

Structures of the catalytic N-acetyltransferase (NAT) domain of the bifunctional N-acetyl-l-glutamate synthase/kinase (NAGS/K) from Xylella fastidiosa bound to N-acetyl-l-glutamate (NAG) with and without an N-terminal His tag have been solved and refined at 1.7 and 1.4 Å resolution, respectively.
Keywords: N-acetylglutamate synthase, N-acetylglutamate kinase, GCN5 N-acetyltransferase, N-acetylglutamate, bifunctional enzymes, arginine biosynthesis
Abstract
Structures of the catalytic N-acetyltransferase (NAT) domain of the bifunctional N-acetyl-l-glutamate synthase/kinase (NAGS/K) from Xylella fastidiosa bound to N-acetyl-l-glutamate (NAG) with and without an N-terminal His tag have been solved and refined at 1.7 and 1.4 Å resolution, respectively. The NAT domain with an N-terminal His tag crystallized in space group P41212, with unit-cell parameters a = b = 51.72, c = 242.31 Å. Two subunits form a molecular dimer in the asymmetric unit, which contains ∼41% solvent. The NAT domain without an N-terminal His tag crystallized in space group P21, with unit-cell parameters a = 63.48, b = 122.34, c = 75.88 Å, β = 107.6°. Eight subunits, which form four molecular dimers, were identified in the asymmetric unit, which contains ∼38% solvent. The structures with and without the N-terminal His tag provide an opportunity to evaluate how the His tag affects structure and function. Furthermore, multiple subunits in different packing environments allow an assessment of the plasticity of the NAG binding site, which might be relevant to substrate binding and product release. The dimeric structure of the X. fastidiosa N-acetytransferase (xfNAT) domain is very similar to that of human N-acetyltransferase (hNAT), reinforcing the notion that mammalian NAGS is evolutionally derived from bifunctional bacterial NAGS/K.
1. Introduction
In most microorganisms and plants, two different enzymes, N-acetyl-l-glutamate (NAG) synthase (NAGS; EC 2.3.1.1) and N-acetyl-l-glutamate kinase (NAGK; EC 2.7.2.8), catalyze the first two reactions in the arginine-biosynthetic pathway (Slocum, 2005 ▶; Cunin et al., 1986 ▶). However, in some bacteria, such as Xanthomonas campestris and Xylella fastidiosa, a single bifunctional enzyme catalyzes both reactions (Shi, Caldovic et al., 2006 ▶; Qu et al., 2007 ▶). Interestingly, the sequences and three-dimensional structures of these bifunctional enzymes have closer evolutionary relationships to mammalian NAGS than to ‘classical’ NAGS enzymes such as those from Escherichia coli and Neisseria gonorrhoeae (Zhao, Jin et al., 2013 ▶; Shi et al., 2008 ▶, 2011 ▶; Qu et al., 2007 ▶). The major function of mammalian NAGS is to provide the obligatory cofactor for carbamylphosphate synthetase 1 (CPS1) in the liver, which catalyzes an early step in the elimination of excess ammonium via the urea cycle (Shigesada & Tatibana, 1978 ▶). Deficiencies in human NAGS cause clinical hyperammonaemia, which may result in death or severe neurological impairment (Caldovic et al., 2007 ▶).
Our previous studies indicated that both ‘classical’ NAGS and vertebrate-like NAGS consist of two independent structural domains linked by a flexible 1–3 amino-acid residue linker (Zhao, Jin et al., 2013 ▶; Shi et al., 2008 ▶, 2011 ▶). The N-terminal domain has a typical amino-acid kinase (AAK) fold with the characteristic α3β8α4 sandwich structure, which is split into N- and C-terminal lobes. The C-terminal domain has a GCN5 N-acetyltransferase fold, which is divided into N- and C-terminal arms. The major role of the N-terminal domain of both ‘classical’ NAGS and mammalian NAGS is to provide the arginine binding site and structural architecture for arginine-induced regulation. However, even though both ‘classical’ and vertebrate-like NAGS have a conserved arginine binding site, their structural architecture and regulatory mechanisms are different. While the ‘classical’ NAGS forms a doughnut-like hexameric structure similar to arginine-sensitive NAGK (Shi et al., 2008 ▶; Ramón-Maiques et al., 2006 ▶), vertebrate-like NAGS has a novel tetrameric architecture, as revealed by the structures of bifunctional NAGS/K and the NAT domain of human NAGS (hNAT) (Fig. 1 ▶; Zhao, Jin et al., 2013 ▶; Shi et al., 2011 ▶). Interestingly, recent structural and functional studies of the arginine-sensitive NAGS/K from Saccharomyces cerevisiae demonstrated that it has a similar tetrameric molecular architecture (de Cima et al., 2012 ▶), reinforcing the notion that the vertebrate-like NAGS, including human NAGS and ascomycetal NAGK, may have evolved from an ancestral bifunctional NAGS/K such as that of X. campestris (xcNAGS/K; Shi, Caldovic et al., 2006 ▶; Qu et al., 2007 ▶). In all NAGS identified to date, the active site is located in the C-terminal domain. Both substrates, AcCoA and glutamate, bind to the central V-shaped cleft of the domain, similar to other members of the GCN5-related N-acetyltransferase family. Since the arginine binding site is far from the NAGS active site, the mechanism of how arginine regulates NAGS activity has been the focal point of our studies (Zhao, Haskins et al., 2013 ▶; Min et al., 2009 ▶). In ‘classical’ bacterial NAGS, arginine binding widens the hexameric ring of the AAK domains, changes their spatial relationship and dramatically alters the interactions between the AAK and NAT domains, eventually affecting the conformation of glutamate-binding loops and inhibiting NAGS activity (Zhao, Haskins et al., 2013 ▶; Min et al., 2009 ▶). In vertebrate-like NAGS, arginine binding changes the relative orientation between the AAK and NAT domains to enhance or inhibit the NAGS activity by affecting AcCoA binding (Zhao, Haskins et al., 2013 ▶; Min et al., 2009 ▶). Even though arginine has disparate effects on the NAGS activity of the bacterial bifunctional NAGS/K and the human NAGS, the arginine-regulatory mechanism seems likely to be similar because both have similar tetrameric architectures.
