

Abstract
Mutations in leucine-rich repeat kinase 2 (LRRK2) are the most common cause of dominant-inherited Parkinson's disease (PD), and yet we do not fully understand the physiological function(s) of LRRK2. Various components of the clathrin machinery have been recently found mutated in familial forms of PD. Here, we provide molecular insight into the association of LRRK2 with the clathrin machinery. We report that through its GTPase domain, LRRK2 binds directly to clathrin-light chains (CLCs). Using genome-edited HA-LRRK2 cells, we localize LRRK2 to endosomes on the degradative pathway, where it partially co-localizes with CLCs. Knockdown of CLCs and/or LRRK2 enhances the activation of the small GTPase Rac1, leading to alterations in cell morphology, including the disruption of neuronal dendritic spines. In Drosphila, a minimal rough eye phenotype caused by overexpression of Rac1, is dramatically enhanced by loss of function of CLC and LRRK2 homologues, confirming the importance of this pathway in vivo. Our data identify a new pathway in which CLCs function with LRRK2 to control Rac1 activation on endosomes, providing a new link between the clathrin machinery, the cytoskeleton and PD.
Keywords: clathrin, Drosophila melanogaster, endosomes, Parkinson's disease, Rac1
Introduction
Parkinson's disease (PD) is the second most common age-related progressive neurodegenerative disorder, and mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are the most common genetic cause of both familial and sporadic PD 1. LRRK2 is a large molecular weight multi-domain protein that includes a kinase domain and a Ras of complex proteins (ROC) GTPase domain 1. LRRK2 functions in at least four different processes related to membrane trafficking; (i) synaptic vesicle (SV) exo/endocytosis 2, (ii) trafficking of the mannose 6-phosphate receptor (MPR) and associated lysosomal hydrolases between the trans-Golgi network (TGN) and the endo-lysosomal system 3,4, (iii) control of epidermal growth factor receptor (EGFR) trafficking and degradation 5, and (iv) regulation of actin dynamics 6–8. The relationship between the structural domains of LRRK2 and the protein's functional activity remains poorly defined.
Clathrin-mediated membrane trafficking provides a major mechanism for protein transport in cells including endocytosis of protein cargo 9, reformation of SVs 10, and trafficking of the MPR 11. Intriguingly, mutations in key components of the clathrin machinery are rare variants in familial forms of PD 12–14. Clathrin coats are assembled from triskelia, composed of three linked clathrin-heavy chain (CHC) proteins and associated clathrin-light chains (CLCs) 15. There are four forms of CLCs, CLCa and b, which are functionally interchangeable and expressed in all tissues, and neuronal CLCa and b (nCLCa/b), which have short splice inserts and are expressed exclusively in neurons 15. CLCs bind to huntingtin-interacting protein 1-related (HIP1R) 16 recruiting it to clathrin coats 17. HIP1R binds actin 18 and functions as a negative regulator of actin assembly 19, and knockdown (KD) of either HIP1R or CLCs causes overly abundant actin assembly in the vicinity of clathrin coats 17,19. CLCs are also components of bilayered clathrin coats on early endosomes that recruit the “endosomal sorting complexes required for transport” machinery, which drives sorting and inward invagination of endocytic cargo, such as EGFR, allowing for formation of multivesicular bodies (MVBs) and degradation of cargo in lysosomes 20–22.
Here, we set out to better define the role of LRRK2 and discovered an interaction of its ROC domain with CLCs. We demonstrate that CLCs and LRRK2 interact biochemically and functionally to negatively regulate Rac1 activation and that disruption of this pathway leads to Rac1 activation and altered function, both in cells and in the Drosophila eye in vivo.
Results and Discussion
CLCs bind directly to the ROC domain of LRRK2
To better understand the cell physiological functions of LRRK2, we screened for ROC domain binding partners. GST-ROC was incubated with soluble rat brain extracts with affinity-selected proteins identified by mass spectrometry. nCLCa/b were detected in this analysis and their binding to GST-ROC was confirmed by blot (Fig1A). nCLCa/b are functionally interchangeable 23 and their binding to GST-ROC is similar (Fig1A). Endogenous LRRK2 from brain extracts binds to GST-nCLCb (Fig1B), and purified full-length nCLCb (residues 1–228) binds robustly to GST-ROC (Fig1C), demonstrating that the interaction is direct.
Figure 1. Identification of CLCs as LRRK2-binding partners.
