

Abstract
Mitochondrial proteostasis is maintained by a network of ATP-dependent quality control proteases including the inner membrane protease YME1L. Here, we show that YME1L is a stress-sensitive mitochondrial protease that is rapidly degraded in response to acute oxidative stress. This degradation requires reductions in cellular ATP and involves the activity of the ATP-independent protease OMA1. Oxidative stress-dependent reductions in YME1L inhibit protective YME1L-dependent functions and increase cellular sensitivity to oxidative insult. Collectively, our results identify stress-induced YME1L degradation as a biologic process that attenuates protective regulation of mitochondrial proteostasis and promotes cellular death in response to oxidative stress.
Keywords: AAA protease, mitochondrial proteostasis, oxidative stress, YME1L
Introduction
The mitochondrial inner membrane is the site of essential mitochondrial functions including electron transport chain activity, phospholipid metabolism, and the regulation of apoptotic signaling. Dysfunction of these processes is intricately associated with the onset and pathology of etiologically diverse human diseases including cancer, diabetes, and neurodegenerative diseases 1–5. Despite the involvement of mitochondrial dysfunction in disease pathogenesis, the underlying molecular mechanisms that lead to altered mitochondrial function in these disorders remain poorly defined. One potential mechanism to explain alterations in mitochondrial function in disease is a decreased capacity to maintain mitochondrial inner membrane protein homeostasis (or proteostasis).
Mitochondrial inner membrane proteostasis is maintained by ATP-dependent AAA+ quality control proteases including YME1L, AFG3L2, and paraplegin. These proteases assemble into homo- or hetero-oligomeric complexes with active sites oriented toward the intermembrane space (IMS; YME1L homooligomers) or the mitochondrial matrix (AFG3L2 homooligomers and AFG3L2/paraplegin heterooligomers) (Fig1A) 6. Mitochondrial inner membrane proteases are involved in nearly all aspects of mitochondrial biology including the maintenance of electron transport chain integrity, regulation of phospholipid metabolism, and the control of mitochondrial morphology and dynamics 7. Alterations in the activity of these proteases significantly impair mitochondrial functions. Genetic depletion of YME1L decreases cellular capacity to regulate mitochondrial inner membrane proteostasis, impairs the regulation of mitochondrial dynamics, alters phospholipid metabolism, attenuates the activity of the electron transport chain, and increases stress sensitivity to apoptotic stimuli 8–12. Similarly, loss of AFG3L2 disrupts mitochondrial proteostasis, mitochondrial protein translation, and trafficking of mitochondria in neuronal axons 13–15. Mutations in AFG3L2 and paraplegin are also associated with the neurodegenerative disorders spinocerebellar ataxia type 28 and hereditary spastic paraplegia type 7, respectively 16,17. Thus, alterations in the activity of these proteases could disrupt mitochondrial inner membrane proteostasis and contribute to the pathologic mitochondrial dysfunction associated with human disease.
Figure 1. YME1L is a stress-sensitive mitochondrial protease.
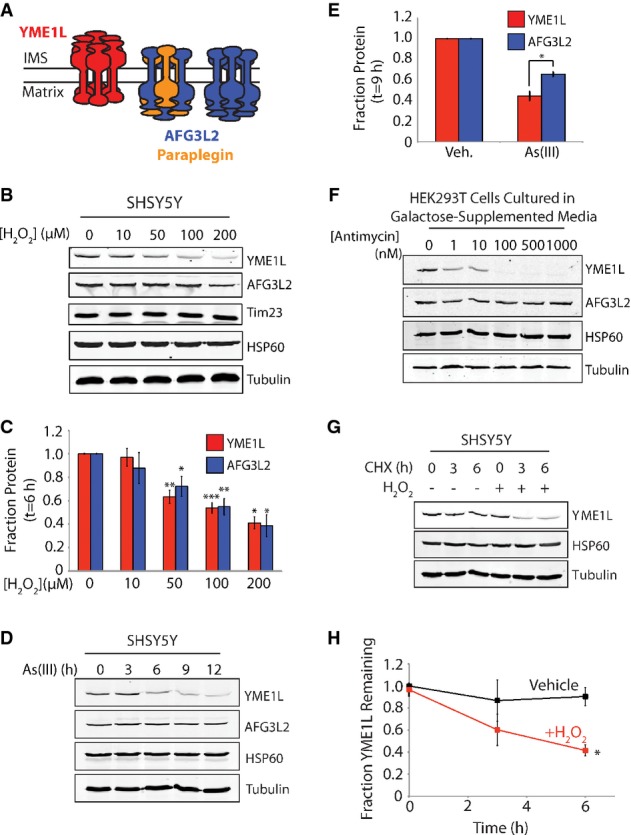
- Illustration showing the oligomer assembly and active site orientation of YME1L (red), AFG3L2 (blue), and paraplegin (orange) in the mitochondrial inner membrane.
- Representative immunoblot of lysates prepared from SHSY5Y cells treated with the indicated concentration of H2O2 for 6 h.
- Quantification of YME1L and AFG3L2 from immunoblots as shown in (B). Error bars show SEM for n ≥ 3. ***P-value < 0.001, **P-value < 0.01, *P-value < 0.05.
- Representative immunoblot of lysates prepared from SHSY5Y cells treated with As(III) (50 μM) for the indicated time.
- Quantification of YME1L and AFG3L2 in SHSY5Y cells treated with As(III) (50 μM; 9 h) from immunoblots as shown in (D). Error bars show SEM for n = 4. *P-value < 0.05.
- Immunoblots of lysates prepared from HEK293T cells cultured in galactose-supplemented media treated with the indicated concentration of antimycin A for 6 h.
- Representative immunoblot of lysates prepared from SHSY5Y cells treated as indicated with cycloheximide (CHX; 50 μg ml−1) and H2O2 (100 μM) for the indicated time.
- Quantification of YME1L protein levels from immunoblots as shown in (G). Error bars show SEM for n = 3. *P-value < 0.05.
Source data are available online for this figure.