Figure 1.

A simplified model showing the different oligomerization states of classical NAGS (a) and vertebrate-like NAGS (b). Different subunits are shown in different colors. The synthase and kinase domains are labeled S and K, respectively.
Since full-length bifunctional NAGS/K can only be crystallized at moderate resolution, in order to produce better quality crystals and improve our understanding of the catalytic mechanism we crystallized the two domains separately. Here, we report the structures of the N-acetyltransferase (NAT) domain of bifunctional NAGS/K from X. fastidiosa (xfNAGS/K) at a higher resolution. The availability of xfNAT domain structures both with and without a His tag (hereafter, these proteins will be abbreviated xfNAT-ht and xfNAT, respectively) also provides an opportunity to assess whether the His tag affects enzyme structure and function. The existence of multiple copies of subunit in the asymmetric unit in both structures allows us to evaluate the plasticity of the structure, which may influence substrate binding and product release.
2. Materials and methods
2.1. Cloning and protein expression and purification
xfNAGS/K was cloned from the genomic DNA (ATCC 35881D). The C-terminal N-acetyltransferase domain (residues Val292–Asp438) was subcloned into pET-28 expression vector. The proteins were expressed in E. coli BL21(DE3) cells (Invitrogen) and purified using nickel-affinity and HiTrap SP-XL columns (GE Healthcare). Protein purity was verified by SDS–PAGE and protein concentration was measured with a NanoDrop 2000 spectrophotometer (Thermo Scientific). The extinction coefficient obtained from the ExPASy web server (http://web.expasy.org/protparam/) was used to calculate protein concentrations. The protein was stored at 253 K in a buffer consisting of 50 mM Tris–HCl pH 7.4, 50 mM NaCl, 10% glycerol, 5 mM β-mercaptoethanol, 1 mM EDTA. xfNAT was prepared by incubating xfNAT-ht (∼10 mg) with 50 units of thrombin at 277 K overnight. The completeness of cleavage was confirmed by SDS–PAGE. No further purification was performed for crystallization.
2.2. Activity assay
Enzymatic activity was assayed using the method described previously (Caldovic et al., 2002 ▶). A stable isotope-dilution method using liquid-chromatography mass spectrometry (LC–MS) to measure NAG production was adopted. Each assay was performed in a 100 µl solution consisting of 50 mM Tris pH 8.5, 10 mM glutamate, 2.5 mM AcCoA. The reaction was initiated by the addition of purified recombinant enzyme (20 µg) and the mixture was incubated at 303 K for 5 min and quenched with 100 µl 30% trichloroacetic acid containing 50 µg N-acetyl-(13C5)-glutamate (13C-NAG) as an internal standard. Precipitated protein was removed by micro-centrifugation. The supernatant (10 µl) was submitted to LC-MS (Agilent) analysis. The mobile phase consisted of 92% solvent A (1 ml trifluoroacetic acid in 1 l water) and 8% solvent B (1 ml trifluoroacetic acid in 1 l 1:9 water:acetonitrile) and the flow rate was 0.6 ml min−1. Glutamate, NAG and 13C-NAG were detected and quantified by selected ion-monitoring mass spectrometry.
2.3. Gel-filtration chromatography
The molecular weight of xfNAT was determined with a Superdex 200 HR 10/30 column (Amersham Biosciences) as described previously (Shi et al., 2008 ▶). The running buffer consisted of 100 mM NaH2PO4 pH 7.4, 150 mM NaCl, 10% glycerol, 1 mM β-mercaptoethanol. Ovalbumin (43 kDa), albumin (67 kDa), chymotrypsinogen A (25 kDa) and ribonuclease (13.7 kDa) were used as protein standards.
2.4. Crystallization
Crystals were grown by the sitting-drop vapor-diffusion method using the Index screening kit from Hampton Research. Before the crystallization experiment, both xfNAT-ht and xfNAT were incubated with the storage solution supplemented with 10 mM CoA and 10 mM NAG. The screenings were carried out using 2 µl protein plus 2 µl crystallization solution. Crystals of xfNAT with His tag were observed in two conditions (G3 and H8). The crystals from condition G3 (0.2 M Li2SO4, 0.1 M Tris pH 6.5, 25% PEG 3350) were used for data collection without further optimization. Crystals of xfNAT without His tag were observed in 11 conditions (A8, B4, C4, C7, D8, D9, G1, G4, G5, G9 and H7). The crystals from condition G5 (0.2 M Li2SO4, 0.1 M Tris pH 8.5, 25% PEG 3350) were used for data collection without further optimization.