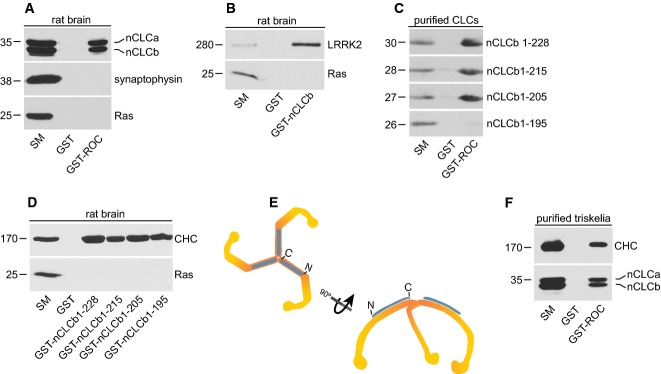
- Equal aliquots (2 mg) of a Triton X-100-solubilized lysate from rat brain were incubated with GST or GST-ROC. Specifically bound proteins were detected by Western blot with antibodies recognizing the indicated proteins.
- Brain lysate as in (A) was incubated with GST or GST-nCLCb. Specifically bound proteins were detected by Western blot with antibodies recognizing the indicated proteins.
- Full-length GST-nCLCb (1-228) and various truncation mutants were purified from bacteria and the GST tag was removed by thrombin cleavage. Equal aliquots of purified protein were incubated with GST or GST-ROC. Specifically bound proteins were detected by Western blot with antibody against CLCs.
- Brain lysate as in (A) was incubated with GST or GST-nCLCb proteins with the indicated boundaries. Specifically bound proteins were detected by Western blot with antibody against CLCs.
- Representation of a triskelia composed of three CHCs (orange) with three associated CLCs (blue).
- Highly enriched triskelia were stripped from purified CCVs. The triskelia were incubated with GST or GST-ROC. Specifically bound proteins were detected by Western blot with antibodies recognizing the indicated proteins.
CLCb and nCLCb bind GST-ROC equally indicating that the neuronal-specific splice insert is not required for binding, whereas a CLC construct encoding residues 1-165 16 does not bind, indicating that the binding site is between residues 166–228 (Supplementary Fig S1). We thus generated C-terminal deletion constructs; whereas purified nCLCb 1-215 and 1-205 bind GST-ROC equivalent to full-length, a 1–195 construct has no binding (Fig1C). However, all constructs bind equally well to CHC (Fig1D), indicating that loss of ROC binding is not due to a major alteration in folding. Thus, the LRRK2-binding site is between residues 195 and 205. This region of nCLCb is identical in all four CLC isoforms, is conserved across species with only one amino acid substitution in Drosophila and has no ascribed binding partner or functional role 24.
Clathrin-light chains interact with CHC as part of triskelia (Fig1E). CLCs are on the outer surface of the triskelia, facing the cytosol in an assembled clathrin coat such that resides 196–205 are accessible to cytosolic proteins 25 (Fig1E). We thus tested whether LRRK2 interacts with CLCs as part of triskelia. GST-ROC was incubated with triskelia stripped from purified CCVs 26 and both CLCs and CHC are detected in the pull down (Fig1F), indicating that CLCs bound to CHC are still accessible to LRRK2. Despite extensive efforts, we were unable to co-immunoprecipitate (co-IP) the two proteins. Like many large multidomain proteins, LRRK2 is predominantly insoluble when generating lysates from cultured cells 27 or tissue 28, and similarly, clathrin triskelia form massive protein complexes when incorporated into coats. Thus, if LRRK2 associates selectively with CLC assembled in coats, this would hinder co-IP. However, we cannot exclude that a transient or low-affinity interaction hampers the ability to observe LRRK2/CLC co-IP. Nevertheless, our discovery that LRRK2 binds directly to CLCs indicates that CLCs have a dual scaffolding function, recruiting LRRK2 and HIP1R via C-terminal and N-terminal regions, respectively.
Endogenous genome-edited LRRK2 localizes to endosomes
A recent systematic analysis of known LRRK2 antibodies shows they are problematic in their recognition of endogenous LRRK2 by immunofluorescence 28. Thus, to assess the localization of endogenous LRRK2, we used CRISPR/Cas9 technology to genome edit LRRK2 in COS-7 cells, adding a triple HA tag between amino acids 1 and 2, downstream of the endogenous promoter (Fig2A and B). Remarkably, LRRK2 co-localizes with internalized EGF (Fig2C), indicating that a significant fraction of the protein is present on membranes of the endosomal system, specifically on the degradative pathway. We also detect partial co-localization with CLCs (Fig2D), likely reflecting bilayered clathrin coats on early endosomes involved in the formation of MVBs during protein degradation 20–22. Consistently, HA-LRRK2 partially co-localizes with the early endosomal marker EEA1 (Fig2E). LRRK2 functions in EGFR trafficking from early endosomes to MVBs and lysosomes, while PD-LRRK2 mutants delay EGFR degradation by trapping the receptor in endosomes 5. Thus, CLCs likely function as a scaffold to recruit LRRK2 to bilayered clathrin coats on early endosomes.
