

Abstract
European red deer (Cervus elaphus elaphus) are susceptible to the agent of bovine spongiform encephalopathy, one of the transmissible spongiform encephalopathies, when challenged intracerebrally but their susceptibility to alimentary challenge, the presumed natural route of transmission, is unknown. To determine this, eighteen deer were challenged via stomach tube with a large dose of the bovine spongiform encephalopathy agent and clinical signs, gross and histological lesions, presence and distribution of abnormal prion protein and the attack rate recorded. Only a single animal developed clinical disease, and this was acute with both neurological and respiratory signs, at 1726 days post challenge although there was significant (27.6%) weight loss in the preceding 141 days. The clinically affected animal had histological lesions of vacuolation in the neuronal perikaryon and neuropil, typical of transmissible spongiform encephalopathies. Abnormal prion protein, the diagnostic marker of transmissible encephalopathies, was primarily restricted to the central and peripheral nervous systems although a very small amount was present in tingible body macrophages in the lymphoid patches of the caecum and colon. Serial protein misfolding cyclical amplification, an in vitro ultra-sensitive diagnostic technique, was positive for neurological tissue from the single clinically diseased deer. All other alimentary challenged deer failed to develop clinical disease and were negative for all other investigations. These findings show that transmission of bovine spongiform encephalopathy to European red deer via the alimentary route is possible but the transmission rate is low. Additionally, when deer carcases are subjected to the same regulations that ruminants in Europe with respect to the removal of specified offal from the human food chain, the zoonotic risk of bovine spongiform encephalopathy, the cause of variant Creutzfeldt-Jakob disease, from consumption of venison is probably very low.
Introduction
Bovine spongiform encephalopathy (BSE) is infectious to, primarily, domestic cattle but also wild bovids, several species of antelope, sheep and felids and is considered to be the cause of variant Creutzfeldt-Jakob disease (vCJD) in humans [1,2]. It is a member of the transmissible spongiform encephalopathies (TSE) group of diseases, also known as prion diseases, which include, amongst others, scrapie in sheep and goats, sporadic, familial and iatrogenic Creutzfeldt-Jakob disease (CJD) and kuru in humans, transmissible mink encephalopathy in ranched mink and chronic wasting disease (CWD) in farmed and free-living cervids [3]. All TSEs are invariably fatal and are characterised by long incubation periods leading to clinical neurological manifestations. The pathology is usually linked to the conversion of the normal host-encoded membrane-bound prion protein (PrPC) to the abnormal disease-associated isoform (PrPd) which accumulates in the nervous system and, depending on the host species and the TSE agent involved, in the lymphoreticular system [4] and also some other viscera including kidney, muscle and adrenal [5]. Definitive diagnosis of TSEs is, at present, dependent on the detection of abnormal PrP in tissues by immunohistochemistry (PrPd) or of proteinase resistant abnormal PrP (PrPres) by a variety of biochemical methods [6]. Susceptibility to natural, presumed orally, acquired TSEs is influenced by the age at exposure and the dose of challenge and epidemiological studies in cattle have suggested younger animals are more susceptible to BSE [7]. Additionally, oral BSE challenge of newborn sheep may be more efficient than sheep exposed to oral BSE as adults [8,9]. Similarly, following scrapie exposure it has been shown experimentally, with respect to both attack rate and incubation period, pre-weaned lambs are at greater risk of infection, possibly due to patent gut epithelium [10].
Polymorphisms in the prion protein gene (PRNP) determine the degree of susceptibility and resistance to TSEs in several species and also frequently influence the length of the incubation period [11]. Development of clinical disease in classical scrapie and BSE in sheep is primarily associated with polymorphisms in three specific codons (136, 154 and 171) of the ovine PRNP gene although other codons may play a role as well [12,13]. No such strong genetic association with susceptibility or resistance to BSE appears to exist for domestic cattle derived from the Bos taurus lineage and most seem at risk (reviewed in [12]). However, in British and German cattle herds more complex variations in the regulatory regions of the PRNP gene may play a role in the control of susceptibility to BSE [14–17]. In humans, codon 129 is strongly influential for susceptibility to both sporadic and variant forms of CJD [18]. In deer, a total of 16 polymorphic codons within PRNP have been reported with a large number of PRNP alleles and amino acid substitutions found in white-tailed deer (Odocoileus virginianus), European red deer (Cervus elaphus elaphus), reindeer (Rangifer tarandus) and mule deer (Odocoileus hemionus) [19]. A degree of genetically conferred resistance to CWD seems to occur in white-tailed deer due to polymorphisms in codons 95 and 96 [20] and in mule deer due to a polymorphism in codon 225 [21] but in neither case is full resistance conferred. No naturally occurring TSE has been diagnosed in reindeer to date but the PRNP sequence suggest they would be susceptible to CWD [22] and recent experimental studies have proven this [23]. With respect to CWD in Rocky mountain elk (wapiti, Cervus canadensis nelsoni) heterozygosity (methionine (M)/leucine (L)) in codon 132 of PRNP, which is the equivalent position to human PRNP codon 129 [24], has been proposed to provide some protection [25] although this has been disputed [26] and other studies suggest this polymorphism primarily alters the length of the incubation period [27].
A PRNP polymorphism in codon 226 (glutamine (Q)/glutamate (E)) has been described for European red deer and Sika deer (Cervus nippon) [28], but not yet for the Rocky mountain elk, and the association of this amino acid change with TSE susceptibility has recently been demonstrated in transgenic mice [29]. Codon 226 glutamine is encoded in PRNP from mule deer [30], white-tailed deer [20], moose (Alces alces), cattle, sheep, goats and nyala (Nyala angasii) [12] whereas glutamate is encoded in PRNP from Rocky mountain elk, kudu (Tragelaphus spp.) and cats all of which have shown natural susceptibility to the CWD or BSE agent [12,31,32]. Retrospective analysis showed European red deer of all codon 226 PRNP genotypes were susceptible to BSE when challenged by the intra-cerebral route [33] M.P. Dagleish pers observation), which is important as they are consumed by humans so if natural transmission takes place they are a potential source of vCJD. However, their susceptibility to oral challenge, the accepted route of natural BSE transmission, is unknown.
The aim of this study was to assess the susceptibility of European red deer to oral challenge with a bovine derived BSE brain homogenate to assess transmission by the presumed natural route and therefore the potential risk of zoonotic transmission of BSE from red deer.
