

Abstract
Tethering factors regulate the targeting of membrane-enclosed vesicles under the control of Rab GTPases. P115, a golgin family tether, has been shown to participate in multiple stages of ER/Golgi transport. Despite extensive study, p115’s mechanism of action is poorly understood. SNARE proteins make up the machinery for membrane fusion, and strong evidence shows that p115’s function is directly linked to its interaction with SNAREs. Using a gel filtration binding assay, we have demonstrated that in solution p115 stably interacts with ER/Golgi SNAREs rbet1 and sec22b, but not membrin and syntaxin 5. These binding preferences stemmed from selectivity of p115 for monomeric SNARE motifs as opposed to SNARE oligomers. Soluble monomeric rbet1 can compete off p115 from COPII vesicles. Furthermore, excess p115 inhibits p115 function in trafficking. We conclude that monomeric SNAREs are a major binding site for p115 on COPII vesicles, and that p115 dissociates from its SNARE partners upon SNAREpin assembly. Our results suggest a model in which p115 forms a mixed p115/SNARE helix bundle with a monomeric SNARE, facilitates the binding activity and/or concentration of the SNARE at pre-fusion sites, and is subsequently ejected as SNARE complex formation and fusion proceed.
Introduction
Protein transport between the endoplasmic reticulum (ER) and the Golgi is one of the most fundamental trafficking pathways in eukaryotic cells, as well as the first step in the secretory pathway. Secretory and membrane proteins are modified and sorted in the ER and translocated to the Golgi in membrane-enclosed vesicles. Vesicular transport from the ER to the Golgi comprises a sequence of budding, fusion, transport, and maturation events. First, COPII coat proteins collect cargo and sculpt the ER membrane into spherical vesicles; these vesicles then undergo homotypic fusion to give rise to an intermediate compartment termed vesicular tubular clusters (VTCs). VTCs sort anterograde cargo from escaped ER proteins and trafficking machinery while undergoing transport via microtubules to the Golgi, where they fuse with the cis-cisternae of the Golgi to deliver their cargo (1, 2).
Fusion of traffic intermediates is carried out by a group of proteins termed soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs). Structurally, a typical SNARE comprises three distinct regions: an amino-terminal domain, of which the structure and function vary, an approximately 60 amino acid conserved amphipathic alpha-helical SNARE motif, and a carboxyl-terminal membrane anchor (3). Usually, a vesicle SNARE (v-SNARE) and its three cognate target SNAREs (t-SNAREs) assemble into a tetrameric SNARE complex, with each SNARE contributing one SNARE motif to a four-helix-bundle called a SNAREpin. The formation of multiple SNAREpins drives the opposing membranes together, overcoming the energetic barriers to membrane fusion (4).
Aside from the requirements for SNAREs, fusion efficiency is largely dependent upon upstream events, including the targeting and tethering of vesicles prior to their fusion (5). A number of protein factors required for these processes have been identified and characterized (6–14). Termed tethering factors, these proteins act either as an individual rod-like tether (6–8), or function as part of a multi-component tethering complex (9–14). Generally effectors of Rab GTPases, tethers stabilize the Golgi cisternae (15–17), form membrane-bridging complexes, and facilitate membrane fusion (18–21). Several tethering factors belong to the Golgin protein family (6–8, 22), the members of which are characterized by structurally predominant alpha-helical coiled-coil domains (6, 8, 23). One of the better-studied Golgins, p115/Uso1p, was first identified as a high molecular weight factor required for the docking/tethering step in intra-Golgi vesicular transport and transcytosis (23–25). Uso1p was also discovered as an essential component for ER-Golgi transport in yeast (24). These early studies placed p115/Uso1p’s action prior to SNARE-mediated membrane fusion (26–29), although later evidence also suggests a downstream role for p115/Uso1p (30, 31). P115 is a direct effector of Rab1 and is recruited to COPII vesicles through transient interactions with Rab1 and perhaps SNAREs (29–33), and is possibly regulated by phosphorylation during mitosis (34–36). Other tethering factors required for ER/Golgi transport include GM130, Giantin, and other Golgins (6, 8), and multisubunit complexes such as TRAPP (transport protein particle) (37) and COG (conserved oligomeric Golgi complex) (38, 39).
Both p115 and its yeast homologue Uso1p exist as homodimers in cells (23, 40, 41), localized primarily to the cis-face of the Golgi (16). The bovine p115 monomer consists of a globular head domain, which spans residues 1-651, an elongated coiled-coil domain (residues 652-909), and an acidic tail domain (residues 910-961) (40). Based on primary sequence comparison, the head domain of p115 contains two highly conserved regions, H1 and H2 (40, 42), which recent X-ray crystallography studies revealed are within a multi-helical armadillo fold. The dimerization interface overlaps with H2, whereas the Rab1 binding site is located to the H1 region (42, 43). In addition, binding sites for the COPI component beta-COP as well as COG2 of the COG complex were also mapped to the head region (42, 43). Following the head domain, the coiled-coil domain of bovine p115 consists of four separate predicted coiled-coils (CC1-CC4), which cover residues 652-694, 732-766, 786-820, and 849-897, respectively (40). The proline and glycine rich interruptions between the coiled-coils are predicted to function as flexible hinges, possibly allowing the normally elongated p115 molecule to adopt a bent conformation during its functional cycle (40, 41). It was proposed that CC1 may contain an alternative Rab1 binding site that is only exposed upon the binding of other Golgi tethers (44), however, the functional significance of this cryptic site remains unclear. Following the coiled-coil domain is a region of highly acidic amino acid content; the extreme C-terminus of p115 forms a very hydrophilic, acidic tail (40). Other members of the Golgin family, such as GM130 and Giantin, can interact with this part of the molecule (16, 34, 36, 45–47), though the functional significance of these interactions is controversial (48–50).
P115’s involvements in mammalian ER to Golgi transport (16, 29, 30), intra-Golgi transport (23, 45, 48, 51, 52), exocytosis (25), as well as Golgi biogenesis (17, 34, 36, 53), have all been clearly demonstrated. Despite these extensive studies, however, the exact roles of p115 in these processes have yet to be established. One theory is that p115, GM130 and Giantin form string-like structures via their coiled-coil domains, in order to secure vesicles prior to fusion (19, 42, 54). There is also evidence suggesting that p115’s function is dependent upon its interactions with specific SNAREs (30, 31, 50, 52, 55). The overexpression of Bet1p or Sec22p, two yeast ER-Golgi SNAREs, suppresses the USO1 deletion in yeast, and certain SNARE complexes cannot accumulate in the absence of Uso1p (26). P115 interacts with SNAREs through two of its SNARE motif homology domains, CC1 and CC4 (50, 56). When coupled to Neutravidin beads, both CC1 and CC4 were able to retain the ER-Golgi SNAREs syntaxin 5, membrin, rSec22p, Bet1p, as well as the Golgi SNAREs Ykt6p and GOS-28, from rat liver Golgi extract (56). Deletion of either CC1 or CC4 impairs p115’s function (50, 52).
It has been proposed that p115 acts as a catalyst during SNARE-mediated fusion, with each subunit of a p115 dimer binding to a subset of SNAREs (56). However, the mechanism by which p115 is recruited to and promotes SNARE assembly remains unknown. Upon blocking SNARE complex disassembly on in vitro budded COPII vesicles with the addition of dominant negative alpha-SNAP, p115 is removed from the vesicle surface, suggesting that p115’s recruitment to COPII vesicles is dependent upon unassembled SNAREs in addition to being Rab1 dependent (55). In vivo expression of another SNARE recycling inhibitor, NSF/E329Q, results in decreased p115 membrane association, further suggesting that the disassembly of SNARE complexes is important for the membrane binding of p115 (31). Although these findings imply that p115 preferentially binds to unassembled SNAREs, neither of these results eliminates the possibility that alpha-SNAP/NSF competes with p115 for binding to assembled SNAREs. Furthermore, it is presently unclear which ER-Golgi SNAREs participate in p115 recruitment, and whether p115 binds to monomeric SNAREs as opposed to SNARE subcomplexes of various stages. It is also possible that p115 binds SNAREs to localize to sites of fusion where it performs other functions not directly related to SNAREs.
We have examined in detail the molecular interactions between the tether protein p115 and ER-Golgi SNAREs. Our findings suggest that monomeric SNARE motifs are the binding sites for p115 on COPII vesicles. In addition, solution binding results imply that whereas p115 weakly promotes the assembly of SNAREs, it is not present in the final SNARE complex. This result was supported by the observation that excess p115 inhibits p115 function in trafficking. Our results suggest a model of p115’s action in which p115 forms a mixed p115/SNARE helix bundle with a monomeric SNARE and that p115 is ejected from the bundle as SNARE complex formation and fusion proceed.
Results
P115’s interactions with individual ER-Golgi SNAREs
Previous studies demonstrated that p115 coprecipitates with syntaxin 5, membrin, bet1p (rbet1), and sec22b (30, 56), yet direct and specific binding interactions between p115 and these SNAREs, individually and as subcomplexes, have not been explored. Detailed knowledge of these interactions is key to understanding the p115 mechanism of action. To study directly p115’s interactions with SNAREs, we first prepared soluble ER-Golgi SNARE proteins. Cytoplamic domains of rat ER/Golgi SNAREs sec22b (residues 2-196), syntaxin 5 (the shorter isoform, representing residues 55-333 of the longer isoform), and rbet1 (residues 1-95) were expressed in E. coli and purified using column chromatography as described before (57). We were unable to express the cytoplamic domain of membrin in E. coli, but a full-length membrin construct (residues 2-212) was well-expressed, soluble in mild detergent and active in binding studies. His6-tagged full-length bovine p115 was also well expressed and freely soluble, and was purified by column chromatography. This p115 preparation has been previously shown to be active in SNARE interaction assays (54). Figure 1 shows an SDS gel analysis of the protein preparations typically used in these studies.
Figure 1. Purified recombinant SNAREs and p115.

Proteins were separated on a 15% SDS-PAGE gel, and stained with Coommassie Blue. Protein loads correspond to the relative amounts used in gel filtration binding assays. Rbet1 (residues 1-95) runs below the 14 kD marker and contains sub-stoichiometric GST contaminants around the 24 kD marker. Syntaxin 5 (residues 55-333) runs between the 29 and 36 kD markers (marked by an asterisk) and contains heavy GST contamination around the 24 kD marker. A C-terminal truncation product of syntaxin 5 lacking the SNARE motif (residues 55-251) runs between the 24 and 29 kD markers. Bands above the 45 kD marker represent uncleaved GST fusion proteins and their C-terminal truncation products. The full-length membrin band between 24 and 29 kD markers is largely free of contaminating GST but contains sub-stoichiometric unknown truncation or degradation products. His6-sec22b between the 20 and 24 kD markers is largely homogeneous. His6-p115 runs above the 97 kD marker and contains various unknown C-terminal truncation products and a substantial contaminant near the 24 kD marker (see Fig. 8B for detail).