Figure 2. Endogenous genome-edited LRRK2 localizes to endosomes.

- A PCR results of LRRK2-WT from clone E1 (1) using primers that detect endogenous LRRK2. Clone E1 is positive for 3× HA-LRRK2 (2) using a primer pair with the antisense in the 3× HA insert and the sense primer in endogenous LRRK2. (3) Control unedited COS-7 cells using the same primer combination as in (2). (4) 1 kb marker.
- B Schematic diagram of the oligonucleotide used to direct insertion of the 3× HA tag into the 5′ end of the human LRRK2 coding sequence and the corresponding coding sequence in the same colors (the LRRK2 start codon is underlined, as is the GGGGS linker).
- C COS-7 clone E1 cells were serum-starved followed by 20-min incubation with Alexa488-EGF, after which the cells were fixed and processed for immunofluorescence using HA antibody. Scale bar, 10 μm for bottom 6 panels and 25 μm for top 3 panels.
- D, E COS-7 clone E1 cells were fixed and processed for immunofluorescence using HA and CLC (D) or HA and EEA1 (E) antibodies. Scale bar, 10 μm (D, bottom 6 panels in E) and 25 μm (top 3 panels in E).
KD of CLCs or LRRK2 activates Rac1 altering cell morphology
Knockdown of CLCs leads to over assembly of actin, 17 and actin was identified in a screen for LRRK2-binding partners 6. Moreover, LRRK2 binds directly to the small GTPase Rac1, which regulates actin assembly 7. Interestingly, Rac1 activation occurs on early endosomes 29. We thus tested whether LRRK2 and CLCs regulate Rac1 activity. We used previously characterized siRNAs for CLCa/b 17 and a smartpool of four LRRK2 siRNAs to efficiently knock down the proteins (Fig3A and B). To measure Rac1 activity, we performed affinity-selection assays with the p21-activated protein kinase Cdc42/Rac1 interactive binding domain (GST-PAK-CRIB), which binds preferentially to the GTP-bound form of Rac1 30. Interestingly, KD of CLCs or LRRK2 causes a > twofold and > threefold activation of Rac1, respectively, compared to control siRNA (Fig3C and D). The simultaneous KD of both does not further increase Rac1 activity, suggesting that CLCs and LRRK2 are on the same pathway for Rac1 regulation. Consistently, expression of myc-LRRK2 rescues the enhanced activation of Rac1 seen upon LRRK2 and CLC KD (Supplementary Fig S2A and B). Activity of the related GTPase Cdc42 is not influenced by CLCs/LRRK2 KD; thus, activation of Rac1 is selective (Supplementary Fig S3). Deconvolution of the LRRK2 smartpool siRNA sequences reveals that two distinct sequences that KD LRRK2 lead to Rac1 activation (Supplementary Fig S4). Thus, CLCs and LRRK2 function to limit Rac1 activity.
Figure 3. KD of LRRK2 and CLCs lead to Rac1 activation.