Materials and Methods
Animals, BSE inocula, challenge procedure and biopsies
Twenty-five European red deer were housed at 1–2 days old and hand reared with milk replacer prior to weaning onto ad libitum hay and water and a weight dependant allocation of proprietary concentrate feed. At 7–10 weeks of age, 18 animals (10 males and 8 females) were each given 25g of a pool of BSE-positive bovine brain material (VLA-Weybridge SE1736 BBP1) [33] diluted 1:4 (w/v) in 0.32M sucrose solution (total volume 100ml) via a stomach tube followed by flushing of the stomach tube with 50ml of water. Unchallenged environmental controls (n = 7, 3 males and 4 females) were kept in separate but adjacent pens to the challenged deer. Animals were examined daily for clinical signs and weighed at regular intervals. Animals were culled at 190 days post-challenge (dpc) (n = 6 BSE-challenged and 1 unchallenged control), 365 dpc (n = 6 BSE-challenged and 2 unchallenged controls) or allowed to progress until clinical signs or the termination of the experiment at 2320 dpc (n = 6 BSE-challenged and 4 unchallenged controls) (Table 1). Four biopsies were taken from the recto-anal mucosa-associated lymphoid tissue (RAMALT) of the group allowed to progress until clinical signs/termination of the experiment and evaluated for the presence of PrPd by immunohistochemistry (IHC, see below) at 511, 706, 855 and 1853 dpc. Biopsies were performed as described previously [34] with the addition of reversible sedation (10mg/kg medetomidine hydrochloride, Domitor, Janssen Animal Health, UK; atipamezole hydrochloride, Antisedan, Janssen Animal Health, UK). All experimental procedures were approved by the Moredun Research Institute Animals Experiments Ethical Committee and authorised under the UK Animals (Scientific Procedures) Act 1986.
Table 1. Summary of animal, immunohistochemical and serial protein misfolding cyclical amplification data.
| Deer ID | Sex | Challenge status | DPC-PME | Genotype (PrP gene, codon 226) | IHC for PrPd | sPMCA 1st Exp. (3rd round) | sPMCA 2nd Exp. (3rd round) | sPMCA 2nd Exp. (4rd round) |
|---|---|---|---|---|---|---|---|---|
| 322 | M | UNC | 190 | N | 0/4 | 0/4 | 0/4 | |
| 319 | M | UNC | 365 | QE | N | - | - | - |
| 334 | M | UNC | 365 | EE | N | 0/4 | 0/4 | 0/4 |
| 032 | F | UNC | 2320 | N | - | - | - | |
| 034 | F | UNC | 2320 | QE | N | - | - | - |
| 039 | F | UNC | 2320 | EE | N | 1/4 | 0/4 | 0/4 |
| 014 | F | UNC | 2320 | EE | N | - | - | - |
| 327 | M | BSE-C | 190 | QE | N | - | - | - |
| 309 | M | BSE-C | 190 | EE | N | - | - | - |
| 031 | F | BSE-C | 190 | N | - | - | - | |
| 320 | M | BSE-C | 190 | QE | N | - | - | - |
| 324 | M | BSE-C | 190 | N | - | - | - | |
| 326 | M | BSE-C | 190 | N | - | - | - | |
| 323 | M | BSE-C | 365 | QE | N | 0/4 | 0/4 | 0/4 |
| 337 | M | BSE-C | 365 | EE | N | 0/4 | 0/4 | 0/4 |
| 314 | M | BSE-C | 365 | N | 0/4 | 0/4 | 0/4 | |
| 318 | M | BSE-C | 365 | QE | N | - | - | - |
| 310 | M | BSE-C | 365 | QE | N | - | - | - |
| 028 | F | BSE-C | 365 | N | 0/4 | 0/4 | 0/4 | |
| 009 | F | BSE-C | 1727 | P | 4/4 | 4/4 | 4/4 | |
| 015 | F | BSE-C | 2320 | QE | N | 0/4 | 0/4 | 0/4 |
| 033 | F | BSE-C | 2320 | EE | N | 0/4 | 0/4 | 0/4 |
| 016 | F | BSE-C | 2320 | EE | N | 0/4 | 0/4 | 0/4 |
| 023 | F | BSE-C | 2320 | QE | N | 0/4 | 0/4 | 0/4 |
| 037 | F | BSE-C | 2320 | EE | N | 0/4 | 0/4 | 0/4 |
Deer identification, sex, challenge status, time of post-mortem examination, genotype at codon 226 of prion protein gene, presence of abnormal protein by immunohistochemistry (PrPd) and western blotting (PrPres) by western blotting after different experiments of serial protein misfolding cyclic amplification (sPMCA). The animal positive for abnormal prion protein is deer 009 and this was by both IHC and sPMCA. Note that for sPMCA a value of 1/4 is considered negative and probably due to contamination of the sample in this exceptionally sensitive technique. DPC-PME = days post challenge of post mortem examination, PrP = prion protein, PrPd/PrPres = abnormal prion protein, IHC = immunohistochemistry, sPMCA = serial protein misfolding cyclic amplification, Exp. = experiment, M = male, F = female, UNC = unchallenged negative control, BSE-C = BSE oral challenged, Q = glutamine, E = glutamic acid, N = negative, P = positive, for sPMCA x/x = number of PrPres positive tubes/number of replicates, – = not examined by sPMCA.
Genotyping
Genotyping was performed as described previously [33], either from blood samples taken from live deer into vacutainers containing EDTA (BD Bioscience, Erembodegem, Belgium) or from frozen brain material collected post-mortem. Briefly, PCR amplification and sequencing were performed as described previously [35], using amplification oligonucleotides DeerPrP-213d (AGGTCAACTTTGTCCTTGGAGGAG) and DeerPrP+139u (TAAGCGCCAAGGGTATTAGCAT) and sequencing oligonucleotides DeerPrP+70u (GCTGCAGGTAGATACTCCCTC) and DeerPrP-86d (CAGTCATTCATTATGCTGCAGACT).
Post-mortem examination
All animals were subjected to full post-mortem examination after euthanasia by intra-venous pentobarbitone after sedation as described above and an extensive range of tissue samples were taken, as described previously [33], and fixed in 10% neutral buffered formalin and/or stored at −80°C. Fixed samples were post-fixed in fresh 10% neutral buffered formalin, processed for histology routinely and then embedded in paraffin wax. Sections (4μm thick) were mounted on glass slides (Superfrost slides, Menzel-Gläser, Braunschweig, Germany) and either stained with haematoxylin and eosin (HE) or subjected to IHC for PrPd (see below).
Immunohistochemical localisation of PrPd
This was performed as described previously [33,36] using two primary antibodies to PrP which have wide inter-species reactivity; F99, clone 97.6.1 (VMRD Inc., Pullman, USA), which binds to amino acid sequence 220–225 of human PrP and BAR224 (CEA, Saclay, France) which recognizes amino acid sequence 141–147 of human PrP. Briefly, antigen retrieval included immersion in 98% formic acid for 5 minutes followed by autoclaving for 30 minutes at 121°C in 0.2% citrate buffer pH6.0. After blocking steps to quench endogenous peroxidase activity and to block reactivity of non-specific tissue antigens, incubation with the primary antibody was carried out overnight at 27°C. The subsequent steps of the immunohistochemical protocol to visualise the bound primary antibodies were performed using a commercial immunoperoxidase technique (Vector-elite ABC kit, goat anti-mouse IgG and DAB chromagen, all Vector Laboratories, Peterborough, UK) at the end of which sections were immersed in 0.5% copper sulphate to enhance immunoperoxidase colour reaction and finally counterstained with Mayer’s haematoxylin.
