinska JB, Yao Y, Fernandes MA, Kalota A, Brooks JSJ, Choi JK, Gewirtz AM, Danet-Desnoyers G-A, Nemiroff RL, et al. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood. 2008;112:4503–4506. doi: 10.1182/blood-2008-05-157859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Cowan G, Mead AJ, Filippi S, Bohn G, Chaidos A, Tunstall O, Chan JKY, Choolani M, Bennett P, et al. Perturbation of fetal liver hematopoietic stem and progenitor cell development by trisomy 21. Proc Natl Acad Sci USA. 2012;109:17579–17584. doi: 10.1073/pnas.1211405109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean GA, Menne TF, Guo G, Sanchez DJ, Park IH, Daley GQ, Orkin SH. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic induced pluripotent cells. Proc Natl Acad Sci USA. 2012;109:17567–17572. doi: 10.1073/pnas.1215468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou ST, Byrska-Bishop M, Tober JM, Yao Y, Vandorn D, Opalinska JB, Mills JA, Choi JK, Speck NA, Gadue P, et al. Trisomy 21-associated defects in human primitive hematopoiesis revealed through induced pluripotent stem cells. Proc Natl Acad Sci USA. 2012;109:17573–17578. doi: 10.1073/pnas.1211175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsammer G, Jilani S, Liu H, Davis E, Gurbuxani S, Le Beau MM, Crispino JD. Highly penetrant myeloproliferative disease in the Ts65Dn mouse model of Down syndrome. Blood. 2008;111:767–775. doi: 10.1182/blood-2007-04-085670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael CL, Majewski IJ, Alexander WS, Metcalf D, Hilton DJ, Hewitt CA, Scott HS. Hematopoietic defects in the Ts1Cje mouse model of Down syndrome. Blood. 2009;113:1929–1937. doi: 10.1182/blood-2008-06-161422. [DOI] [PubMed] [Google Scholar]
- Alford KA, Slender A, Vanes L, Li Z, Fisher EM, Nizetic D, Orkin SH, Roberts I, Tybulewicz VL. Perturbed hematopoiesis in the Tc1 mouse model of Down syndrome. Blood. 2010;115:2928–2937. doi: 10.1182/blood-2009-06-227629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, Crispino JD. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest. 2012;122:948–962. doi: 10.1172/JCI60455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vita S, Canzonetta C, Mullighan C, Delom F, Groet J, Baldo C, Vanes L, Dagna-Bricarelli F, Hoischen A, Veltman J, et al. Trisomic dose of several chromosome 21 genes perturbs haematopoietic stem and progenitor cell differentiation in Down's syndrome. Oncogene. 2010;29:6102–6114. doi: 10.1038/onc.2010.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down's syndrome. Lancet. 2000;355:165–169. doi: 10.1016/S0140-6736(99)05264-2. [DOI] [PubMed] [Google Scholar]
- Hasle H. Pattern of malignant disorders in individuals with Down's syndrome. Lancet Oncol. 2001;2:429–436. doi: 10.1016/S1470-2045(00)00435-6. [DOI] [PubMed] [Google Scholar]
- Hermon C, Alberman E, Beral V, Swerdlow AJ. Mortality and cancer in persons with Down's syndrome, their parents and siblings. Ann Hum Genet. 2001;65:167–176. doi: 10.1017/S0003480001008508. [DOI] [PubMed] [Google Scholar]
- Yang Q, Rasmussen SA, Friedman JM. Mortality associated with Down's syndrome in the USA from 1983 to 1997: a population-based study. Lancet. 2002;359:1019–1025. doi: 10.1016/s0140-6736(02)08092-3. [DOI] [PubMed] [Google Scholar]
- Hill DA, Gridely G, Cnattingius S, Mellemkjaer L, Linet M, Adami HQ, Olsen JH, Nyren O, Fraumeni JF., Jr Mortality and cancer incidence among individuals with Down syndrome. Arch Intern Med. 2003;163:705–711. doi: 10.1001/archinte.163.6.705. [DOI] [PubMed] [Google Scholar]
- Goldacre M, Wotton CJ, Seagroate V, Yeates D. Cancers and immune related diseases associated with Down's syndrome: a record linkage study. Arch Dis Child. 2004;89:1014–1017. doi: 10.1136/adc.2003.046219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patja K, Pukkala E, Sund R, Iivanainen M, Kaski M. Cancer incidence of persons with down syndrome in Finland: a population-based study. Int J Cancer. 2006;118:1769–1772. doi: 10.1002/ijc.21518. [DOI] [PubMed] [Google Scholar]
- Gilbert D, Rapley E, Shipley J. Testicular germ cell tumours: predisposition genes and the male germ cell nice. Nat Rev Cancer. 2011;11:278–288. doi: 10.1038/nrc3021. [DOI] [PubMed] [Google Scholar]
- Malinge S, Izraeli S, Crispino JD. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood. 2009;113:2619–2628. doi: 10.1182/blood-2008-11-163501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Roberts I, Vyas P. Biology and management of transient abnormal myelopoiesis (TAM) in children with Down syndrome. Semin Fetal Neonatal Med. 2012b;17:196–201. doi: 10.1016/j.siny.2012.02.010. [DOI] [PubMed] [Google Scholar]
- Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, Crispino JD. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002;32:148–152. doi: 10.1038/ng955. [DOI] [PubMed] [Google Scholar]
- Rainis L, Bercovich D, Strehl S, Teigler-Schlegel A, Stark B, Trka J, Amariglio N, Biondi A, Muler I, Rechavi G, et al. Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood. 2003;102:981–986. doi: 10.1182/blood-2002-11-3599. [DOI] [PubMed] [Google Scholar]