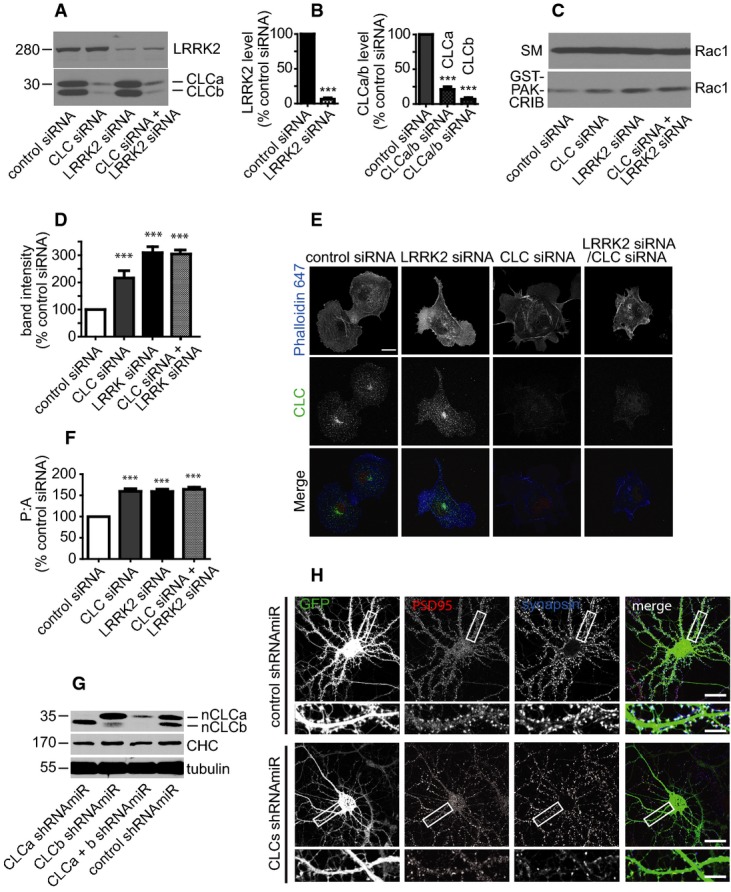
- A, B Lysates from COS-7 cells transfected with siRNA as indicated were processed for Western blot with antibodies recognizing the indicated proteins (A). Band intensities are presented in (B) as % control siRNA. Two-tailed Student's t-test, Mann–Whitney post hoc test, ***P < 0.001, N = 3.
- C, D NP-40 soluble lysates were prepared from COS-7 cells transfected with siRNA as indicated and equal protein aliquots (1.5 mg) were incubated with GST-PAK-CRIB domain. Specifically bound proteins were detected by Western blot with antibody recognizing Rac1 (C). An aliquot of the starting material (SM) equal to 5% of that added to GST-PAK-CRIB was analyzed in parallel. Band intensities are presented in (D) as % control siRNA. Bars represent mean ± s.e.m. One-way ANOVA, Bonferroni's post hoc test, ***P < 0.001, N = 3.
- E, F COS-7 cells transfected with siRNA as indicated were fixed and processed for immunofluorescence using Phalloidin-647 and antibody recognizing CLCs (E). Scale bar, 10 μm. Perimeter and area of cells were measured (F), and a perimeter:area ratio was plotted as a % of the control siRNA-treated cells. One-way ANOVA, Bonferroni's post hoc test, ***P < 0.001, N = 3.
- G Lysates from 21-day in vitro cultured hippocampal neurons transduced with lentivirus (at 7 days in vitro) driving expression of control shRNAmiR or shRNAmiRs specific for CLCa, CLCb, or both viruses in combination. The lysates were processed for Western blot with antibody that recognizes all forms of CLCs.
- H Hippocampal neurons transduced as in (G) were fixed at 21 days in vitro and processed for immunofluorescence with polyclonal antibody specific for the pre-synaptic protein synapsin and monoclonal antibody specific for the post-synaptic protein PSD95. The lentivirus drives expression of GFP. The areas indicated by white boxes in the low power images (squares) are shown enlarged immediately below. The scale bars represents 10 μm for the low magnification and 2.5 μm for the high magnification.
Consistent with the Rac1 activation phenotype, KD of either CLCa/b or LRRK2 leads to alterations in cell morphology, with cells appearing more irregularly shaped with variable protrusions (Fig3E). These changes were quantified as an increase in the perimeter:area ratio (Fig3F). KD of CLCs, LRRK2, or both led to quantitatively similar phenotypes, suggesting that the proteins function in a common pathway. Consistently, the enhanced perimeter:area resulting from CLC KD is rescued by myc-LRRK2 expression (Supplementary Fig S2C and D). Moreover, the Rac1 inhibitor NSC-23766 31 significantly decreases changes in cell morphology resulting from LRRK2 KD, indicating that they result from Rac1 activation (Supplementary Fig S5).
Our results are consistent with a previous study reporting an increased perimeter:area following LRRK2 KD in NIH3T3 cells 6. Moreover, LRRK2 loss of function leads to neurite over-branching phenotypes at the neuromuscular junction in Drosophila 32, and in primary neuronal culture 8. Additionally, there is a decrease in the number of mature dendritic spines in LRRK2−/− neurons, which is accompanied by altered synaptic transmission 33. We therefore examined whether KD of CLCs through lentivirus-driven expression of shRNAmiRs 34 would result in a similar actin-dependent phenotype. KD of nCLCa/b in cultured neurons was confirmed by Western blot (Fig3G), and we observe a loss of mature dendritic spines (Fig3H). Although the underlying molecular mechanisms for these various alterations in cell morphology remain undefined, they may result from hyper activation of Rac1 at early endosomes. Once active on endosomes, Rac1 recycles back to the plasma membrane to regulate actin cytoskeleton dynamics 29. Thus, it appears that CLCs interact with LRRK2 at endosomes to limit actin assembly by inhibiting the activation of Rac1. Our finding that LRRK2 KD activates Rac1 seems contrary to Chan et al 7, who observe that LRRK2 overexpression activates Rac1 resulting in decreased neurite outgrowth. However, Matta et al 2 report that both overexpression and KD of LRRK2 negatively affects SV endocytosis. Thus, it appears that a fine balance in the level of LRRK2 is required for normal function, which could explain the apparent discrepancy between our results and those of Chan and colleagues 7.
