

Abstract
Inactivation of APC is a strongly predisposing event in the development of colorectal cancer1,2, prompting us to search for vulnerabilities specific to cells that have lost APC function. Signalling through the mTOR pathway is known to be required for epithelial cell proliferation and tumour growth3-5 and the current paradigm suggests that a critical function of mTOR activity is to upregulate translational initiation through phosphorylation of 4EBP16,7. This model predicts that the mTOR inhibitor rapamycin, which does not efficiently inhibit 4EBP18, would be ineffective in limiting cancer progression in APC deficient lesions. Here we show that mTORC1 activity is absolutely required for the proliferation of APC deficient (but not wild type) enterocytes, revealing an unexpected opportunity for therapeutic intervention. Although APC deficient cells show the expected increases in protein synthesis, our studies reveals that it is translation elongation, and not initiation, which is the rate-limiting component. Mechanistically, mTORC1 mediated inhibition of eEF2 kinase is required for the proliferation of APC deficient cells. Importantly, treatment of established APC deficient adenomas with rapamycin (which can target eEF2 through the mTORC1 – S6K – eEF2K axis) causes tumour cells to undergo growth arrest and differentiation. Taken together our data suggest that inhibition of translation elongation using existing, clinically approved drugs such as the Rapalogs, would provide clear therapeutic benefit for patients at high-risk of developing colorectal cancer.
The ability of the intestinal epithelium to regenerate following challenge has been well described9-11. We have shown that this is a Wnt-driven process that mimics the proliferation observed following Apc deletion11,12 and is a valuable model of the early stages of intestinal cancer. However, the underlying mechanisms controlling these processes are largely unknown. The serine/threonine kinase mTOR, particularly the mTOR Complex 1 (mTORC1), is a known mediator of cell growth and proliferation13. Previous studies have suggested that mTORC1 may be important in both the intestinal stem-cell niche and for intestinal tumourigenesis4,5,14. We therefore queried the role of mTORC1 in intestinal proliferation following Wnt activation. Following Apc deletion there was an increase in the phosphorylation status of mTORC1 effectors rpS6 and 4EBP1 that was dependent on MYC expression. Increased phosphorylation of these proteins was also seen during crypt regeneration (Fig. 1a,b,c, Extended Data Fig. 1a). Importantly, the mTOR inhibitor rapamycin blocked intestinal regeneration, demonstrating that mTOR signalling is required for this process (Fig. 1d,e). Given that rapamycin did not affect apoptosis nor proliferation in the normal intestine (Extended Data Fig. 1b,c), these data suggest that there may be a potential therapeutic window, between normal intestinal enterocytes and those with a high level of Wnt activity. Therefore Raptor (an essential component of mTORC1) was deleted in the intestinal epithelium (Extended Data Fig. 1d). Surprisingly, normal gut homeostasis was unaffected by Raptor loss 4 days post-Cre induction, when using an epithelium-specific Cre-Recombinase (VillinCREER Rptorfl/fl) (Extended Data Fig. 1e,f). Furthermore, 400 days after induction, no phosphorylation of rpS6 or 4EBP1 was observed, showing that Raptor deletion was sustained (Extended Data Fig. 2a). Raptor loss caused no change in either levels of mitosis or apoptosis (Extended Data Fig. 2b,c) but proved to be essential for the proliferative phenotype observed during regeneration or following Apc deletion (Fig. 1a,b,d,e,f). Nuclear localisation of β-catenin and high levels of MYC could be demonstrated by IHC, showing that Wnt-activation is still present (Extended Data Fig. 3a,b).
Figure 1. mTORC1 is essential for Wnt-driven proliferation in a MYC-dependent manner.

a+b) Representative IHC of phospho-rpS6 and phospho-4EBP1 showing increased staining 96hrs following Apc deletion. Raptor deletion caused a loss of positivity in both, whereas 10mg/kg rapamycin treatment (beginning at 24hrs) specifically disrupts rpS6 phosphorylation (representative of 6 biological replicates); c) Representative IHC of phospho-rpS6 96hrs after Cre induction showing that Wnt-driven rpS6 phosphorylation is MYC dependent (representative of 3 biological replicates); d) Animals were exposed to 14Gy γ-irradiation and intestinal regeneration was measured 72hrs later, by counting the number of viable crypts and multiplying that by the average size of the regenerating crypts. Boxplot shows that 10mg/kg rapamycin treatment and Raptor deletion significantly decrease intestinal regeneration. Whiskers are max and min, black line is median (n=5 biological replicates per group; **p-value<0.02, Mann-Whitney U test); e) Representative H+E staining of regenerating intestines 72hrs after exposure to 14Gy γ-irradiation. Arrowheads indicate regenerating crypt; (representative of 5 biological replicates).; f) Representative H+E staining 96hrs after Apc loss, showing that 10mg/kg rapamycin treatment or Raptor deletion prevent Wnt-driven proliferation (representative of 6 biological replicates). Treatment began 24hrs after Apc deletion. Red bar is graphical representation of crypt size. Scale bar = 100μm.