2.5. Data collection and structure determination
Crystals were transferred from the crystallization plate to well solution supplemented with 25% glycerol and then cooled directly in liquid nitrogen. Diffraction data were collected on beamline 22-ID equipped with a MAR300 CCD detector at the Advanced Photon source (APS), Argonne National Laboratory, USA. All data were processed using the HKL-2000 package (Otwinowski & Minor, 1997 ▶); statistics are summarized in Table 1 ▶. The structure was solved by molecular replacement using Phaser (Read, 2001 ▶; Storoni et al., 2004 ▶) with the dimer (subunit AB) of the human NAT-domain structure (PDB entry 4k30; Zhao, Jin et al., 2013 ▶) as a search model. After several cycles of refinement with PHENIX (Adams et al., 2010 ▶) and model adjustments with Coot (Emsley & Cowtan, 2004 ▶), NAG was visible in the electron-density map and was built into the model. In the last run of the refinement, the translation/liberation/screw parameters were included and refined with one group per subunit (Winn et al., 2001 ▶). The final R and R free values were 18.5 and 22.0%, respectively, for xfNAT-ht and 17.9 and 19.9%, respectively, for xfNAT. Refinement statistics for the final refined model are given in Table 1 ▶. The final refined coordinates for NAG-bound xfNAT-ht and xfNAT and their structure factors have been deposited in the Protein Data Bank with accession codes 4nf1 and 4nex, respectively.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Sample | xfNAT-ht | xfNAT |
|---|---|---|
| Data collection | ||
| Bound ligands | NAG | NAG |
| Space group | P41212 | P21 |
| Wavelength () | 1.0 | 1.0 |
| Resolution () | 501.70 (1.731.70) | 501.40 (1.421.40) |
| Unit-cell parameters (, ) | a = b = 51.7, c = 242.3 | a = 63.9, b = 123.4, c = 76.7, = 107.6 |
| Measurements | 485488 | 1477630 |
| Unique reflections | 37347 (1820) | 209796 (10725) |
| Multiplicity | 13.0 (10.9) | 7.1 (6.0) |
| Completeness (%) | 98.4 (97.2) | 94.5 (96.5) |
| I/(I) | 55.8 (3.4) | 32.9 (2.0) |
| R merge † (%) | 7.9 (69.0) | 8.4 (78.4) |
| Refinement | ||
| Resolution range () | 401.70 (1.741.70) | 401.40 (1.431.40) |
| No. of protein atoms | 2817 | 11717 |
| No. of waters | 230 | 1691 |
| No. of heteroatoms | 41 | 119 |
| R.m.s.d., bond lengths () | 0.007 | 0.006 |
| R.m.s.d., bond angles () | 1.1 | 1.1 |
| R work ‡ (%) | 18.5 (25.2) | 17.9 |
| R free § (%) | 22.0 (31.5) | 19.9 |
| Ramachandran plot (%) | ||
| Favored | 97.29 | 97.38 |
| Allowed | 2.37 | 2.18 |
| Outliers | 0.34 | 0.44 |
R
merge = 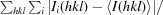
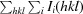 , where I
i(hkl) is the intensity of the ith observation of reflection hkl and I(hkl) is the average intensity of redundant measurements of reflection hkl.
, where I
i(hkl) is the intensity of the ith observation of reflection hkl and I(hkl) is the average intensity of redundant measurements of reflection hkl.
R
work = 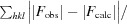
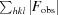 .
.
R
free =
for a reserved 5% of the reflections.
3. Results
3.1. Enzymatic activity of xfNAT-ht and xfNAT
The specific NAGS activities of xfNAT-ht and xfNAT are 0.56 ± 0.01 and 0.53 ± 0.06 µmol min−1 mg−1, respectively. These values are approximately twofold lower than the specific NAGS activity of full-length wild-type xfNAGS/K in the absence of l-arginine (Table 2 ▶). As expected, the presence of arginine does not inhibit the NAGS activity of xfNAT, but rather enhances the NAGS activity slightly. In contrast, 1 mM arginine significantly inhibits the NAGS activity of full-length xfNAGS/K. These results further confirm that the NAGS active site is located solely in the NAT domain. The NAGK domain hosts the arginine binding site and provides the structural machinery for arginine-induced regulation of NAGS activity. Relative to other members of the vertebrate-like NAGS family, the specific activity of xfNAGS/K is low (Table 2 ▶). In addition, the effect of arginine on NAGS activity changes from inhibition in bifunctional bacterial NAGS/K to neutral in frog NAGS and to enhancement in mammalian NAGS. The varied specific NAGS activities and different arginine responses for the different species may reflect to some extent their differing enzyme stabilities and different assays. The differences may also reflect the changing biological roles of NAGS from arginine biosynthesis in lower organisms to ammonium removal via the urea cycle in mammals. This change may be related to the migration of tetrapods from the sea to land (Haskins et al., 2008 ▶).
Table 2. Specific activity.
Means standard errors of means (n = 3) are shown.
| Activity (molmin1mg1) | |||
|---|---|---|---|
| Enzyme | Without 1mM Arg | With 1mM Arg | References |
| xfNAT-ht | 0.56 0.01 | 0.67 0.02 | Present work |
| xfNAT | 0.53 0.06 | 0.60 0.01 | Present work |
| xfNAGS/K with His tag | 1.31 0.01 | 0.48 0.07 | Present work |
| mmNAGS/K with His tag | 6.81 0.23 | Zhao, Haskins et al. (2013 ▶) | |
| xcNAGS/K with His tag | 44.05 0.21 | Zhao, Haskins et al. (2013 ▶) | |
| hNAT | 1.05 0.01 | 1.04 0.01 | Zhao, Jin et al. (2013 ▶) |
| hNAGS | 19.29 0.38 | 29.28 0.21 | Haskins et al. (2008 ▶) |
| African clawed frog NAGS | 0.44 0.01 | 0.528 0.004 | Haskins et al. (2008 ▶) |
| Western clawed frog NAGS | 0.098 0.002 | 0.136 0.005 | Haskins et al. (2008 ▶) |
| Zebrafish NAGS | 10.64 0.09 | 5.48 0.12 | Haskins et al. (2008 ▶) |
| Pufferfish NAGS | 20.89 0.28 | 15.90 0.14 | Haskins et al. (2008 ▶) |
3.2. Structure of xfNAT-ht bound to NAG
The structure of xfNAT-ht was determined at 1.7 Å resolution and refined to R work and R free values of 18.5 and 22.0%, respectively (Table 1 ▶). The model has good geometry, with 97.29% of the residues located in the most favored area of the Ramachandran plot. Only residue Ser283 in the A chain is located in the disallowed region. Two copies of each subunit that form one biological relevant dimer were identified in the asymmetric unit. Each subunit has a typical central seven-stranded β-sheet arranged as a V-shaped structure, with three antiparallel β-strands in the C-terminal arm and four antiparallel β-strands in the N-terminal arm (Figs. 2 ▶ a and 2 ▶ b). The central β-sheet is flanked by five helices, with three helices on one side and two helices on the other. The structure has the typical fold of GCN5-related N-acetyltransferases (Dyda et al., 2000 ▶) and is similar to the NAT-domain structure of the bifunctional NAGS/K from Maricaulis maris and human (Zhao, Jin et al., 2013 ▶; Shi et al., 2011 ▶).