Ovine BSE shows reduced patterns of intracellular labelling with antibodies that recognise the extreme N-terminus of PrP and that are characteristically different from most scrapie sources [5]. To confirm the BSE nature of the infection, serial sections of selected brain areas were incubated with the N-terminal 12B2 monoclonal antibody (kindly provided by Jan Langeveld, Central Institute for Animal Disease Control, Netherlands), which recognizes amino acid sequence 93–97 of ovine PrP [37], and subjected to the same IHC protocol as described above.
Protein Misfolding Cyclic Amplification (PMCA)
Serial rounds of PMCA were performed as described previously [38]. Briefly, 50μl of cattle brain homogenate, confirmed to be BSE negative, was supplemented with 0,5% sulfated dextran (Mr ~70,000) from Leuconostoc spp. (Sigma-Aldrich) and seeded with 5μl of red deer brain samples (10% brain homogenate in phosphate buffered saline) from 13 of the animals (n = 6 BSE challenged animals that either developed clinical signs or were culled at the end of the experiment at 2320 dpc, n = 4 culled at 365 dpc and n = 3 environmental controls also culled at the end of the experiment at 2320 dpc). To ensure bovine brain homogenate was a suitable substrate for European red deer samples with respect to serial PMCA (sPMCA) an initial titration (10−1 to 10−11) was performed using red deer brain material that was known to be positive for PrPd by IHC (deer ID 009, see below).
All 13 red deer brain samples (in quadruplicate) were subjected to two independent experiments of sPMCA. All resultant samples were analysed for PrPres (proteinase resistant PrP which is another disease associated form of PrPC) by western blotting using monoclonal antibody 2A11 diluted 1:2000 [39]; for the first experiment after round 3 and for the second experiment after rounds 3 and 4. Unseeded tubes (n = 4) containing only the BSE negative cattle brain homogenate were included as a negative control preparation.
Results
Clinical signs
None of the animals subjected to post-mortem examinations at either 190 or 365 dpc showed any clinical signs of disease. Of the six remaining BSE challenged deer, a single animal (ID 009, female) developed acute clinical signs at 1726 dpc. These included fear of people (prior to this the animal was very tame and actively approached people in the pen), restlessness, pacing, stereotypic head movements and low head carriage, abnormal flicking of the ears, laboured and audible mouth breathing and ptyalism. The animal was euthanased the following day (1727 dpc) on welfare grounds and subjected to full post-mortem examination. At post-mortem examination the animal weighed 57.6 Kg and had lost 27.6% (21.9 Kg) of its body weight compared to its peak weight of 79.5 Kg recorded at 1586 dpc. None of the five remaining BSE challenged deer, nor any of the negative control animals, developed clinical signs or weight loss, other than small seasonal variations, by the termination of the experiment at day 2320 dpc.
Genotype
All animals were genotyped at polymorphic codons 132 and 226 of PRNP, other amino acid changes were not deduced from the gene sequences. All deer in the study were homozygous for methionine at codon 132 whereas they were of mixed genotypes at codon 226 (Table 1). The only animal which developed clinical disease was homozygous for glutamine (QQ) at codon 226 and this was the only animal of that genotype in the BSE-challenged group of longest duration which was comprised of six deer allowed to progress to clinical signs or termination of the experiment (Table 1).
Histological lesions and immunohistochemical labelling of PrPd
PrPd was not detected by IHC within any of the RAMALT biopsy samples from any of the deer over the whole course of the experiment.
Microscopical examination of HE stained sections of brain from the single clinically affected deer (ID 009) revealed widespread spongiform change with vacuolation seen in the neuronal perikaryon and neuropil of the brainstem (Fig. 1A), the molecular layer of the cerebellum (Fig. 1B) and thalamus (Fig. 1C). No such lesions were observed in any of the other 24 deer either BSE-challenged or unchallenged controls and no significant lesions were present in any of the other tissues examined.
Figure 1. Vacuolation of neuronal perikarya and neuropil in the dorsal motor nucleus of the vagus nerve (DMNV) in the medulla oblongata (a), the molecular layer of the cerebellum (b) and the thalamus (c) of the only clinically affected deer (ID 009).
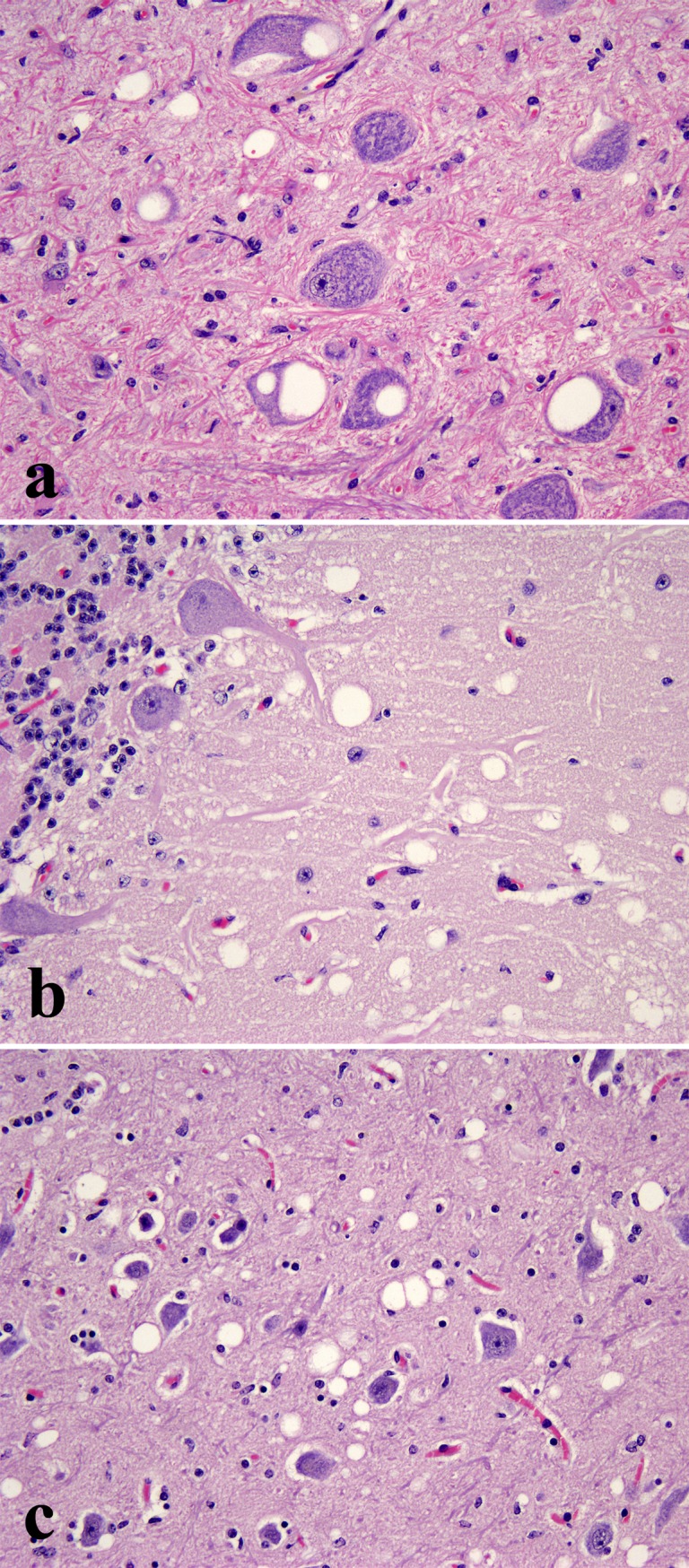
HE, x200.