- Hitzler JK, Cheung J, Li Y, Scherer SW, Zipursky A. GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood. 2003;101:4301–4304. doi: 10.1182/blood-2003-01-0013. [DOI] [PubMed] [Google Scholar]
- Groet J, McElwaine S, Spinelli M, Rinaldi A, Burtscher I, Mulligan C, Mensah A, Cavani S, Dagna-Bricarelli F, Basso G, et al. Acquired mutations in GATA1 in neonates with Down's syndrome with transient myeloid disorder. Lancet. 2003;361:1617–1620. doi: 10.1016/S0140-6736(03)13266-7. [DOI] [PubMed] [Google Scholar]
- Ahmed M, Sternberg A, Hall G, Thomas A, Smith O, O'Marcaigh A, Wynn R, Stevens R, Addison M, King D, et al. Natural history of GATA1 mutations in Down syndrome. Blood. 2004;103:2480–2489. doi: 10.1182/blood-2003-10-3383. [DOI] [PubMed] [Google Scholar]
- Hollanda LM, Lima CSP, Cunha AF, Albuquerque DM, Vassallo J, Ozelo MC, Joazeiro PP, Saad STO, Costa FF. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet. 2006;38:807–812. doi: 10.1038/ng1825. [DOI] [PubMed] [Google Scholar]
- Buitenkamp TD, Izraeli S, Zimmermann M, Forestier E, Heerema NA, van den Heuvel-Eibrink MM, Pieters R, Korbijn CM, Silverman LB, Schmiegelow K, et al. Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood. 2014;123:70–77. doi: 10.1182/blood-2013-06-509463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, Shochat C, Cazzaniga G, Biondi A, Basso G, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet. 2008;372:1484–1492. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- Hertzberg L, Vendramini E, Ganmore I, Cazzaniga G, Schmitz M, Chalker J, Shiloh R, Iacobucci I, Shochat C, Zeligson S, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood. 2010;115:1006–1017. doi: 10.1182/blood-2009-08-235408. [DOI] [PubMed] [Google Scholar]
- Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, Ma J, Coustan-Smith E, Harvey RC, Willman CL, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41:1243–1246. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell LJ, Capasso M, Vater I, Akasaka T, Bernard OA, Calasanz MJ, Chandrasekaran T, Chapiro E, Gesk S, Griffiths M, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114:2688–2698. doi: 10.1182/blood-2009-03-208397. [DOI] [PubMed] [Google Scholar]
- Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, te Kronnie G, Cario G, Cazzaniga G, Kulozik AE, Stanulla M, et al. Gain-of-function mutations in interleukin-7 receptor-{alpha} (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med. 2011;208:901–908. doi: 10.1084/jem.20110580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal N, Shochat C, Geron I, Bercovich D, Izraeli S. Interleukin 7 and thymic stromal lymphopoietin: from immunity to leukemia. Cell Mol Life Sci. 2014;71:365–378. doi: 10.1007/s00018-013-1337-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev SI, Garieri M, Santoni F, Falconnet E, Ribaux P, Guipponi M, Murray A, Groet J, Giarin E, Basso G, et al. Frequent cases of Ras-mutated Down syndrome acute lymphoblastic leukaemia lack JAK2 mutations. Nat Commun. 2014;5:4654–4659. doi: 10.1038/ncomms5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher VP. Screening of medical problems in adults with Down syndrome. Downs Syndr Res Pract. 1994;2:59–66. [Google Scholar]
- Zana M, Szecsenyi A, Czibula A, Bjelik A, Juhasz A, Rimanoczy A, Szabo K, Vetro A, Szucs P, Varkonyi A, et al. Age-dependent oxidative stress-induced DNA damage in Down's lymphocytes. Biochem Biophys Res Commun. 2006;345:726–733. doi: 10.1016/j.bbrc.2006.04.167. [DOI] [PubMed] [Google Scholar]
- Pellegrini FP, Marinoni M, Frangione V, Tedeschi A, Gandini V, Ciglia F, Mortara L, Accolla RS, Nespoli L. Down syndrome, auto-immunity and T regulatory cells. Clin Exp Immunol. 2012;169:238–243. doi: 10.1111/j.1365-2249.2012.04610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean S, McHale C, Enright H. Hematological abnormalities in adult patients with Down's syndrome. Ir J Med Sci. 2009;178:35–38. doi: 10.1007/s11845-008-0223-2. [DOI] [PubMed] [Google Scholar]
- Zipursky A. Susceptibility to leukemia and resistance to solid tumors in Down syndrome. Pediatr Res. 2000;47:704. doi: 10.1203/00006450-200006000-00002. [DOI] [PubMed] [Google Scholar]
- Whitlock JA, Sather HN, Gaynon P, Robison LL, Wells RJ, Trigg M, Heerema NA, Bhatia S. Clinical characteristics and outcome of children with Down syndrome and acute lymphoblastic leukemia: a Children's Cancer Group study. Blood. 2005;106:4043–4049. doi: 10.1182/blood-2003-10-3446. [DOI] [PubMed] [Google Scholar]