Disruption of Clc or dlrrk enhances morphogenetic eye phenotypes resulting from Rac1 overexpression
Drosophila melanogaster provides a powerful model system to examine cell physiological pathways in vivo. We therefore adopted Drosophila to demonstrate that the LRRK2/CLC interaction is physiologically relevant and phylogenetically conserved. Overexpression of Rac1 under control of the glass multiple reporter (GMR-GAL4) promoter, which is expressed predominantly in the eye, leads to rough eye phenotypes 35. We thus tested for Clc (Drosophila CLC) and dLRRK (Drosophila LRRK2)-dependent regulation of Rac1 in vivo. We generated a UAS-Rac1 line driven by longGMR-GAL4, an eye-specific promoter, and observed a mild rough eye phenotype of several fused ommatidia patches (Fig4Ai, arrows). When this line is crossed with two separate UAS-RNAi lines for dLRRK and two UAS-RNAi lines for Clc, there is a dramatically enhanced rough eye phenotype with most ommatidia fused and smaller overall eye size (Fig4Aii-v). KD efficiency was verified by immunofluorescence on eye disks and Western blot of protein levels on adult head with a previously validated dlrrk antibody 36 (Supplementary Fig S6). Additionally, we used a previously established dlrrk UAS-RNAi line from Imai and colleagues 36 to show similar KD efficiency (Supplementary Fig S6B and C). No phenotype is seen with the four UAS-RNAi lines alone, even when driven off of the strong GMR-GAL4 promoter (FigAvii-x). Moreover, the mild rough eye phenotype with Rac1 overexpression is not enhanced with KD of an inward rectifying potassium channel (UAS-irk3-RNAi) as a control (compare Fig4Avi with Ai), but is largely rescued with KD of Slipper (Supplementary Fig S7), a Drosophila homologue of the mammalian mixed lineage kinase family shown to rescue the rough eye phenotype resulting from Rac1 overexpression 37. To further verify that these findings were not due to off-target effects, we used a null allele line for dLRRK (dLRRKe03680), which when crossed with the Rac1 overexpressing line showed a similar enhanced eye phenotype as the KD lines (compare Fig4Bi/ii with Aii/iii).
Figure 4. KD of dLRRK or Clc enhances eye morphogenetic defects caused by Rac1 overexpression in the Drosophila eye.

- Ai-x Lateral view of the adult fly head. Scale bar, 100 μm. (i) longGMR,UAS-Rac1W/TM6B, Hu, Tb causes a mild rough eye phenotype with fused (arrows) and disorganized but still separated ommatidia (arrowheads). (ii, iii) Rac1 overexpression with KD of dLRRK [UAS-dLRRK-RNAi (v22139 or v22140)/longGMR-GAL4, UAS-Rac1W], results in smaller eyes where most ommatidia are fused. (iv, v) Rac1 overexpression with KD of Clc [UAS-Clc-RNAi (v22318 or v106632)/+;longGMR-GAL4,UAS-Rac1W/+], results in smaller eyes with some ommatidia fused. (vi) Rac1 overexpression with KD of the inwardly rectifying potassium channel 3 (irk3), a non-relevant RNAi control [UAS-irk3-RNAi(v101174)/+; longGMR-GAL4,UAS-Rac1W/+], has no effect. (vii, viii) KD of dLRRK with GMR-GAL4/+; UAS-dLRRK-RNAi(v22139 or 22140)/+, respectively as control have no effect on eye morphology. (ix, x) KD of Clc with GMR-GAL4/UAS-Clc-RNAi (v22318 or v106632), respectively, as control has no effect on eye morphology.
- Bi-ii Lateral view of the adult fly head. Scale bar, 100 μm. (i) Rac1 overexpression with removing one copy of dlrrk (dLRRKe03680/longGMR-GAL4,UAS-Rac1W) results in smaller eyes where most ommatidia are fused together. (ii) Removing one copy of dLRRKe03680 in GMR-GAL4 background, as control has no effect on eye morphology (GMR-GAL4/+;dLRRKe03680/+).