Our binary binding studies utilized a gel filtration assay that only detects stable complexes formed in solution. Purified p115 was passed through a Superdex 200 column. Due to its elongated shape, the peak of p115 elution was in fraction 10 (Fig. 2A), at a predicted size of 700 kD, much larger than the expected 230 kD dimer, as shown previously (23). This unusual feature of p115 made a gel filtration SNARE binding assay feasible since the p115 elution did not overlap with SNARE complexes or homo-oligomers. Next, soluble syntaxin 5, membrin, rbet1, and sec22b were passed through the Superdex column separately, fractions were collected and analyzed by Western blotting. Rbet1, sec22b, syntaxin 5 and membrin were eluted in fractions 28–34, 26–36, 18–30, and 16–30, respectively (Fig. 2B–E, top panels, slightly different fractions are shown to optimize display of each protein). The SNAREs were then each incubated separately with p115 overnight on ice, the reactions were gel-filtered and analyzed by Western using anti-SNARE antibody. The elution of each mixed incubation (Fig. 2B–E, bottom panels) was compared to the SNARE-only elution (top panels). In the presence of p115, a significant portion of both rbet1 and sec22b shifted to higher molecular weight fractions, indicating the formation of a complex. Furthermore, the elution peaks of the complexes overlapped the p115 peak, suggesting the presence of p115 in these complexes. Since the p115/SNARE complexes overlapped the void volume of the column, especially in the case of sec22b (part C), we independently verified the presence of freely soluble p115-sec22b complexes by centrifuging the co-incubated proteins at 100,000 × g to remove high molecular weight aggregates followed immediately by co-immunoprecipitation of the two proteins using anti-p115 antibody (Figure 2J).
Figure 2. Binary interactions of p115 with ER-Golgi SNAREs rbet1 and sec22b, but not syntaxin 5, membrin, or SNARE N-terminal domains.

Soluble recombinant p115, syntaxin 5, membrin, rbet1, and sec22b were passed through a Superdex 200 column separately, fractions were collected and analyzed by Western blotting. The SNAREs were then each incubated separately with 1.3 μM p115 overnight on ice, the reactions were gel-filtered and analyzed by Western using anti-SNARE antibody. (A) P115 only elution. (B) Rbet1 only (top) and rbet1-p115 reaction mix (bottom) elutions. (C) Sec22b only (top) and sec22b-p115 reaction mix (bottom) elutions. (D) Syntaxin 5 only (top) and syntaxin 5-p115 reaction mix (bottom) elutions. (E) Membrin only (top) and membrin-p115 reaction mix (bottom) elutions. (F) Syntaxin 5 (residues 55-333) was subjected to thrombin cleavage. The SNARE motif (residues 251-333) was purified by velocity sedimentation on 5–30% glycerol gradients. Top panel: Syntaxin 5 SNARE motif only elution. Bottom panel: Elution of syntaxin 5 SNARE motif-p115 reaction mix. Hexahistidine-tagged N-terminal domain constructs of sec22b, membrin, and Ykt6 were expressed in E. coli and purified by nickel-affinity chromatography (see Materials and Methods). Sec22b NT (G), membrin NT (H), and Ykt6 NT (I) were passed through gel filtration column separately (top panels). Each protein was then incubated with p115 and then the mixed reactions were gel-filtered and immunoblotted with anti-SNARE antibodies (bottom panels). Gel filtration fraction numbers are shown above top panels. Elution positions of Dextran 2000 (void), native thyroglobulin (669 kDa), ferritin (440 kDa), catalase (232 kDa), and BSA (67 kDa) are indicated with arrows above the fraction numbers. (J) sec22b and p115 were co-incubated as in part C, centrifuged at 100,000 × g to remove potential aggregates, and then precipitated using α-p115 antibody beads. The presence of specifically co-precipitated sec22b demonstrates the presence of a soluble p115-sec22b complex.”*” marks IgG heavy chain; “**” marks a contaminant in the p115 preparation that cross-reacts with the anti-sec22b antibody.
SNARE motif is the binding site for p115
Structurally, a typical SNARE consists of an N-terminal domain, a SNARE motif, and a membrane anchor (3). Most SNARE-binding tethering complexes such as the Dsl1 complex, Golgi-associated retrograde protein (GARP) complex and homotypic vacuole fusion and protein sorting (HOPS) complex, primarily interact with the N-terminal domain of SNAREs (21, 58–61), whereas the conserved oligomeric Golgi (COG) complex preferentially interacts with the SNARE motif of Sed5p (62). The CC1 and CC4 domains of p115 share weak sequence homology to the SNARE motif (56, 63), and it has been demonstrated that p115 interacts with SNAREs through these two domains (50, 52, 56). The binding site for p115 on a SNARE has not been mapped before, but we expected that p115 binds the SNARE motif through coiled-coil interactions, similarly to SNARE-SNARE binding. As one demonstration of this principle, rbet1 is a SNARE that lacks the N-terminal domain, meaning that the cytoplasmic portion of rbet1 (residues 1-95) employed in our binding studies consists purely of the rbet1 SNARE motif. Since p115 and the SNARE motif of rbet1 formed a stable complex that can be resolved by gel filtration (Fig. 2B), the SNARE motif must be the binding site for p115.
However, these results do not exclude the possibility that p115 can also interact with a SNARE’s N-terminal domain. Sec22b has an unusual N-terminal structure composed of a β-sheet and several α-helices termed the longin domain (64). Similar structures of other SNAREs have been reported to bind SNARE motifs and exert regulatory effects on SNARE activities (65–67) and localization (68). The longin domain of sec22b contains two potential binding grooves for the SNARE motif, although intramolecular binding has not been clearly demonstrated. To test possible interactions between p115 and the sec22b longin domain, we expressed and purified the N-terminal longin domain of sec22b. We did not detect any binding between the sec22b longin domain and p115 using the gel filtration assay (Fig. 2G), which provides additional support that p115 interacts with the SNARE motif. In addition, we also purified the N-terminal regions of membrin, that possesses a three-helix N-terminal domain (69, 70), and Ykt6, another ER-Golgi SNARE that possesses a longin domain that has been shown to interact with SNARE motifs (65, 68) and tested their binding to p115. No binding was detected between p115 and either construct (Fig. 2H, I).
P115 interacts with monomeric SNAREs, but not SNARE complexes
We noted that membrin and syntaxin 5, two SNAREs that have been shown to interact with p115 using a bead-binding format (30, 56), did not form stable complexes with p115 under our binding conditions (Fig. 2, D and E.). One obvious possibility is that the syntaxin 5 and membrin N-terminal domains bound their respective SNARE motifs, precluding p115 access. The syntaxin 5 N-terminal domain has been shown to reduce other SNARE binding (57). To test for potential interference by the syntaxin 5 N-terminal domain, a highly active construct consisting of only the SNARE motif of syntaxin 5 (residues 252-333) was generated by thrombin cleavage of the cytoplasmic domain (residues 55-333), and purified by velocity sedimentation. This fragment of syntaxin 5 was demonstrated to be at least an order of magnitude more efficient in SNARE complex formation than the full cytoplasmic form (57). However, when we used the truncated construct in gel filtration studies, we were unable to detect binding between p115 and the syntaxin 5 SNARE motif (Fig. 2F).
Perhaps relevant to their lack of binding, we also noticed that both membrin and syntaxin 5 eluted in fractions in the 150–200 kD range, much larger than their expected monomer molecular weights of 25 and 34 kD, respectively. For reference, the syntaxin 5-membrin-rbet1-sec22b 1:1:1:1 complex in this system elutes in fraction 17 with a calibrated molecular weight of ~300 kD (57). Apparently under our purification conditions, syntaxin 5 and membrin form homo-oligomeric coiled-coils in solution. Furthermore, p115’s binding could have been prevented by this oligomerization.
These results left two possibilities to explain the lack of p115 binding to syntaxin 5 and membrin; 1) p115 does not form stable complexes with SNARE oligomers or 2) p115 is selective for binding certain SNAREs, and does not interact with syntaxin 5 and membrin. However the latter possibility seems unlikely since interactions between p115, syntaxin 5, and membrin have been observed (30, 56). More likely, our particular protein preparations and/or gel filtration binding assay are able to detect a binding preference for monomeric SNAREs that had not been apparent before.
To further analyze possible interactions between p115 and membrin, and to understand the basis for the potential discrepancy with the literature, GST-tagged full-length membrin was immobilized on Glutathione-Sepharose beads, along with GST or GST-rbet1 for negative and positive controls, respectively. These beads were then incubated with p115, washed, and the levels of bound p115 were analyzed by SDS-PAGE and immunoblotting (Fig. 3A). By using this bead-binding assay, we were able to demonstrate specific binding between p115 and membrin beads (Fig. 3B). However, unlike GST-rbet1, GST-membrin bound only at higher p115 concentrations at which nonspecific binding was also evident. Since a higher affinity interaction is more likely to be solution-stable, this likely explains why membrin and syntaxin 5 failed to produce complexes in the gel filtration assay, and suggests that the discrepancy we saw was due to different experimental conditions.
Figure 3. P115 binds to bead-immobilized GST-rbet1 with higher affinity than GST-membrin.

(A) GST, GST-rbet1, or GST-membrin were immobilized on glutathione-Sepharose (GSH-Seph.) beads. 100 μl GST or SNARE beads each containing 0.5 mg/ml protein were incubated for 4 hrs at 4°C, in the presence of 20 nM and 60 nM of soluble p115, respectively, in a reaction volume of 200 μl. An additional 1 mg/ml of BSA was added to all reactions to reduce non-specific binding. After incubation with p115, beads were washed and subjected to SDS-PAGE and Western analysis with anti-p115 antibody. (B) P115 binding to GST, GST-rbet1, and GST-membrin beads, at the two assayed concentrations. (C) Ponceau S staining of membrane demonstrate that similar amounts of bead-immobilized fusion proteins were present in each reaction.
In summary, p115 bound stably to rbet1 and sec22b in solution, but did not form stable complexes with membrin or syntaxin 5 (Fig. 2). The low affinity interactions observed with membrin in the bead format, together with the fact that p115 did not stably interact with the highly active syntaxin 5 SNARE motif led us to hypothesize that oligomerization was the main reason for p115’s apparent selectivity in the gel filtration binding assay. If this is true, then the formation of a hetero-oligomeric SNARE complex, which is structurally even more robust than SNARE homo-oligomers, is predicted to also block the binding of p115.
Since the formation of a SNARE complex does not involve the SNARE N-terminal domains, it is not surprising that tethers associating with SNARE N-terminal domains can remain bound to the SNARE complex after its completion (21). However, it is harder to imagine p115, which interacts with SNARE motifs, not being displaced after the SNARE motifs assemble into a four-helix-bundle. Studies have demonstrated that despite its involvement in SNARE assembly, p115 can be washed off with high salt from formed SNARE complexes, indicating that p115 is not integrated into these complexes (56). P115/Uso1p-facilitated SNARE assembly yields SNARE complexes lacking p115 (26, 56). Furthermore, blocking the recycling of SNAREs decreases the amount of membrane-associated p115 (55), suggesting SNARE complexes cannot recruit p115 as efficiently as SNAREs in monomeric form.
To test whether p115 can interact with SNARE complexes, we performed a solution binding study using rbet1 in its hetero-oligomeric vs. its monomeric state. Recombinant rbet1, syntaxin 5 and membrin were incubated together prior to fractionation by velocity gradient sedimentation. The presence of rbet1-containing SNARE complexes in fraction 7 was detected by anti-rbet1 immunoblotting and then confirmed by gel filtration followed by immunoblotting. The velocity gradient-purified rbet1-containing t-SNARE complex was found in gel filtration fractions 16, 17, and 18 (Fig. 4, step 2, upper right panel). As was established in previous studies, these fractions contain predominantly rbet1-syntaxin 5-membrin ternary complexes, as well as some rbet1-membrin and rbet1-syntaxin 5 binary complexes. (57). As a control, soluble rbet1 alone was also subjected to velocity sedimentation, and fractions containing the monomeric rbet1 were pooled and adjusted to a comparable rbet1 concentration as in the SNARE complex fraction. Gel filtration of this fraction confirmed its monomeric state (Fig. 4, step 2, upper left panel). Next, the velocity gradient-purified ternary SNARE complex and monomeric rbet1 were separately incubated with p115 prior to gel filtration chromatography. As expected, an rbet1-p115 complex was detected in fractions 10–14 (Fig. 4, step 2, middle left panel) when p115 was incubated with monomeric rbet1. However, there was little detectable binding between p115 and the t-SNARE complexes containing rbet1 of similar concentration (Fig. 4, step 2, middle right panel). This is the first direct demonstration that p115 binds stably to a monomeric SNARE but not to the same SNARE when integrated into a SNARE bundle.