Given that rapamycin treatment and Raptor deletion had similar effects, we examined whether rapamycin treatment was sufficient to modify intestinal tumourigenesis, either prophylactically, or chemotherapeutically. First we assessed whether rapamycin could suppress a model of intestinal tumourigenesis, in which Apc deletion is targeted to LGR5-positive stem cells using the LGR5CREER (LGR5CREER Apcfl/fl). Mice were treated starting 10 days post-Cre induction, and in contrast to controls, remained tumour free for the duration of the experiment (Fig. 2a,b). Next we treated mice (ApcMin/+ or LGR5CREER Apcfl/fl) with established adenomas. Remarkably, the mice lost their clinical symptoms of disease and survived significantly longer than controls (Fig. 2c,d; Extended Data Fig. 3c). We next analysed the tumours from these mice over a time-course post-rapamycin treatment. Treatment caused a loss of proliferation specifically within the tumours by 72-hours, and an increase in the number of Lysozyme-positive paneth cells (Fig. 2e; Extended Data Fig. 3d,e). By 30 days, most tumours had shrunk considerably to small non-proliferative lesions that no longer contained paneth cells (Fig. 2f; Extended Data Fig. 3f). Within the normal intestine there are two main cell populations that show high levels of Wnt-signalling; the label-retaining/progenitor population and the paneth cell population15. Our data suggest that treatment of mice with rapamycin causes the differentiation of the tumour’s Wnt-high progenitor cells into the other Wnt-high fate in the intestine; namely non-proliferative paneth-like cells. The cell-cycle arrest in these cells was examined by staining for p21, p16, p53. No increase in these markers was observed, suggesting a classical cell cycle arrest pathway had not been engaged (Extended Data Fig. 4a). We reasoned that if mice were removed from rapamycin the tumours would regain proliferative capacity. Indeed, when rapamycin treatment was halted, signs of intestinal neoplasia were observed approximately 40–60 days later (Extended Data Fig. 3c). This suggested that intestinal adenoma stem cells were still present. Tumours from LGR5-GFP-CREER mice were stained to detect LGR5-GFP positivity. We found that following rapamycin treatment, numerous LGR5-positive cells were still present, indicating that, while rapamycin treatment causes a regression of the lesions, the tumour initiating cells remain (Extended Data Fig. 4b).
Figure 2. Apc-driven tumourigenesis requires mTORC1 activation.

a+b) Graphical representation of prophylactic rapamycin treatment strategy and Kaplan-Meyer survival curve showing that prophylactic rapamycin treatment prevents tumourigenesis. 10mg/kg rapamycin treatment began at Day 10 post-Apc deletion, and lasted 30 days, after which mice were sampled. Area highlighted by red indicates duration of rapamycin treatment (n=8, vehicle; n=13 rapamycin; ***p-value≤0.001, Log Rank test); c+d) Graphical representation of chemotherapeutic rapamycin treatment strategy and Kaplan-Meyer survival curve showing that rapamycin treatment can regress established intestinal tumours. 10mg/kg rapamycin treatment started when mice showed signs of intestinal disease, and lasted 30 days, after which mice were sampled. Graph represents survival while on rapamycin treatment (n=5, vehicle; n= 16, rapamycin; ***p-value≤0.001, Log Rank test); e) Representative IHC of phospho-rpS6, BrdU and Lysozyme, showing that 72hrs of 10mg/kg rapamycin treatment causes a loss in rpS6 phosphorylation and BrdU positivity, and an increase in lysozyme staining in intestinal tumours (representative of 5 biological replicates); f) Representative H+E and IHC for BrdU showing that small, non-proliferative lesions remain after 30 days of 10mg/kg rapamycin treatment (representative of 5 biological replicates). Scale bar = 100μm
We next examined the mechanism of mTORC1 requirement following Apc loss. mTORC1 is known to regulate protein synthesis on multiple levels and most research has focused on two downstream effectors: 4EBP1 and S6K. A number of studies have suggested that translation initiation, via the 4EBP1 – eIF4E axis, is the critical effector of mTOR in cancer6,16. However, it has been shown that rapamycin preferentially inhibits the phosphorylation of S6K over 4EBP18, suggesting that 4EBP1-mediated inhibition of translation initiation may not be limiting in the context of Apc loss. To assess the changes in translational control in response to mTORC1 inhibition, we measured the polysomal distribution in WT, Apc-deficient and Apc/Raptor-deficient intestinal epithelial cells 4-days post gene deletion. Apc deletion resulted in a decrease in the number of polysomes, whereas Apc/Raptor co-deletion reversed this effect (Fig. 3a). The decrease in the number of polysomes following Apc deletion could suggest either reduced translation initiation (and consequently a lower overall level of translation) and/or a faster rate of translational elongation. Global translation rates were measured using an in vitro intestinal crypt 3d-culture model17. The Apc-deficient cells were shown to have increased 35S-labelled methionine/cysteine incorporation compared to WT, showing higher overall levels of protein synthesis (Fig. 3b). Unfortunately, Raptor deletion prevented the growth of crypts in vitro so this could not be assayed (Extended Data Fig. 5).
Figure 3. mTORC1 drives increased translational elongation.

a) Representative polysome profiles of intestinal epithelial cells showing altered RNA distribution 96hrs after Apc deletion. Bar graph represents the ratio of sub-polysomes compared to polysomes (S:P). Data are average ± s.e.m. (n=3 per group; *p-value≤0.05, Mann-Whitney U test); b) Intestinal crypt culture was pulsed for 30 min with 35S-labelled methionine/cysteine. Incorporation of 35S into protein was quantified by scintillation counting and normalized to total protein. Apc deletion increases 35S incorporation. Data are average ± s.e.m. (n=3 biological replicates per group; *p-value≤0.05, Mann-Whitney U test); c) The ribosome run-off rate was measured by addition of the initiation inhibitor harringtonine to ex vivo crypts from wild-type and Apc deleted mice. Harringtonine was added for 0 or 180 seconds and the increase in sub-polysomes (S) relative to polysome (P) calculated. This run-off rate represents the shift in S:P between the two time points, which is proportional to elongation speed. Data are average ± s.e.m. (n=3 biological replicates per group; *p-value≤0.05, Mann Whitney U test). Also see Supplemental Figure 7.