Here, we identify YME1L as a stress-sensitive mitochondrial inner membrane protease. We show that YME1L is degraded in response to acute oxidative stress in a mechanism involving reductions in cellular ATP and the activity of the ATP-independent mitochondrial inner membrane protease OMA1. Furthermore, we show that stress-induced YME1L degradation impairs YME1L-dependent regulation of mitochondrial inner membrane proteostasis, sensitizing cells to oxidative insult. Collectively, our work reveals stress-induced YME1L degradation as a mechanism that can contribute to the pathologic mitochondrial dysfunction associated with many human diseases.
Results and Discussion
YME1L is degraded in response to oxidative stress
In order to identify stress-induced alterations in inner membrane protease composition, we monitored YME1L and AFG3L2 protein levels by quantitative immunoblotting in cells treated with various oxidative stressors. H2O2 decreased YME1L and AFG3L2 protein levels in SHSY5Y cells (Fig1B and C). Other mitochondrial inner membrane proteins such as Tim23 were not affected by H2O2 (Fig1B, Supplementary Fig S1A). Similar results were observed in all cell lines tested including N2a, HEK293T, and HeLa cells—the latter, a cell line that lacks PARKIN 18–20, indicating that the loss of YME1L and AFG3L2 is independent of mitophagy (Supplementary Fig S1B–D). The superoxide generator paraquat also reduced YME1L and AFG3L2 protein levels in SHSY5Y cells (Supplementary Fig S1E). Alternatively, the environmental toxin As(III) selectively reduced YME1L protein levels relative to AFG3L2 (FigD and E). YME1L was also substantially decreased in HEK293T cells cultured in galactose-supplemented media treated with the complex III inhibitor antimycin A, while AFG3L2 was not affected in these cells (Fig1F). The addition of antimycin A to cells cultured in glucose-containing media did not influence YME1L protein levels (Supplementary Fig S1F). These results show that YME1L is preferentially sensitive to oxidative stress relative to AFG3L2.
We next evaluated whether reduced YME1L results from a stress-induced decrease in YME1L stability. We employed quantitative immunoblotting to measure YME1L stability in SHSY5Y cells treated with cycloheximide (CHX) for 0, 3, or 6 h in the presence or absence of H2O2 (Fig1G and H). In the absence of H2O2, YME1L is a stable protein, demonstrating near complete recovery following a 6-h CHX treatment. In contrast, co-administration of CHX and H2O2 significantly reduced YME1L protein levels, indicating that H2O2 increases YME1L targeting to proteolytic degradation.
YME1L degradation involves the activity of the ATP-independent mitochondrial protease OMA1
We characterized the molecular mechanism of YME1L degradation in isolated mitochondria. YME1L is degraded in isolated mitochondria incubated in the absence of ATP, while AFG3L2 is not affected under these conditions (Fig2A). The addition of ATP inhibits YME1L degradation in isolated mitochondria (Fig2A). Identical results were observed in mitochondria purified from mouse livers (Supplementary Fig S2A). Incubating isolated mitochondria with H2O2 does not induce YME1L degradation in the presence of ATP, suggesting that ATP depletion as a consequence of oxidative insult, and not the oxidative insult itself, is necessary to induce YME1L degradation (Supplementary Fig S2B). The degradation of YME1L in isolated mitochondria is also inhibited by ADP, but not AMP (Fig2B). The stabilization of YME1L afforded by ADP cannot be attributed to conversion of ADP to ATP by mitochondrial ATP synthase, as the addition of oligomycin A did not influence ADP-dependent YME1L stabilization (Supplementary Fig S2C). The non-hydrolyzable ATP analog AMP-PNP also inhibits YME1L degradation in isolated mitochondria (Fig2B). These results suggest that binding of nucleotide stabilizes YME1L against degradation independent of ATP hydrolysis.
Figure 2. YME1L degradation involves the activity of the ATP-independent mitochondrial protease OMA1.
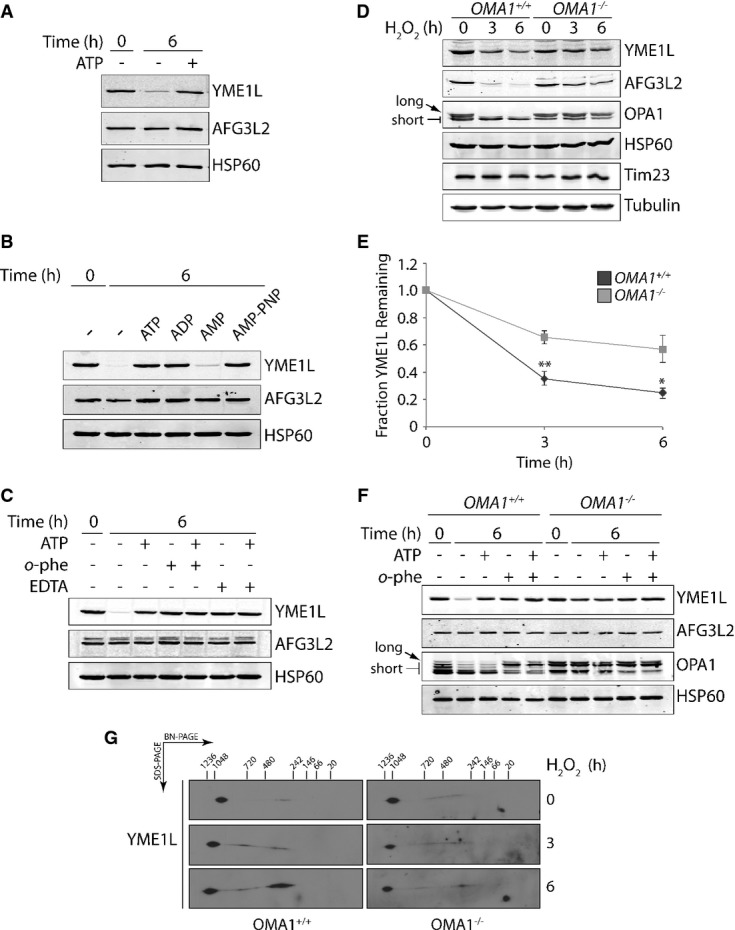
- Immunoblot of mitochondria purified from SHSY5Y cells and incubated at 37°C in the presence or absence of ATP (5 mM) for 0 or 6 h, as indicated.