Immunohistochemically PrPd was found in the clinically affected deer (ID 009) only and was largely restricted to the central and peripheral nervous systems (CNS and PNS respectively). Antibodies F99 and BAR224 were both equally effective at labelling PrPd in deer tissue with no discernible differences. In the CNS, PrPd accumulated predominantly in the brainstem and cerebellum with widespread diffuse particulate labelling of the neuropil and conspicuous intra-neuronal accumulation (Fig. 2). Peri-neuronal labelling was prominent in the dorsal motor nucleus of the vagus nerve (DMNV) and in the striatum. Intraneuronal granular accumulations were prominent in nuclei of the medulla, such as the accessory cuneate, spinal trigeminal (Fig. 2A) and posterior olivary nucleus (Fig. 2B), but were also present elsewhere in the brain with the exception of the Purkinje cells of the cerebellum. Intraneuronal PrPd aggregates were greatly diminished or even absent when serial sections were incubated with the PrP N-terminal 12B2 antibody (Fig. 2C & 2D). Such a marked decrease in intracellular signal with preservation of the extracellular signal following labelling with PrP N-terminal specific antibodies is a consistent feature of BSE infections in several other species [5]. PrPd was also detected in all segments of the spinal cord, in autonomic ganglia, cranial and peripheral nerves (Fig. 3A), sensory retina (Fig. 3B) and ganglion cells (myenteric plexus) throughout the enteric nervous system (Fig. 3C) which were frequently in close proximity to nearby lymphoid follicles, the majority of which were negative. However, sparse deposits of PrPd were also detected within tingible body macrophages in a very small number of the lymphoid follicles within the caecum (Fig. 4A) and colon (Fig. 4B) but not in any other lymphoid tissue examined. All other organs examined including skin, cardiac and skeletal muscles, lung, liver, rumen, abomasum, small intestine, kidney, pancreas and mammary gland were negative for PrPd.
Figure 2. Accumulation of PrPd (brown pigment) in the brain of the only clinically affected deer (ID 009).
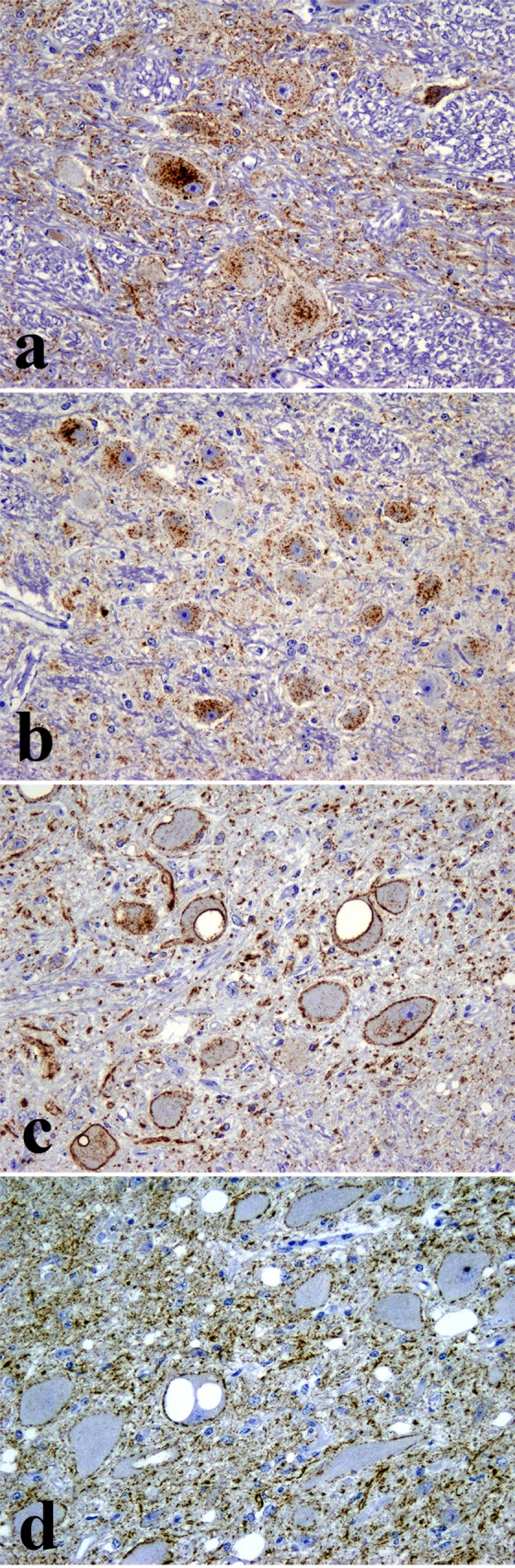
Diffuse particulate and punctate intraneuronal PrPd in the spinal tract nucleus of the trigeminal nerve (a) and inferior olivary nucleus (b); IHC with antibody F99, clone 97.6.1 x200. (c) Diffuse particulate, peri-neuronal and punctate intraneuronal PrPd in the dorsal motor nucleus of the vagus nerve with antibody F99, clone 97.6.1 (x200). Only the diffuse particulate and peri-neuronal PrPd types remain within the dorsal motor nucleus of the vagus nerve while the punctate intraneuronal PrPd type is unlabelled after incubation with the PrP N-terminal specific 12B2 antibody (d; x200).
Figure 3. Accumulation of PrPd (brown pigment) in the sympathetic chain ganglion cells (a), the plexiform layers of the retina (b) and myenteric plexus (c) of the single clinically affected deer (ID 009).