To measure the rate of translational elongation, an in vitro harringtonine run-off assay was performed18, as described in Methods. There was a >2.5-fold increase in ribosome run-off in crypts with Apc deletion compared to wild-type (Fig. 3c; Extended Data Fig. 6a-d). This suggests that following Wnt activation, elongation, rather than initiation, is rate limiting for protein synthesis and mTORC1 must be activated to overcome this.
Cycloheximide (an inhibitor of elongation19) reduced proliferation associated with Apc deletion, to a similar level to rapamycin (Extended Data Fig. 6e,f). While cycloheximide is acknowledged to inhibit elongation19, it must be emphasised that 72hr treatment could result in broad alterations in protein synthesis. However, the Apcfl/fl-specific loss of proliferation observed here provides “proof of principle” to demonstrate that modulation of protein synthesis may be useful as a chemotherapeutic strategy.
As most previous work has suggested translation initiation downstream of 4EBP1 is limiting to cancer20, it was important to probe known effectors of mTORC1 in this system. Given the alteration of elongation rates, the elongation factor 2 kinase (eEF2K), a known target of S6K21,22 was of particular interest. eEF2K is a negative regulator of the elongation factor 2 (eEF2), giving mTORC1 the ability to promote translational elongation via S6K23. Using multiple mouse knockout and knock-in alleles, we further dissected the downstream effectors of mTORC1 in intestinal regeneration. S6k1/2 knockout decreased intestinal regeneration, whilst knockout of Eif4ebp1/2 had no effect. As the 4EBP proteins are negative regulators of eIF4E, an increase (rather than decrease) in regeneration may have been predicted, but this was not found. Moreover these intestines were still sensitive to rapamycin, demonstrating that rapamycin was acting via the mTORC1-S6K branch rather than 4EBP1-eIF4E (Extended Data Fig. 7). We then used an Eef2k-null mouse and showed that following irradiation and treatment with rapamycin, these mice were now resistant to mTORC1 inhibition, confirming the importance of translational elongation (Fig. 4a). To ensure that S6K was not also acting through its more established effector, rpS6, we used an rpS6 phospho-mutant that cannot be phosphorylated by S6K. This was unable to phenocopy Raptor deletion (Fig. 4a), showing that Wnt-driven regeneration requires increased translational elongation, mediated through mTORC1.
Figure 4. mTORC1 signalling via eEF2K controls intestinal proliferation following Wnt-signalling.

a) Graphical representation of findings and boxplots showing that murine intestinal regeneration following irradiation implicates the mTORC1-S6K-eEF2K axis in Wnt-driven proliferation. Animals were exposed to 14Gy γ-irradiation, and intestinal regeneration was calculated 72hrs after exposure, by examining the number and size of regenerating crypts, relative to WT regenerating intestines. Whiskers are max and min, black line is median (n=6 per group; *p-value<0.05, NS: not significant, Mann-Whitney U test); b+c) Representative H+E and boxplot showing Eef2k deletion confers resistance to 10mg/kg rapamycin treatment, 96hrs after Apc deletion. Treatment began 24hrs after induction. Red bar is graphical representation of crypt size. Whiskers are max and min, black line is median (n=3 biological replicates per group; *p-value<0.05, Mann-Whitney U test); d) qRT-PCR of intestinal epithelial cells using primers for Cdk4, Cdk6, Ccnd1, Ccnd2 and Ccnd3. Ccnd3 is not regulated at the transcriptional level. Data were normalised to Gapdh. Data are average ± s.e.m. (n=3 biological replicates per group, *p-value≤0.05, NS: not significant, Mann Whitney U test); e) Western blot analysis of intestinal epithelial cells from each group. Antibodies to CDK4, CDK6, Cyclin D1, Cyclin D2, Cyclin D3 and β-actin are shown. Each well represents a different mouse from the relevant group, and are the same samples used for the qRT-PCR. Scale bar = 100μm.
To prove that inhibition of eEF2K by S6K is required to allow increased eEF2 activity following Apc loss, we intercrossed VillinCREER Apcfl/fl to Eef2k−/− mice and treated these with rapamycin. In contrast to VillinCREER Apcfl/fl mice, these intestines were now resistant to growth inhibitory effects of rapamycin (Fig. 4b,c). Tellingly, these mice no longer show an increase in the inhibitory phosphorylation of eEF2 following rapamycin treatment (Extended Data Fig. 8).
To assess whether the increased elongation following Apc deletion had differential affects on cell cycle regulating proteins, RNA and protein levels of several key cell cycle regulators were tested (Fig. 4d,e). This analysis revealed that whilst there were increased RNA and protein levels of Cyclin D1, D2, CDK4 and CDK6, Cyclin D3 had increased protein levels in the absence of increased mRNA levels. Cyclin D3 protein levels were sensitive to rapamycin exposure, and this sensitivity depends on eEF2K (Extended Data Fig. 9). Additionally, ribosomes were shown to elongate approximately 4-times faster on Cyclin D3 messages in Apc-deficient cells than WT cells. No differences were detected in other messages tested (Extended Data Fig. 10). Taken together, these data suggest that Cyclin D3 is translationally regulated at the level of elongation, consistent with previous reports24,25. The contribution of Cyclin D3 to the proliferative phenotype remains to be elucidated.