- Immunoblot of mitochondria purified from SHSY5Y cells and incubated at 37°C for 0 or 6 h in the presence of ATP (5 mM), ADP (5 mM), AMP (5 mM), or AMP-PNP (5 mM), as indicated.
- Immunoblot of mitochondria purified from SHSY5Y cells and incubated at 37°C in the presence of ATP (5 mM), o-phenanthroline (o-phe; 1 mM), or EDTA (5 mM) for 0 or 6 h, as indicated.
- Representative immunoblot of lysates prepared from OMA1+/+ or OMA1−/− MEFs treated with H2O2 (200 μM) for the indicated time. The long and short isoforms of OPA1 are indicated by the arrows.
- Quantification of normalized YME1L protein levels from immunoblots as shown in (D). Error bars show SEM for n = 4. **P-value < 0.01, *P-value < 0.05.
- Immunoblot of mitochondria purified from OMA1+/+ or OMA1−/− MEFs incubated at 37°C in the presence of ATP (5 mM) or o-phenanthroline (o-phe; 1 mM) for 0 or 6 h, as indicated. The long and short isoforms of OPA1 are indicated by the arrows.
- Immunoblot of BN-PAGE/SDS–PAGE of mitochondria isolated from OMA1+/+ and OMA1−/− from cells treated with H2O2 (200 μM) for 0, 3, or 6 h, as indicated. These immunoblots were overexposed to observe alterations in YME1L oligomeric structure in these cells despite reductions in YME1L protein levels.
Source data are available online for this figure.
YME1L degradation in isolated mitochondria incubated in the absence of ATP is also inhibited by the zinc chelator o-phenanthroline (o-phe) and the divalent cation chelator EDTA (Fig2C). Inhibitors of serine, cysteine, and aspartic acid proteases did not inhibit YME1L degradation in isolated mitochondria (Supplementary Fig S2D). This suggests that YME1L degradation involves the activity of an ATP-independent zinc metalloprotease. Specifically, a metalloprotease with an active site oriented toward the intermembrane space (IMS), as EDTA cannot cross the inner mitochondrial membrane and only inhibits metalloproteases with IMS-oriented active sites 21. Currently, the only known ATP-independent zinc metalloprotease embedded in the mitochondrial inner membrane is OMA1. OMA1 is activated by depolarization of the mitochondrial membrane and functions to maintain mitochondrial proteostasis through the degradation of misfolded proteins within the inner membrane 22 and the regulation of mitochondrial dynamics through the stress-induced proteolytic processing of the dynamin-like GTPase OPA1 23,24.
To interrogate the role of OMA1 in stress-induced YME1L degradation, we measured H2O2-dependent YME1L degradation in wild-type (OMA1+/+) mouse embryonic fibroblasts (MEFs) and MEFs homozygous for a null allele of OMA1 (OMA1−/−) 25. YME1L is efficiently degraded in H2O2-treated OMA1+/+ MEFs (Fig2D and E, and Supplementary Fig S2E). The activation of OMA1 in OMA1+/+ MEFs was confirmed by the H2O2-dependent increase in OPA1 processing from long to short isoforms (arrows in Fig2D and Supplementary Fig S2E). Alternatively, H2O2-dependent YME1L degradation is attenuated, but not completely inhibited, in OMA1−/− MEFs. AFG3L2 degradation was likewise reduced in H2O2-treated OMA1−/− MEFs (Fig2D). In vitro YME1L degradation was also attenuated, but not completely inhibited, in mitochondria purified from OMA1−/− MEFs incubated in the absence of ATP (Fig2F). The activation of OMA1 in these experiments is evident in mitochondria purified from OMA1+/+, but not OMA1−/−, MEFs by OPA1 processing. This inability to completely inhibit YME1L degradation in OMA1−/− MEFs or OMA1−/− isolated mitochondria suggests that another mitochondrial protease(s) also contributes to stress-induced YME1L degradation. Regardless, our results implicate OMA1 activity in the regulation of YME1L stability during oxidative stress.
Oxidative stress could promote YME1L degradation through alterations in its oligomeric structure. YME1L exists as an ∼1 MDa i-AAA protease complex in mammals, comparable to the Yme1-Mgr1/3 complex in yeast 8,26,27. We monitored the YME1L complexes in OMA1+/+ and OMA1−/− MEFs treated with or without H2O2 using Blue Native polyacrylamide gel electrophoresis (BN-PAGE). YME1L primarily forms an ∼1 MDa oligomeric complex in both OMA1+/+ and OMA1−/− MEFs, with a small population of smaller YME1L complexes between 720 kDa and 242 kDa (Fig2G). The addition of H2O2 increases the populations of these smaller complexes, indicating that H2O2 affects YME1L complex stability (Fig2G). This suggests that YME1L degradation may proceed through dissociation of the YME1L oligomer, although further mechanistic studies are required to define a specific role for YME1L dissociation in this process.