- Ci-viii Lateral view of the adult fly head. Scale bar, 100 μm. (i) longGMR,UAS-Rac1W/TM6B, Hu, Tb causes a mild rough eye phenotype with fused and disorganized but still separated ommatidia (as explained in Ai). (ii) Rac1 co-expression with dLRRK-WT using UAS-dLRRK-WT/+;longGMR-GAL4,UAS-Rac1W/+, results in rescued eye morphology. (iii) Rac1 co-expression with Clc using UAS-GFP-Clc/+;longGMR-GAL4,UAS-Rac1W/+, results in rescued eye morphology. (iv) Rac1 co-expression with dLRRK-WT-3KD using UAS-dLRRK-3KD/+;longGMR-GAL4,UAS-Rac1W/+, results in rescued eye morphology. (v) A wild-type eye of the longGMR-GAL4 promoter as a control. (vi–viii) Overexpression of dLRRK-WT, dLRRK-3KD, or Clc alone with GMR-GAL4 as control has no effect on eye morphology.
We next examined whether overexpression of dLRRK or Clc could rescue the Rac1-mediated rough eye phenotype. We co-expressed Rac1 and either dLRRK or Clc in the Drosophila eye and saw a dramatic improvement in the rough eye phenotype resulting from Rac1 overexpression (Fig4Ci-iii). A kinase-dead dlrrk mutant (dlrrk-3KD) 36 yielded similar rescue (Fig4Civ). No phenotype is seen with Clc or dLRRK, WT or 3KD alone (Fig4Cv-viii). These observations demonstrate that dLRRK and Clc regulate Rac1 activity in vivo.
Enhanced intrinsic kinase activity of LRRK2 pathogenic mutants 38 is correlated to increased neurotoxicity 39, suggesting that aberrant enzymatic activity of LRRK2 underlies neuropathogenesis in LRRK2-PD. The LRRK2 field has thus focused on gain-of-function effects of its kinase activity, and the fundamental physiological role(s) of LRRK2 remain understudied. By investigating a basic biological function of LRRK2 in a loss-of-function paradigm, we have uncovered a novel CLC/LRRK2-dependent pathway regulating actin dynamics that functions both in vitro and in vivo. This newfound link between the clathrin machinery and a major PD gene, coupled with the observation that mutations in key components of the clathrin machinery are rare variants in PD 12–14, provides undeniable evidence of the importance of clathrin-mediated membrane trafficking in PD.
Materials and Methods
Affinity-selection and Rac1 activation assays
Brain extract or purified proteins were incubated for 30 min with GST fusion proteins pre-coupled to glutathione–Sepharose beads and washed three times with buffer A (20 mM HEPES, pH 7.4, 0.83 mM benzamidine, 0.23 mM PMSF, 0.5 μg/ml aprotinin/leupeptin) containing 5 mM CaCl2 and 1% Triton X-100. For Rac1 activation assays, 96 h post-transfection with siRNA, COS-7 cells lysates were incubated for 40–50 min at 4°C with ∼28 μg GST-PAK-CRIB fusion proteins pre-coupled to glutathione–Sepharose beads and washed 3 times with ice-cold buffer B (2.5 mM HEPES, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 2% glycerol, 1% NP-40, 0.5 μg/ml aprotinin/leupeptin). Samples were eluted, resolved by SDS–PAGE, and processed for Western blotting.
Microscopy
Cells were imaged on a Zeiss LSM710 confocal microscope using a plan-apochromat 63× oil objective and 20× objective (Zeiss). Perimeter:area ratio was determined using NIH ImageJ software. Fly eye pictures were acquired with a Canon EOS 1000D DSLR (rebel XS) camera mounted on a Zeiss Axioskop 40 microscope with 10× objective (0.25 = N.A).
Statistical analysis
All statistical analyses were performed by one-way, parametric analysis of variance (ANOVA) or two-tailed Student's t-tests, using GraphPad Prism 5 software. Error bars represent the mean ± s.e.m. Differences were considered significant if P < 0.05. For the NSC-23766 experiments, images were randomized and an independent, blinded observer counted normal and elongated cell morphology. Percentage of elongated cells was determined for each image and analyzed by one-way ANOVA.
Other materials and methods can be found in the Supplementary Information.