In summary, we report that mTORC1 is an essential downstream effector of Wnt-signalling in the intestine. We show that intestinal proliferation associated with Wnt-signalling requires the mTORC1 – S6K – eEF2K – eEF2 axis, and that the resulting increase in the rate of elongation of specific polypeptides overcomes a limiting translational step. Our work highlights key functional roles for eEF2K and translational elongation in the control of the initiation of cancer and adenomatous proliferation. The importance of elongation in this context has been suggested in a small number of publications, but this study provides definitive in vivo evidence26-28. Finally, we have also shown that targeting mTOR and translational control may be a viable strategy for chemoprevention of CRC in high risk patients, and treatment of early stage disease. Indeed, recent studies have suggested that the chemopreventative agents aspirin and mesalazine also target mTOR29,30.
METHODS
Mouse colonies
All experiments were performed according with UK Home Office regulations (licence 60/4183) which undergoes local ethical review at Glasgow University. Outbred male mice from 6 to 12 weeks of age were used. The majority of the work was performed on C57BL/6 mice: AhCre, Apcfl/fl, ApcMin/+, Rptorfl/fl, S6k1/2 knockout and Rps6mut mice were all C57BL/6J. Some treatment experiments were performed on mice that were only three generations C57BL/6 (Lgr5CreER Apcfl/fl).
The alleles used were as follows: VillinCreER (ref. 31). AhCre (ref. 11), Lgr5CreER (ref. 32), Apc580 (ref. 33), ApcMin/+ (ref. 34), Mycfl/fl (ref. 35), Rptor fl/fl (ref. 36), ROSA-tdRFP (ref. 37), Eif4ebp1 knockout (ref. 38), Eif4ebp2 knockout (ref. 39), S6k1 knockout (ref. 40), S6k2 knockout (ref. 41), Eef2k knockout (ref. 42), Rps6mut (ref. 43). Recombination by VillinCreER was induced with one intraperitoneal (i.p.) injection of 80 mg kg−1 tamoxifen on day 0 and day 1. Analysis of VillinCreER-induced mice was at day 4 after induction. Red fluorescent protein (RFP) analysis was carried out by inducing recombination by AhCre using a single i.p. injection of 80 mg kg−1 β-napthoflavone. RFP visualization was carried out on day 4. Mice carrying the Lgr5CreER transgene were given one i.p. injection of 120 mg kg−1 tamoxifen.
For regeneration experiments, mice were exposed to γ-irradiation from caesium-137 sources. This delivered γ-irradiation at 0.423 Gy min−1.
Rapamycin treatment was performed using a daily i.p. injection of 10 mg kg−1 (refs 43, 44) in 5% Tween80 and 5% polyethylene glycol in PBS. Cycloheximide treatment was performed using a daily i.p. injection of 35 mg kg−1 in PBS.
In accordance with the 3Rs, the smallest sample size was chosen that could give a significant difference. Given the robust phenotype of the Apcfl/fl, and our prediction that mTOR was essential, the minimum sample size assuming no overlap in control versus experimental is three animals.
No randomization was used and the experimenter was blinded to drugs and genotypes.
IHC
Standard IHC techniques were used throughout this study. Antibody concentrations used were as follows: phospho-rpS6Ser235/236 (1:800; Cell Signaling 4858), phospho-4EBP1Thr37/46 (1:500; Cell Signaling 2855), phospho-eEF2Thr56 (1:500; Novus Biologicals NB100-92518), c-MYC (1:200; Santa Cruz sc-764), β-catenin (1:50; BD Biosciences 610154), BrdU (1:200; BD Biosciences 347580), lysozyme (1:150; Dako A099), GFP (1:1,000; Abcam ab6556), p21 (1/4; CNIO Madrid), p16 (1:400; Santa Cruz sc1661), p53 (1/150; Vector Laboratories VPP956). For each antibody, staining was performed on at least three mice of each genotype. Representative images are shown for each staining.
Assaying apoptosis, mitosis and proliferation in vivo
Apoptosis and mitotic index were scored from H&E-stained sections as previously described11. Proliferation levels were assessed by measuring BrdU incorporation. Mice were injected with 250 μl of BrdU (Amersham Biosciences) 2 h before being killed. Immunohistochemical staining for BrdU was then performed using an anti-BrdU antibody. For each analysis, 25 full crypts were scored from at least three mice of each genotype.
Intestinal epithelium extraction
To generate tissue for polysomal profile analysis, 10-cm portions of intestine were flushed with 0.1 mg ml−1 cycloheximide (Sigma) in PBS and inverted over a glass rod to expose the epithelial surface. Intestines were incubated in 0.1 mg ml−1 cycloheximide in HBSS (Gibco) with 10 mM EDTA for 5 min at 37 °C followed by 5 min of vigorous shaking. Intestines were transferred to 0.1 mg ml−1 cycloheximide in PBS and incubated for a further 5 min at 4 °C, followed by 5 min of vigorous shaking. This fraction contained intestinal crypts and was used for downstream analysis.
Sucrose density ultracentrifugation
Intestinal epithelial cells were lysed in ice-cold 300 mM NaCl, 15 mM MgCl2, 15 mM Tris (pH 7.5) containing 500 units ml−1 RNAsin, 1 mg ml−1 heparin sulphate and 0.1 mg ml−1 cycloheximide supplemented with 0.1% (v/v) Triton X-100. Post-nuclear lysates were layered on 10 ml 10–50% (w/v) sucrose gradients of the same buffer omitting Triton X-100. Gradients were centrifuged at 38,000 r.p.m. for 2 h at 4 °C in a SW40Ti rotor (Beckman Coulter) and separated through a live OD254nm ultraviolet spectrometer (Isco). Comparison of peak abundance was based on the area under the curve.