- Kusters MA, Verstegen RH, Gemen EF, de Vries E. Intrinsic defect of the immune system in children with Down syndrome: a review. Clin Exp Immunol. 2009;156:189–193. doi: 10.1111/j.1365-2249.2009.03890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, Purton LE. A Microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor γ deficiency. Cell. 2007a;129:1097–1110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007b;129:1081–1095. doi: 10.1016/j.cell.2007.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause DS, Fulzele K, Catic A, Sun CC, Dombkowski D, Hurley MP, Lezeau S, Attar E, Wu JY, Lin HY, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med. 2013;19:1513–1517. doi: 10.1038/nm.3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R, et al. Leukemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature. 2014;506:240–244. doi: 10.1038/nature12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klusmann J-H, Creutzig U, Zimmermann M, Dworzak M, Jorch N, Langebrake C, Pekrun A, Macakova-Reinhardt K, Reinhardt D. Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood. 2008;111:2991–2998. doi: 10.1182/blood-2007-10-118810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamis AS, Woods WG, Alonzo TA, Buxton A, Lange B, Barnard DR, Gold S, Smith FO Children's Cancer Group Study 2891. Increased age at diagnosis has a significantly negative effect on outcome in children with Down syndrome and acute myeloid leukemia: a report from the Children's Cancer Group Study 2891. J Clin Oncol. 2003;21:3415–3422. doi: 10.1200/JCO.2003.08.060. [DOI] [PubMed] [Google Scholar]
- Klusmann J-H, Godinho FJ, Heitmann K, Maroz A, Koch ML, Reinhardt D, Orkin SH, Li Z. Developmental stage-specific interplay of GATA1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis. Genes Dev. 2010;24:1659–1672. doi: 10.1101/gad.1903410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng AP, Hyland CD, Metcalf D, Carmichael CL, Loughran SJ, Di Rago L, Kile BT, Alexander WS. Trisomy of Erg is required for myeloproliferation in a mouse model of Down syndrome. Blood. 2010;115:3966–3969. doi: 10.1182/blood-2009-09-242107. [DOI] [PubMed] [Google Scholar]
- Birchler JA. Reflections on studies of gene expression in aneuploids. Biochem J. 2010;426:119–123. doi: 10.1042/BJ20091617. [DOI] [PubMed] [Google Scholar]
- Letourneau A, Santoni FA, Bonilla X, Sailani MR, Gonzalez D, Kind J, Chevalier C, Thurman R, Sandstrom RS, Hibaoui Y, et al. Domains of genome-wide gene expression dysregulation in Down's syndrome. Nature. 2014;508:345–350. doi: 10.1038/nature13200. [DOI] [PubMed] [Google Scholar]
- Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441:595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- Emmrich S, Rasche M, Schoening J, Reimer C, Keihani S, Maroz A, Xie Y, Li Z, Schambach A, Reinhardt D, et al. miR99a/100∼125b tricistrons regulate hematopoietic stem and progenitor cell homeostasis by shifting the balance between TGFβ and Wnt signalling. Genes Dev. 2014;28:858–874. doi: 10.1101/gad.233791.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klusmann JH, Li Z, Bohmer K, Maroz A, Koch ML, Emmrich S, Godinho FJ, Orkin SH, Reinhardt D. miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukemia. Genes Dev. 2010b;24:478–490. doi: 10.1101/gad.1856210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birger Y, Goldber L, Chlon TM, Goldenson B, Muler I, Schiby G, Jacob-Hirsch J, Rechavi G, Crispino JD, Izraeli S. Perturbation of fetal hematopoiesis in a mouse model of Down syndrome's transient myeloproliferative disorder. Blood. 2013;122:988–998. doi: 10.1182/blood-2012-10-460998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AA, Chapuy B, Lin CY, Tivey T, Li H, Townsend EC, van Bodegom D, Day TA, Wu SC, Liu H, et al. Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat Genet. 2014;46:618–623. doi: 10.1038/ng.2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salek-Ardakani S, Smooha G, de Boer J, Sebire NJ, Morrow M, Rainis L, Lee S, Williams O, Izraeli S, Brady HJ. ERG is a megakaryocytic oncogene. Cancer Res. 2009;69:4665–4673. doi: 10.1158/0008-5472.CAN-09-0075. [DOI] [PubMed] [Google Scholar]
- Bourquin JP, Subramanian A, Langebrake C, Reinhardt D, Bernard O, Ballerini P, Baruchel A, Cave H, Dastugue N, Hasle H, et al. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci USA. 2006;103:3339–3344. doi: 10.1073/pnas.0511150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein K, Bock O, Theophile K, Schultz-Bischof K, Porwit A, Schlue J, Jonigk D, Kreipe H. MPLW515L mutation in acute megakaryoblastic leukaemia. Leukemia. 2009;23:852–855. doi: 10.1038/leu.2008.371. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Toki T, Okuno Y, Kanezaki R, Shiraishi Y, Sato-Otsubo A, Sanada M, Park MJ, Terui K, Suzuki H, et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet. 2013;45:1293–1299. doi: 10.1038/ng.2759. [DOI] [PubMed] [Google Scholar]
- Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, et al. MPLW515L is a novel somatic mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet G, David G, Cassiman JJ. Cellular ageing of Alzheimer's disease and Down syndrome cells in culture. Mutat Res. 1991;256:221–231. doi: 10.1016/0921-8734(91)90013-2. [DOI] [PubMed] [Google Scholar]
- Kimura M, Cao X, Skurnick J, Cody M, Soteropoulos P, Aviv A. Proliferation dynamics in cultured skin fibroblasts from Down syndrome subjects. Free Radic Biol Med. 2005;39:374–380. doi: 10.1016/j.freeradbiomed.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, Lieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi regulates cell proliferation and senescence through the ink4 locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Gil J, O'Loghlen A. PRC1 complex diversity: where is it taking us? Trends Cell Biol. 2014;24:632–641. doi: 10.1016/j.tcb.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Jacobsen GK, Henriques UV. A fetal testis with intratubular germ cell neoplasia (ITGCN) Mod Pathol. 1992;5:547–549. [PubMed] [Google Scholar]