Acknowledgments
We thank Jacynthe Philie, Martine Girard, Ambika Srinivasan, Vincent Soubannier, and Martin Loignon for excellent experimental support. We are grateful to Shawn Ferguson for assistance with CRISPR/Cas9 experiments, and we thank Mark Cookson for the myc-LRRK2 plasmid, Feng Zhang for the plasmid encoding human optimized Cas9, and Bingwei Lu for dlrrk antibody and various fly lines. We also thank Tommy Nilsson and the Clinical Proteomics Centre. This work was supported by a grant from the Canadian Institutes of Health Research (MOP-13461) to P.S.M. A.M.A.S. was supported by a fellowship from Parkinson Society of Canada. E.A.F. is a Chercheur National of the FRQS. P.A.B. and P.S.M. are James McGill Professors.
Author contributions
AMAS, EAF, and PSM conceived the experiments. AMAS, EAF, PSM, MC, WR, and PAB designed the experiments. AMAS, MC, WR, and SL performed the experiments. AMAS, EAF, and PSM wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Review Process File
References
- Gandhi PN, Chen SG, Wilson-Delfosse AL. Leucine-rich repeat kinase 2 (LRRK2): a key player in the pathogenesis of Parkinson's disease. J Neurosci Res. 2009;87:1283–1295. doi: 10.1002/jnr.21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock PJ, Morais VA, Vilain S, Haddad D, et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75:1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson MW, Zhang T, Jiang C, Chen S, Guo M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet. 2012;21:1350–1363. doi: 10.1093/hmg/ddr573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Suaga P, Rivero-Rios P, Fdez E, Blanca Ramirez M, Ferrer I, Aiastui A, Lopez De Munain A, Hilfiker S. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu395. doi: 10.1093/hmg/ddu395. [DOI] [PubMed] [Google Scholar]
- Meixner A, Boldt K, Van Troys M, Askenazi M, Gloeckner CJ, Bauer M, Marto JA, Ampe C, Kinkl N, Ueffing M. A QUICK screen for Lrrk2 interaction partners–leucine-rich repeat kinase 2 is involved in actin cytoskeleton dynamics. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.001172. M110 001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D, Citro A, Cordy JM, Shen GC, Wolozin B. Rac1 protein rescues neurite retraction caused by G2019S leucine-rich repeat kinase 2 (LRRK2) J Biol Chem. 2011;286:16140–16149. doi: 10.1074/jbc.M111.234005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habig K, Gellhaar S, Heim B, Djuric V, Giesert F, Wurst W, Walter C, Hentrich T, Riess O, Bonin M. LRRK2 guides the actin cytoskeleton at growth cones together with ARHGEF7 and Tropomyosin 4. Biochim Biophys Acta. 2013;1832:2352–2367. doi: 10.1016/j.bbadis.2013.09.009. [DOI] [PubMed] [Google Scholar]
- Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- Saheki Y, De Camilli P. Synaptic vesicle endocytosis. Cold Spring Harb Perspect Biol. 2012;4:a005645. doi: 10.1101/cshperspect.a005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochim Biophys Acta. 2009;1793:605–614. doi: 10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat. 2013;34:1200–1207. doi: 10.1002/humu.22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S, Jalas C, Lesage S, Brice A, Taraboulos A, et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE. 2012;7:e36458. doi: 10.1371/journal.pone.0036458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilarino-Guell C, Rajput A, Milnerwood AJ, Shah B, Szu-Tu C, Trinh J, Yu I, Encarnacion M, Munsie LN, Tapia L, et al. DNAJC13 mutations in Parkinson disease. Hum Mol Genet. 2014;23:1794–1801. doi: 10.1093/hmg/ddt570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky FM, Chen CY, Knuehl C, Towler MC, Wakeham DE. Biological basket weaving: formation and function of clathrin-coated vesicles. Annu Rev Cell Dev Biol. 2001;17:517–568. doi: 10.1146/annurev.cellbio.17.1.517. [DOI] [PubMed] [Google Scholar]
- Legendre-Guillemin V, Metzler M, Lemaire JF, Philie J, Gan L, Hayden MR, McPherson PS. Huntingtin interacting protein 1 (HIP1) regulates clathrin assembly through direct binding to the regulatory region of the clathrin light chain. J Biol Chem. 2005;280:6101–6108. doi: 10.1074/jbc.M408430200. [DOI] [PubMed] [Google Scholar]