Crypt culture
Mouse small intestines were opened longitudinally and washed with PBS. Crypts were isolated as previously described17. Isolated crypts were mixed with 50 μl of Matrigel (BD Bioscence), plated in 24-well plates in Advanced DMEM/F12 with Noggin (100 ng ml−1, Peprotech). Wild-type crypts were also supplemented with R-spondin (500 ng ml−1; R&D Systems). Growth factors were added every other day. Sphere formation was scored 7 days after plating, by counting the number of spheres present per well.
Determination of protein synthesis rates
Cells were treated with 30 μCi ml−1 35S-methionine label (Hartmann Analytic) for 30 min then harvested and lysed. Protein was precipitated onto filter paper (Whatmann) by addition of trichloroacetic acid to 12.5% and washed with 70% ethanol then acetone. Scintillation was read from dried filter paper in triplicate for each experimental condition (National Diagnostics). Total protein content was determined by bicinchoninic acid (BCA) assay (Pierce) for standardization between conditions.
Harringtonine run-off assay
Harringtonine inhibits de novo translational initiation, allowing ribosomes engaged in elongation to run-off their messages while limiting re-initiation post-termination. Harringtonine was added for 0 or 180 s and the increase in sub-polysomes relative to polysomes was calculated. This run-off rate represents the shift in S:P between the two time points, which is proportional to elongation speed. Crypt cultures were treated with 2 μg ml−1 Harringtonine (Insight Biotechnology) and at set time periods (0 and 180 s) 0.1 mg ml−1 cycloheximide was added. Cells were scraped into PBS at 4 °C and prepared for sucrose gradient ultracentrifugation as previously described.
Western blotting
Snap-frozen intestinal epithelial tissue (50–100 mg) was homogenized using the Precellys 24 (Stretton Scientific) in 500 μl of Ripa-lysis buffer. Protein concentrations were determined using a BCA Protein Assay Kit (Pierce). Equal amounts of cellular protein (30 μg) were separated on a 4–12% gradient gel (Novex) and subsequently transferred to a PVDF membrane (Amersham). Total protein was visualized with Poinceau (Sigma). After blocking the membranes in TBS containing 5% BSA (Sigma), 0.02% Triton X-100 for 1 h, primary antibodies were added in block solution at the following dilutions: CDK4 (Santa Cruz SC-260, 1:1,000), CDK6 (Cell Signaling 3136, 1:1,000), cyclin D1 (Cell Signaling 2926, 1:2,000), cyclin D2 (Cell Signaling 3741, 1:1,000), cyclin D3 (Cell Signaling 2936, 1:2,000), eEF2K (Cell Signaling 3692, 1:1,000) and β-actin (Sigma A2228, 1:5,000). After washing, the appropriate HRP-conjugated secondary goat antibodies (Dako) were added diluted 1:10,000 in block for 1 h. Antibody binding was detected using ECL Western Blotting Substrate (Pierce). Primary antibody incubations were carried out at 4 °C overnight. Remaining incubations were carried out at room temperature.
RNA isolation
Snap-frozen intestinal epithelial tissue was homogenized and RNA was extracted using the TRIzol method (Ambion).
qPCR
One microgram of RNA was reverse transcribed to cDNA using a Quantitect Reverse Transcription Kit (Qiagen) in a reaction volume of 20 μl. qPCR was performed on each sample in triplicate in a 20 μl reaction mixture containing 10 μl of 2× DyNAmo HS master mix (Thermo Scientific), 0.5 μM of each of the primers (detailed later) and 2 μl cDNA generated previously. The reaction mixture without a template was run in triplicate as a control. The reaction conditions were as follows: 95 °C for 15 min, followed by 40 cycles of three steps consisting of denaturation at 94 °C for 15 s, primer annealing at 60 °C for 30 s, and primer extension at 72 °C for 30 s. A melting curve analysis was performed from 70 °C to 95 °C in 0.3 °C intervals. Gapdh was used to normalize for differences in RNA input.
qRT–PCR primers
qRT–PCR primers were as follows. Ccnd1 forward, 5′-GAGA AGTTGTGCATCTACACTG-3′; Ccnd1 reverse, 5′-AAATGAACTTCACATCTGTGGC-3′; Ccnd2 forward, 5′-CTACCGACTTCAAGTTTGCC-3′; Ccnd2 reverse, 5′-GCTTTGAGACAATCCACATCAG-3′; Cdk4 forward, 5′-AATGTTGTACGGCTGATGGA-3′; Cdk4 reverse, 5′-AGAAACTGACGCATTAGATCCT-3′; Cdk6 forward, 5′-GGCGTACCCACAGAAACCATA-3′; Cdk6 reverse, 5′-AGGTAAGGGCCATCTGAAAACT-3′; Ccnd3 forward, 5′-CGAGCCTCCTACTTCCAGTG-3′; Ccnd3 reverse, 5′-GGACAGGTAGCGATCCAGGT-3′; Rps6 forward, 5′-AGCTCCGCACCTTCTATGAGA-3′; Rps6 reverse, 5′-GGGAAAACCTTGCTTGTCATTC-3′;Rps21 forward,5′-GTCCATCCAGATGAACGTGG-3′;Rps21 reverse, 5′-CCATCAGCCTTAGCCAATCGG-3′.
Extended Data
Extended Data Fig. 1. mTORC1 is activated following Wnt-signal and its inhibition does not affect homeostasis.