Figure 4. P115 binds monomeric rbet1, but not t-SNARE complexes containing rbet1.
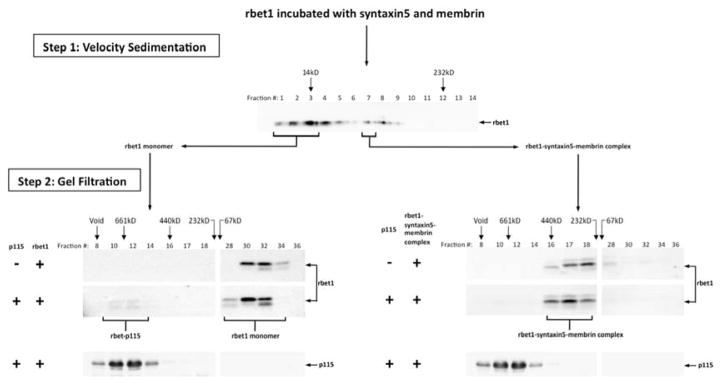
Recombinant rbet1 was incubated with recombinant syntaxin 5 and membrin overnight on ice to allow the formation of SNARE complexes. Reaction mix was loaded onto a 5–30% glycerol gradient, and subjected to velocity sedimentation (Step 1). The presence of t-SNARE complexes in fraction 7 was confirmed by Western blotting (data not shown). Recombinant rbet1 alone was also subjected to velocity sedimentation, and fractions containing monomeric rbet1 were pooled and adjusted to a similar rbet1 concentration as the SNARE complex (for simplicity this is depicted as a pool of those fractions from the complexes gradient). Both monomeric rbet1 and SNARE complex were passed through a gel filtration column, with fractions analyzed by Western blotting using an anti-rbet1 antibody (Step 2, top panels). Monomeric rbet1 and the SNARE complex were incubated with p115 separately, the mixed reactions were gel-filtered and analyzed by Western blotting using the same antibody (Step 2, middle panels). p115 in the mixed reaction fractionation was also detected by immunoblot (Step 2, bottom panels).
Although we cannot exclude that there was a small amount of binding between p115 and the rbet1-syntaxin 5-membrin complex, two experimental factors argue that any such binding was orders of magnitude lower than binding to rbet1 monomers. First, there was some monomer present in the purified complex preparation (Fig. 4, step 2, upper right panel, fraction 32). This could account for a slight binding of that preparation to p115 based on a p115-rbet1 binary interaction. Second, the ternary complex preparation is composed primarily of the more binding-active form of rbet1, which is the more rapidly migrating form on SDS-PAGE. Although the structural basis of the two closely-migrating forms is unclear, they are observed in both E. coli-produced as well as endogenous cell extracts (71), with the faster-migrating form exhibiting greater SNARE binding and recruitment to COPII vesicles. The relative efficiency of binding to p115 of the monomer preparation is vastly greater than that of the ternary preparation when only the lower rbet1 band is considered.
Membrin displaces p115 from a pre-formed rbet1-p115 complex
Because p115 is not part of the final SNARE complex, we hypothesized that in order for the SNARE assembly to complete, p115 would need to dissociate from its bound SNAREs. To confirm this prediction, GST-rbet1 or control GST were immobilized on Glutathione-Sepharose beads, then incubated with p115 to allow the formation of a GST-rbet1-p115 complex (Fig. 5A). After several rounds of washing to rinse off unbound p115, the beads were incubated either with an excess amount of soluble membrin, or with bovine serum albumin (BSA) as a control. After this incubation, supernatants were collected and the amount of dissociated p115 found in the supernatant was analyzed by immunoblotting. p115 was eluted from the GST-rbet1-p115 beads by membrin in a dose-dependent manner; furthermore, this elution effect pertained to p115 specifically bound to GST-rbet1 beads and not to GST beads (Figure 5B top vs. bottom panels, and quantitated in C). On the other hand BSA had only a small, presumably nonspecific effect on the rbet1-p115 complex that appeared to pertain to non-specifically bound p115 present on either GST or GST-rbet1 beads (Figure 5B, left panels, and quantitated in C).
Figure 5. Soluble membrin displaces p115 from a pre-formed p115-rbet1 complex.

(A) GST or GST-rbet1 was immobilized on glutathione-Sepharose beads. Beads containing 0.5 mg/ml GST-rbet1 or GST were then incubated with 600 nM p115 at 4°C overnight to allow the formation of p115-rbet1 complexes. After thorough washing, increasing concentrations of membrin or control BSA were added to the p115-treated beads. Following an incubation of 1 hr at 4°C, beads were centrifuged, supernatants were harvested, and p115 present was analyzed by SDS-PAGE and Western blotting. (B) Amounts of p115 present in the supernatants from GST beads (bottom panels) or GST-rbet1 beads (top panels) incubated with BSA or membrin as indicated. (C) Quantitation of blots from panel B.
Monomeric SNAREs are a required binding site for p115 on COPII vesicles
p115 is still associated with membrane in the absence of active Rab1 GTPase (31), suggesting an additional binding site for p115 on the COPII vesicle membrane. SNAREs have been shown to play an essential role in the recruitment of p115 to COPII vesicles (55), yet it is unclear which SNARE species are responsible for this recruitment. So far we have demonstrated that p115 selectively binds to monomeric SNAREs but not SNARE complexes using solution-binding assays, and here we test the hypothesis that p115 requires monomeric SNAREs for its retention on in vitro generated COPII vesicles. This assay utilizes the vesicular stomatitis virus glycoprotein (VSV-G) ts045 as a model cargo. Normal rat kidney (NRK) cells were transfected with myc-VSV-G DNA and cultured at 37°C. Following a subsequent incubation at 40°C that allows VSV-G to accumulate in the ER, cells were permeablized and incubated in the presence of rat liver cytosol, an ATP regenerating system, and buffer, allowing them to produce COPII coated vesicles with a cytoplasmically accessible myc tag. These vesicles were incubated in the presence or absence of recombinant soluble rbet1, then immuno-isolated using anti-myc antibody beads (Fig. 6A). The amount of p115 present on the vesicles, as well as internal membrane markers, was analyzed by immunoblotting. Compared to the control, the addition of excess soluble rbet1 removed all detectable p115 from the COPII vesicles (Fig. 6B). The fact that a soluble SNARE alone can compete p115 off vesicle membranes suggests that monomeric SNAREs account for the p115 interactions with COPII vesicles—or at least for those ongoing p115 interactions strong enough to withstand the immuno-isolation of vesicles. This finding is consistent with, but more direct than previous studies showing that an inhibition of SNARE complex disassembly impairs p115-membrane association (31, 55).
Figure 6. Excess soluble rbet1 removes p115 from COPII vesicle membranes.

(A) Normal rat kidney (NRK) cells were transfected with myc-VSV-G DNA, infected with vaccinia virus to drive over-expression (see Methods), and incubated at 40°C to allow the accumulation of myc-VSV-G in the ER. The cells were then permeabilized at 40°C and incubated with rat liver cytosol in the presence of an ATP regeneration system. Generated COPII vesicles were separated from cells by centrifugation. 0.3 μM of soluble rbet1 was added to the vesicles followed by an incubation of 1 hr. Vesicles were then immunoisolated using the myc tag. The recombinant rbet1 used in this experiment was dialyzed extensively in Buffer A. To eliminate any potential effects caused by the buffer, similar amount of Buffer A was also added to the vesicles as control. (B) Isolated vesicles were subjected to SDS-PAGE and amounts of p115 (top panel), cargo myc-VSV-G (middle panel), and vesicle membrane marker p24 (bottom panel) were determined by immunoblotting. Vesicles generated by untransfected NRK cells served as negative control; vesicles generated by myc-VSV-G transfected cells without rbet1 or Buffer A addition served as positive control.
P115’s role in SNARE complex assembly
The formation of a Sed5p/Sec22p/Betlp SNARE complex in yeast requires the presence of the yeast p115 homologue Uso1p (26). Also, p115 stimulates the assembly of GOS-28–syntaxin-5 SNAREpin in vitro (56). Since p115 bound to monomeric SNAREs in our studies, we wondered whether this binding had a detectable effect on SNARE complex assembly.
Soluble recombinant syntaxin 5, membrin, rbet1 and sec22b were incubated on ice either in the presence or absence of p115 for four hours. Reaction mixtures were gel-filtered, and the fractions analyzed by immunoblotting. Quaternary SNARE complexes are eluted from the gel filtration column in fractions 16, 17, 18 (57). To distinguish between the complete quaternary complex and ternary or binary SNARE complexes whose elutions overlapped the same fractions, an antibody against the v-SNARE sec22b was used, for sec22b only enters into a SNARE complex when all three of its cognate binding partners are present in a single complex (57). As is typical, only a small fraction of sec22b was incorporated into quaternary complexes. Unexpectedly, we were not able to detect a stimulatory effect of p115 on the SNARE assembly, though p115-sec22b complexes were clearly present in fractions 8–10 (Fig. 7A).
Figure 7. P115 chaperone effect on SNARE complex assembly.

(A) Recombinant syntaxin 5, membrin, rbet1, and sec22b were incubated in the presence or absence of 0.9 μM His6-p115 for 4 hrs on ice. The reactions were passed through a gel filtration column. Fractions were analyzed with SDS-PAGE and immunoblotting using anti-sec22b antibody. The sec22b-syntaxin 5-membrin-rbet1 complex elutes in fractions 16–18. (B) P115 was incubated with sec22b overnight on ice. The next day, other SNAREs were added to the reaction mixes and incubated for 4 hrs on ice. Reactions were then gel-filtered and analyzed as described in (A). Top panel in (B) represents the elution of a SNARE assembly reaction carried out in the presence of a similar concentration of p115 but without any overnight pre-incubation between p115 and sec22b. The absence of a p115-sec22b binary complex in the top panel of (B) was likely due to relative short incubation time (4 hrs as opposed to overnight, as in the bottom panel). (C) Quantitation of A (left panel) and B (right panel) fractions 6 through 26. Beyond fraction 26, the chemiluminescence signal saturated the camera pixels and quantitative data was not possible.
One possibility that could explain the lack of stimulatory effects was that p115 binding has a stabilizing effect on SNARE conformation rather than an immediate catalytic effect. We therefore tried a different approach. Sec22b was pre-incubated overnight with or without p115, before the addition of other SNAREs. After a second incubation of four hours to allow the formation of SNARE complexes, both reactions were passed through a gel filtration column and SNARE complexes were quantified and compared. As shown in Fig. 7B, the long pre-incubation of sec22b with p115 enhanced the formation of SNARE complex by at least two-fold. Adding rbet1 to the pre-incubation mixture of sec22b and p115 did not cause any additional enhancement of the complex formation (data not shown). Our findings suggest that p115 may have a mild ‘chaperone’ effect on the SNAREs, possibly by binding to the SNARE motif and keeping it in an optimal conformation for complex assembly. This prediction does not necessarily contradict the catalyst model, for p115 could still function as a SNARE catalyst downstream. Three caveats of this purified experimental system that could also explain the lack of apparent catalytic activity are the lack of cofactors required for p115’s function, the lack of membrane topological constraints, and the possibility that purified recombinant p115 was not fully functional.