Figure 2.

Structure of xfNAT-ht. (a) Ribbon diagram of subunit A of xfNAT-ht, showing the extra β-strand in its N-terminal arm in blue. The sulfates and the modeled Thr in the active site are shown as sticks. (b) Ribbon diagram of subunit B of xfNAT-ht. The bound NAG is shown as sky-blue sticks. The electron-density map (2F o − F c) around the bound NAG (contoured at 1.0σ) is shown as a blue cage. (c) Superimposition of subunits A (in red ribbons) and B (in green ribbons) of xfNAT with a His tag. NAG bound in subunit B is shown as sky-blue sticks. The side chains of Arg317, Arg385 and Arg387 in subunits A and B are shown as sky-blue and magenta sticks, respectively. The conformations of the side chains of Arg387 are significantly different between subunits A and B. The largest difference in the main-chain atoms between subunits A and B was found in the β18–β19 loop, with an r.m.s. deviation of 4.56 Å
Superposition of the two subunits results in an r.m.s. deviation of 0.86 Å. The most significant differences are located in the loop regions, such as the β18–β19, β16–α13 and β17–α14 loops, consistent with the hNAT structure (Zhao, Jin et al., 2013 ▶). Interestingly, the electron density corresponding to the whole thrombin-recognition site after the His tag is visible for subunit A. Modeling indicates that there is an extra β-strand in the N-terminal arm, producing a five-stranded antiparallel β-sheet (Figs. 2 ▶ a and 2 ▶ b). No electron density was identified in the equivalent region in subunit B.
3.3. Structure of xfNAT with NAG bound
The structure of xfNAT (residues 293–438) was determined at 1.4 Å resolution and refined to R work and R free values of 17.9 and 19.9%, respectively (Table 1 ▶). The model has good geometry, with 97.38% of the residues located in the most favored area of the Ramachandran plot. Five residues [Ala362, Gln363, Asn390 (chain B), His417 (chain D) and Asp361 (chain F)] are in the disallowed region because of their weak electron density. Eight copies of the subunit form four biologically relevant dimers in the asymmetric unit. Most subunits are well defined, except for subunit H, in which the electron density of part of the structure is weak relative to the other subunits. Subunit H has the highest average temperature factor of 29.1 Å2, while subunit A has the lowest value of 15.2 Å2 (Supplementary Table S1). The structures of all subunits are similar. Superimpositions among different subunits result in r.m.s. deviations in the range 0.50–1.00 Å. As observed in the structure of xfNAT with the His tag, most differences are located in the loop regions, reflecting the influence of local environments on these highly flexible regions (Supplementary Fig. S1a).
Since the structure of xfNAT was determined at a higher resolution and its sequence is closer to the native protein, the following results and discussion are based on the xfNAT structure unless mentioned otherwise.
3.4. Dimerization
Even though multiple subunits were identified in the asymmetric units of both xfNAT-ht and xfNAT, the PISA server (Krissinel & Henrick, 2007 ▶) indicated that the stable molecule is a dimer. This observation is consistent with the gel-filtration studies in solution. The observed molecular weight of xfNAT is 37.4 kDa in the gel-filtration experiments, while the calculated molecular weight from the protein sequence of xfNAT is 38.8 kDa for a dimer, clearly demonstrating that xfNAT exists as a dimer in solution (Fig. 3 ▶), as do the NAT domains of human and mouse NAGS (Zhao, Jin et al., 2013 ▶). The dimer interface is formed by the C-terminal arm of one subunit interacting with the C-terminal arm of the other subunit to form a continuous six-stranded antiparallel β-sheet across the dimer interface, similar to the NAT–NAT domain interaction observed in the M. maris NAGS/K (mmNAGS/K) structure and the human NAT-domain structure (Fig. 4 ▶ a). This dimer interface has a buried interface in the range 1659–2034 Å2, slightly larger than the dimer interface in the human NAT-domain structure, which has a buried area of 1477 Å2. The interactions in this interface involve extensive main-chain (Asp401, Tyr403 and Lys405) and side-chain hydrogen-bonding interactions (Tyr403–Ser400′) and other hydrophobic interactions (Val426, Tyr397, Phe419, Gln423 and Tyr403) (Fig. 4 ▶ b).
Figure 3.

Oligomeric structure of xfNAT in solution. Analytic gel chromatography of xfNAT demonstrated that xfNAT is a dimer in solution. Inset: plot of sigma factor versus molecular weight of standard proteins (filled circles) and xfNAT (filled square). K av was calculated using the formula K av = (V e − V o)/(V i − V o), where V e is the elution volume of the standard proteins and xfNAT, V o is the void volume and V i is the included volume.
Figure 4.

xfNAT dimer. (a) The dimer formed by subunits A and B in xfNAT is shown as a ribbon diagram. A central continuous six-stranded antiparallel β-sheet was found across the dimer interface. (b) Detailed interactions across the dimer interface. Subunits A and B are colored red and green, respectively. Only partial residues are labeled for clarity.