a) Representative IHC of phospho-rpS6 and phospho-4EBP1 show mTORC1 activity during intestinal regeneration, 72hrs after 14Gy γ-irradiation (representative of 5 biological replicates); b+c) boxplots demonstrating that 72hrs of 10mg/kg rapamycin treatment does not alter mitosis or apoptosis in normal intestinal crypts. Whiskers are max and min, black line is median (n=4 per group; NS: not significant, Mann-Whitney U test); d) Intestines imaged on OV100 microscope, 96hrs post induction, for RFP. Tissue without the ROSA-tdRFP reporter (labelled Neg Control) show no RFP positivity, while the positive control and Raptor fl/fl intestines show high RFP positivity (representative of 3 biological replicates).; e+f) boxplot showing that Raptor deletion does not affect mitosis or apoptosis rates in intestinal crypts, 96hrs after induction. Whiskers are max and min, black line is median (n=4 per group; NS: not significant, Mann-Whitney U test). Scale bar = 100μm
Extended Data Fig. 2. Raptor deletion is maintained in the small intestine.

a) Representative IHC of phospho-rpS6 and phospho-4EBP1 shows maintained loss of mTORC1 signalling 400+ days after Raptor deletion. Arrows indicate unrecombined escaper crypts that still show active mTORC1 signalling (Representative of 5 biological replicates).; b+c) boxplots showing that mitosis and apoptosis are unchanged 400+ days after Raptor deletion. Mitosis and apoptosis were counted on H+E sections and are quantified as percent mitosis or apoptosis per crypt. Whiskers are max and min, black line is median (n=5 per group; NS: not significant, Mann-Whitney U test). Scale bar = 100μm
Extended Data Fig. 3. Wnt-signalling is still active after Raptor deletion and Rapamycin treatment causes regression of established tumours.

a+b) Representative IHC of MYC and β-catenin showing high MYC levels and nuclear localisation of β-catenin 96hrs after Apc and Apc/Raptor deletion, demonstrating active Wnt-signalling. Nuclear staining (as opposed to membranous staining) of β-catenin is indicative of active Wnt-signalling; Scale bar = 100μm (representative of 3 biological replicates). c) Kaplan-Meyer survival curve of ApcMin/+ mice treated with rapamycin when showing signs of intestinal neoplasia. 10mg/kg rapamycin treatment started when mice showed signs of intestinal disease, and was withdrawn after 30 days. Animals continued to be observed until signs of intestinal neoplasia. Death of animals in the rapamycin group almost always occurred following rapamycin withdrawal. (n=8 per group, ***p-value≤0.001, Log Rank test); d) boxplot showing that 72hr 10mg/kg rapamycin treatment causes an increase in Lysozyme positive cells in tumours. Percent Lysozyme positivity within tumours was calculated using Image J software (http://imagej.nih.gov/ij/). Whiskers are max and min, black line is median (10 tumours from each of 5 mice per group were measured, **p-value≤0.014, Mann-Whitney U test); e) boxplot showing that 72hrs 10mg/kg rapamycin treatment causes a decrease in BrdU positivity within tumours. Percent BrdU positivity within tumours was calculated using Image J software. Whiskers are max and min, black line is median (10 tumours from each of 5 mice per group were measured, **p-value≤0.021, Mann-Whitney U test); f) Representative IHC of Lysozyme, showing a lack of Lysozyme positive paneth cells in remaining cystic tumours after 30 days of 10mg/kg rapamycin treatment; Scale bar = 100μm (representative of 5 biological replicates).
Extended Data Fig. 4. IHC following rapamycin treatment.

a) Representative IHC of p21, p16, and p53 after 6hrs and 72hrs of 10mg/kg rapamycin treatment. Staining shows no induction of these proteins in tumours following rapamycin treatment (representative of 5 biological replicates).; b) Representative IHC for LGR5 GFP showing high numbers of LGR5-positive cells after 7 and 30 days of 10mg/kg daily rapamycin treatment. (representative of 5 biological replicates). Scale bar = 100μm.
Extended Data Fig. 5. Raptor deletion in the intestinal crypt is lethal in vitro.
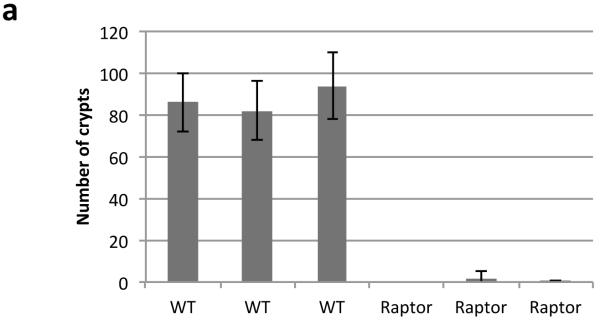
a) Graph showing that Raptor deletion prevents intestinal crypts from growing ex vivo. Intestinal crypts were isolated and cultured as previously described17, 96hrs after Cre induction. Number of viable organoids was counted by eye 72hrs after crypt isolation. Data are average ± s.d. (n=3 biological replicates per group)
Extended Data Fig 6. Apc deletion increases translational elongation rates and Cycloheximide treatment phenocopies rapamycin treatment.