P115’s effects on COPII vesicle tethering and fusion in vitro
A requirement for p115 in COPII vesicle transport has been demonstrated before (29, 30, 72), but the precise steps at which p115 is involved in the tethering and fusion of COPII vesicles has not been resolved. We have developed in vitro assays of COPII vesicle tethering and fusion in which two distinctly labeled, fusogenic COPII vesicle populations are generated from permeabilized normal rat kidney (NRK) cells, one carrying the myc tag, the other radiolabeled by 35S (55, 73). Notably, if the two vesicle populations are co-incubated at 32°C, fusion of the differently labeled vesicles results in a heterotrimer containing both a myc-labeled VSV-G subunit and an 35S labeled VSV-G* subunit, due to the dynamic equilibrium between VSV-G trimers and monomers (74). To detect fusion, the mixtures are detergent-solubilized and VSV trimers are immunoprecipitated using the myc tag. In this protocol, fusion is a function of the 35S labeled VSV-G trimers present in the solubilized vesicles. To detect tethering, the two populations of COPII vesicles are incubated together at 32°C and immuno-isolated intact without solubilization using anti-myc antibody beads. The beads are then analyzed by SDS-PAGE and autoradiography for the presence of 35S-VSV-G, which represents co-isolated, physically associated vesicles.
To test p115’s effects on vesicle fusion, we first generated a p115-free cytosol to use in the assay. Rat liver cytosol was incubated with a p115 antibody before depletion with fixed S. aureus cells. The depletion of p115 was complete as shown in Fig. 8A, top left panel. Amounts of several known p115-interacting partners were also analyzed by Western. With the exception of GM130, none were affected. GM130 is a primarily Golgi-localized tether and was implicated to function at a later step in ER-Golgi anterograde transport independently of p115 (75). There has been no evidence suggesting GM130 may affect homotypic COPII vesicle fusion. Thus we did not expect a partial co-depletion of GM130 to have a significant effect on the COPII vesicle tethering/fusion assay. A ‘mock’ cytosol treated with non-immune rabbit IgG was used as control.
Figure 8. Recombinant full-length p115 inhibits in vitro COPII vesicle tethering and fusion.

(A) Depletion of p115 from rat liver cytosol. Rat liver cytosol was treated with rabbit anti-p115 antibody (p115 dpl.), or purified rabbit IgG (Mock) for 2 hrs on ice, and depleted with fixed S. aureus cells for 15 min. (Ctrl.) represents RLC without any treatment. Amounts of p115, GM130, sec23, beta-COP, sec31, and GBF1 were analyzed after depletion. (B) Ni-NTA purified recombinant His6-tagged p115 (left lane) was further purified on a Mono Q column (middle lane). The small truncation fragment of p115 was purified from the full-length protein preparation by velocity sedimentation (right lane). (C) Both p115 depletion and recombinant p115 addition inhibits in vitro COPII vesicle fusion. Myc-tagged and 35S-tagged VSV-G COPII vesicles were incubated together at 32°C for 1 hr, either in the presence of mock-depleted cytosol (blue curve) or p115-depleted cytosol (red curve), and the indicated concentrations of recombinant p115 ranging from 0.1–1 μM (x-axis) was added to the reactions. After adding Triton X-100, VSV-G trimers were immunoprecipitated using the myc tag and heterotrimers containing 35S were quantitated as a measure of fusion. Fusion was normalized to percentage of the positive control reaction in which untreated RLC was used and no additional protein was added. Ice control (green) indicates fusion reactions incubated on ice. Data points at zero p115 concentration are representative of triplicate reactions, with error bars representing S. D. The remaining data points were obtained from singlicate reactions. The experiment was repeated three times with qualitatively similar outcomes. (D) COPII vesicle fusion in the presence or absence of recombinant Mono Q purified p115. Myc-tagged and 35S-tagged VSV-G COPII vesicles were incubated together at 32°C for 1 hr in the presence of untreated RLC with normal level of endogenous p115 and the indicated concentrations of Mono Q p115 (red curve) or p115 small fragment (green curve) ranging from 0.002–0.09 μM. Triton X-100 was added to the reactions and VSV-G heterotrimers were immunoprecipitated and quantitated as described above. Due to the high KCl content of the Mono Q column elution buffer, we also tested Mono Q elution buffer (blue curve). Fusion was normalized to percentage of the buffer control with no additional p115. The no-bio control represents signal obtained from pellets isolated with non-biotinylated myc antibody. Data points are duplicates, with error bars representing S.E.M. The asterisk marks a control data point that resulted in p=.027 in a two-tailed T-test with the corresponding full-length p115 data point. (E) COPII vesicle tethering in the presence or absence of recombinant p115. Myc-tagged and 35S-tagged VSV-G COPII vesicles were incubated together at 32°C for 1 hr with untreated RLC and the indicated concentrations of Mono Q purified p115 (red curve), p115 small fragment (green curve), or Mono Q buffer control (blue curve). Intact vesicles were immunoisolated in the absence of detergent using the myc tag and recovered 35S-VSV-G quantitated as a measure of tethering. Tethering was normalized to percentage of the buffer control with no additional p115. Data points at 0.002 μM protein concentration were obtained from quadruplicate reactions, other data points are representative of duplicates. Error bars represent S.E.M. The experiments in D and E were repeated twice with similar outcomes.
To test the functional effects of p115 depletion and the back-addition of recombinant p115, myc-VSV-G vesicles and 35S-VSV-G vesicles were mixed and incubated in either p115-free cytosol or mock-treated cytosol, with or without additional purified recombinant p115, and fusion was monitored. Fusion levels were normalized to the level in untreated RLC. Depletion of p115 reduced vesicle fusion by 70% of mock (Fig. 8C, red versus blue curves at zero point on x-axis). This extends previous antibody inhibition studies (55) implying that p115 is required at the first fusion step in the secretory pathway, leading to VTC biogenesis. Recombinant p115 failed to restore fusion activity, although we observed a small stimulation at a concentration of 0.1 μM (Fig. 8C, red curve). The reasons for the lack of rescue are not clear. It is possible that the optimal dose range for p115’s function is very narrow (see Discussion), and we were not able to pinpoint the concentration at which sufficient p115 is provided yet the ratio and/or access of p115 to its cofactors (e.g. Rab1) is close to physiological. Surprisingly, though, recombinant p115 seemed to have a strong inhibitory effect on vesicle fusion when added in excess to the endogenous protein in mock-depleted cytosol (Fig. 8C, blue curve). The p115 preparation employed in these studies was the Ni-NTA fraction shown in Fig. 8B.
We wanted to understand the unexpected inhibitory effect of wild type recombinant p115. To eliminate the possibility that a truncated, dominant-negative form of p115 was present in the protein preparation, we further purified the Ni-NTA enriched p115 with Mono Q anion exchange chromatography (Fig. 8B, also see Materials and Methods). Although many partial products were removed by the additional step, the purified fraction still contained a low molecular weight degradation/truncation fragment of p115. Since we were unable to prepare the full-length protein without this contaminant, we purified the small fragment to near-homogeneity by velocity sedimentation (Figure 8B, third lane) and used it as a control in subsequent experiments. When titrated in the presence of untreated cytosol, the Mono Q-purified full-length p115 inhibits both fusion (Fig. 8D) and tethering (Fig. 8E) of COPII vesicles in a dose dependent manner. Neither buffer control nor the p115 small fragment had any effects on fusion or tethering activities. Thus, inhibitory activity appears to reside with the full-length protein. We observed a small stimulation of vesicle tethering by recombinant p115 in low excesses (0.002 μM, Fig. 8E). This effect was not present in the fusion experiment (Fig. 8D). On the other hand, we noted that compared to the tethering assay, the fusion assay required lower concentrations of excess p115 to reach the maximum inhibitory effect. It is possible that the fusion of vesicles is more sensitive to changes in p115 concentration than vesicle tethering. In summary, recombinant full-length p115 had very slight stimulatory effects at very low concentrations that were overcome by potent inhibitory activity in both the presence and absence of endogenous p115.
To test whether excess p115 also inhibited p115 trafficking function in vivo, we over-expressed full-length, functional p115 in HeLa cells. As shown in Fig. 9A–C, overexpression of a myc-tagged p115 construct in vivo causes disruption of HeLa cell Golgi structure monitored with GM130 labeling. Golgi disruption clearly correlated with p115 expression level, since moderately expressing cells displayed morphologically normal Golgis. We know that the myc-tagged construct was intrinsically functional, since when expressed at moderate levels it restored the normal GM130 pattern to p115-depleted cells with otherwise dispersed Golgis (Fig. 9D–F). Phenotypes of siRNA depletion of p115 in HeLa cells have been characterized earlier (50).
Figure 9. Effects of full-length myc-tagged p115 in vivo.

HeLa cells overexpressing myc-p115 labeled with anti-myc antibodies (A) displayed disrupted Golgi morphology, as indicated by labeling of the Golgi marker GM130 (B). The same myc-p115 construct, when expressed at moderate levels in p115-knockdown cells (D), rescued the Golgi fragmentation phenotype caused by p115 depletion, as indicated by GM130 labeling (E).
It would appear that p115 has a tight functional dose range. A certain level of p115 is required for COPII vesicle tethering and fusion, yet these events are inhibited when the tether is present in excess. These findings are in line with our SNARE-binding results, which suggest that p115 needs to be removed from SNARE complexes prior to their completion. One would expect that excess p115 might inhibit SNARE-mediated fusion by slowing down or preventing this removal process.
Binding properties of purified recombinant p115 recapitulate endogenous rat liver p115
We wanted to validate the most important conclusions from purified SNARE binding studies in Figures 2–6 using functionally robust, endogenous p115. A partially purified liver p115 could potentially provide optimal functional intactness and yet also likely reflect the intrinsic binding properties of p115. Toward this end, we prepared rat p115 at three increasing levels of enrichment. First was crude NRK cytosol from cells expressing rat his-p115 (referred to as “NRK cytosol”; immunnoblot analysis not shown). Second was endogenous rat liver p115 purified by ammonium sulfate fractionation and Superdex 200 chromatography (referred to as “rat liver Superdex”), enriched approximately 18-fold relative to crude rat liver cytosol. This fraction is characterized by immunoblotting in Figure 10D. Third was endogenous rat liver p115 purified by several conventional chromatography steps to yield a simplified protein pattern with p115 representing a major band (referred to as “rat liver Mono Q”). This fraction is characterized by Coommassie gel in Figure 10B, left panel. Next we evaluated whether rat p115 at different purity levels would behave similarly in SNARE bead binding assays. As shown in Figure 10A and B, when tested at a limiting concentration, crude His-p115 in NRK cytosol and endogenous rat liver p115 in the Mono Q fraction bound specifically to SNARE beads with similar characteristics--weak binding to membrin and strong binding to rbet1, as observed in Figure 3 with purified recombinant His-p115 expressed in bacteria. SNAREs immobilized on the beads are characterized by Coommassie gel in Figure 10B, right panel.
Figure 10. Binding properties of endogenous rat liver p115.