To assess the variation in the relative orientation of the subunits within the dimer, the equivalent subunits of two different dimers were superimposed and the movement required to superimpose the other equivalent subunits was calculated. For example, to determine whether there is any difference in the orientation of subunits in dimer AB and dimer CD, subunits A and C were first superimposed and the angle required to superimpose subunit D on subunit B was then calculated (Supplementary Fig. S2). The differences in angles among the four different dimers in xfNAT and the dimer in xfNAT-ht are in the range 2–3.5°, implying that the orientation of the two subunits in the dimer is quite rigid. However, the differences for the dimers in xfNAT from the NAT-domain dimer in the mmNAGS/K structure (PDB entry 3s6g; dimers AX and BY) or from the dimer in the human NAT-domain structure (PDB entry 4k30; dimer AB) can be as large as 10°. Even though the relative domain orientation between the N-terminal amino-acid kinase domain and the C-terminal synthase domain among the four subunits in the asymmetric unit of the mmNAGS/K structure can be as large as 19° (Shi et al., 2011 ▶), the relative orientation difference of the NAT–NAT domains in the AX and BY dimers was only 3.8°, suggesting that during the arginine-induced relative domain movement the NAT–NAT dimer rotates as a rigid unit with the linker between the amino-acid kinase domain and the NAT domain as the pivot point.
3.5. NAG binding in two different conformations of xfNAT
NAG binding was easily identified at the enzyme active site, where it binds in a cavity similar to that in the human NAT-domain structure (Fig. 5 ▶; Supplementary Fig. S3). Interestingly, the side chain of NAG exists in two different conformations in the eight subunits of the xfNAT structure. NAG binds in one conformation in subunits A, C and G (Figs. 5 ▶ a and 5 ▶ c; Supplementary Fig. S3) and in another conformation in the remaining subunits (Figs. 5 ▶ b and 5 ▶ d). In both conformations the residues involved in hydrogen-bond interactions with the α carboxyl group, the α amino N atom and the acetyl O atom of NAG are the same. The main-chain O atoms of Asp354 and Arg385 help to anchor the α amino N atom of NAG, and the main-chain N atom of Phe356 fixes the acetyl group. The side chains of two residues, Lys355 from strand β16 and Arg385 from strand β17, bind the α-carboxyl group in place (Table 3 ▶). Interestingly, the hydrogen-bonding interactions with the γ-carboxyl group of NAG are different for the two conformations, which may reflect two different states for substrate binding and product release. In the first conformation, the side chain of Arg317 from the loop connecting helices α11 and α12 swings into the active site to form a salt bridge with the γ-carboxyl group of NAG (Fig. 5 ▶ c, Table 3 ▶). This conformation appears to represent the substrate-binding conformation, which is found in subunits C and G. In the second conformation, the side chain of Asn390 from the loop connecting β17 and α14 and the main-chain N atom of Arg387 are involved in binding to one of the carboxyl O atoms of the γ-carboxyl group of NAG (Figs. 5 ▶ b and 5 ▶ d). The NAGs in subunits B, D, E, F and H are in this conformation. Even though the NAG binding in subunit A is close to the first conformation, no direct interactions between the protein and the γ-carboxyl group of NAG are identified. All interactions are mediated by solvent (Table 3 ▶ and Fig. 5 ▶ a). This NAG-binding conformation possibly represents the NAG-releasing conformation. In accordance with the two NAG-binding conformations, the conformation of the side chain of Arg317 could be in two different conformations, with one swinging into the active site and the other swinging out from the active site. The side chain of Arg317, together with that of Arg387 (for which two conformations could be observed in the structure of xfNAT-ht; Fig. 2 ▶ c), may form a gate for substrate binding and product release.
Figure 5.

NAG binding site. (a) NAG binding site of subunit A of xfNAT. The side chain of NAG interacts with protein residues via waters only and therefore probably represents the product-releasing conformation. (b) NAG binding site of subunit B of xfNAT, representing one of the possible NAG-binding conformations. (c) NAG binding site of subunit C of xfNAT, representing another of the possible NAG-binding conformations. Even though this conformation is close to that of subunit A, because the side chains of Arg317 and Arg387 are in a closed conformation and thus able to interact with NAG, this conformation may represent the substrate-binding conformation. (d) NAG binding site of subunit D of xfNAT, similar to that in subunit B. The bound NAG is shown as sky-blue sticks. The side chains involved in hydrogen-bonding interactions with NAG are shown as sky-blue sticks. The electron-density map (2F o − F c) around bound NAG (contoured at 1.0σ) is shown as a blue cage. Potential hydrogen-bonding interactions are shown as red dashed lines.
Table 3. Interactions between N-acetyl-L-glutamate and protein atoms in the xfNAT structure.
| Distance () | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| NAG | Protein | A | B | C | D | E | F | G | H |
| N2 | Arg385O | 3.52 | 3.30 | 3.43 | 3.35 | 3.36 | 3.35 | 3.37 | 3.34 |
| Ap354O | 3.21 | 3.23 | 3.32 | 3.22 | 3.28 | 3.26 | 3.35 | 3.25 | |
| O7 | Phe356N | 2.98 | 2.95 | 2.89 | 2.99 | 2.92 | 2.97 | 2.90 | 3.00 |
| OXT | Lys355NZ | 2.76 | 2.77 | 2.79 | 2.79 | 2.84 | 2.80 | 2.74 | 2.80 |
| O | Arg385NE | 2.96 | 3.07 | 3.09 | 3.04 | 3.08 | 3.08 | 3.03 | 3.15 |
| OE1 | Asn390ND | 2.92 | 2.94 | 2.87 | 3.02 | ||||
| Arg387N | 3.07 | 3.01 | 2.95 | 3.04 | 2.92 | ||||
| Arg317NH2 | 2.82 | 2.96 | |||||||
| OE2 | Arg317NE | 3.00 | 2.89 | ||||||
| Arg317NH1 | 3.59 | ||||||||
| Arg317NH2 | 2.96 | ||||||||
| Arg387NE | 3.30 | ||||||||
Since the structures were determined at high resolution, the water molecules in the NAG binding site were well defined. One particular water, w1409 in subunit A, could be identified in all subunits and was located at a position to hydrogen bond to the α-amino N atom of NAG, the side chain of Tyr352, the main-chain N atom of Arg385 and the main-chain O atom of Leu353 (Fig. 6 ▶). This water, in combination with residue Tyr352, seems to be in a position to act as a proton shuttle to facilitate the deprotonation of the amino N atom of NAG via a water wire during the catalytic reaction.