a) Representative polysome profiles from wild-type ex vivo crypts incubated with harringtonine for 0 seconds (left) and 180 seconds (right) prior to harvest. (n=3 per time point) b) The areas under the sub-polysome (40S, 60S and 80S) and polysome sections as indicated by the dashed lines in a) were quantified and expressed as a percentage of their sum. Data in the bar graph is the average ± s.e.m (n=3 per time point). c) and d) show data for Apc deleted crypts as for wild-type in b) and c) (n=3 biological replicates). e) Representative H+E showing that 35mg/kg cycloheximide treatment phenocopies rapamycin treatment 96hrs after Apc deletion. Treatment began 24hrs after induction (n=3 biological replicates); f) Representative IHC for BrdU showing a loss of proliferation in tumours after 72hrs of 35mg/kg cycloheximide treatment. (n=3 biological replicates). Arrow highlights normal proliferating crypts. Scale bar = 100μm
Extended Data Fig. 7. S6k deletion decreases intestinal regeneration.

Graphical representation of findings, and boxplot showing that murine intestinal regeneration following irradiation is dependent on S6K. Animals were exposed to 14Gy γ-irradiation, and intestinal regeneration was calculated 72hrs after exposure by counting the number of viable crypts and multiplying that by the average size of the regenerating crypts. Relative regeneration was calculated by comparing each group to WT regeneration. Rapamycin treatment arm is reproduced from Fig.4 for visual clarity. Whiskers are max and min, black line is median (n=6 per group, *p-value=0.034, Mann Whitney U test).
Extended Data Fig. 8. Eef2k deletion drives resistance to rapamycin.

a) Representative IHC of phospho-eEF2 and phospho-S6 in WT, Apc-deficient and Apc- and Eef2k-deficient (with and without 72hrs 10mg/kg rapamycin treatment) shows that rapamycin is unable to induce eEF2 phosphorylation in the absence of eEF2K (n=6 biological replicates). Scale bar = 100μm
Extended Data Fig. 9. Cyclin D3 is regulated at the level of elongation.

a) Representative IHC of Apcfl/fl intestines with and without Eef2k deletion. Antibodies to eEF2K, phospho-S6, and Cyclin D3 are shown (representative of 3 biological replicates). Following Eef2k KO, Cyclin D3 levels are no longer decreased upon 10mg/kg rapamycin treatment; b) boxplot showing the number of Cyclin D3 positive cells per crypt, 96hrs after Apc deletion, with and without 10mg/kg rapamycin treatment. Graph shows that in Eef2k KO animals, rapamycin no longer reduced Cyclin D3 levels (n=3 biological replicates per group; *p-value≤0.05, Mann Whitney U test); c) western blot analysis of intestinal epithelial cells from Apcfl/fl and Apcfl/fl Eef2k KO, with and without 10mg/kg rapamycin. Antibodies to eEF2K, phospho-S6, Cyclin D3 and β-actin are shown. Each well represents a different mouse from the relevant group. Cyclin D3 levels are no longer reduced following Eef2k deletion. Scale bar = 100μm.
Extended Data Fig. 10. Ribosomes elongate faster on Ccnd3 following Apc deletion.

The ribosome run-off rate of various messages was measured as in Figure 3. Elongation of Ccnd3 was significantly increased, while Actb, Rps21, Rps6 and Ccnd1 remain unchanged. Data are average ± s.e.m. (n=3 biological replicates per group; *p-value≤0.05, Mann Whitney U test).
Supplementary Material
Acknowledgements
WJF is funded by AICR. OJS is funded by CRUK, ERC Investigator Grant (COLONCAN) and the European Union Seventh Framework Programme FP7/2007-2013 under grant agreement number 278568. MB is an MRC Senior Fellow. Authors acknowledge Patrizia Cammareri, Jennifer Morton and Claudio Murgia for proof reading of the manuscript.
Footnotes
Extended Data is linked to the online version of the paper at www.nature.com/nature.
Competing financial interests
The Authors declare no competing financial interests.
References
- 1.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 2.Korinek V, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 3.Ashton GH, et al. Focal adhesion kinase is required for intestinal regeneration and tumorigenesis downstream of Wnt/c-Myc signaling. Developmental cell. 2010;19:259–269. doi: 10.1016/j.devcel.2010.07.015. doi:10.1016/j.devcel.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujishita T, Aoki K, Lane HA, Aoki M, Taketo MM. Inhibition of the mTORC1 pathway suppresses intestinal polyp formation and reduces mortality in ApcDelta716 mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13544–13549. doi: 10.1073/pnas.0800041105. doi:10.1073/pnas.0800041105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gulhati P, et al. Targeted inhibition of mammalian target of rapamycin signaling inhibits tumorigenesis of colorectal cancer. Clin Cancer Res. 2009;15:7207–7216. doi: 10.1158/1078-0432.CCR-09-1249. doi:10.1158/1078-0432.CCR-09-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pourdehnad M, et al. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11988–11993. doi: 10.1073/pnas.1310230110. doi:10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martineau Y, et al. Pancreatic tumours escape from translational control through 4E-BP1 loss. Oncogene. 2013 doi: 10.1038/onc.2013.100. doi:10.1038/onc.2013.100. [DOI] [PubMed] [Google Scholar]
- 8.Jiang YP, Ballou LM, Lin RZ. Rapamycin-insensitive regulation of 4e-BP1 in regenerating rat liver. J Biol Chem. 2001;276:10943–10951. doi: 10.1074/jbc.M007758200. doi:10.1074/jbc.M007758200. [DOI] [PubMed] [Google Scholar]
- 9.Bach SP, Renehan AG, Potten CS. Stem cells: the intestinal stem cell as a paradigm. Carcinogenesis. 2000;21:469–476. doi: 10.1093/carcin/21.3.469. [DOI] [PubMed] [Google Scholar]