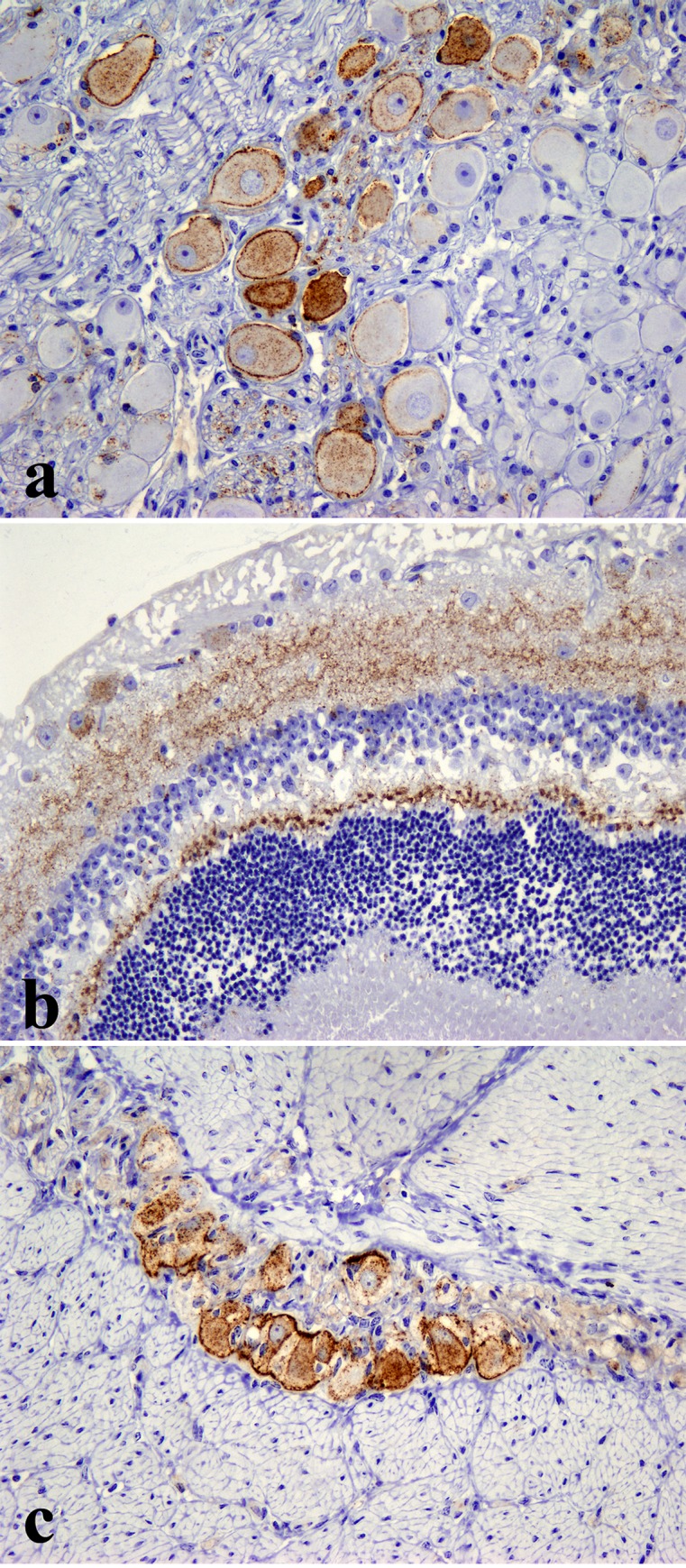
IHC with antibody Bar224, x200.
Figure 4. Accumulation of PrPd (brown pigment) in tingible body macrophages present in the lymphoid follicles of the caecum (a) and colon (b) of the only clinically affected deer (ID 009).

IHC with antibody Bar224, x200.
Protein Misfolding Cyclic Amplification
The initial titration to determine the suitability of bovine brain homogenate for sPMCA with a European red deer brain sample (the single clinically positive animal, ID009) gave a positive result down to a sample dilution of 10−11 after 3 rounds of sPMCA (Fig. 5) confirming its suitability for use in this study.
Figure 5. Western immnoblot for PrPres, using monoclonal antibody 2A11, of brain homogenate from initial titration experiment to determine the feasibility and sensitivity of serial protein misfolding cyclical amplification (sPMCA) using cattle brain homogenate for the detection of BSE agent in tissues from European red deer.
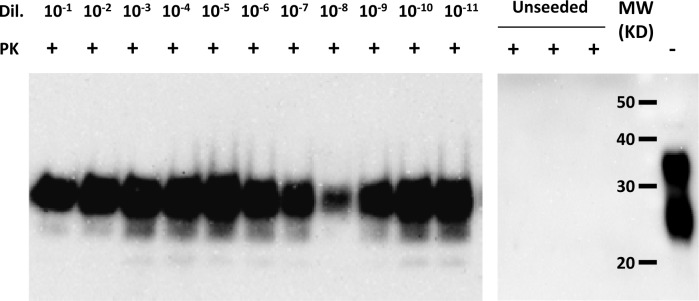
Brain tissue from the single European red deer that developed clinical signs (009) used. Note the presence of PrPres at a dilution of 10−11 after only three rounds of sPMCA showing exceptional sensitivity and complete lack of PrPres in negative control samples showing lack of spontaneous conversion of PrPC to PrPres. Dil = dilution, PK = proteinase-K treated (+) or not (−), MW = molecular weight markers.
All European red deer brain samples for both independant experiments of sPMCA from the single clinically positive animal (ID 009) were consistently positive for PrPres in quadruplicate at all rounds examined (third round in the first experiment and third and fourth rounds in the second experiment, Table 1 and Fig. 6). All the rest of the samples were negative except a single tube of one of the quadruplicate samples in the third round from experiment one from a negative control animal (039) but which was negative in all rounds throughout experiment two (see Table 1).
Figure 6. Western immunoblot for PrPres, using monoclonal antibody 2A11, of brain homogenates from orally challenged European red deer (n = 10) and unchallenged negative controls (n = 3) subjected to serial protein misfolding cyclical amplification (sPMCA), experiment two, round four.

Note PrPres is present only in brain homogenate from deer 009, the only animal that developed clinical signs which appeared at 1726 days after oral challenge with BSE, and the positive control samples (BSE positive bovine, scrapie positive ovine and chronic wasting disease (CWD) positive cervine brain homogenates). PME = post-mortem examination, dpc = days post-challenge, Neg Control = negative control, ID = deer identification number, PK = proteinase-K treated (+) or not (−), MW = molecular weight markers, KD = kilo Daltons.
Discussion
This investigation resulted in the first and only known case, to date, of clinical disease or accumulation of abnormal PrPd in any cervid species due to oral challenge with BSE. The increase in incubation period compared to European red deer challenged with BSE intra-cerebrally (1060 days) [33] compared to oral challenge (1727 days) is approximately 60% and similar to the differences observed in incubation periods for sheep or goats when challenged with TSE agents by these two routes [40,41]. The neurological clinical signs observed could be broadly related to the spongiform encephalopathy and the accumulation of PrPd in that the restlessness, stereotypic head movements and pacing may be due to compromise of the nucleus accumbens [42], found in the striatum, and the laboured breathing due to the lesions in the medulla, where the respiratory centre is located [43]. Alternatively, the laboured and audible mouth breathing may have been due to, or contributed to by, compromise of either of the recurrent laryngeal nerves resulting in some degree of laryngeal paralysis but we were unable to determine this. Apart from the gradual loss of body weight, the speed of onset of clinical signs and progression was very rapid but animal welfare requirements precluded any further longitudinal study of these. The clinical signs described for this animal are broadly similar to those reported for clinical BSE in European red deer challenged via the intracerebral route [33], clinical cases of CWD in deer [44] and clinical cases of BSE in cattle [45].