(A) GST, GST-rbet1, or GST-membrin were immobilized on glutathione-Sepharose beads. Beads were incubated with crude or partially purified fractions, “NRK cytosol” and “rat liver MonoQ”, respectively, such that the final p115 concentration was approximately 20 nM by Western blot. After binding and washing reactions similarly to as in Figure 3, the bead-bound p115 was determined by Western blots and quantitated. (B) Coommassie-stained gels showing the Mono Q p115 preparation used in the binding experiment with a rat his-p115 standard (left panel) and the proteins on each type of beads at the concentration employed (right panel). (C) ER-to-Golgi transport reconstitution in semi-intact NRK cells, where VSV-G transport is indicated by conversion of VSV G cargo from the endoglycosidase H-sensitive to –resistant form. The partially purified rat liver Superdex fraction fully reconstituted the assay in the presence of p115-depleted cytosol (closed circles); the Superdex fraction lost its restorative activity when it was depleted with anti-p115 (open circles). (D) Immunoblot analysis of the Superdex fraction, with or without immunodepletion, relative to the starting cytosol and ammonium sulfate cut, and equal protein loading in each lane. Blotting for other species demonstrates selectivity in the purification; rsly1 shows an overall lack of enrichment over the two-step purification, while a 55 kD degradation/truncation product of p115 visible in cytosol on over-exposed blots was removed during the purification. (E) Coommassie–stained gel documenting proteins on the beads for binding studies in part (F) and (G). Stoichiometry of the membrin and rbet1 bands indicated on the gels on the membrin+rbet1 beads was calculated by quantitation relative to BSA standards. (F) Rat liver Superdex fraction was incubated with the beads shown in (E) at approximately 30 nM p115, then unbound p115 was removed by washing and bound p115 determined by Western blotting and quantitation. The graph shows beads used along the bottom and bound p115 on the y-axis. (G) Membrin beads and soluble rbet1 can form a potentiated ternary SNARE complex. Presence of different beads and soluble proteins are indicated below and syntaxin 5 binding is shown on the y axis. Error bars represent standard error of duplicate determinations.
Given the apparent robustness of the SNARE bead binding assay with regard to p115 purity, we chose to confirm and extend our main conclusion, that p115 prefers to bind SNARE monomers and not complexes, using the endogenous rat liver Superdex fraction, since it represented a compromise between biochemical purity and likely functional intactness. Importantly, when tested in an ER-to-Golgi transport reconstitution in semi-intact NRK cells (76), the Superdex p115 fully reconstituted transport activity to p115-depleted cytosol, indicating that it was functionally intact (Figure 10C, solid circles). This was important since we found that further purified p115 preparations from various sources only partially restored activity and/or lost activity readily (not shown), perhaps due to the lack of cofactors or stabilizing interactions. The p115 in the Superdex fraction was required for the reconstituting activity, as opposed to other rate-limiting factors, since p115 antibody depletion blocked the reconstitution (Figure 10C, open circles).
To test whether the rat liver p115 bound to SNAREs but not SNARE complexes, we prepared GST-rbet1 beads, GST-membrin beads, and beads that contained a stoichiometric complex of GST-membrin and rbet1 (Figure 10E. The Superdex p115 fraction was incubated with each of the beads and unbound or loosely bound proteins were removed by washing. As shown in Figure 10F, the p115 bound most strongly to the rbet1 beads, but at the concentration tested also significantly interacted with membrin beads. Strikingly, no specific binding was detected to the membrin•rbet1 beads, despite the presence of stoichiometric amounts of two proteins it interacted with separately. The rbet1 preparation on these beads was identical to the soluble rbet1 used for Figures 2 and 4, which bound p115 very well. Additionally, the membrin and rbet1 employed in the binary complex were fully capable of forming higher order complexes, since together they exhibited potentiated binding with soluble syntaxin 5, indicating formation of a ternary SNARE complex (Figure 10G) (57). In conclusion, functionally intact endogenous rat liver p115 bound to individual SNAREs but not SNARE complexes, extending and validating the conclusions from purified recombinant proteins.
Discussion
p115’s SNARE binding preferences in vitro
Using a gel filtration assay, we have for the first time provided direct evidence that p115 preferentially interacts with monomeric SNARE motifs. Our results demonstrated that p115 binds mammalian ER-Golgi v-SNARE sec22b and t-SNARE rbet1. The fact that p115 can interact with the ΔTM recombinant rbet1 led us to believe that the SNARE motif is the binding site for p115. Further studies with the N-terminal longin domain of sec22b showed that p115 does not interact with this region, suggesting that the SNARE motif accounts for the interactions between p115 and the recombinant ΔTM sec22b. Structural similarities between p115’s coiled-coil domain and the SNARE motif suggest that they could form a coiled-coil structure.
It was unexpected that our solution binding method did not detect p115-membrin or p115-syntaxin 5 interactions, which have been demonstrated before using other methods. One possibility is that the highly oligomeric state of syntaxin 5 and membrin blocked p115’s access to the SNARE motif. Due to lower affinity binding, p115’s interactions with these SNAREs may be transient, and the resulting complexes would be too unstable to be detected by the gel filtration assay.
Since SNARE motifs are the binding sites for p115, and since these motifs are joined into a four-helix-bundle during fusion, p115 might have to dissociate from its bound partner during the formation of a SNARE complex. In support of this hypothesis, our results showed that p115 interacts with rbet1 alone, but not SNARE complexes containing rbet1. In addition, membrin is able to displace p115 from a pre-formed rbet1-p115 complex. Furthermore, in vitro studies show that soluble monomeric rbet1 can compete p115 off COPII vesicles, suggesting a free monomeric SNARE motif is a major binding site for p115 on membrane.
We have also demonstrated that excess p115 inhibits COPII vesicle tethering and fusion in vitro and Golgi homeostasis in vivo. These findings are consistent with our binding studies, from which we have concluded that p115 needs to dissociate from SNARE complexes before their formation is complete. Excess p115 could potentially impair fusion efficiency by slowing down this removal process. However, it is worth noting that the inhibitory effect of p115 on vesicle fusion has not been observed in previous studies using in vitro systems. p115 purified from rat liver showed no inhibitory effects on the transport of VSV-G to the medial Golgi at 160% of cytosolic level (29), and recombinant bovine p115 purified from insect cells showed no such effects on VSV-G processing at high concentrations, either (30). One potential explanation for the apparent discrepancy is that our assay specifically measures the homotypic fusion aspect of the VSV-G transport pathway, and employs vesicles in very dilute suspension; this could make it more sensitive to changes in p115 on-off dynamics than a more intact subcellular reconstitution containing local concentration gradients of cofactors and a cytoskeleton. In our hands the in vitro ER-to-Golgi assay was also inhibited by excess p115, but at concentrations exceeding those that inhibited in the vesicle fusion assay. Both assays contained about 30 nM of rat liver cytosolic p115 under normal conditions. Addition of an additional 20 nM of purified recombinant p115 inhibited the fusion assay (representing ~166% of cytosolic, Figure 8D) whereas inhibition of the ER-to-Golgi transport assay didn’t occur until addition of an additional 60 nM of Superdex p115 (representing ~300% of cytosolic, data not shown). Fully purified p115 also inhibited the ER-to-Golgi assay, but to a much lesser degree than in the fusion assay. We believe that the fusion assay helped reveal an inhibitory activity of excess p115 not apparent until higher concentrations in other systems.
We did not see as high a level of stimulation by p115 on SNARE complex formation as was observed in previous studies performed with Golgi detergent extract (56). Many factors could have contributed to this difference. First, our binding studies were carried out with purified proteins as opposed to Golgi extracts. It is possible that certain p115 accessory factors and/or SNARE destabilizing factors are absent in the purified system, resulting in p115 appearing to lose some SNARE assembly function. Another factor is that the embedding of SNAREs in a membrane is important for p115’s catalytic action. We did notice, however, that with the addition of p115, the elution of some monomer rbet1 shifted from fractions 32–34 to fractions 26–32 (Fig. 2B), suggesting that the SNARE might have undergone conformational changes in the presence of p115. Furthermore, whereas p115 appears to have no instantaneous effect on the formation of SNARE complexes, its pre-incubation with sec22b increases the complex yield by 2~3 fold (Fig. 7B). This increase is more or less in agreement with p115’s published effects on purified syntaxin 5-GOS-28 complex formation (56). Based on the evidence, we suspect that instead of a SNARE catalyst, p115 may act as a SNARE chaperone by modifying the SNARE motifs and/or keeping them in an optimal conformation, at least in soluble SNARE assembly reactions. Whether p115 also assembles SNAREs catalytically when embedded in membranes remains to be experimentally addressed.
Model of p115’s mechanism of Action
Taken together, our results support a model in which p115 binds monomeric SNAREs on the membrane to stabilize their conformation and possibly facilitate fusion by catalyzing SNARE-SNARE interactions, and then is displaced as the SNAREs come together. This potential mechanism of action would explain the observed inhibition of trafficking by excess p115 in vitro and in vivo. For a monomeric SNARE, the binding of p115 and the binding of another SNARE could be mutually exclusive, and excess p115 would slow down p115’s removal from the forming SNARE complex. The model also provides insights into the observed highly dynamic state of membrane-bound p115 (31) that could indicate synchronization between p115 recruitment/dissociation and the fusion process.
A possible model for molecular events during p115-mediated docking and fusion of vesicles is that p115 is recruited to the membrane by the dimer’s globular head and/or CC1 domains via binding to Rab1 or SNAREs (Fig. 11A). P115 could then use a free SNARE-interacting domain (either CC1 or CC4) or the head domain to initiate interactions with SNAREs or Rab1 on the opposing membrane (green arrows), thus tethering the vesicles together. Subsequently, p115 could align the SNAREs on juxtaposed membranes by conformational changes within the p115 molecule. It is also possible that p115 never directly bridges the two membranes, but works solely on the vesicle membrane to enhance the efficiency of SNARE assembly by maintaining their reactivity and concentrating monomeric SNAREs at fusion sites where Rab1 is active (Fig. 11B). This would make p115 less of a traditional ‘tether’, and more of a chaperone and/or catalyst for SNARE-mediated fusion. Further studies will be essential to define the details of molecular tethering and SNARE coupling by p115.
Figure 11. Possible mechanisms of action of p115.

(A) Two membrane-bridging models for p115. P115 dimer (green) is recruited to membranes by interacting with monomeric SNAREs (yellow and red, gray and cyan) through the CC1 and/or CC4 domains, and/or by binding to Rab1 (dark blue) through the globular head domain. In the upper version, p115 also binds a monomeric SNARE on an opposing membrane using its free CC4 domain. The binding of p115’s globular head domain to Rab1 on the same or opposing membrane causes a conformational change in the tail region of p115, which shortens the distance between membranes and places the N-termini of the SNARE motifs in close proximity. The formation of the four-helix bundle expels p115. (B) P115 dimer captures and concentrates the SNAREs on the same membrane (yellow and cyan) using both the CC1 and CC4 domains. Conformational changes bring the juxtaposed SNAREs together, as well as prime them to interact with SNAREs on the opposing membrane. Our data does not distinguish between the displayed scenarios and several similar variations. The cartoon representation of p115 was adapted from the p115 structure with the acidic C-terminus omitted (43).