YME1L stability is sensitive to reduced ATP levels and mitochondrial membrane depolarization
The results shown in Fig2 suggest that YME1L stability is sensitive to intracellular nucleotide levels and OMA1 activation. In order to separate the contributions of these two parameters on YME1L stability, we treated HEK293T cells with 2-deoxyglucose (2-DG; an inhibitor of glycolysis) and/or carbonyl cyanide m-chlorophenyl hydrazone (CCCP; an uncoupler that depolarizes the mitochondrial membrane and activates OMA1). We then measured cellular ATP levels, OMA1 activation (via OPA1 processing), and YME1L protein levels. The addition of 2-DG reduces cellular ATP by 60%, but does not increase OPA1 processing and only modestly decreases YME1L (Fig3A). The addition of CCCP decreases ATP levels by ∼20% and increases OPA1 processing, but again only modestly decreases YME1L. The co-administration of 2-DG and CCCP reduces ATP levels > 90%, increases OPA1 processing, and decreases YME1L by 85%. The addition of 2-DG and/or CCCP does not affect AFG3L2 protein levels in these cells (Fig3A). These results demonstrate that neither depletion of ATP nor OMA1 activation alone recapitulates the extent of YME1L degradation observed during oxidative stress. Only a combination of these two treatments induces a robust decrease in YME1L. The inability of OMA1 activation alone to induce YME1L degradation is also shown by the ATP-dependent stabilization of YME1L in isolated mitochondria where OMA1 is active (Fig2F).
Figure 3.
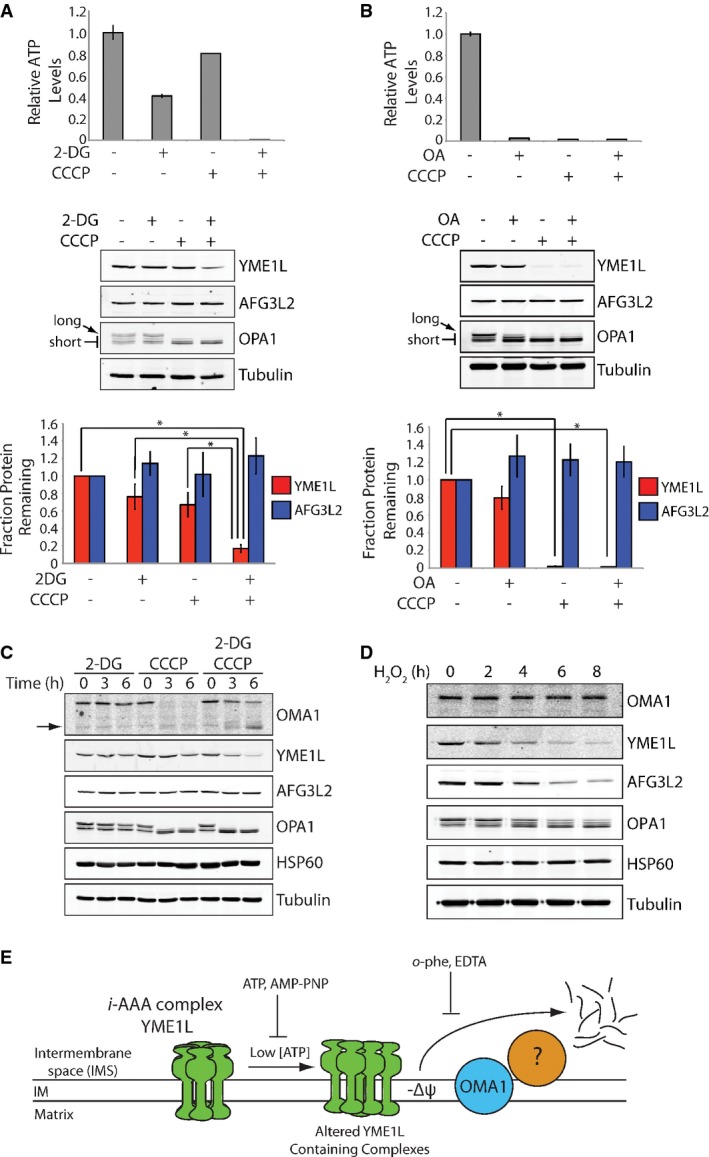
YME1L degradation is initiated by reductions in intracellular ATP and OMA1 activation
- Top, bar graph showing the intracellular ATP levels in HEK293T cells incubated with 2-deoxyglucose (2-DG; 10 mM) and/or CCCP (50 μM) for 6 h, as indicated. Error bars show SEM for n = 3. Middle, representative immunoblot of lysates prepared from HEK293T cells incubated with 2-deoxyglucose (2-DG; 10 mM) and/or CCCP (50 μM) for 6 h, as indicated. The long and short isoforms of OPA1 are indicated by the arrows. Bottom, quantification of YME1L (red) and AFG3L2 (blue) from immunoblots as shown above. Error bars show SEM for n = 3. *P-value < 0.05.
- Top, bar graph showing the intracellular ATP levels in HEK293T cells cultured in galactose-supplemented media incubated with oligomycin A (OA; 5 nM) and/or CCCP (50 μM) for 6 h, as indicated. Error bars show SEM for n = 3. Middle, representative immunoblot of lysates prepared from HEK293T cells cultured in galactose-supplemented media incubated with oligomycin A (OA; 5 nM) and/or CCCP (50 μM) for 6 h, as indicated. The long and short isoforms of OPA1 are indicated by the arrows. Bottom, quantification of YME1L (red) and AFG3L2 (blue) from immunoblots as shown above. Error bars show SEM for n = 3. *P-value < 0.05.
- Immunoblot of lysates from HEK293T cells incubated with 2-deoxyglucose (2-DG; 10 mM) and/or CCCP (50 μM) for the indicated times. Full-length OMA1 (FL) and a cleaved, proteolytically active OMA1 fragment are shown by the arrows.
- Immunoblot of lysates from SHSY5Y cells treated with H2O2 (200 μM) for the indicated times.
- Illustration depicting the predicted mechanism for stress-induced YME1L degradation.
Source data are available online for this figure.
We further explored the relative dependence of YME1L stability on nucleotide depletion and OMA1 activation by treating HEK293T cells cultured in galactose-supplemented media with the ATP synthase inhibitor oligomycin A and/or CCCP. Under these culture conditions, oligomycin A reduces cellular ATP levels, as the majority of ATP is produced through mitochondrial oxidative phosphorylation. Alternatively, CCCP both activates OMA1 and reduces cellular ATP through disruption of the mitochondrial inner membrane potential required for oxidative phosphorylation. The addition of oligomycin A reduces ATP levels > 90%, but does not significantly activate OMA1 or reduce YME1L (Fig3B). Alternatively, the addition of CCCP alone or in combination with oligomycin A depletes ATP levels > 90%, significantly activates OMA1, and reduces YME1L > 90%. Again, AFG3L2 is not decreased in cells treated with oligomycin A and/or CCCP (Fig3B).