- Satge D, Jacobsen GK, Cessot F, Raffi F, Vekemans M. A fetus with Down syndrome and intratubular germ cell neoplasia. Pediatr Pathol Lab Med. 1996;16:107–112. [PubMed] [Google Scholar]
- Sussan TE, Yang A, Li F, Ostrowski MC, Reeves RH. Trisomy represses Apc(Min)-mediated tumours in mouse models of Down's syndrome. Nature. 2008;451:73–75. doi: 10.1038/nature06446. [DOI] [PubMed] [Google Scholar]
- Baek KH, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, Lensch MW, Park IH, Yoon SS, Minami T, et al. Down's syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature. 2009;459:1126–1130. doi: 10.1038/nature08062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Reeves RH. Increased survival following tumorigenesis in Ts65Dn mice that model Down syndrome. Cancer Res. 2011;71:3573–3581. doi: 10.1158/0008-5472.CAN-10-4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richtsmeier JT, Baxter LL, Reeves RH. Parallels of craniofacial maldevelopment in Down syndrome and Ts65Dn mice. Dev Dyn. 2000;217:137–145. doi: 10.1002/(SICI)1097-0177(200002)217:2<137::AID-DVDY1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Richtsmeier JT, Zumwalt A, Carlson EJ, Epstein CJ, Reeves RH. Craniofacial phenotypes in segmentally trisomic mouse models for Down syndrome. Am J Med Genet. 2002;107:317–324. doi: 10.1002/ajmg.10175. [DOI] [PubMed] [Google Scholar]
- Trainor PA. Specification and patterning of neural crest cells during craniofacial development. Brain Behav Evol. 2005;66:266–280. doi: 10.1159/000088130. [DOI] [PubMed] [Google Scholar]
- Roper RJ, VanHorn JF, Cain CC, Reeves RH. A neural crest deficit in Down syndrome mice is associated with deficient mitotic response to Sonic hedgehog. Mech Dev. 2009;126:212–219. doi: 10.1016/j.mod.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trazzi S, Mitrugno VM, Valli E, Fuchs C, Rizzi S, Guidi S, Perini G, Bartesaghi R, Ciani E. APP-dependent up-regulation of Ptch1 underlies proliferation impairment of neural precursors in Down syndrome. Hum Mol Genet. 2011;20:1560–1573. doi: 10.1093/hmg/ddr033. [DOI] [PubMed] [Google Scholar]
- Roper RJ, Baxter LL, Saran NG, Klinedinst DK, Beachy PA, Reeves RH. Defective cerebellar response to mitogenic Hedgehog signaling in Down's syndrome mice. Proc Natl Acad Sci USA. 2006;103:1452–1456. doi: 10.1073/pnas.0510750103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley CN, Allen JR, Baumann DD, Deitz SL, Blazek JD, Newbauer A, Darrah A, Long BC, Young B, Clement M, et al. Non-trisomic homeobox gene expression during craniofacial development in the Ts65Dn mouse model of Down syndrome. Am J Med Genet A. 2013;161:1866–1874. doi: 10.1002/ajmg.a.36006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo I, Kuratani S, Kimura C, Takeda N, Aizawa S. Mouse Otx2 functions in the formation and patterning of rostral head. Genes Dev. 1995;9:2646–2658. doi: 10.1101/gad.9.21.2646. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Rentschler S, Fishman G, Sassoon DA. Cellular and cis-regulation of En-2 expression in the mandibular arch. Mech Dev. 2002;111:125–136. doi: 10.1016/s0925-4773(01)00618-9. [DOI] [PubMed] [Google Scholar]
- Sumarsono SH, Wilson TJ, Tymms MJ, Ventner DJ, Corrick CM, Kola R, Lahoud MH, Papas TS, Seth A, Kola I. Down's syndrome-like skeletal abnormalities in Ets2 transgenic mice. Nature. 1996;379:534–537. doi: 10.1038/379534a0. [DOI] [PubMed] [Google Scholar]
- Hammerle B, Lizalde C, Tejedor FJ. The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. Eur J Neurosci. 2008;27:1061–1074. doi: 10.1111/j.1460-9568.2008.06092.x. [DOI] [PubMed] [Google Scholar]
- Altafaj X, Dierssen M, Baamonde C, Marti E, Visa J, Guimera J, Oset M, Gonzalez JR, Florez J, Fillat C, et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down's syndrome. Hum Mol Genet. 2001;10:1915–1923. doi: 10.1093/hmg/10.18.1915. [DOI] [PubMed] [Google Scholar]
- Olson LE, Richtsmeier J, Leszl J, Reeves RH. A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science. 2004;306:687–690. doi: 10.1126/science.1098992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko PV, Masliah E, Kleschevnikov AM, Villar AJ, Epstein CJ, Salehi A, Mobley WC. Synaptic structural abnormalities in the Ts65Dn mouse model of Down syndrome. J Comp Neurol. 2004;480:281–298. doi: 10.1002/cne.20337. [DOI] [PubMed] [Google Scholar]