Materials and Methods
Protein purification
Recombinant SNARE proteins were expressed and purified as previously described (57). Hexahistidine-tagged full-length bovine p115 was expressed in E. coli strain NM522 using a pQE9 construct obtained from the Lowe lab (54). Bacteria cultures were grown at 37°C to an A600 of 0.4, before induction with 1 mM isopropyl-1-thio-β-D-galactopyranoside at the same temperature for 2–3 hours. Bacteria were pelleted and resuspended in French Press buffer (50 mM Tris, pH 8.0, 0.1 M NaCl, 1 mM EDTA, 0.05% Tween 20, 2 μg/ml leupeptin, 4 μg/ml aprotinin, 1 μg/ml pepstatin A, and 1 mM phenylmethylsulfonyl fluoride). After 2 rounds of French Press, cell lysates were centrifuged at 20,000 g for 20 minutes, the supernatant was then collected and centrifuged again at 100,000 g for 45 minutes. Supernatant from the second centrifigation was passed through a Ni-NTA column equilibrated with 50 mM Tris, pH 8.0, 0.3 M NaCl, 0.05% Tween 20, and 0.025 M imidazole, and eluted with the same buffer containing a 0.025–0.25 M gradient of imidazole. Purified His-tagged p115 was dialyzed into protease inhibitors (1 mM DTT, 2 μg/ml leupeptin, 4 μg/ml aprotinin, and 1 μg/ml pepstatin A) supplemented Buffer A (20 mM Hepes, pH 7.2, 0.15 M KCl, 2 mM EDTA, 5% glycerol), and quantitated according to A280. Sec22b N-terminal domain was present as a major degradation product in the sec22b preparation, and was separated from the full-length protein by gel filtration. Hexahistidine-tagged membrane N-terminal domain and hexahistidine-tagged Ykt6 N-terminal domain were expressed using pQE9 constructs and purified in the same way as full-length sec22b.
For the in vitro tethering and fusion assay, Ni-NTA purified p115 was further purified on an ion exchange column (FPLC-Mono Q, Amersham Pharmacia Biotech). The column was washed abundantly with running buffer (20 mM Tris, 1mM EGTA, pH 7.6). After loading p115, the column was washed with 15 ml running buffer, then eluted with the same buffer containing a 0–1 M gradient of KCl. Purified p115 was dialyzed into 25 mM Hepes, 125 mM potassium acetate, pH 7.2.
p115 from rat liver cytosol was partially purified by two procedures. The first procedure essentially followed (25), with minor modifications; this generated a MonoQ fraction with a simple protein pattern in which p115 is a major band by SDS gel and Coommassie staining. A second procedure involved ammonium sulfate fractionation as in Barroso et al., followed directly by Superdex 200 gel filtration. The peak Superdex 200 fraction was quite complex, but was enriched for p115 by 18-fold relative to cytosol.
Gel filtration binding studies
Approximately 20 μg of each full-length SNARE protein or NT domain was incubated with or without ~1.3 μM of p115 in Buffer A supplemented with 0.1 % Triton X-100, in a reaction volume of 300 μl. After an overnight incubation, 250 μl of each protein mix was fractionated on a Superdex 200 column (Amersham Pharmacia Biotech) equilibrated in Buffer A containing 0.5 mg/ml BSA and 0.1% Triton X-100. Fractions were acetone-precipitated and subjected to SDS-PAGE and Western analysis.
Bead-binding studies
For the SNARE-p115 binary binding experiments, for example in Figure 3, 100 μl of 50% glutathione-Sepharose beads containing 0.5 mg/ml membrin, rbet1, or GST was each incubated with 20 nM and 60 nM of p115, respectively, in the presence of Buffer A containing 1 mg/ml BSA and 0.1% Triton X-100. Reaction volume was 200 μl. All reactions were incubated at 4°C overnight with agitation. Beads were then pelleted and washed with 3 × 1 ml of the same reaction buffer, after which 50 μl of SDS-PAGE sample buffer was added to each pellet prior to SDS-PAGE and Western analysis. For bead binding assays shown in Figure 10, only 20 μl of beads were used per 200 μl reaction, but the other parameters were similar.
For the p115 displacement experiment, reactions were conducted in the same buffer as the binary binding experiments. 100 μl of 50% glutathione-Sepharose beads containing 0.5 mg/ml rbet1 or GST was incubated with 600 nM p115 at 4°C overnight with agitation. The next day, after washing the beads with 3 × 1ml of reaction buffer, 0.025, 0.05, or 0.075 mg/ml of membrin or BSA was added to the reactions. All reactions were then incubated at 4°C for 4 hrs with agitation. The supernatants were collected and supplemented with equal volume of sample buffer, before subjected to SDS-PAGE and Western analysis for p115.
SNARE monomer-p115 vs. SNARE complex-p115 binding studies
All reactions were conducted in the same buffer used in other gel filtration experiments. Purified recombinant rbet1, syntaxin 5, and membrin were incubated together on ice for 48 hrs, before loading onto 5–30% glycerol gradients run in Buffer A supplemented with 0.1% Triton X-100, then centrifuged at 200,000 × g for 24 hrs in a Beckman MLS-50 rotor. Rbet1 alone was also subjected to velocity sedimentation under the same conditions. Fractions from both gradients were analyzed and quantitated with anti-rbet1 Western analysis, complex and monomer fractions were each pooled and dialyzed into Buffer A containing 0.1% Triton X-100.
Monomeric rbet1 and SNARE complex fractions were each incubated with 2.0 μM of p115 overnight on ice. The reactions were gel-filtered as described above and the fractions were acetone-precipitated, dissolved in sample buffer and analyzed with Western blotting for rbet1.
SNARE complex assembly assay
For the instantaneous effects of p115, 50 μl of purified recombinant syntaxin 5, membrin, rbet1, and sec22b were incubated together on ice for 4 hrs, in the presence or absence of 0.9 μM of p115. The reactions were gel-filtered as described above. The fractions were acetone-precipitated, dissolved in sample buffer and analyzed with Western blotting for sec22b.
For the chaperone effects studies, 50 μl of purified recombinant sec22b was incubated with p115 overnight on ice, before the addition of syntaxin 5, membrin, and rbet1. The final p115 concentration in the reaction mix was 0.9 μM. For the control reaction, sec22b was incubated alone on ice overnight, before the addition of other SNAREs and 0.9 μM of p115. Both reactions were incubated on ice for 4 hrs, then subjected to gel filtration, SDS-PAGE, and Western analysis for sec22b as described above.
Immunodepletion of p115 from rat liver cytosol
Pansorbin beads (Calbiochem) were washed five times with 50 × volume 25/125 buffer (25 mM Hepes, 125 mM potassium acetate, pH 7.2), and re-suspended at a concentration of 10%. Rat liver cytosol was dialyzed into 25/125 buffer, then supplemented with 0.05 × volume of affinity-purified rabbit anti-p115 (see below) and incubated at 4°C for 2 hrs. Immediately after the incubation, Pansorbin suspension was added to the cytosol at a concentration of 0.06 × volume. The mixture was incubated with agitation at 4°C for 15 minutes, before centrifugation at 20,000 × g. The supernatant was centrifuged again at 20,000 × g. Supernatant from the second centrifugation was then collected. In addition, a mock-depleted RLC was produced using identical methods and non-immune rabbit IgG.
COPII vesicle immunoisolation, tethering, and fusion assays
COPII vesicles used in rbet1 binding studies were generated in vitro as previously described in the one stage assay (55, 73), with small modifications. In short, NRK cells were transfected with myc-VSV-G DNA. After 24 hrs, cells were infected with vaccinia virus, followed by a 5 hr incubation at 40°C. The cells were then permeabilized by scraping and suspended in 25/125 buffer. Myc-labeled vesicles were produced in a buffer consisting of 75 μl of the cell suspension, 96 μl of water, 65 μl of 25/125 buffer, 15 μl of 0.1 M magnesium acetate, 30 μl of ATP-regenerating system, 9 μl of 1 M HEPES, pH 7.2, 60 μl of Ca/EGTA buffer (20 mM HEPES, pH 7.2, 1.8 mM CaCl2, and 5 mM EGTA), and 150 μl of rat liver cytosol dialyzed in 25/125 buffer. After incubation at 32°C for 30 minutes, cells were removed from the reactions by centrifigation. 160 μl of recombinant rbet1 (~2.5 μg) in Buffer A, 160 μl of Buffer A, or 160 μl of 25/125 buffer (positive control) was then added to the vesicles. Reactions were incubated on ice for 20 minutes prior to another 1 hr incubation at 32°C, after which vesicles were immunoisolated with 5 μg of anti-myc mAb, and subjected to SDS-PAGE and Western analysis.
COPII vesicle co-isolation and heterotrimerization assays were also carried out as previously described. Two distinctly labeled COPII vesicle populations were generated for these assays. Myc-VSV-G-containing vesicles were prepared as described above. To prepare the 35S-labeled vesicles, NRK cells were transfected with vesicular stomatitis virus strain ts045 and incubated at 32°C for 4 hrs, before labeling with 500 μCi of 35S-cysteine/methionine at 40°C, in a cysteine/methionine-free medium. Cells were then permeabilized and suspended in 25/125 buffer. Both types of vesicles were produced in a reaction mix consists of 337.5 μl of either cell suspension, 432 μl of water, 67.5 μl of 0.1 M magnesium acetate, 135 μl of ATP-regenerating system, 40.5 μl of 1 M HEPES, pH 7.2, 270 μl of Ca/EGTA buffer, and 675 μl of rat liver cytosol (either untreated, mock-depleted, or p115-depleted, all dialyzed in 25/125 buffer). Supernatants containing COPII vesicles were collected by centrifigation. 72.5 μl of myc-VSV-G vesicle suspension and 72.5 μl of 35S-VSV-G vesicle suspension were added to each reaction tube. For certain reactions, recombinant p115 dialyzed in 25/125 was also added. The final volume of all reactions was brought up to 200 μl with 25/125 buffer. Reacion tubes were first incubated on ice for 20 minutes, then shifted to a 32°C water bath and incubated for another 1 hr.
Treatments for fusion versus tethering assays differ from this point on. For the tethering/co-isolation assay, the reactions were chilled and centrifuged at 20,000 × g for 5 minutes to remove large vesicles, then immunoisolated with 5 μg of biotinylated anti-myc mAb and 10 μl 50% streptavidin-Sepharose (GE Healthcare). The isolates were dissolved in sample buffer, run on 12% SDS-PAGE gels, and transferred to nitrocellulose membrane. The amount of myc-VSV-G in each isolate was quantitated by Western analysis. The membrane was then dried and analyzed by phosphorimaging. Amounts of 35S were measured, and normalized based on the myc-VSV-G level, which reflects the amount of vesicle present. For the vesicle fusion assay, 2% Triton X-100 was added to the reactions following the 1 hr incubation at 32°C. Reactions were then incubated with agitation at 4°C for 20 minutes, before centrifugation at 100,000 × g for 30 min. Immunoisolation and analysis were performed on the supernatants as described for the tethering assay.
ER to Golgi transport assay
Adapted from the original semi-intact CHO cell assay [76] that we conduct using VSV-G ts045-infected NRK cells (29, 77). Briefly, NRK cells were electroporated with p115 siRNA (Ambion, Silencer Select; 5′-CUAUUGACGCAACAGGUAAtt-3′) twice over five days prior to assays. This procedure reduced the cell-associated p115 “background” activity so that transport was dependent upon added soluble p115 activity. NRK cells were then infected with VSV-G, shifted to 40 °C and pulse-radiolabeled with 35S-labeled amino acids. The cells were permeabilized by scraping with a rubber policeman and washed to remove fully soluble proteins. These semi-intact cells were then reconstituted with rat liver cytosol, various other soluble proteins, and an ATP-regenerating system, and incubated at 32 °C to allow ER to Golgi transport to occur. Incubated semi-intact cells were dissolved in detergent and proteins were digested with or without endoglycosidase H. The endoglycosidase H-resistant VSV-G in each sample was determined following gel electrophoresis and autoradiography. Percent endoglycosidase H resistance of VSV-G, the measure of ER to Golgi transport, was defined as the intensity of the endoglycosidase H resistant band divided by the sum of the resistant and sensitive bands × 100%.