OMA1 undergoes rapid degradation in response to stresses that depolarize the mitochondrial membrane such as CCCP 22,28. Thus, we evaluated the relationship between OMA1 and YME1L degradation induced by CCCP in the presence or absence of 2-DG. As observed previously, CCCP induces a reduction in total OMA1 protein levels (Fig3C) 22,28. Alternatively, the co-addition of 2-DG with CCCP slows OMA1 degradation and results in the accumulation of a proteolytically processed OMA1 isoform previously shown to retain protease activity (arrows in Fig3C) 28. The addition of 2-DG alone does not influence OMA1 stability. These results suggest that CCCP-dependent OMA1 degradation is sensitive to cellular ATP levels. Consistent with this prediction, efficient OMA1 degradation in isolated mitochondria requires the addition of exogenous ATP (Supplementary Fig S3A). Other stresses that induce YME1L degradation such as H2O2 and antimycin A in galactose-cultured cells also do not induce OMA1 degradation (Fig3D and Supplementary Fig S3B), although antimycin A promotes OMA1 cleavage into the active proteolytic fragment. The activation of OMA1 in antimycin-treated cells cultured in galactose is consistent with the depolarization of the mitochondrial membrane in these cells, as shown by reduction in TMRE fluorescence (Supplementary Fig S3C). These results show that OMA1 and YME1L degradation are differentially sensitive to cellular stresses, potentially yielding distinctive mitochondrial inner membrane proteolytic activities in response to specific pathologic insults.
The results shown in Fig3 demonstrate that YME1L intracellular stability is sensitive to reductions in cellular ATP and inner membrane depolarization, the latter of which activates OMA1. Combined with the biochemical characterization of YME1L degradation shown in Fig2, these results suggest a model for the mechanism of stress-induced YME1L degradation whereby a reduction in cellular ATP alters YME1L conformational stability through reduced nucleotide binding, increasing the targeting of YME1L to proteolytic degradation mediated through a process involving active OMA1 and at least one additional mitochondrial protease(s) (Fig3E).
Loss of YME1L decreases the regulation of mitochondrial proteostasis and sensitizes cells to oxidative insult
Stress-induced YME1L degradation could represent an early step in apoptotic signaling. To test this prediction, we monitored apoptotic signaling by caspase-3 activation and PARP cleavage in cells treated with stresses that induce YME1L degradation. Despite inducing robust YME1L degradation, we observe no caspase-3 activation or PARP cleavage in N2a cells treated with H2O2 (Fig4A) or HEK293T cells cultured in galactose-supplemented media and treated with antimycin A (Supplementary Fig S3B). Conversely, staurosporine robustly activates apoptosis without significantly affecting YME1L stability (Fig4A). Alternatively, YME1L degradation, caspase-3 activation, and PARP cleavage are all observed in As(III)-treated SHSY5Y cells (Supplementary Fig S4A). These results indicate that stress-induced YME1L degradation does not correspond with the initiation of apoptosis. Furthermore, YME1L protein levels recover over a 24-h period following a H2O2 insult, mirroring the recovery of full-length OPA1 (Fig4B) 29. This suggests that YME1L degradation does not represent a ‘point of no return’ for cell death.
Figure 4.
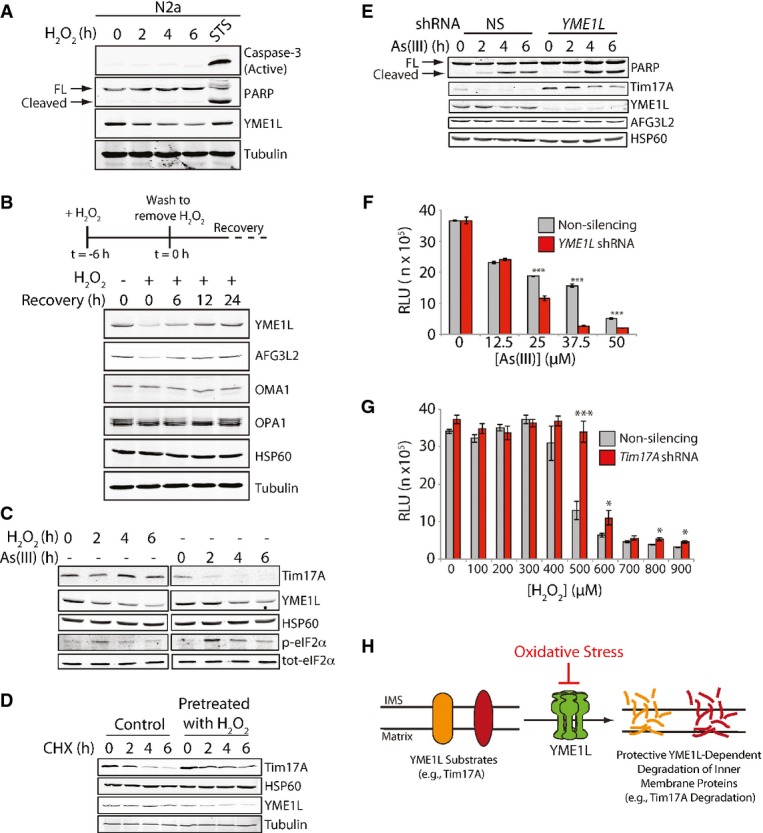
Loss of YME1L decreases cellular capacity to regulate mitochondrial proteostasis and sensitizes cells to oxidative insult
- Immunoblot of lysates prepared from N2a cells treated with H2O2 (200 μM) for the indicated times. Lysates prepared from N2a cells treated with staurosporine (STS; 1 μM, 6 h) are shown as a control for apoptosis. Full-length (FL) and cleaved PARP are shown by the arrows.