The predominant hindbrain vacuolation pattern in this clinically affected European red deer challenged orally with BSE was similar to the same species challenged intra-cerebrally with the same BSE inoculum [33], cattle naturally infected with BSE [46], sheep challenged orally with the BSE agent [41] and deer and elk naturally infected with CWD [47]. Additionally, the tissue distribution of PrPd in the clinically affected European red deer, which was restricted primarily to the central and peripheral nervous systems, was similar to that seen in the same species challenged intracerebrally with the same BSE inoculum [33]. However, this was in contrast to deer and elk naturally infected with CWD [44] and sheep naturally [48] or experimentally challenged via the oral route with scrapie [5] or BSE [41] which all have extensive peripheral accumulation of PrPd in the lymphoid tissues and supports the body of work that states that both the host species and the TSE agent both play roles in determining the pattern of accumulation of PrPd [4,5]. The PrPd labelling in the brain of the clinically affected European red deer was predominantly punctate and granular and this pattern, along with the sites listed, is typical of accumulations in the brains of other ruminant sources of BSE infection [41].
The presence of PrPd in a small number of tingible body macrophages in gut-associated lymphoid follicles in the clinically affected European red deer was not present when the same species was challenged by the intracerebral route with the same inoculum [33]. The mechanism of spread of accumulation of PrPd in the intracerebrally challenged European red deer was probably incubation-period related centrifugal spread from the brain to the spinal column, cranial and other peripheral nerves [33]. This is thought also to be the major mechanism of dissemination in cattle infected by the oral route with the BSE agent after initial haematogenous spread from the intestine to the brain [49]. Studies have examined, in detail, the anatomy of initial translocation of both the BSE [50] and scrapie [10] agents across the intestinal mucosal barrier of sheep and concluded that the villous lacteals of the lamina propria and submucosal lymphatics are the key route in the first two to three and a half hours of exposure. PrPd was not found in draining lymph nodes until 24 hours post-challenge and not before 30 days in Peyer’s patches [10] suggesting that the lymph node accumulation was probably residual inoculum and that in the Peyer’s patches was probably de novo PrPd. As PrPd was detected only in the lymphoid tissue of the clinically affected European red deer challenged orally with BSE after an incubation period of 1727 days, and then only in very small amounts, and not those examined at 180 and 360 dpc nor those challenged intracerebrally which were sampled at 794–1290 dpc [33] it would appear that this is de novo PrPd rather than residual inoculum. This being the case, European red deer challenged orally with BSE appear very similar to cattle with respect to organ distribution of PrPd [51,52]. These findings further support the belief that the accumulation and distribution of PrPd are influenced by a combination of the TSE agent and the host species [4,5].
The only animal to develop clinical disease in this study was homozygous for glutamine at PRNP codon 226 and, unfortunately, this was the only animal of this genotype in the group allowed to progress to clinical disease or termination of the experiment (Table 1). At the commencement of this study the only known genetic variation in the PRNP of cervid species closely related to European red deer and known to influence TSE susceptibility was codon 132 in elk where heterozygosity (methionine/leucine) was thought to confer a degree of resistance/lengthening of the incubation period in CWD infections [25–27]. This was supported by codon 132 in elk being the equivalent of human PRNP codon 129 which is highly influential in susceptibility to both sporadic and variant CJD [18,53]. Therefore, the European red deer in this study were initially subjected to evaluation of PRNP codon 132 only and as all were homozygous for methionine they were randomly assigned to the four experimental groups (6 month cull, 12 month cull, progression to clinical signs/termination of experiment and unchallenged negative control). At a later date it was shown that codon 226 of deer PRNP contained a polymorphism [28] that may be associated with TSE susceptibility [29] hence this was determined retrospectively after the animals had been assigned to their groups and already challenged with the BSE agent. Although it is tempting to speculate that European red deer homozygous for glutamine at PRNP codon 226 may be genetically more susceptible to BSE by the oral route than deer carrying glutamate at codon 226, one clinical animal cannot be statistically sufficient to make this association and further trials are required. Similar problems have been encountered in other long-term TSE studies where variations in previously unexamined PRNP codons have subsequently been shown to significantly affect the clinical disease outcome [11]. The only published assessments of the frequency of the various genotypes of PRNP codon 226 for European red deer were from those in Scotland were the QQ genotype was found in only 12.9% of animals over the whole country compared to 50% which were EE and 37.1% EQ [28]. However, one well studied island population within Scotland, which has no exchange with the mainland, had a prevalence for the QQ genotype of only 6.0% and the whole study examined 132 animals only [28]. If this study is representative of the European red deer population then the prevalence of the PRNP codon 226 QQ genotype is the lowest.
The presence of possible infectivity in both the single clinically affected animal and the non-clinically affected animals would have ideally been assessed by subjecting the resultant European red deer brain material to in vivo infectivity studies in a rodent model of shorter incubation period and/or further passage in European red deer but this was out-with the scope of this study. Fortunately, sPMCA was available and due to its exceptionally high sensitivity it has been proposed as a credible replacement for bioassay [54]. Our initial trials using bovine brain tissue as a substrate for detecting BSE by sPMCA showed that it was highly suitable for testing tissue from European red deer being able to detect a positive result to a sample dilution of 10−11 and, therefore, would probably be suitable for testing other closely related deer species, if not all cervid species. The consistent presence of PrPres in 4/4 tubes at all rounds in both experiments of sPMCA is strongly suggestive that infectivity was present in the single clinically affected European red deer. The single positive tube, one of four replicates, from a negative control animal (039) in round three of the first experiment of sPMCA was considered negative and to be due to contamination of the sample at some point as all subsequent rounds from this animal in experiment two were negative for PrPres. Additionally, as all replicates at all rounds of the unseeded BSE negative bovine brain homogenate were negative it is unlikely this was a stochastic event.
The results of this study show that alimentary transmission of BSE to European red deer is possible but the transmission rate is low. However, culling of red deer in the UK, particularly males, occurs mostly when they are 8–10 years (2920–3650 days) old and therefore well within the possible incubation period for BSE [55]. Altogether our data suggest that when deer carcases for human consumption are subjected to the same regulations as other ruminant carcases (The Specified Bovine Offal Order 1990 [56] and its amendments) the zoonotic risk of BSE from consuming muscle from European red deer is probably very low.
Acknowledgments
We would like to thank members of the MRI Clinical Department, especially Mr. R. Todd for care and maintenance of the animals and Dr. T. John Fletcher for expert husbandry advice.
Data Availability
All relevant data are within the paper.
Funding Statement
These studies were funded by the UK Food Standards Agency grant M03024 and three Spanish grants (AGL2012-37988-C04-01, CTP2013-P05 and EFA282/11).