Antibody production and purification
Hexahistidine-tagged rat p115 construct was expressed in E. coli with pET-21b vector. Cultures were grown at 37°C to an A600 of 0.5, before induction with 0.5 mM isopropyl-1-thio-β-D-galactopyranoside at the same temperature for 3 hours. Harvested cells were subjected to two rounds of French Press and centrifuged at 20,000 g for 20 minutes. The supernatant was then collected and centrifuged again at 100,000 g for 45 minutes. Pellets from the second centrifigation were collected and dissolved in sample buffer and loaded onto SDS-PAGE gels. Gels were stained with 0.1% Commassie in water, and the resolved p115 bands were excised from the gel and subjected to electroelution in 25 mM Tris, 191 M glycine, 0.1% SDS, and 1 mM DTT. The eluted protein solution was concentrated and injected subcutaneously into a rabbit using Freund’s adjuvant. Collected sera were supplemented with equal volume of 10 mM Tris, pH 7.5, filtered with syringe filter, and passed through a CNBr-Sepharose column conjugated with GST as nonspecific control. The flowthrough was then loaded onto another CNBr-Sepharose column conjugated with rat p115. After sample loading, columns were washed with 3 × 5ml of 10 mM Tris, pH 7.5, then washed with the same buffer containing 0.5M NaCl, once again with 10 mM Tris, pH 7.5, and finally eluted with 0.1M glycine, pH 2.5. Fractions were neutralized with 2M Tris, pH 8.0, and quantitated at A280. Peak fractions were pooled and dialyzed into 25/125 buffer.
P115 depletion and overexpression in HeLa cells
For the overexpression of p115, HeLa cells were transfected with full-length myc-tagged p115. For the depletion of p115 and subsequent rescue, Hela cells were transfected with anti-human p115 siRNA for 3 days and then transfected with myc-tagged rat full-length p115. SiRNA and construct design, transfection, and immunofluorescence microscopy were all performed as previously described (50).
Synopsis.
Binding studies employing purified proteins reveal that tethering factor p115 selectively binds monomeric SNAREs, and is excluded from the completed 4-helix bundle SNARE complex. Excess p115 inhibits p115 function in trafficking, supporting a model in which p115 facilitates the assembly of monomeric SNAREs by maintenance of binding activity and/or concentration at focal pre-fusion sites, but is itself ejected from the complex concurrently with SNAREpin formation.
Acknowledgments
This work was supported by NIH grants GM59378 and GM106323 (to JCH) and by the University of Montana Center for Structural and Functional Neuroscience.
References
- 1.Bannykh SI, Rowe T, Balch WE. The organization of endoplasmic reticulum export complexes. J Cell Biol. 1996;135(1):19–35. doi: 10.1083/jcb.135.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scales SJ, Pepperkok R, Kreis TE. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 1997;90(6):1137–1148. doi: 10.1016/s0092-8674(00)80379-7. [DOI] [PubMed] [Google Scholar]
- 3.Ungar D, Hughson FM. SNARE protein structure and function. Annu Rev Cell Dev Biol. 2003;19:493–517. doi: 10.1146/annurev.cellbio.19.110701.155609. [DOI] [PubMed] [Google Scholar]
- 4.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92(6):759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 5.Waters MG, Pfeffer SR. Membrane tethering in intracellular transport. Curr Opin Cell Biol. 1999;11(4):453–459. doi: 10.1016/s0955-0674(99)80065-9. [DOI] [PubMed] [Google Scholar]
- 6.Linstedt AD, Hauri HP. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol Biol Cell. 1993;4(7):679–693. doi: 10.1091/mbc.4.7.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mu FT, Callaghan JM, Steele-Mortimer O, Stenmark H, Parton RG, Campbell PL, McCluskey J, Yeo JP, Tock EP, Toh BH. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine “fingers” and contains a calmodulin-binding IQ motif. J Biol Chem. 1995;270(22):13503–13511. doi: 10.1074/jbc.270.22.13503. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131(6 Pt 2):1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.TerBush DR, Maurice T, Roth D, Novick P. The Exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBO J. 1996;15(23):6483–6494. [PMC free article] [PubMed] [Google Scholar]
- 10.Sacher M, Jiang Y, Barrowman J, Scarpa A, Burston J, Zhang L, Schieltz D, Yates JR, 3rd, Abeliovich H, Ferro-Novick S. TRAPP, a highly conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion. EMBO J. 1998;17(9):2494–2503. doi: 10.1093/emboj/17.9.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conibear E, Stevens TH. Vps52p, Vps53p, and Vps54p form a novel multisubunit complex required for protein sorting at the yeast late Golgi. Mol Biol Cell. 2000;11(1):305–323. doi: 10.1091/mbc.11.1.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seals DF, Eitzen G, Margolis N, Wickner WT, Price A. A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc Natl Acad Sci U S A. 2000;97(17):9402–9407. doi: 10.1073/pnas.97.17.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ungar D, Oka T, Brittle EE, Vasile E, Lupashin VV, Chatterton JE, Heuser JE, Krieger M, Waters MG. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J Cell Biol. 2002;157(3):405–415. doi: 10.1083/jcb.200202016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kraynack BA, Chan A, Rosenthal E, Essid M, Umansky B, Waters MG, Schmitt HD. Dsl1p, Tip20p, and the novel Dsl3(Sec39) protein are required for the stability of the Q/t-SNARE complex at the endoplasmic reticulum in yeast. Mol Biol Cell. 2005;16(9):3963–3977. doi: 10.1091/mbc.E05-01-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barr FA, Puype M, Vandekerckhove J, Warren G. GRASP65, a protein involved in the stacking of Golgi cisternae. Cell. 1997;91(2):253–262. doi: 10.1016/s0092-8674(00)80407-9. [DOI] [PubMed] [Google Scholar]
- 16.Nelson DS, Alvarez C, Gao YS, Garcia-Mata R, Fialkowski E, Sztul E. The membrane transport factor TAP/p115 cycles between the Golgi and earlier secretory compartments and contains distinct domains required for its localization and function. J Cell Biol. 1998;143(2):319–331. doi: 10.1083/jcb.143.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shorter J, Warren G. A role for the vesicle tethering protein, p115, in the post-mitotic stacking of reassembling Golgi cisternae in a cell-free system. J Cell Biol. 1999;146(1):57–70. doi: 10.1083/jcb.146.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zolov SN, Lupashin VV. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J Cell Biol. 2005;168(5):747–759. doi: 10.1083/jcb.200412003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orci L, Perrelet A, Rothman JE. Vesicles on strings: morphological evidence for processive transport within the Golgi stack. Proc Natl Acad Sci U S A. 1998;95(5):2279–2283. doi: 10.1073/pnas.95.5.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walter DM, Paul KS, Waters MG. Purification and characterization of a novel 13 S hetero-oligomeric protein complex that stimulates in vitro Golgi transport. J Biol Chem. 1998;273(45):29565–29576. doi: 10.1074/jbc.273.45.29565. [DOI] [PubMed] [Google Scholar]
- 21.Ren Y, Yip CK, Tripathi A, Huie D, Jeffrey PD, Walz T, Hughson FM. A structure-based mechanism for vesicle capture by the multisubunit tethering complex Dsl1. Cell. 2009;139(6):1119–1129. doi: 10.1016/j.cell.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lupashin V, Sztul E. Golgi tethering factors. Biochim Biophys Acta. 2005;1744(3):325–339. doi: 10.1016/j.bbamcr.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 23.Waters MG, Clary DO, Rothman JE. A novel 115-kD peripheral membrane protein is required for intercisternal transport in the Golgi stack. J Cell Biol. 1992;118(5):1015–1026. doi: 10.1083/jcb.118.5.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakajima H, Hirata A, Ogawa Y, Yonehara T, Yoda K, Yamasaki M. A cytoskeleton-related gene, uso1, is required for intracellular protein transport in Saccharomyces cerevisiae. J Cell Biol. 1991;113(2):245–260. doi: 10.1083/jcb.113.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barroso M, Nelson DS, Sztul E. Transcytosis-associated protein (TAP)/p115 is a general fusion factor required for binding of vesicles to acceptor membranes. Proc Natl Acad Sci U S A. 1995;92(2):527–531. doi: 10.1073/pnas.92.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sapperstein SK, Lupashin VV, Schmitt HD, Waters MG. Assembly of the ER to Golgi SNARE complex requires Uso1p. J Cell Biol. 1996;132(5):755–767. doi: 10.1083/jcb.132.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barlowe C. Coupled ER to Golgi transport reconstituted with purified cytosolic proteins. J Cell Biol. 1997;139(5):1097–1108. doi: 10.1083/jcb.139.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cao X, Ballew N, Barlowe C. Initial docking of ER-derived vesicles requires Uso1p and Ypt1p but is independent of SNARE proteins. EMBO J. 1998;17(8):2156–2165. doi: 10.1093/emboj/17.8.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarez C, Fujita H, Hubbard A, Sztul E. ER to Golgi transport: Requirement for p115 at a pre-Golgi VTC stage. J Cell Biol. 1999;147(6):1205–1222. doi: 10.1083/jcb.147.6.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allan BB, Moyer BD, Balch WE. Rab1 recruitment of p115 into a cis-SNARE complex: programming budding COPII vesicles for fusion. Science. 2000;289(5478):444–448. doi: 10.1126/science.289.5478.444. [DOI] [PubMed] [Google Scholar]
- 31.Brandon E, Szul T, Alvarez C, Grabski R, Benjamin R, Kawai R, Sztul E. On and off membrane dynamics of the endoplasmic reticulum-golgi tethering factor p115 in vivo. Mol Biol Cell. 2006;17(7):2996–3008. doi: 10.1091/mbc.E05-09-0862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lupashin VV, Hamamoto S, Schekman RW. Biochemical requirements for the targeting and fusion of ER-derived transport vesicles with purified yeast Golgi membranes. J Cell Biol. 1996;132(3):277–289. doi: 10.1083/jcb.132.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gmachl MJ, Wimmer C. Sequential involvement of p115, SNAREs, and Rab proteins in intra-Golgi protein transport. J Biol Chem. 2001;276(21):18178–18184. doi: 10.1074/jbc.M101513200. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura N, Lowe M, Levine TP, Rabouille C, Warren G. The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell. 1997;89(3):445–455. doi: 10.1016/s0092-8674(00)80225-1. [DOI] [PubMed] [Google Scholar]
- 35.Sohda M, Misumi Y, Yano A, Takami N, Ikehara Y. Phosphorylation of the vesicle docking protein p115 regulates its association with the Golgi membrane. J Biol Chem. 1998;273(9):5385–5388. doi: 10.1074/jbc.273.9.5385. [DOI] [PubMed] [Google Scholar]