- Immunoblot of lysates from SHSY5Y cells pretreated with H2O2 (200 μM) for 6 h, as indicated, then washed, and incubated in fresh media for the indicated times. An illustration of the experimental design for this experiment is shown above.
- Immunoblot of lysates prepared from HEK293T cells treated with the H2O2 (100 μM) or As(III) (50 μM) for the indicated time. Note the H2O2-dependent increase in eIF2α phosphorylation, but lack of Tim17A degradation in these cells.
- Immunoblot of lysates prepared from HEK293T cells treated with cycloheximide (CHX; 50 μg ml−1) in the presence or absence of an H2O2 pretreatment (100 μM; 3 h).
- Immunoblot of lysates prepared from SHSY5Y cells stably expressing non-silencing (NS) shRNA or YME1L shRNA. Cells were treated with As(III) (50 μM) for the indicated time. Full-length (FL) and cleaved PARP are shown by the arrows.
- Graph showing the viability of SHSY5Y cells expressing non-silencing (NS) shRNA or YME1L shRNA treated with increasing concentrations of As(III) for 24 h. Cell viability was measured using CellTiter-Glo. Error bars show SEM for n = 3. ***P-value < 0.001.
- Graph showing the viability of HEK293T cells expressing non-silencing (NS) shRNA or TIM17A shRNA treated with increasing concentrations of H2O2 for 24 h. Cell viability was measured using CellTiter-Glo. Error bars show SEM for n = 3. ***P-value < 0.001, *P-value < 0.05. These results are representative of 3 independent experiments.
- Illustration showing that oxidative insults that induce YME1L degradation decrease protective YME1L-dependent, stress-regulated degradation of inner mitochondrial membrane proteins. This decreases cellular capacity to maintain critical mitochondrial inner membrane functions during stress and promotes cellular death.
Source data are available online for this figure.
Alternatively, YME1L degradation could decrease the capacity for cells to regulate inner membrane proteostasis. To explore this potential consequence of YME1L degradation, we monitored the impact of oxidative stress on YME1L-mediated regulation of the TIM23 mitochondrial protein import complex 30. Mammalian TIM23 forms two exclusive complexes containing distinct core interactions between the subunit Tim23 and one of the two mammalian paralogs of yeast Tim17, Tim17A, or Tim17B 31. Previous work showed that Tim17A is a stress-regulated TIM23 subunit that is rapidly degraded by YME1L in response to eIF2α phosphorylation-dependent translational attenuation 30. However, Tim17B is not subject to this regulation. Thus, YME1L-mediated degradation of Tim17A reduces the population of active TIM23 complexes containing a core Tim23–Tim17A interaction without impacting TIM23 complexes containing a core Tim23–Tim17B interaction 30. This provides a mechanism for cells to sensitively attenuate, but not completely inhibit, TIM23-dependent protein import in response to pathologic insult 30. The attenuation in mitochondrial protein import afforded by Tim17A degradation is predicted to promote mitochondrial proteostasis through mechanisms such as reducing the population of newly synthesized unfolded proteins entering mitochondrial during stress and by promoting transcriptional remodeling of mitochondrial proteostasis pathways 30,32. Consistent with a protective role for Tim17A degradation, reducing Tim17A levels increases cellular viability in response to mitochondrial insults such as paraquat 30. The benefits attributed to YME1L-dependent Tim17A degradation are in contrast to those observed for the degradation of Tim23, the core channel forming subunit of TIM23 complexes, which is observed during cell death in the presence of caspase inhibitors 33. Since Tim23 is an essential subunit for all TIM23 complexes, reductions in Tim23 will decrease the cellular populations of all active TIM23 complexes, completely inhibiting TIM23-dependent mitochondrial protein import 33. Consistent with the differential impacts of reducing Tim17A or Tim23 on mitochondrial protein import, RNAi depletion of TIM23 inhibits cellular proliferation, while TIM17A depletion does not impact cellular growth 30,33. These results serve to further highlight the protective role for selective, YME1L-dependent Tim17A degradation in regulating TIM23-dependent mitochondrial protein import, promoting mitochondrial proteostasis and increasing cellular viability in response to stress 30.
H2O2 is known to induce stress-regulated eIF2α phosphorylation 34 and thus should induce Tim17A degradation. Interestingly, despite inducing eIF2α phosphorylation, Tim17A is not degraded in H2O2-treated SHSY5Y cells where YME1L is rapidly degraded (Fig4C). This is in contrast to the degradation of Tim17A observed in As(III)-treated cells. The capacity for As(III) to induce Tim17A degradation despite also promoting YME1L degradation likely reflects the slower rate of As(III)-induced YME1L degradation relative to H2O2 (c.f. Fig1D and G). The inability for H2O2 to induce Tim17A degradation could be attributed to lower levels of eIF2α phosphorylation induced by this treatment, as compared to As(III). In order to better define the capacity for H2O2 to inhibit YME1L-dependent Tim17A degradation, we measured Tim17A levels in SHSY5Y cells pretreated with H2O2 and subjected to CHX-dependent translational attenuation—a treatment that induces Tim17A degradation independent of eIF2α phosphorylation 30. Pretreatment with H2O2 attenuates CHX-dependent Tim17A degradation, consistent with H2O2-induced YME1L degradation inhibiting Tim17A regulation (Fig4D). H2O2-dependent YME1L degradation is not affected by TIM17A depletion, showing that YME1L degradation occurs upstream of Tim17A (Supplementary Fig S4B). Importantly, the addition of H2O2 to isolated mitochondria does not inhibit the YME1L-mediated, ATP-dependent degradation of Tim17A (Supplementary Fig S4C). This shows that H2O2 does not antagonize the ability for YME1L to degrade Tim17A, but instead reflects the predicted reduction in YME1L activity in cells treated with H2O2. Collectively, these results indicate that H2O2 inhibits YME1L proteolytic activity, compromising the capacity for cells to regulate mitochondrial protein import and global mitochondrial inner membrane proteostasis during oxidative stress.