Figure 6.

Stereo diagram of the conserved water w1409 and the ‘water wire’ channel. The bound NAG is shown as thick green sticks. Water molecules are shown as red balls. Residues involved in hydrogen-bonding interactions are shown as thin orange sticks. Potential hydrogen-bonding interactions are shown as red dashed lines.
3.6. NAG binding sites in xfNAT-ht
NAG binding could be identified in only one of the subunits (subunit B) in the xfNAT-ht structure (Fig. 2 ▶ b; Supplementary Table S2). The conformation of NAG bound to subunit B is similar to the second conformation found in the xfNAT structure. The NAG binding site in subunit A seems to be filled with solvent molecules such as water, sulfate and others (tentatively modeled as the amino acid threonine). Comparison of NAG binding between subunits A and B indicated that the cleft in subunit A is slightly wider than that in subunit B (Fig. 2 ▶ c), and the conformations of the β18–β19 and β17–α14 loops and the side chain of Arg387 are significantly different. The formation of the extra β-strand in the N-terminal arm may also be a factor in the widening of the NAG binding site in subunit A. Since both subunits have His tags in their sequences, these differences are most likely to be caused by different packing environments in the crystal structure. As a result, slight differences in the NAG binding site lead to subunit B being able to bind NAG while subunit A cannot.
3.7. CoA binding site
To determine how CoA binds, 10 mM CoA was included in the crystallization buffer. However, no continuous electron density corresponding to CoA was observed in the ‘V-shaped’ groove where the pantetheine moiety of CoA usually binds. Even though extensive efforts have been made, to date no complex structure of vertebrate-like NAGS bound to AcCoA or CoA has been determined, in contrast to the bacterial-like ‘classical’ NAGS, for which AcCoA-bound and CoA-bound structures were easily obtained. This may imply generally weaker binding of AcCoA to vertebrate-like NAGS relative to the bacterial-like ‘classical’ NAGS. This situation is not uncommon, since CoA-bound or AcCoA-bound structures were also difficult to obtain for some other GCN5-related acetyltransferases (Vetting et al., 2008 ▶), although the unambiguous identification of NAG in the expected site suggested that CoA was likely to bind in a site similar to those found in other GCN5-related acetyltransferases (Vetting et al., 2005 ▶; Dyda et al., 2000 ▶). As expected, the pyrophosphate binding region, Gln363-Gly364-Glu365-Gly366-Leu367-Gly368, which conforms to the (Arg/Gln)-Xaa-Xaa-Gly-Xaa-(Gly/Ala) motif for CoA recognition in known GCN5-related N-acetyltransferases (Neuwald & Landsman, 1997 ▶), is located at a position equivalent to those of other N-acetyltransferases. In the absence of CoA or AcCoA binding, this part of the structure varies significantly among the different subunits. Therefore, the exact binding mode of the CoA moiety, the amino-acid residues involved in binding, and how and whether AcCoA binding induces dramatic conformational changes remain to be determined.
3.8. Comparison with the NAT-domain structures of human NAGS
The overall xfNAT structure is similar to that of the hNAT structure (Fig. 7 ▶) and can be aligned with an r.m.s. deviation of ∼1.2 Å. The sequence of xfNAT has 38% identity to that of hNAT. All active-site residues involved in hydrogen bonding to NAG are conserved, except for Arg317, which is equivalent to Lys401 in human NAGS. This structural similarity strongly supports a close evolutionary relationship between human NAGS and the bifunctional NAGS/K.
Figure 7.

Stereo diagrams of the superimposition of xfNAT with hNAT. (a) Superimposition of subunit B of xfNAT with subunit A of hNAT. The structure of xfNAT (subunit B) is shown as magenta ribbons, while the structure of hNAT (subunit A) is shown as green ribbons. The conformations of bound NAGs in xfNAT and hNAT are different and are shown as magenta and green sticks, respectively. (b) Superimposition of subunit C of xfNAT with subunit A of hNAT. The structure of xfNAT (subunit B) is shown as yellow ribbons. The structure of hNAT (subunit A) is shown as green ribbons. The conformations of bound NAGs in xfNAT and hNAT are similar and are shown as yellow and green sticks, respectively. Residues that are mentioned in the text are shown as sticks.
In addition to major structural differences in the loop regions (the β15–β16, β16–α13, β19–α15 and β18–β19 loops), some significant differences are observed in the conformation of the active-site residues Lys401 and Arg476. In the hNAT structure, both residues hydrogen-bond to the γ-carboxyl group of NAG, even though the side chains of Arg476 can be in two different conformations (in one conformation in subunits A, B and Y and in another conformation in subunit X). In the xfNAT structure, two conformations were also observed for the equivalent residues Arg317 and Arg387, which may be related to substrate binding and product release.