- Hill CA, Sussan TE, Reeves RH, Richtsmeier JT. Complex contributions of Ets2 to craniofacial and thymus phenotypes of trisomic “Down syndrome” mice. Am J Med Genet A. 2009;149:2158–2165. doi: 10.1002/ajmg.a.33012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trazzi S, Fuchs C, Valli E, Perini G, Bartesaghi R, Ciani E. The amyloid precursor protein (APP) triplicated gene impairs neuronal precursor differentiation and neurite development through two different domains in the Ts65Dn mouse model for down syndrome. J Biol Chem. 2013;288:20817–20829. doi: 10.1074/jbc.M113.451088. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Haydar TF, Reeves RH. Trisomy 21 and early brain development. Trends Neurosci. 2012;35:81–91. doi: 10.1016/j.tins.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contestabile A, Fila T, Ceccarelli C, Bonasoni P, Bonapace L, Santini D, Bartesaghi R, Ciani E. cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus. 2007;17:665–678. doi: 10.1002/hipo.20308. [DOI] [PubMed] [Google Scholar]
- Guidi S, Bonasoni P, Ceccarelli C, Santini D, Gualtieri F, Ciani E, Bartesaghi R. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 2008;18:180–197. doi: 10.1111/j.1750-3639.2007.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi S, Caina E, Bonasoni P, Santini D, Bartesaghi R. Widespread proliferation impairment and hypocellularity in the cerebellum of fetuses with down syndrome. Brain Pathol. 2011;21:361–373. doi: 10.1111/j.1750-3639.2010.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter LL, Moran TH, Richtsmeier JT, Troncoso J, Reeves RH. Discovery and genetic localization of Down syndrome cerebellar phenotypes using the Ts65Dn mouse. Hum Mol Genet. 2000;9:195–202. doi: 10.1093/hmg/9.2.195. [DOI] [PubMed] [Google Scholar]
- Lorenzi HA, Reeves RH. Hippocampal hypocellularity in the Ts65Dn mouse originates early in development. Brain Res. 2006;1104:153–159. doi: 10.1016/j.brainres.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Chakrabarti L, Galdzicki Z, Haydar TF. Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J Neurosci. 2007;27:11483–11495. doi: 10.1523/JNEUROSCI.3406-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi P, Ciani E, Guidi S, Trazzi S, Felice D, Grossi G, Fernandez M, Giuliani A, Calza L, Bartesaghi R. Early pharmacotherapy restores neurogenesis and cognitive performance in the Ts65Dn mouse model for Down syndrome. J Neurosci. 2010;30:8769–8779. doi: 10.1523/JNEUROSCI.0534-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contestabile A, Fila T, Bartesaghi R, Ciani E. Cell cycle elongation impairs proliferation of cerebellar granule cell precursors in the Ts65Dn mouse, an animal model for Down syndrome. Brain Pathol. 2009;19:224–237. doi: 10.1111/j.1750-3639.2008.00168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altafaj X, Martin ED, Ortiz-Abalia J, Valderrama A, Lao-Peregrin C, Dierssen M, Fillat C. Normalization of Dyrk1a expression by AAV2/1-shDyrk1A attenuates hippocampal-dependent defects in the Ts65Dn mouse model of Down syndrome. Neurobiol Dis. 2013;52:117–127. doi: 10.1016/j.nbd.2012.11.017. [DOI] [PubMed] [Google Scholar]
- Olson LE, Roper RJ, Sengstaken CL, Peterson EA, Aquino V, Galdzicki Z, Siarey R, Pletnikov M, Moran TH, Reeves RH. Trisomy for the Down syndrome ‘critical region’ is necessary but not sufficient for brain phenotypes of trisomic mice. Hum Mol Genet. 2007;16:774–782. doi: 10.1093/hmg/ddm022. [DOI] [PubMed] [Google Scholar]
- Das I, Park JM, Shin JH, Jeon SK, Lorenzi H, Linden DJ, Worley PF, Reeves RH. Hedgehog agonist therapy corrects structural and cognitive defects in a Down syndrome mouse model. Sci Transl Med. 2013;5:201ra120. doi: 10.1126/scitranslmed.3005983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi S, Stagni F, Bianchi P, Ciani E, Giacomini A, De Franceschi M, Moldrich R, Kurniawan N, Mardon K, Giulani A, et al. Prenatal pharmacotherapy rescues brain development in a Down's syndrome mouse model. Brain. 2014;137:380–401. doi: 10.1093/brain/awt340. [DOI] [PubMed] [Google Scholar]
- Trazzi S, Fuchs C, De Franceschi M, Mitrugno VM, Bartesaghi R, Ciani E. APP-dependent alteration of GSK3β activity impairs neurogenesis in the 65Dn mouse model of Down syndrome. Neurobiol Dis. 2014;67:24–36. doi: 10.1016/j.nbd.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539–551. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S, Schwalbe J, Stasko MR, Yarowsky PJ, Costa AC. Fluoxetine rescues deficient neurogenesis in hippocampus of the Ts65Dn mouse model for Down syndrome. Exp Neurol. 2006;200:256–261. doi: 10.1016/j.expneurol.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Guedi F, Pereira PL, Najas S, Barallobre MJ, Chabert C, Souchet B, Sebrie C, Verney C, Herault Y, Arbones M, et al. DYRK1A: a master regulatory protein controlling brain growth. Neurobiol Dis. 2012;46:190–203. doi: 10.1016/j.nbd.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Thomazeau A, Lassalle O, Iafrati J, Souchet B, Guedi F, Janel J, Chavis P, Delabar J, Manzoni OJ. Prefrontal deficits in a murine model overexpressing the down syndrome candidate gene dyrk1a. J Neurosci. 2014;34:1138–1147. doi: 10.1523/JNEUROSCI.2852-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souchet B, Guedi F, Sahun I, Duchon A, Daubigny F, Badel A, Yanagawa Y, Barallaobre MJ, Dierssen M, Yu E, et al. Excitation/inhibition balance and learning are modified by Dyrk1a gene dosage. Neurobiol Dis. 2014;69:65–75. doi: 10.1016/j.nbd.2014.04.016. [DOI] [PubMed] [Google Scholar]