- 36.Dirac-Svejstrup AB, Shorter J, Waters MG, Warren G. Phosphorylation of the vesicle-tethering protein p115 by a casein kinase II-like enzyme is required for Golgi reassembly from isolated mitotic fragments. J Cell Biol. 2000;150(3):475–488. doi: 10.1083/jcb.150.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sacher M, Barrowman J, Wang W, Horecka J, Zhang Y, Pypaert M, Ferro-Novick S. TRAPP I implicated in the specificity of tethering in ER-to-Golgi transport. Mol Cell. 2001;7(2):433–442. doi: 10.1016/s1097-2765(01)00190-3. [DOI] [PubMed] [Google Scholar]
- 38.Wuestehube LJ, Duden R, Eun A, Hamamoto S, Korn P, Ram R, Schekman R. New mutants of Saccharomyces cerevisiae affected in the transport of proteins from the endoplasmic reticulum to the Golgi complex. Genetics. 1996;142(2):393–406. doi: 10.1093/genetics/142.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.VanRheenen SM, Cao X, Lupashin VV, Barlowe C, Waters MG. Sec35p, a novel peripheral membrane protein, is required for ER to Golgi vesicle docking. J Cell Biol. 1998;141(5):1107–1119. doi: 10.1083/jcb.141.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sapperstein SK, Walter DM, Grosvenor AR, Heuser JE, Waters MG. p115 is a general vesicular transport factor related to the yeast endoplasmic reticulum to Golgi transport factor Uso1p. Proc Natl Acad Sci U S A. 1995;92(2):522–526. doi: 10.1073/pnas.92.2.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamakawa H, Seog DH, Yoda K, Yamasaki M, Wakabayashi T. Uso1 protein is a dimer with two globular heads and a long coiled-coil tail. Journal of Structural Biology. 1996;116(3):356–365. doi: 10.1006/jsbi.1996.0053. [DOI] [PubMed] [Google Scholar]
- 42.An Y, Chen CY, Moyer B, Rotkiewicz P, Elsliger MA, Godzik A, Wilson IA, Balch WE. Structural and functional analysis of the globular head domain of p115 provides insight into membrane tethering. J Mol Biol. 2009;391(1):26–41. doi: 10.1016/j.jmb.2009.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Striegl H, Roske Y, Kummel D, Heinemann U. Unusual armadillo fold in the human general vesicular transport factor p115. PLoS One. 2009;4(2):e4656. doi: 10.1371/journal.pone.0004656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beard M, Satoh A, Shorter J, Warren G. A cryptic Rab1-binding site in the p115 tethering protein. J Biol Chem. 2005;280(27):25840–25848. doi: 10.1074/jbc.M503925200. [DOI] [PubMed] [Google Scholar]
- 45.Sonnichsen B, Lowe M, Levine T, Jamsa E, Dirac-Svejstrup B, Warren G. A role for giantin in docking COPI vesicles to Golgi membranes. J Cell Biol. 1998;140(5):1013–1021. doi: 10.1083/jcb.140.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lesa GM, Seemann J, Shorter J, Vandekerckhove J, Warren G. The amino-terminal domain of the golgi protein giantin interacts directly with the vesicle-tethering protein p115. J Biol Chem. 2000;275(4):2831–2836. doi: 10.1074/jbc.275.4.2831. [DOI] [PubMed] [Google Scholar]
- 47.Linstedt AD, Jesch SA, Mehta A, Lee TH, Garcia-Mata R, Nelson DS, Sztul E. Binding relationships of membrane tethering components. The giantin N terminus and the GM130 N terminus compete for binding to the p115 C terminus. J Biol Chem. 2000;275(14):10196–10201. doi: 10.1074/jbc.275.14.10196. [DOI] [PubMed] [Google Scholar]
- 48.Puthenveedu MA, Linstedt AD. Evidence that Golgi structure depends on a p115 activity that is independent of the vesicle tether components giantin and GM130. J Cell Biol. 2001;155(2):227–238. doi: 10.1083/jcb.200105005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Satoh A, Warren G. In situ cleavage of the acidic domain from the p115 tether inhibits exocytic transport. Traffic. 2008;9(9):1522–1529. doi: 10.1111/j.1600-0854.2008.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grabski R, Balklava Z, Wyrozumska P, Szul T, Brandon E, Alvarez C, Holloway ZG, Sztul E. Identification of a functional domain within the p115 tethering factor that is required for Golgi ribbon assembly and membrane trafficking. J Cell Sci. 2012;125(Pt 8):1896–1909. doi: 10.1242/jcs.090571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kondylis V, Rabouille C. A novel role for dp115 in the organization of tER sites in Drosophila. J Cell Biol. 2003;162(2):185–198. doi: 10.1083/jcb.200301136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puthenveedu MA, Linstedt AD. Gene replacement reveals that p115/SNARE interactions are essential for Golgi biogenesis. Proc Natl Acad Sci U S A. 2004;101(5):1253–1256. doi: 10.1073/pnas.0306373101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levine TP, Rabouille C, Kieckbusch RH, Warren G. Binding of the vesicle docking protein p115 to Golgi membranes is inhibited under mitotic conditions. J Biol Chem. 1996;271(29):17304–17311. doi: 10.1074/jbc.271.29.17304. [DOI] [PubMed] [Google Scholar]
- 54.Diao A, Frost L, Morohashi Y, Lowe M. Coordination of golgin tethering and SNARE assembly: GM130 binds syntaxin 5 in a p115-regulated manner. J Biol Chem. 2008;283(11):6957–6967. doi: 10.1074/jbc.M708401200. [DOI] [PubMed] [Google Scholar]
- 55.Bentley M, Liang Y, Mullen K, Xu D, Sztul E, Hay JC. SNARE status regulates tether recruitment and function in homotypic COPII vesicle fusion. J Biol Chem. 2006;281(50):38825–38833. doi: 10.1074/jbc.M606044200. [DOI] [PubMed] [Google Scholar]
- 56.Shorter J, Beard MB, Seemann J, Dirac-Svejstrup AB, Warren G. Sequential tethering of Golgins and catalysis of SNAREpin assembly by the vesicle-tethering protein p115. J Cell Biol. 2002;157(1):45–62. doi: 10.1083/jcb.200112127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu D, Joglekar AP, Williams AL, Hay JC. Subunit structure of a mammalian ER/Golgi SNARE complex. J Biol Chem. 2000;275(50):39631–39639. doi: 10.1074/jbc.M007684200. [DOI] [PubMed] [Google Scholar]
- 58.Conibear E, Cleck JN, Stevens TH. Vps51p mediates the association of the GARP (Vps52/53/54) complex with the late Golgi t-SNARE Tlg1p. Mol Biol Cell. 2003;14(4):1610–1623. doi: 10.1091/mbc.E02-10-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siniossoglou S, Pelham HR. An effector of Ypt6p binds the SNARE Tlg1p and mediates selective fusion of vesicles with late Golgi membranes. EMBO J. 2001;20(21):5991–5998. doi: 10.1093/emboj/20.21.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stroupe C, Collins KM, Fratti RA, Wickner W. Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J. 2006;25(8):1579–1589. doi: 10.1038/sj.emboj.7601051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laage R, Ungermann C. The N-terminal domain of the t-SNARE Vam3p coordinates priming and docking in yeast vacuole fusion. Mol Biol Cell. 2001;12(11):3375–3385. doi: 10.1091/mbc.12.11.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shestakova A, Suvorova E, Pavliv O, Khaidakova G, Lupashin V. Interaction of the conserved oligomeric Golgi complex with t-SNARE Syntaxin5a/Sed5 enhances intra-Golgi SNARE complex stability. J Cell Biol. 2007;179(6):1179–1192. doi: 10.1083/jcb.200705145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weimbs T, Low SH, Chapin SJ, Mostov KE, Bucher P, Hofmann K. A conserved domain is present in different families of vesicular fusion proteins: a new superfamily. Proc Natl Acad Sci U S A. 1997;94(7):3046–3051. doi: 10.1073/pnas.94.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gonzalez LC, Jr, Weis WI, Scheller RH. A novel snare N-terminal domain revealed by the crystal structure of Sec22b. J Biol Chem. 2001;276(26):24203–24211. doi: 10.1074/jbc.M101584200. [DOI] [PubMed] [Google Scholar]
- 65.Tochio H, Tsui MM, Banfield DK, Zhang M. An autoinhibitory mechanism for nonsyntaxin SNARE proteins revealed by the structure of Ykt6p. Science. 2001;293(5530):698–702. doi: 10.1126/science.1062950. [DOI] [PubMed] [Google Scholar]
- 66.Martinez-Arca S, Alberts P, Zahraoui A, Louvard D, Galli T. Role of tetanus neurotoxin insensitive vesicle-associated membrane protein (TI-VAMP) in vesicular transport mediating neurite outgrowth. J Cell Biol. 2000;149(4):889–900. doi: 10.1083/jcb.149.4.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martinez-Arca S, Rudge R, Vacca M, Raposo G, Camonis J, Proux-Gillardeaux V, Daviet L, Formstecher E, Hamburger A, Filippini F, D’Esposito M, Galli T. A dual mechanism controlling the localization and function of exocytic v-SNAREs. Proc Natl Acad Sci U S A. 2003;100(15):9011–9016. doi: 10.1073/pnas.1431910100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hasegawa H, Zinsser S, Rhee Y, Vik-Mo EO, Davanger S, Hay JC. Mammalian ykt6 is a neuronal SNARE targeted to a specialized compartment by its profilin-like amino terminal domain. Mol Biol Cell. 2003;14(2):698–720. doi: 10.1091/mbc.E02-09-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Antonin W, Dulubova I, Arac D, Pabst S, Plitzner J, Rizo J, Jahn R. The N-terminal domains of syntaxin 7 and vti1b form three-helix bundles that differ in their ability to regulate SNARE complex assembly. J Biol Chem. 2002;277(39):36449–36456. doi: 10.1074/jbc.M204369200. [DOI] [PubMed] [Google Scholar]
- 70.Mossessova E, Bickford LC, Goldberg J. SNARE selectivity of the COPII coat. Cell. 2003;114(4):483–495. doi: 10.1016/s0092-8674(03)00608-1. [DOI] [PubMed] [Google Scholar]
- 71.Bentley M, Nycz DC, Joglekar A, Fertschai I, Malli R, Graier WF, Hay JC. Vesicular calcium regulates coat retention, fusogenicity, and size of pre-Golgi intermediates. Mol Biol Cell. 2010;21(6):1033–1046. doi: 10.1091/mbc.E09-10-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seemann J, Jokitalo EJ, Warren G. The role of the tethering proteins p115 and GM130 in transport through the Golgi apparatus in vivo. Mol Biol Cell. 2000;11(2):635–645. doi: 10.1091/mbc.11.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu D, Hay JC. Reconstitution of COPII vesicle fusion to generate a pre-Golgi intermediate compartment. J Cell Biol. 2004;167(6):997–1003. doi: 10.1083/jcb.200408135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zagouras P, Rose JK. Dynamic equilibrium between vesicular stomatitis virus glycoprotein monomers and trimers in the Golgi and at the cell surface. J Virol. 1993;67(12):7533–7538. doi: 10.1128/jvi.67.12.7533-7538.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moyer BD, Allan BB, Balch WE. Rab1 interaction with a GM130 effector complex regulates COPII vesicle cis--Golgi tethering. Traffic. 2001;2(4):268–276. doi: 10.1034/j.1600-0854.2001.1o007.x. [DOI] [PubMed] [Google Scholar]
- 76.Schwaninger R, Beckers CJ, Balch WE. Sequential transport of protein between the endoplasmic reticulum and successive Golgi compartments in semi-intact cells. J Biol Chem. 1991;266(20):13055–13063. [PubMed] [Google Scholar]
- 77.Williams AL, Ehm S, Jacobson NC, Xu D, Hay JC. rsly1 binding to syntaxin 5 is required for endoplasmic reticulum-to-Golgi transport but does not promote SNARE motif accessibility. Mol Biol Cell. 2004;15(1):162–175. doi: 10.1091/mbc.E03-07-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]