The decreased capacity for YME1L to regulate mitochondrial inner membrane proteostasis should sensitize cells to stress. As reported previously 8, YME1L depletion sensitized SHSY5Y cells to apoptosis induced by staurosporine (STS) shown by increased PARP cleavage (Supplementary Fig S4D). Likewise, PARP cleavage was enhanced in YME1L-depleted cells following As(III) treatment (Fig4E). YME1L depletion also reduced cell viability in response to As(III) (Fig4F). This suggests that the loss of YME1L-dependent regulation of mitochondrial inner membrane proteostasis through activities such as Tim17A degradation sensitizes cells to oxidative insult. Consistent with this prediction, depletion of TIM17A in HEK293T cells—mimicking stress-regulated Tim17A degradation—increases cellular viability in response to H2O2, suggesting that inhibition of YME1L-dependent Tim17A degradation reduces cellular capacity to regulate mitochondria in response to oxidative insult (Fig4G). Collectively, the results shown in Fig4C–G indicate that loss of YME1L activity reduces cellular capacity to regulate mitochondrial inner membrane proteostasis during stress and increases cellular sensitivity to pathologic insult.
Conclusions
YME1L is a critical regulator of essential mitochondrial functions 8,9,11. Here, we show that YME1L is rapidly degraded through a mechanism involving the mitochondrial protease OMA1 in response to oxidative insults that deplete cellular ATP and depolarize the mitochondrial inner membrane. Reducing YME1L activity compromises the stress-dependent degradation of YME1L substrates such as Tim17A, impairing the capacity to regulate inner mitochondrial membrane proteostasis and function during stress (Fig4G). The deficiencies in mitochondrial proteostasis and quality control caused by loss of YME1L increase cellular sensitivity to subsequent oxidative stress, as indicated by the increased stress sensitivity of YME1L-depleted cells. While our results show that oxidative stress-induced YME1L degradation is a mechanism that can contribute to the pathologic mitochondrial dysfunction involved in human diseases, YME1L degradation could also play a regulatory role in adapting mitochondrial function during conditions of stress. For example, it was recently shown that YME1L activity is needed for the de novo cleavage of OPA1 to mediate fusion events 10. Therefore, YME1L degradation may provide a mechanism to sequester terminally damaged mitochondria away from the healthy pool thereby restricting them to clearance via mitophagy, although additional studies will be required to define these potentially protective consequences of stress-induced YME1L degradation.
Oxidative stress and dysregulation of mitochondrial inner membrane proteins are intricately associated in the onset and pathology of human diseases including cancer, diabetes, and neurodegenerative disorders 35,36,37,38. Despite this association, the relationship between mitochondrial dysfunction and oxidative insults remains poorly defined. Our identification of YME1L as an oxidative stress-sensitive mitochondrial protease reveals a mechanism that can contribute to pathologic dysregulation of mitochondrial inner membrane functions involved in the pathogenesis of these diseases.
Materials and Methods
Cell lines and culture conditions
SHSY5Y cells were grown in Dulbecco's modified Eagle's medium/Ham's F-12 Nutrient Mixture (DMEM/F12) supplemented with 4 mM L-glutamine, 10% FBS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin at 37°C and 5% CO2. HEK293T, N2a, HeLa, and mouse embryonic fibroblasts (MEFs) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM L-glutamine, 10% FBS, 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin at 37°C and 5% CO2. For cells grown in galactose and 2-deoxy-D-glucose treatments, see Supplementary Methods. Cell lines with stable shRNA knockdown of YME1L and Tim17A were previously described 30.
Lysate preparation and immunoblot analysis
Lysate preparation and immunoblotting were performed as described previously 30. Primary antibodies were obtained commercially and used as described in Supplementary Methods.
Mitochondrial isolation and assays
Mitochondrial were purified as described previously 39. Isolated mitochondria were handled as described previously and treated as indicated 30.
Blue Native PAGE
Blue Native PAGE was performed as described previously 30,40. Membranes were visualized using HRP-conjugated secondary antibody (Cell Signaling) and detected by enhanced chemiluminescence with Luminata Forte (Millipore) using film.
Quantification of relative ATP levels and determination of cell viability
Cells were plated in poly-D-lysine-coated 12-well plates and treated as indicated for ATP depletion or OMA1 activation. Relative ATP levels were quantified using CellTiter-Glo (Promega). Briefly, treated cells were washed into DPBS and lysed by the addition of CellTiter-Glo reagent. Lysate was transferred to a flat black 96-well plate and incubated in the dark for 10 min to stabilize signal. For cell viability assays, cells were directly seeded into flat black 96-well plates and treated as indicated before lysis with CellTiter-Glo reagent and handled similarly. Luminescence intensity was read in an Infinite F200 PRO plate reader (Tecan) and background corrected. All measurements were made in biologic triplicate.
Acknowledgments
We thank Carlos López-Otín and Pedro Moral Quirós (University of Oviedo) for providing the OMA1+/+ and OMA1−/− MEFs. We thank Chris Vickers and Dennis Wolan (TSRI) for advice and critical comments. We also thank Michael Petrascheck, Jeff Kelly (TSRI), and Cole Haynes (MSKCC) for fruitful discussions. This work was supported by the Ellison Medical Foundation, Arlene and Arnold Goldstein, and NIH (R21NS079882).