4. Discussion
4.1. Effects of the His tag on crystallization, structure and activity
His tags are commonly used to prepare large amounts of pure recombinant proteins since His-tagged proteins can easily be purified using immobilized metal ion-affinity chromatography (Smith et al., 1988 ▶; Xiao et al., 2010 ▶). However, many studies have suggested that the His tag might affect the expression, the biochemical properties and the structures of proteins (Smith et al., 1988 ▶; Khan et al., 2012 ▶; Sayari et al., 2007 ▶; Song & Markley, 2007 ▶). Recent surveys and comparisons between crystal structures with and without His tags have indicated that His tags generally have no significant effect on the structure of native proteins (Carson et al., 2007 ▶). The availability of both xfNAT-ht and xfNAT structures provided us with one more opportunity to evaluate the effect of the His tag on the crystallization, biological function and structure of a protein. Our crystallization screening of xfNAT-ht and xfNAT demonstrated that the His tag affected protein crystallization significantly. There were only two conditions that generated crystals of xfNAT-ht from the 96 Index screen conditions; in contrast, more than ten conditions were able to produce crystals of xfNAT. These crystals were also of better quality than those with the His tag. We have also found that the His tag affects the crystallization and crystal quality of other proteins. In our structural studies of N-succinylornithine carbamyltransferase, crystals without substrate or substrate analogue bound were obtained from protein with a C-terminal His tag (Shi et al., 2002 ▶). However, extensive efforts to produce crystals with the substrate or a substrate analogue bound using the C-terminally His-tagged protein were not successful. Crystals of the substrate-bound protein could only be obtained when the His tag was moved to the N-terminus (Shi, Morizono et al., 2006 ▶). In determination of structures of N-acetylcitrulline deacetylase, high-quality crystals could only be obtained using protein without His tag (Qu et al., 2007 ▶). Since the His tag is an important parameter that affects protein crystallization, screening of crystallization conditions should be performed on protein both with and without His tag, and for proteins with the His tag on different termini.
In our previous studies of NAGS, the His tag generally did not affect the function of the protein significantly (Caldovic et al., 2006 ▶). Our present studies also demonstrate that the His tag does not significantly affect the NAGS activity of xfNAT. However, the effect of His tags on biological activity is likely to vary from protein to protein, and should be tested individually.
Our current studies confirmed that the His tag does not generally affect the structure. The structural differences between xfNAT-ht and xfNAT are comparable to the structural differences among the different subunits in the crystallized forms of both xfNAT-ht and xfNAT. It is interesting to note that the partial tag sequence could be fitted into density in only one of the two subunits in the xfNAT-ht structure. Even though significant differences could be observed in the active-site structure, with one having NAG bound and one with solvent molecules, the differences were more likely to be created by the differences in the packing environments of the different subunits in an asymmetric unit than by the presence or absence of the His tag.
4.2. Catalytic mechanism, substrate binding and product release
GCN5-related acetyltranferases are generally believed to use a direct attack mechanism for the catalytic reaction. In this mechanism, a residue that functions as a general base helps to deprotonate the attacking amino group of the attacking substrate, such as l-glutamate in NAGS. A residue that functions as a general acid facilitates the protonation of the thiol group of the leaving group, CoA. It has been suggested that Tyr485 in human NAGS, Tyr397 in mmNAGS/K and Tyr405 in xcNAGS/K act as a catalytic acid that donates a proton to the thiol group of CoA. Previous mutagenesis studies on these residues indicated that they are critical for the NAGS activity (Zhao, Haskins et al., 2013 ▶). An equivalent tyrosine can be identified in most GCN5-related acetyltransferases (He et al., 2003 ▶). In the present structure, Tyr396, which is located in the equivalent position, appears to play a similar role as a general acid.
The residues that function as a general base are quite varied in the GCN5-related acetyltransferases. Histidine, glutamate and tyrosine have all been proposed to play the role of a general base (Dyda et al., 2000 ▶; Vetting et al., 2005 ▶). In the human NAT-domain structure, a highly conserved Tyr441 was proposed to act as the catalytic base in proton removal from the amino group of l-glutamate (Zhao, Jin et al., 2013 ▶). In the present structure, Tyr352, which is located at the equivalent position, can bind to the α-amino group of l-glutamate via a water molecule, form a water channel that links to solvent on the surface of the protein and play the role of a general base in the catalytic reaction.
In contrast to the human NAT-domain structure, in which NAG binds to the active site in a similar way in all four subunits in the asymmetric unit, NAG binding and the conformations of the residues involving in binding to the γ-carboxyl group of NAG vary significantly in the different subunits in the current xfNAT structures. The variability in the structures of the different subunits in the asymmetric unit provides a snapshot of the possible different binding states that may be related to substrate binding and product release. Since NAG binds to subunits C and G in a conformation in which the side chain of Arg317 has swung into the active site, similar to NAG binding in human NAT-domain structures, this conformation may represent the binding state of the substrate, l-glutamate, before the catalytic reaction. The conformation in subunit A may represent a binding state for the product, NAG, before release, since only solvent molecules are involved in binding the γ-carboxyl group of NAG and the side chain of Arg317 swings towards the protein surface to open the exit channel.