- Poupon V, Girard M, Legendre-Guillemin V, Thomas S, Bourbonniere L, Philie J, Bright NA, McPherson PS. Clathrin light chains function in mannose phosphate receptor trafficking via regulation of actin assembly. Proc Natl Acad Sci USA. 2008;105:168–173. doi: 10.1073/pnas.0707269105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett TJ, Legendre-Guillemin V, McPherson PS, Fremont DH. Structural definition of the F-actin-binding THATCH domain from HIP1R. Nat Struct Mol Biol. 2006;13:121–130. doi: 10.1038/nsmb1043. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein AE, Zhang CX, Carreno S, Barroso C, Heuser JE, Drubin DG. RNAi-mediated Hip1R silencing results in stable association between the endocytic machinery and the actin assembly machinery. Mol Biol Cell. 2004;15:1666–1679. doi: 10.1091/mbc.E03-09-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachse M, Urbe S, Oorschot V, Strous GJ, Klumperman J. Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol Biol Cell. 2002;13:1313–1328. doi: 10.1091/mbc.01-10-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Bache KG, Mehlum A, Stang E, Stenmark H. Hrs recruits clathrin to early endosomes. EMBO J. 2001;20:5008–5021. doi: 10.1093/emboj/20.17.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Parkinson MD, Gray SR, Bright NA. The delivery of endocytosed cargo to lysosomes. Biochem Soc Trans. 2009;37:1019–1021. doi: 10.1042/BST0371019. [DOI] [PubMed] [Google Scholar]
- Acton SL, Brodsky FM. Predominance of clathrin light chain LCb correlates with the presence of a regulated secretory pathway. J Cell Biol. 1990;111:1419–1426. doi: 10.1083/jcb.111.4.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky FM. Diversity of clathrin function: new tricks for an old protein. Annu Rev Cell Dev Biol. 2012;28:309–336. doi: 10.1146/annurev-cellbio-101011-155716. [DOI] [PubMed] [Google Scholar]
- Wilbur JD, Hwang PK, Ybe JA, Lane M, Sellers BD, Jacobson MP, Fletterick RJ, Brodsky FM. Conformation switching of clathrin light chain regulates clathrin lattice assembly. Dev Cell. 2010;18:841–848. doi: 10.1016/j.devcel.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard M, Allaire PD, Blondeau F, McPherson PS. Isolation of clathrin-coated vesicles by differential and density gradient centrifugation. Curr Protoc Cell Biol. 2005 doi: 10.1002/0471143030.cb0313s26. Chapter 3, Unit 3 13. [DOI] [PubMed] [Google Scholar]
- Hatano T, Kubo S, Imai S, Maeda M, Ishikawa K, Mizuno Y, Hattori N. Leucine-rich repeat kinase 2 associates with lipid rafts. Hum Mol Genet. 2007;16:678–690. doi: 10.1093/hmg/ddm013. [DOI] [PubMed] [Google Scholar]
- Davies P, Hinkle KM, Sukar NN, Sepulveda B, Mesias R, Serrano G, Alessi DR, Beach TG, Benson DL, White CL, et al. Comprehensive characterization and optimization of anti-LRRK2 (leucine-rich repeat kinase 2) monoclonal antibodies. Biochemical Journal. 2013;453:101–113. doi: 10.1042/BJ20121742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamidessi A, Frittoli E, Garre M, Faretta M, Mione M, Testa I, Diaspro A, Lanzetti L, Scita G, Di Fiore PP. Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell. 2008;134:135–147. doi: 10.1016/j.cell.2008.05.034. [DOI] [PubMed] [Google Scholar]
- Teo M, Manser E, Lim L. Identification and molecular cloning of a p21cdc42/rac1-activated serine/threonine kinase that is rapidly activated by thrombin in platelets. J Biol Chem. 1995;270:26690–26697. doi: 10.1074/jbc.270.44.26690. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Liu HP, Lin WY, Guo H, Lu B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J Neurosci. 2010;30:16959–16969. doi: 10.1523/JNEUROSCI.1807-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L, Gu XL, Lin X, Crowley NA, Lovinger DM, et al. LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci. 2014;17:367–376. doi: 10.1038/nn.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter B, Murphy S, Dokainish H, Girard M, Gudheti MV, Kozlov G, Halin M, Philie J, Jorgensen EM, Gehring K, et al. NECAP 1 regulates AP-2 interactions to control vesicle size, number, and cargo during clathrin-mediated endocytosis. PLoS Biol. 2013;11:e1001670. doi: 10.1371/journal.pbio.1001670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan KM, Barrett K, Lu Y, Hu KQ, Vincent S, Settleman J. Myoblast city, the Drosophila homolog of DOCK180/CED-5, is required in a Rac signaling pathway utilized for multiple developmental processes. Genes Dev. 1998;12:3337–3342. doi: 10.1101/gad.12.21.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stronach B, Perrimon N. Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev. 2002;16:377–387. doi: 10.1101/gad.953002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Review Process File