- 10.Bernal NP, et al. Evidence for active Wnt signaling during postresection intestinal adaptation. Journal of pediatric surgery. 2005;40:1025–1029. doi: 10.1016/j.jpedsurg.2005.03.021. discussion 1029, doi:10.1016/j.jpedsurg.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 11.Ireland H, et al. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of beta-catenin. Gastroenterology. 2004;126:1236–1246. doi: 10.1053/j.gastro.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 12.Muncan V, et al. Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Molecular and cellular biology. 2006;26:8418–8426. doi: 10.1128/MCB.00821-06. doi:10.1128/MCB.00821-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. doi:10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yilmaz OH, et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. 2012;486:490–495. doi: 10.1038/nature11163. doi:10.1038/nature11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farin HF, Van Es JH, Clevers H. Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology. 2012;143:1518–1529. e1517. doi: 10.1053/j.gastro.2012.08.031. doi:10.1053/j.gastro.2012.08.031. [DOI] [PubMed] [Google Scholar]
- 16.She QB, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18:39–51. doi: 10.1016/j.ccr.2010.05.023. doi:10.1016/j.ccr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. doi:10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 18.Fresno M, Jimenez A, Vazquez D. Inhibition of translation in eukaryotic systems by harringtonine. European journal of biochemistry / FEBS. 1977;72:323–330. doi: 10.1111/j.1432-1033.1977.tb11256.x. [DOI] [PubMed] [Google Scholar]
- 19.Schneider-Poetsch T, et al. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6:209–217. doi: 10.1038/nchembio.304. doi:10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh AC, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. doi:10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–480. doi: 10.1038/nature03205. doi:10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 22.Browne GJ, Proud CG. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Molecular and cellular biology. 2004;24:2986–2997. doi: 10.1128/MCB.24.7.2986-2997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ryazanov AG, Shestakova EA, Natapov PG. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature. 1988;334:170–173. doi: 10.1038/334170a0. doi:10.1038/334170a0. [DOI] [PubMed] [Google Scholar]
- 24.Gorshtein A, et al. Mammalian target of rapamycin inhibitors rapamycin and RAD001 (everolimus) induce anti-proliferative effects in GH-secreting pituitary tumor cells in vitro. Endocr Relat Cancer. 2009;16:1017–1027. doi: 10.1677/ERC-08-0269. doi:10.1677/ERC-08-0269. [DOI] [PubMed] [Google Scholar]
- 25.Gutzkow KB, et al. Cyclic AMP inhibits translation of cyclin D3 in T lymphocytes at the level of elongation by inducing eEF2-phosphorylation. Cell Signal. 2003;15:871–881. doi: 10.1016/s0898-6568(03)00038-x. [DOI] [PubMed] [Google Scholar]
- 26.Firczuk H, et al. An in vivo control map for the eukaryotic mRNA translation machinery. Mol Syst Biol. 2013;9:635. doi: 10.1038/msb.2012.73. doi:10.1038/msb.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hussey GS, et al. Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Mol Cell. 2011;41:419–431. doi: 10.1016/j.molcel.2011.02.003. doi:10.1016/j.molcel.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakamura J, et al. Overexpression of eukaryotic elongation factor eEF2 in gastrointestinal cancers and its involvement in G2/M progression in the cell cycle. Int J Oncol. 2009;34:1181–1189. [PubMed] [Google Scholar]
- 29.Din FV, et al. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology. 2012;142:1504–1515. e1503. doi: 10.1053/j.gastro.2012.02.050. doi:10.1053/j.gastro.2012.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baan B, et al. 5-Aminosalicylic acid inhibits cell cycle progression in a phospholipase D dependent manner in colorectal cancer. Gut. 2012;61:1708–1715. doi: 10.1136/gutjnl-2011-301626. doi:10.1136/gutjnl-2011-301626. [DOI] [PubMed] [Google Scholar]
Methods Reference
- 31.El Marjou F, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. doi:10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 32.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. doi:nature06196 [pii] 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 33.Shibata H, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997;278:120–123. doi: 10.1126/science.278.5335.120. [DOI] [PubMed] [Google Scholar]
- 34.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 35.de Alboran IM, et al. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity. 2001;14:45–55. doi: 10.1016/s1074-7613(01)00088-7. [DOI] [PubMed] [Google Scholar]
- 36.Polak P, et al. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell metabolism. 2008;8:399–410. doi: 10.1016/j.cmet.2008.09.003. doi:10.1016/j.cmet.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur J Immunol. 2007;37:43–53. doi: 10.1002/eji.200636745. doi:10.1002/eji.200636745. [DOI] [PubMed] [Google Scholar]
- 38.Tsukiyama-Kohara K, et al. Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nature medicine. 2001;7:1128–1132. doi: 10.1038/nm1001-1128. doi:10.1038/nm1001-1128. [DOI] [PubMed] [Google Scholar]
- 39.Banko JL, et al. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. doi:10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shima H, et al. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. The EMBO journal. 1998;17:6649–6659. doi: 10.1093/emboj/17.22.6649. doi:10.1093/emboj/17.22.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryazanov AG. Elongation factor-2 kinase and its newly discovered relatives. FEBS Lett. 2002;514:26–29. doi: 10.1016/s0014-5793(02)02299-8. [DOI] [PubMed] [Google Scholar]
- 42.Ruvinsky I, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes & development. 2005;19:2199–2211. doi: 10.1101/gad.351605. doi:10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarbassov DD, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Molecular cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. doi:10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 44.Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010;468:1100–1104. doi: 10.1038/nature09584. doi:10.1038/nature09584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.