Supplementary Material
Supplementary Tables S1 and S2 and Supplementary Figures S1-S3.. DOI: 10.1107/S2053230X14026788/hv5267sup1.pdf
Acknowledgments
This work was supported by Public Health Service grant DK-DK064913 (MT). We thank Dr David Davies for facilitating the use of the diffraction equipment in the Molecular Structure Section of the National Institutes of Health and Dr Fred Dyda for help in data collection. High-resolution data were collected on the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science and Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Caldovic, L., Lopez, G. Y., Haskins, N., Panglao, M., Shi, D., Morizono, H. & Tuchman, M. (2006). Mol. Genet. Metab. 87, 226–232. [DOI] [PubMed]
- Caldovic, L., Morizono, H. & Tuchman, M. (2007). Hum. Mutat. 28, 754–759. [DOI] [PubMed]
- Caldovic, L., Morizono, H., Yu, X., Thompson, M., Shi, D., Gallegos, R., Allewell, N. M., Malamy, M. H. & Tuchman, M. (2002). Biochem. J. 364, 825–831. [DOI] [PMC free article] [PubMed]
- Carson, M., Johnson, D. H., McDonald, H., Brouillette, C. & DeLucas, L. J. (2007). Acta Cryst. D63, 295–301. [DOI] [PubMed]
- Cima, S. de, Gil-Ortiz, F., Crabeel, M., Fita, I. & Rubio, V. (2012). PLoS One, 7, e34734. [DOI] [PMC free article] [PubMed]
- Cunin, R., Glansdorff, N., Piérard, A. & Stalon, V. (1986). Microbiol. Rev. 50, 314–352. [DOI] [PMC free article] [PubMed]
- Dyda, F., Klein, D. C. & Hickman, A. B. (2000). Annu. Rev. Biophys. Biomol. Struct. 29, 81–103. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Haskins, N., Panglao, M., Qu, Q., Majumdar, H., Cabrera-Luque, J., Morizono, H., Tuchman, M. & Caldovic, L. (2008). BMC Biochem. 9, 24. [DOI] [PMC free article] [PubMed]
- He, H., Ding, Y., Bartlam, M., Sun, F., Le, Y., Qin, X., Tang, H., Zhang, R., Joachimiak, A., Liu, J., Zhao, N. & Rao, Z. (2003). J. Mol. Biol. 325, 1019–1030. [DOI] [PubMed]
- Khan, F., Legler, P. M., Mease, R. M., Duncan, E. H., Bergmann-Leitner, E. S. & Angov, E. (2012). Biotechnol. J. 7, 133–147. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Min, L., Jin, Z., Caldovic, L., Morizono, H., Allewell, N. M., Tuchman, M. & Shi, D. (2009). J. Biol. Chem. 284, 4873–4880. [DOI] [PMC free article] [PubMed]
- Neuwald, A. F. & Landsman, D. (1997). Trends Biochem. Sci. 22, 154–155. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Qu, Q., Morizono, H., Shi, D., Tuchman, M. & Caldovic, L. (2007). BMC Biochem. 8, 4. [DOI] [PMC free article] [PubMed]
- Ramón-Maiques, S., Fernández-Murga, M. L., Gil-Ortiz, F., Vagin, A., Fita, I. & Rubio, V. (2006). J. Mol. Biol. 356, 695–713. [DOI] [PubMed]
- Read, R. J. (2001). Acta Cryst. D57, 1373–1382. [DOI] [PubMed]
- Sayari, A., Mosbah, H., Verger, R. & Gargouri, Y. (2007). J. Colloid Interface Sci. 313, 261–267. [DOI] [PubMed]
- Shi, D., Caldovic, L., Jin, Z., Yu, X., Qu, Q., Roth, L., Morizono, H., Hathout, Y., Allewell, N. M. & Tuchman, M. (2006). Acta Cryst. F62, 1218–1222. [DOI] [PMC free article] [PubMed]
- Shi, D., Gallegos, R., DePonte, J. III, Morizono, H., Yu, X., Allewell, N. M., Malamy, M. & Tuchman, M. (2002). J. Mol. Biol. 320, 899–908. [DOI] [PubMed]
- Shi, D., Li, Y., Cabrera-Luque, J., Jin, Z., Yu, X., Zhao, G., Haskins, N., Allewell, N. M. & Tuchman, M. (2011). PLoS One, 6, e28825. [DOI] [PMC free article] [PubMed]
- Shi, D., Morizono, H., Cabrera-Luque, J., Yu, X., Roth, L., Malamy, M. H., Allewell, N. M. & Tuchman, M. (2006). J. Biol. Chem. 281, 20623–20631. [DOI] [PubMed]
- Shi, D., Sagar, V., Jin, Z., Yu, X., Caldovic, L., Morizono, H., Allewell, N. M. & Tuchman, M. (2008). J. Biol. Chem. 283, 7176–7184. [DOI] [PMC free article] [PubMed]
- Shigesada, K. & Tatibana, M. (1978). Eur. J. Biochem. 84, 285–291. [DOI] [PubMed]
- Slocum, R. D. (2005). Plant Physiol. Biochem. 43, 729–745. [DOI] [PubMed]
- Smith, M. C., Furman, T. C., Ingolia, T. D. & Pidgeon, C. (1988). J. Biol. Chem. 263, 7211–7215. [PubMed]
- Song, J. & Markley, J. L. (2007). Protein Pept. Lett. 14, 265–268. [DOI] [PubMed]
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed]
- Vetting, M. W., Bareich, D. C., Yu, M. & Blanchard, J. S. (2008). Protein Sci. 17, 1781–1790. [DOI] [PMC free article] [PubMed]
- Vetting, M. W. S. de, Carvalho, L. P., Yu, M., Hegde, S. S., Magnet, S., Roderick, S. L. & Blanchard, J. S. (2005). Arch. Biochem. Biophys. 433, 212–226. [DOI] [PubMed]
- Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001). Acta Cryst. D57, 122–133. [DOI] [PubMed]
- Xiao, R. et al. (2010). J. Struct. Biol. 172, 21–33. [DOI] [PMC free article] [PubMed]
- Zhao, G., Haskins, N., Jin, Z., Allewell, N. M., Tuchman, M. & Shi, D. (2013). Biochem. Biophys. Res. Commun. 437, 585–590. [DOI] [PMC free article] [PubMed]
- Zhao, G., Jin, Z., Allewell, N. M., Tuchman, M. & Shi, D. (2013). PLoS One, 8, e70369. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables S1 and S2 and Supplementary Figures S1-S3.. DOI: 10.1107/S2053230X14026788/hv5267sup1.pdf