Author contributions
RLW guided the project. TKR, JMS, and RLW designed and carried out the experiments, and analyzed the data. TKR and RLW prepared the figures, discussed the data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Review Process File
Source Data for Figure 1D, G
Source Data for Figure 2A, B
Source Data for Figure 3A, B, C
Source Data for Figure 4A, B
References
- Parish R, Petersen KF. Mitochondrial dysfunction and type 2 diabetes. Curr Diab Rep. 2005;5:177–183. doi: 10.1007/s11892-005-0006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal BE. Role of mitochondrial mutations in cancer. Endocr Pathol. 2006;17:203–212. doi: 10.1385/ep:17:3:203. [DOI] [PubMed] [Google Scholar]
- Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–4662. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther. 2012;342:619–630. doi: 10.1124/jpet.112.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klanner C, Prokisch H, Langer T. MAP-1 and IAP-1, two novel AAA proteases with catalytic sites on opposite membrane surfaces in mitochondrial inner membrane of Neurospora crassa. Mol Biol Cell. 2001;12:2858–2869. doi: 10.1091/mbc.12.9.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiburek L, Cesnekova J, Kostkova O, Fornuskova D, Vinsova K, Wenchich L, Houstek J, Zeman J. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol Biol Cell. 2012;23:1010–1023. doi: 10.1091/mbc.E11-08-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014;19:630–641. doi: 10.1016/j.cmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potting C, Tatsuta T, Konig T, Haag M, Wai T, Aaltonen MJ, Langer T. TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab. 2013;18:287–295. doi: 10.1016/j.cmet.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Claypool SM, Whited K, Srijumnong S, Han X, Koehler CM. Barth syndrome mutations that cause tafazzin complex lability. J Cell Biol. 2011;192:447–462. doi: 10.1083/jcb.201008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonn F, Tatsuta T, Petrungaro C, Riemer J, Langer T. Presequence-dependent folding ensures MrpL32 processing by the m-AAA protease in mitochondria. EMBO J. 2011;30:2545–2556. doi: 10.1038/emboj.2011.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondadi AK, Wang S, Montagner S, Kladt N, Korwitz A, Martinelli P, Herholz D, Baker MJ, Schauss AC, Langer T, et al. Loss of the m-AAA protease subunit AFG3L2 causes mitochondrial transport defects and tau hyperphosphorylation. EMBO J. 2014;33:1011–1026. doi: 10.1002/embj.201387009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltecca F, Casari G. In vivo detection of oxidized proteins: a practical approach to tissue-derived mitochondria. Methods Mol Biol. 2010;648:257–267. doi: 10.1007/978-1-60761-756-3_17. [DOI] [PubMed] [Google Scholar]
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, Pastore A, Finardi A, Cagnoli C, Tempia F, Frontali M, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 2010;42:313–321. doi: 10.1038/ng.544. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlyk AC, Giasson BI, Sampathu DM, Perez FA, Lim KL, Dawson VL, Dawson TM, Palmiter RD, Trojanowski JQ, Lee VM. Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J Biol Chem. 2003;278:48120–48128. doi: 10.1074/jbc.M306889200. [DOI] [PubMed] [Google Scholar]
- Denison SR, Wang F, Becker NA, Schule B, Kock N, Phillips LA, Klein C, Smith DI. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene. 2003;22:8370–8378. doi: 10.1038/sj.onc.1207072. [DOI] [PubMed] [Google Scholar]
- Kaser M, Kambacheld M, Kisters-Woike B, Langer T. Oma1, a novel membrane-bound metallopeptidase in mitochondria with activities overlapping with the m-AAA protease. J Biol Chem. 2003;278:46414–46423. doi: 10.1074/jbc.M305584200. [DOI] [PubMed] [Google Scholar]
- Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, Langer T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014;33:578–593. doi: 10.1002/embj.201386474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. 2014;204:919–929. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirós PM, Ramsay AJ, Sala D, Fernández-Vizarra E, Rodríguez F, Peinado JR, Fernández-García MS, Vega JA, Enríquez JA, Zorzano A, et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012;31:2117–2133. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CD, Lee MS, Spencer FA, Jensen RE. A genomewide screen for petite-negative yeast strains yields a new subunit of the i-AAA protease complex. Mol Biol Cell. 2006;17:213–226. doi: 10.1091/mbc.E05-06-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CD, Tamura Y, Sesaki H, Jensen RE. Mgr3p and Mgr1p are adaptors for the mitochondrial i-AAA protease complex. Mol Biol Cell. 2008;19:5387–5397. doi: 10.1091/mbc.E08-01-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Li H, Song Z. Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep. 2014;15:576–585. doi: 10.1002/embr.201338240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainbolt TK, Atanassova N, Genereux JC, Wiseman RL. Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 2013;18:908–919. doi: 10.1016/j.cmet.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer MF, Gempel K, Reichert AS, Rappold GA, Lichtner P, Gerbitz KD, Neupert W, Brunner M, Hofmann S. Genetic and structural characterization of the human mitochondrial inner membrane translocase. J Mol Biol. 1999;289:69–82. doi: 10.1006/jmbi.1999.2751. [DOI] [PubMed] [Google Scholar]
- Rainbolt TK, Saunders JM, Wiseman RL. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol Metab. 2014;25:528–537. doi: 10.1016/j.tem.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Goemans CG, Boya P, Skirrow CJ, Tolkovsky AM. Intra-mitochondrial degradation of Tim23 curtails the survival of cells rescued from apoptosis by caspase inhibitors. Cell Death Differ. 2008;15:545–554. doi: 10.1038/sj.cdd.4402290. [DOI] [PubMed] [Google Scholar]
- Dunand-Sauthier I, Walker CA, Narasimhan J, Pearce AK, Wek RC, Humphrey TC. Stress-activated protein kinase pathway functions to support protein synthesis and translational adaptation in response to environmental stress in fission yeast. Eukaryot Cell. 2005;4:1785–1793. doi: 10.1128/EC.4.11.1785-1793.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Supale S, Li N, Brun T, Maechler P. Mitochondrial dysfunction in pancreatic beta cells. Trends Endocrinol Metab. 2012;23:477–487. doi: 10.1016/j.tem.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25:4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick DU, Dennerlein S, Wiese H, Reinhold R, Pacheu-Grau D, Lorenzi I, Sasarman F, Weraarpachai W, Shoubridge EA, Warscheid B, et al. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell. 2012;151:1528–1541. doi: 10.1016/j.cell.2012.11.053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Review Process File
Source Data for Figure 1D, G
Source Data for Figure 2A, B
Source Data for Figure 3A, B, C
Source Data for Figure 4A, B