- Garcia-Cerro S, Martinez P, Vidal V, Corrales A, Florez J, Vidal R, Rueda N, Arbones M, Martinez-Cue C. Overexpression of Dyrk1A is implicated in several cognitive, electrophysiological and neuromorphological alterations found in a mouse model of Down syndrome. PLoS ONE. 2014;9:e106572. doi: 10.1371/journal.pone.0106572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Abalia J, Sahun I, Altafaj X, Andreu N, Estivill X, Dierssen M, Fillat C. Targeting Dyrk1A with AAVshRNA attenuates motor alterations in TgDyrk1A, a mouse model of Down syndrome. Am J Hum Genet. 2008;83:479–488. doi: 10.1016/j.ajhg.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibaoui Y, Grad I, Letourneau A, Sailani MR, Dahoun S, Santoni FA, Gimelli S, Guipponi M, Pelte MF, Bene F, et al. Modelling and rescuing neurodevelopmental defect of Down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. PLoS Genet. 2013;9:e1003515. doi: 10.1002/emmm.201302848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzonetta C, Mulligan C, Deutsch S, Ruf S, O'Doherty A, Lyle R, Borel C, Lin-Marq N, Delom F, Groet J, et al. DYRK1A-dosage imbalance perturbs NRSF/REST levels, deregulating pluripotency and embryonic stem cell fate in Down syndrome. Am J Hum Genet. 2008;83:388–400. doi: 10.1016/j.ajhg.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijazi M, Fillat C, Medina JM, Velasco A. Overexpression of DYRK1A inhibits choline acetyltransferase induction by oleic acid in cellular models of Down syndrome. Exp Neurol. 2013;239:229–234. doi: 10.1016/j.expneurol.2012.10.016. [DOI] [PubMed] [Google Scholar]
- Williams AD, Mjaatvedt CH, Moore CS. Characterization of the cardiac phenotype in neonatal Ts65Dn mice. Dev Dyn. 2008;237:426–435. doi: 10.1002/dvdy.21416. [DOI] [PubMed] [Google Scholar]
- Dunlevy L, Bennett M, Slender A, Lana-Elola E, Tybulewicz VL, Fisher EM, Mohun T. Down's syndrome-like cardiac developmental defects in embryos of the transchromosomic Tc1 mouse. Cardiovasc Res. 2010;88:287–295. doi: 10.1093/cvr/cvq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Morishima M, Jiang X, Yu T, Meng K, Ray D, Pao A, Ye P, Parmacek MS, Yu YE. Engineered chromosome-based genetic mapping establishes a 3.7 Mb critical genomic region for Down syndrome-associated heart defects in mice. Hum Genet. 2014;133:743–753. doi: 10.1007/s00439-013-1407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs C, Ciani E, Guidi S, Trazzi S, Bartesaghi R. Early-occurring proliferation defects in peripheral tissues of the Ts65Dn mouse model of Down syndrome are associated with patched1 over expression. Lab Invest. 2012;92:1648–1660. doi: 10.1038/labinvest.2012.117. [DOI] [PubMed] [Google Scholar]
- Conti A, Fabbrini F, D'Agostino P, Negri R, Greco D, Genesio R, D'Armiento M, Olla C, Paladini D, Zannini M, et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genom. 2007;8:268. doi: 10.1186/1471-2164-8-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand N, Roux M, Ryckebusch L, Niederreither K, Dolle P, Moon A, Capecchi M, Zaffran S. Hox genes define distinct progenitor sub-domains within the second heart field. Dev Biol. 2011;353:266–274. doi: 10.1016/j.ydbio.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforest B, Bertrand N, Zaffran S. Genetic lineage tracing of anterior Hox expressing cells. Methods Mol Biol. 2014;1196:37–48. doi: 10.1007/978-1-4939-1242-1_3. [DOI] [PubMed] [Google Scholar]
- Goddeeris MM, Rho S, Petiet A, Davenport CL, Jihnson GA, Meyers EN, Klingensmith J. Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart. Development. 2008;135:1887–1895. doi: 10.1242/dev.016147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AD, Peterson MA, Friedland-Little JM, Anderson SA, Moskowitz IP. Sonic hedgehog is required in pulmonary endoderm for atrial septation. Development. 2009;136:1761–1770. doi: 10.1242/dev.034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y, Camenisch TD, Itin A, Fishman GI, McDonald JA, Carmeliet P, Kesheti E. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. 2001;128:1531–1538. doi: 10.1242/dev.128.9.1531. [DOI] [PubMed] [Google Scholar]
- Yuan S, Zaidi S, Brueckman M. Congenital heart disease: emerging themes linking genetics and development. Curr Opin Genet Dev. 2013;23:352–359. doi: 10.1016/j.gde.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman C, Locke AE, Feingold E, Reshey B, Espana K, Thusberg J, Mooney S, Bean LJ, Dooley KJ, Cua CL, et al. An excess of deleterious variants in VEGF-A pathway genes in Down-syndrome-associated atrioventricular septal defects. Am J Hum Genet. 2012;91:646–659. doi: 10.1016/j.ajhg.