References
- 1. Wells GAH, McGill IS (1992) Recently described scrapie-like encephalopathies of animals-case definitions. Res Vet Sci 53: 1–10. [DOI] [PubMed] [Google Scholar]
- 2. Foster JD, Hope J, Fraser H (1993) Transmission of Bovine Spongiform Encephalopathy to Sheep and Goats. Vet Rec 133: 339–341. [DOI] [PubMed] [Google Scholar]
- 3. Collinge J (2001) Prion diseases of humans and animals: Their causes and molecular basis. Annu Rev Neurosci 24: 519–550. [DOI] [PubMed] [Google Scholar]
- 4. Collins SJ, Lawson VA, Masters CL (2004) Transmissible spongiform encephalopathies. Lancet 363: 51–61. [DOI] [PubMed] [Google Scholar]
- 5. Jeffrey M, Gonzalez L (2007) Classical sheep transmissible spongiform encephalopathies: pathogenesis, pathological phenotypes and clinical disease. Neuropath Appl Neuro 33: 373–394. [DOI] [PubMed] [Google Scholar]
- 6. Gavier-Widen D, Stack MJ, Baron T, Balachandran A, Simmons M (2005) Diagnosis of transmissible spongiform encephalopathies in animals: a review. J Vet Diagn Invest 17: 509–527. [DOI] [PubMed] [Google Scholar]
- 7. Ferguson NM, Donnelly CA, Woolhouse MEJ, Anderson RM (1997) The epidemiology of BSE in cattle herds in Great Britain. 2. Model construction and analysis of transmission dynamics. Philos Trans R Soc Lond Ser B-Biol Sci 352: 803–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Foster JD, Goldmann W, McKenzie C, Smith A, Parnham DW, et al. (2004) Maternal transmission studies of BSE in sheep. J Gen Virol 85: 3159–3163. [DOI] [PubMed] [Google Scholar]
- 9. Hunter N, Houston F, Foster J, Goldmann W, Drummond D, et al. (2012) Susceptibility of Young Sheep to Oral Infection with Bovine Spongiform Encephalopathy Decreases Significantly after Weaning. J Virol 86: 11856–11862. 10.1128/JVI.01573-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jeffrey M, Gonzalez L, Espenes A, Press CM, Martin S, et al. (2006) Transportation of prion protein across the intestinal mucosa of scrapie-susceptible and scrapie-resistant sheep. J Pathol 209: 4–14. [DOI] [PubMed] [Google Scholar]
- 11. González L, Jeffrey M, Dagleish MP, Goldman W, Sisó S, et al. (2012) Susceptibility to scrapie and disease phenotype in sheep: cross-PRNP genotype experimental transmissions with natural sources. Vet Res 43: 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goldmann W (2008) PrP genetics in ruminant transmissible spongiform encephalopathies. Vet Res 39 10.1051/vetres:2008038 [DOI] [PubMed] [Google Scholar]
- 13. Goldmann W, Houston F, Stewart P, Perucchini M, Foster J, et al. (2006) Ovine prion protein variant A(136) R(154)L(168)Q(171) increases resistance to experimental challenge with bovine spongiform encephalopathy agent. J Gen Virol 87: 3741–3745. [DOI] [PubMed] [Google Scholar]
- 14. Houston F, Goldmann W, Chong A, Jeffrey M, Gonzalez L, et al. (2003) Prion diseases: BSE in sheep bred for resistance to infection. Nature 423: 498 [DOI] [PubMed] [Google Scholar]
- 15. Haase B, Doherr MG, Seuberlich T, Droegemueller C, Dolf G, et al. (2007) PRNP promoter polymorphisms are associated with BSE susceptibility in Swiss and German cattle. BMC Genet 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Castilla J, Gutierrez-Adan A, Brun A, Pintado B, Parra B, et al. (2004) Different behavior toward bovine spongiform encephalopathy infection of bovine prion protein transgenic mice with one extra repeat octapeptide insert mutation. Journal of Neuroscience 24: 2156–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brun A, Gutierrez-Adan A, Castilla J, Pintado B, az-San Segundo F, et al. (2007) Reduced susceptibility to bovine spongiform encephalopathy prions in transgenic mice expressing a bovine PrP with five octapeptide repeats. J Gen Virol 88: 1842–1849. [DOI] [PubMed] [Google Scholar]
- 18. Windl O, Dempster M, Estibeiro JP, Lathe R, Desilva R, et al. (1996) Genetic basis of Creutzfeldt-Jakob disease in the United Kingdom: A systematic analysis of predisposing mutations and allelic variation in the PRNP gene. Hum Genet 98: 259–264. [DOI] [PubMed] [Google Scholar]
- 19. Robinson SJ, Samuel MD, O’Rourke KI, Johnson CJ (2012) The role of genetics in chronic wasting disease of North American cervids. Prion 6: 153–162. 10.4161/pri.19640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson C, Johnson J, Vanderloo JP, Keane D, Aiken JM, et al. (2006) Prion protein polymorphisms in white-tailed deer influence susceptibility to chronic wasting disease. J Gen Virol 87: 2109–2114. [DOI] [PubMed] [Google Scholar]
- 21. Jewell JE, Conner MM, Wolfe LL, Miller MW, Williams ES (2005) Low frequency of PrP genotype 225SF among free-ranging mule deer (Odocoileus hemionus) with chronic wasting disease. J Gen Virol 86: 2127–2134. [DOI] [PubMed] [Google Scholar]
- 22. Happ GM, Huson HJ, Beckmen KB, Kennedy LJ (2007) Prion protein genes in caribou from Alaska. J Wildl Dis 43: 224–228. [DOI] [PubMed] [Google Scholar]
- 23. Mitchell GB, Sigurdson CJ, O’Rourke KI, Algire J, Harrington NP, et al. (2012) Experimental Oral Transmission of Chronic Wasting Disease to Reindeer (Rangifer tarandus tarandus). PloS One 7: e39055 10.1371/journal.pone.0039055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Palmer MS, Dryden AJ, Hughes JT, Collinge J (1991) Homozygous Prion Protein Genotype Predisposes to Sporadic Creutzfeldt-Jakob Disease. Nature 352: 340–342. [DOI] [PubMed] [Google Scholar]
- 25. O’Rourke KI, Besser TE, Miller MW, Cline TF, Spraker TR, et al. (1999) PrP genotypes of captive and free-ranging Rocky Mountain elk (Cervus elaphus nelsoni) with chronic wasting disease. J Gen Virol 80: 2765–2769. [DOI] [PubMed] [Google Scholar]
- 26. Perucchini M, Griffin K, Miller MW, Goldmann W (2008) PrP genotypes of free-ranging wapiti (Cervus elaphus nelsoni) with chronic wasting disease. J Gen Virol 89: 1324–1328. 10.1099/vir.0.83424-0 [DOI] [PubMed] [Google Scholar]
- 27.Williams ES, Miller MW (2004) Epidemilogy and control of chronic wasting disease [Abstract]. Proceedings of the American Association of Veterinary Laboratory Diagnosticians 47th Annual Conference, Greensboro, North Carolina, USA.