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsa E, Burridge PW, Wu JC. Human stem cells for modelling heart disease and for drug discovery. Sci Trans Med. 2014;6:239ps. doi: 10.1126/scitranslmed.3008921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Habibollah S, Tilgner K, Collin J, Barta T, Al-Aama JY, Tesarov L, Hussain R, Trafford AW, Kirkwood G, et al. An induced pluripotent stem cell model of hypoplastic left heart syndrome (HLHS) reveals multiple expression and functional defects in HLHS-derived cardiac myocytes. Stem Cells Trans Med. 2014;3:416–423. doi: 10.5966/sctm.2013-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copplola A, Romito A, Borel C, Gehrig C, Gagnebin M, Falconnet E, Izzo A, Altucci L, Banfi S, Antonarakis SE, et al. Cardiomyogenesis is controlled by the miR99a/let-7c cluster and epigenetic modification. Stem Cell Res. 2014;12:323–337. doi: 10.1016/j.scr.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Nikolaev SI, Santoni F, Vannier A, Falconnet E, Giarin E, Basso G, Hoischen A, Veltman JA, Groet J, Nizetic D, et al. Exome sequencing identifies putative drivers of progression of transient myeloproliferative disorder to AMKL in infants with Down syndrome. Blood. 2013;122:554–561. doi: 10.1182/blood-2013-03-491936. [DOI] [PubMed] [Google Scholar]
- Veitia RA, Bottani S, Birchler JA. Gene dosage effects: nonlinearities, genetic interactions, and dosage compensation. Trends Genet. 2013;29:385–393. doi: 10.1016/j.tig.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Sheltzer JM, Torres EM, Dunham MJ, Amon A. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci USA. 2012;109:12644–12649. doi: 10.1073/pnas.1209227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA. Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc Natl Acad Sci USA. 2012;109:14746–14753. doi: 10.1073/pnas.1207726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Jing Y, Cost GJ, Chiang JC, Kolpa HJ, Cotton AM, Carone DM, Carone BR, Shivak DA, Guschin DY, et al. Translating dosage compensation to trisomy 21. Nature. 2013;500:296–300. doi: 10.1038/nature12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granese B, Scala I, Spatuzza C, Vlaentino A, Coletta M, Vacca RA, De Luca P, Andria G. Validation of microarray data in human lymphoblasts shows a role of the ubiquitin-proteasome system and NF-kB in the pathogenesis of Down syndrome. BMC Med Genomics. 2013;6:24. doi: 10.1186/1755-8794-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LB, Chang KH, Wang PR, Hirata RK, Papayannopoulou T, Rissell DW. Trisomy correction in Down syndrome induced pluripotent stem cells. Cell Stem Cell. 2012;11:615–619. doi: 10.1016/j.stem.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CY, Liu LY, Chen CY, Tsai CH, Hwa HL, Chang LY, Lin YS, Hsieh FJ. Gene expression variation increase in trisomy 21 tissues. Mamm Genome. 2008;19:398–405. doi: 10.1007/s00335-008-9121-1. [DOI] [PubMed] [Google Scholar]
- Slonim DK, Koide K, Johnson KL, Tantravahi U, Cowan JM, Jarrah Z, Bianchi DW. Functional genomic analysis of amniotic fluid cell-free mRNA suggests that oxidative stress is significant in Down syndrome fetuses. Proc Natl Acad Sci USA. 2009;106:9425–9429. doi: 10.1073/pnas.0903909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk M, Mayer A, Lovrecic L, Luvan P, Peterlin B. Expression signature as a biomarker for prenatal diagnosis of trisomy 21. PLoS ONE. 2013;8:e74184. doi: 10.1371/journal.pone.0074184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altug-Teber O, Bonin M, Walter M, Mau-Holzmann UA, Dufke A, Stappert H, Tekesin I, Heilbronner H, Nieselt K, Reiss O. Specific transcriptional changes in human fetuses with autosomal trisomies. Cytogenet Genome Res. 2007;119:171–184. doi: 10.1159/000112058. [DOI] [PubMed] [Google Scholar]
- Li C, Jin L, Bai Y, Chen Q, Fu L, Yang M, Xiao H, Zhao G, Wang S. Genome-wide expression analysis in Down syndrome: onsight into immunodeficiency. PLoS ONE. 2012;7:e49130. doi: 10.1371/journal.pone.0049130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loudin MG, Wang J, Leung HC, Gurusiddappa S, Meyer J, Condos G, Morrison D, Tsimelzon A, Devidas M, Heerema NA, et al. Genomic profiling in Down syndrome acute lymphoblastic leukemia identifies histone gene deletions associated with altered methylation profiles. Leukemia. 2011;25:1555–1563. doi: 10.1038/leu.2011.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElwaine S, Mullighan C, Groet J, Spinelli M, Rinaldi A, Denyer G, Mensah A, Cavani S, Baldo C, Dagna-Bricarelli F, et al. Microarray transcript profiling distinguishes the transient from the acute type of megakaryoblastic leukemia (M7) in Down's syndrome, revealing PRAME as a specific discriminating marker. Br J Haematol. 2004;125:729–742. doi: 10.1111/j.1365-2141.2004.04982.x. [DOI] [PubMed] [Google Scholar]
- Xavier AC, Edawards H, Dombkowski AA, Balci TB, Berman JN, Dellaire G, Xie C, Buck SA, Matherly LH, Ge Y, et al. A unique role of Gata1s in Down syndrome acute megakaryocytic leukemia biology and therapy. PLoS ONE. 2011;6:e27486. doi: 10.1371/journal.pone.0027486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroz A, Stachorski L, Emmrich S, Reinhardt K, Xu J, Shao Z, Käbler S, Dertmann T, Hitzler J, Roberts I, et al. GATA1s induces hyperproliferation of eosinophil precursors in Down syndrome transient leukemia. Leukemia. 2014;28:1259–1270. doi: 10.1038/leu.2013.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao R, Wang X, Spitznagel EL, Frelin LP, Ting JC, Ding H, Kim JW, Ruczinski I, Downey TJ, Pevsner J. Primary and secondary transcriptional effects in the developing human Down syndrome brain and heart. Genome Biol. 2005;6:R107. doi: 10.1186/gb-2005-6-13-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JA, Sun J, Shepherd J, Ovchinnikov DA, Chung TL, Nayler SP, Kao LP, Morrow CA, Thakar NY, Soo SY, et al. Integration-free induced pluripotent stem cells model genetic and neural developmental features of down syndrome etiology. Stem Cells. 2013;31:467–478. doi: 10.1002/stem.1297. [DOI] [PubMed] [Google Scholar]
- Helguera P, Seiglie J, Rodriguez J, Hanna M, Helguera G, Busciglio J. Adaptive downregulation of mitochondrial function in down syndrome. Cell Metab. 2013;17:132–140. doi: 10.1016/j.cmet.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, Bahn S. Gene expression profiling in the adult Down syndrome brain. Genomics. 2007;90:647–660. doi: 10.1016/j.ygeno.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Weick JP, Held DL, Bonadurer GF, Doers ME, Liu Y, Maguire C, Clark A, Knackert JA, Molinarolo K, Musser M, et al. Deficits in human trisomy 21 iPSCs and neurons. Proc Natl Acad Sci USA. 2013;110:9962–9967. doi: 10.1073/pnas.1216575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Lee YK, Lim YC, Zheng Z, Lin XM, Ng DP, Holbrook JD, Law HY, Kwek KY, Yeo GS, et al. Global DNA hypermethylation in down syndrome placenta. PLoS Genet. 2013;9:e1003515. doi: 10.1371/journal.pgen.1003515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffaire J, Rivals I, Dauphinot L, Pasteau F, Wherle R, Larrat B, Vitalis T, Moldrich RX, Rossier J, Sinkus R, et al. Gene expression signature of cerebellar hypoplasia in a mouse model of Down syndrome during postnatal development. BMC Genom. 2009;10:138. doi: 10.1186/1471-2164-10-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurs-Hoeijmakers JH, Vulto-van Silfhout AT, Vissers LE, van de Vondervoort II, van Bon BW, de Ligt J, Gilissen C, Hehir-Kwa JY, Neveling K, del Rosario M, et al. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J Med Genet. 2013;50:802–811. doi: 10.1136/jmedgenet-2013-101644. [DOI] [PubMed] [Google Scholar]
- Guihard-Costa AM, Khung S, Delbecque K, Menez F, Delezoide AL. Biometry of face and brain in fetuses with trisomy 21. Pediatr Res. 2006;59:33–38. doi: 10.1203/01.pdr.0000190580.88391.9a. [DOI] [PubMed] [Google Scholar]
- Allanson JE, O'Hara P, Farkas LG, Nair RC. Anthropometric craniofacial pattern profiles in Down syndrome. Am J Med Genet. 1993;47:748–752. doi: 10.1002/ajmg.1320470530. [DOI] [PubMed] [Google Scholar]
- Hawli Y, Nasrallah M, El-Hajj Fuleihan G. Endocrine and musculoskeletal abnormalities in patients with Down syndrome. Nat Rev Endocrinol. 2009;5:327–334. doi: 10.1038/nrendo.2009.80. [DOI] [PubMed] [Google Scholar]
- Patterson T, Rapsey CM, Glue P. Systematic review of cognitive development across childhood in Down syndrome: implications for treatment interventions. J Intellect Disab Res. 2013;57:306–318. doi: 10.1111/jir.12037. [DOI] [PubMed] [Google Scholar]
- Margallo-Lana ML, Moore PB, Kay DW, Perry RH, Reid BE, Berney TP, Tyrer SP. Fifteen-year follow up of 92 hospitalized adults with Down's syndrome: incidence of cognitive decline, its relationship to age and neuropathology. J Intellect Disabil Res. 2007;51:463–477. doi: 10.1111/j.1365-2788.2006.00902.x. [DOI] [PubMed] [Google Scholar]
- Lockrow JP, Fortress AM, Granholm AC. Age-related neurodegeneration and memory loss in down syndrome. Curr Gerontol Geriatr Res. 2012;2012:463909. doi: 10.1155/2012/463909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll KN, Arbogast PG, Dudley JA, Cooper WO. Increase in incidence of medically treated thyroid disease in children with Down syndrome after release of American Academy of Pediatrics Health Supervision guidelines. Pediatrics. 2008;122:e493–e498. doi: 10.1542/peds.2007-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber E, Chacko E, Regelmann MO, Costin G, Rapaport R. Down syndrome and thyroid function. Endocrinol Metab Clin North Am. 2012;41:735–745. doi: 10.1016/j.ecl.2012.08.008. [DOI] [PubMed] [Google Scholar]
- Freeman SB, Taft LF, Dooley KJ, Allran K, Sherman SL, Hassold TJ, Khoury MJ, Saker DM. Population-based study of congenital heart disease in Down syndrome. Am J Hum Genet. 1998;80:213–217. [PubMed] [Google Scholar]
- Weijerman ME, van Furth AM, van der Mooren MD, van Weissenbruch MM, Rammeloo L, Broers CJ, Gemke RJ. Prevalence of congenital heart disease and persistent pulmonary hypertension of the neonate with Down syndrome. Eur J Pediatr. 2010;169:1195–1199. doi: 10.1007/s00431-010-1200-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizen NJ, Magyar CI, Kuschner ES, Sulkes SB, Druschel C, van Wijngaarden E, Rodgers L, Diehl A, Lowry R, Hyman SL. A community cross-sectional survey of medical problems in 440 children with Down syndrome in New York State. J Pediatr. 2014;164:871–875. doi: 10.1016/j.jpeds.2013.11.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1
Supplementary Table S2