- 28. Peletto S, Perucchini M, Acin C, Dalgleish MP, Reid HW, et al. (2009) Genetic variability of the prion protein gene (PRNP) in wild ruminants from Italy and Scotland. J Vet Sci 10: 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vickery CM, Lockey R, Holder TM, Thorne L, Beck KE, et al. (2014) Assessing the susceptibility of transgenic mice over-expressing deer prion protein to bovine spongiform encephalopathy. J Virol 88: 1830–1833. 10.1128/JVI.02762-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brayton KA, O’Rourke KI, Lyda AK, Miller MW, Knowles DP (2004) A processed pseudogene contributes to apparent mule deer prion gene heterogeneity. Gene 326: 167–173. [DOI] [PubMed] [Google Scholar]
- 31. Kirkwood JK, Cunningham AA (1994) Epidemiologic observations on spongiform encephalopathies in captive wild animals in the British-Isles. Vet Rec 135: 296–303. [DOI] [PubMed] [Google Scholar]
- 32. Wyatt JM, Pearson GR, Smerdon TN, Gruffyddjones TJ, Wells GAH, et al. (1991) Naturally-Occurring Scrapie-Like Spongiform Encephalopathy in 5 Domestic Cats. Vet Rec 129: 233–236. [DOI] [PubMed] [Google Scholar]
- 33. Dagleish MP, Martin S, Steele P, Finlayson J, Siso S, et al. (2008) Experimental transmission of bovine spongiform encephalopathy to European red deer (Cervus elaphus elaphus). BMC Vet Res 4 10.1186/1746-6148-4-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gonzalez L, Dagleish MP, Martin S, Dexter G, Steele P, et al. (2008) Diagnosis of preclinical scrapie in live sheep by the immunohistochemical examination of rectal biopsies. Vet Rec 162: 397–403. [DOI] [PubMed] [Google Scholar]
- 35. Tan BC, Blanco ARA, Houston EF, Stewart P, Goldmann W, et al. (2012) Significant differences in incubation times in sheep infected with bovine spongiform encephalopathy result from variation at codon 141 in the PRNP gene. J Gen Virol 93: 2749–2756. 10.1099/vir.0.039008-0 [DOI] [PubMed] [Google Scholar]
- 36. Gonzalez L, Martin S, Begara-McGorum I, Hunter N, Houston F, et al. (2002) Effects of agent strain and host genotype on PrP accumulation in the brain of sheep naturally and experimentally affected with scrapie. J Comp Pathol 126: 17–29. [DOI] [PubMed] [Google Scholar]
- 37. Jeffrey M, Gonzaalez L, Chong A, Foster J, Goldmann W, et al. (2006) Ovine infection with the agents of scrapie (CH1641 isolate) and bovine spongiform encephalopathy: Immunochemical similarities can be resolved by immunohistochemistry. J Comp Pathol 134: 17–29. [DOI] [PubMed] [Google Scholar]
- 38. Castilla J, Saa P, Hetz C, Soto C (2005) In vitro generation of infectious scrapie prions. Cell 121: 195–206. [DOI] [PubMed] [Google Scholar]
- 39. Brun A, Castilla J, Ramirez MA, Prager K, Salquerro FJ, et al. (2004) Proteinase K enhanced immunoreactivity of the prion protein-specific monoclonal antibody 2A11. Neuroscience Research 48: 75–83. [DOI] [PubMed] [Google Scholar]
- 40. Gonzalez L, Chianini F, Martin S, Siso S, Gibbard L, et al. (2007) Comparative titration of experimental ovine BSE infectivity in sheep and mice. J Gen Virol 88: 714–717. [DOI] [PubMed] [Google Scholar]
- 41. Jeffrey M, Ryder S, Martin S, Hawkins SAC, Terry L, et al. (2001) Oral inoculation of sheep with the agent of bovine spongiform encephalopathy (BSE). 1. Onset and distribution of disease- specific PrP accumulation in brain and viscera. J Comp Pathol 124: 280–289. [DOI] [PubMed] [Google Scholar]
- 42. McBride SD, Parker MO (2014) The disrupted basal ganglia and behavioural control: An intergrative cross-domain perspective of spontaneous stereotypy. Behav Brain Res 10.1016/j.bbr.2014.05.057 [DOI] [PubMed] [Google Scholar]
- 43. Carlson NR (2007) Structures of the Nervous System. In: Carlson. N.R., editors. Physiology of Behaviour. London: Pearson; pp. 69–101. [Google Scholar]
- 44. Williams ES (2005) Chronic wasting disease. Vet Pathol 42: 530–549. [DOI] [PubMed] [Google Scholar]
- 45. Konold T, Bone G, Ryder S, Hawkins SAC, Courtin F, et al. (2004) Clinical findings in 78 suspected cases of bovine spongiform encephalopathy in Great Britain. Vet Rec 155: 659–666. [DOI] [PubMed] [Google Scholar]
- 46. Wells GAH, Wilesmith JW (1995) The Neuropathology and Epidemiology of Bovine Spongiform Encephalopathy. Brain Pathol 5: 91–103. [DOI] [PubMed] [Google Scholar]
- 47. Williams ES, Young S (1993) Neuropathology of chronic wasting disease of mule deer (Odocoileus-hemionus) and elk (Cervus-elaphus-nelsoni). Vet Pathol 30: 36–45. [DOI] [PubMed] [Google Scholar]
- 48.Jeffrey M, McGovern G, Martin S, Goodsir CM, Brown KL (2000) Cellular and sub-cellular localisation of PrP in the lymphoreticular system of mice and sheep. Arch Virol 23–38. [DOI] [PubMed]
- 49. Siso S, Jeffrey M, Gonzalez L (2009) Neuroinvasion in sheep transmissible spongiform encephalopathies: the role of the haematogenous route. Neuropath Appl Neuro 35: 232–246. [DOI] [PubMed] [Google Scholar]
- 50. Dagleish MP, Hamilton S, Gonzalez L, Eaton SL, Steele P, et al. (2010) Digestion and transportation of bovine spongiform encephalopathy-derived prion protein in the sheep intestine. J Gen Virol 91: 3116–3123. 10.1099/vir.0.025049-0 [DOI] [PubMed] [Google Scholar]
- 51. Terry LA, Marsh S, Ryder SJ, Hawkins SAC, Wells GAH, et al. (2003) Detection of disease-specific PrP in the distal ileum of cattle exposed orally to the agent of bovine spongiform encephalopathy. Vet Rec 152: 387–392. [DOI] [PubMed] [Google Scholar]
- 52. Espinosa JC, Morales M, Castilla J, Rogers M, Torres JM (2007) Progression of prion infectivity in asymptomatic cattle after oral bovine spongiform encephalopathy challenge. J Gen Virol 88: 1379–1383. [DOI] [PubMed] [Google Scholar]
- 53. Fox KA, Jewell JE, Williams ES, Miller MW (2006) Patterns of PrP(CWD) accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus). J Gen Virol 87: 3451–3461. [DOI] [PubMed] [Google Scholar]
- 54. Saa P, Castilla J, Soto C (2006) Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem 281: 35245–35252. [DOI] [PubMed] [Google Scholar]
- 55.Watson P, Williams B (2009) Deer Culling on Exmoor. Exmoor national Park Authority.
- 56.Anonymous (1990) The Specified Bovine Offal Order 1990. Available: www.legislation.gov.uk/en/uksi/1995/1928/introduction/made.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.