

Abstract
HIV-associated neurocognitive disorders (HAND) afflict approximately half of HIVinfected patients. HIV-infected cells within the CNS release neurotoxic viral proteins such as the transactivator of transcription (Tat). Tat caused a biphasic change in NMDAR function; NMDA-evoked increases in intracellular Ca2+ were initially potentiated following 16 h exposure to Tat and then adapted by gradually returning to baseline by 24 h. Following Tat-induced NMDAR potentiation, a RhoA/Rho-associated protein kinase (ROCK) signaling pathway was activated; a subsequent remodeling of the actin cytoskeleton reduced NMDA-evoked increases in intracellular Ca2+. Pharmacologic or genetic inhibition of RhoA or ROCK failed to affect potentiation but prevented adaptation of NMDAR function. Activation of RhoA/ROCK signaling increases the formation of filamentous actin. Drugs that prevent changes to filamentous actin blocked adaptation of NMDAR function following Tat-induced potentiation, while stimulating either depolymerization or polymerization of actin attenuated NMDAR function. These findings indicate that Tat activates a RhoA/ROCK signaling pathway resulting in actin remodeling and subsequent reduction of NMDAR function. Adaptation of NMDAR function may be a mechanism to protect neurons from excessive Ca2+ influx and could reveal targets for the treatment of HAND.
Introduction
Approximately half of HIV-infected patients in the U.S. are affected by HIVassociated neurocognitive disorders (HAND) (Tozzi et al. 2005, Cysique et al. 2004, Heaton et al. 2011) and the prevalence is likely higher in areas of the world where effective pharmacotherapy is uncommon. HAND ranges in severity from asymptomatic to severely debilitating (Heaton et al. 2004, Antinori et al. 2007). Multiple reports during the past decade indicate that cognitive dysfunction remains a significant and persistent problem despite effective management of viral load with combination anti-retroviral therapy (cART) (Heaton et al. 2010, Antinori et al. 2007, Heaton et al. 2011). Current cART regimens prolong the lifespan of patients by effectively managing viral load; unfortunately, HAND remains common among HIV-infected patients (Heaton et al. 2010) and the therapeutic approaches to combat HIV-associated cognitive impairment remain ineffective (Uthman & Abdulmalik 2008).
Penetrance of HIV into the CNS occurs as early as 2 weeks post infection (Resnick et al. 1988, Davis et al. 1992, An et al. 1999). Once inside the CNS, HIV infects microglial cells and perivascular macrophages (Albright et al. 1999, Williams et al. 2001), which serve as reservoirs for viral replication. HAND is thought to arise, in part, from latent viral reservoirs that are not entirely accessible to cART. Because HIV does not infect neurons, neuronal injury leading to cognitive impairment results from indirect mechanisms. Namely, HIV-infected cells within the CNS release agents that are neurotoxic including inflammatory cytokines (Genis et al. 1992), nitric oxide (Eugenin et al. 2007), and glutamate (Jiang et al. 2001), as well as the viral proteins, gp120 (Schneider et al. 1986) and the transactivator of transcription (Tat) (Chang et al. 1997). While HIV-induced neuropathogenesis likely involves a combination of these agents to create a neurotoxic environment, Tat is particularly important because current antiretroviral therapy is unable to halt the production of this potent neurotoxin once the viral genome integrates into cellular DNA (Li et al. 2009).
Tat is a non-structural viral protein that is responsible for initiating transcription of the HIV genome (Sodroski et al. 1985b, Sodroski et al. 1985a). In addition to its role as a transcriptional transactivator, Tat is released from HIV-infected cells (Chang et al. 1997) and is detected in the CNS and sera of HIV-infected patients (Hudson et al. 2000, Del Valle et al. 2000). Anti-Tat antibody titers in the cerebrospinal fluid of HIV+ patients are approximately 30% lower in cognitively impaired patients relative to unimpaired patients, suggesting that antibody responses against Tat may be neuroprotective and preserve cognitive function (Bachani et al. 2013). Cognitive decline in HAND correlates with damage to synaptodendritic structures, including axonal disruptions and pruning of dendritic spines (Ellis et al. 2007). In a transgenic mouse model, induction of Tat expression results in a loss of dendritic spines resulting in learning and memory impairment (Fitting et al. 2010, Fitting et al. 2013, Carey et al. 2012). Similarly, exposure of primary hippocampal neurons to Tat in vitro causes loss of excitatory synapses (Kim et al. 2008) and presynaptic terminals (Shin & Thayer 2013), and simultaneous gain of inhibitory synapses (Hargus & Thayer 2013). Importantly, almost all instances of Tatinduced neurotoxicity are prevented by pharmacologic inhibition of the NMDA receptor (NMDAR) (Eugenin et al. 2007, Kim et al. 2008, Shin et al. 2012, Hargus & Thayer 2013) indicating that Tat-induced neurotoxicity requires NMDAR-mediated Ca2+ influx.
Tat potentiates NMDA-evoked responses (Haughey et al. 2001, Chandra et al. 2005) which then adapt by gradually returning to and eventually dropping below basal responses (Krogh et al. 2014). Reduction of NMDAR function and a decrease in the ratio of excitatory-to-inhibitory synapses may be neuroprotective responses orchestrated by the cell to prevent excessive Ca2+ influx and excitotoxicity. Thus, these adaptive changes may improve neuronal survival at the cost of impaired network function. The signaling pathway responsible for adaptation of NMDA-evoked [Ca2+]i responses following prolonged exposure to Tat is therefore the focus of this study.
Here we examined NMDAR function during 24 h exposure to Tat. Previously, we found that Tat evoked a biphasic change in NMDAR function (Krogh et al. 2014). As illustrated in figure 1, Tat potentiated NMDA-evoked increases in intracellular Ca2+ concentration ([Ca2+]i) via the low-density lipoprotein receptor-related protein (LRP) and activation of Src tyrosine kinase. Subsequently, NMDA-evoked responses adapted by gradually returning to basal levels after 24 h exposure to Tat and eventually dropping below baseline responses by 48 h. Adaptation resulted from activation of a nitric oxide synthase (NOS), soluble guanylate cyclase (sGC), cGMP-dependent protein kinase (PKG) signaling pathway (Krogh et al. 2014). However, effectors downstream of PKG responsible for attenuating the NMDA-evoked response have not been identified. Here we show that Tat activates a signaling pathway that includes the small GTPase RhoA and Rho-associated protein kinase (ROCK). Activation of RhoA/ROCK results in reorganization of the actin cytoskeleton leading to attenuated NMDA-evoked increases in [Ca2+]i. Abnormal activation of RhoA/ROCK has been observed in various models of CNS disorders including Alzheimer's disease, stroke, and multiple sclerosis (Mueller et al. 2005). Thus, RhoA/ROCK are promising drug targets for the treatment of various neurological conditions; however, little is known about this signaling pathway in HAND. This study suggests that following Tat-induced potentiation, NMDA-evoked responses are reduced following RhoA/ROCK-mediated reorganization of the actin cytoskeleton.
Fig. 1. HIV-1 Tat causes a biphasic change in NMDA-evoked increases in [Ca2+]i.
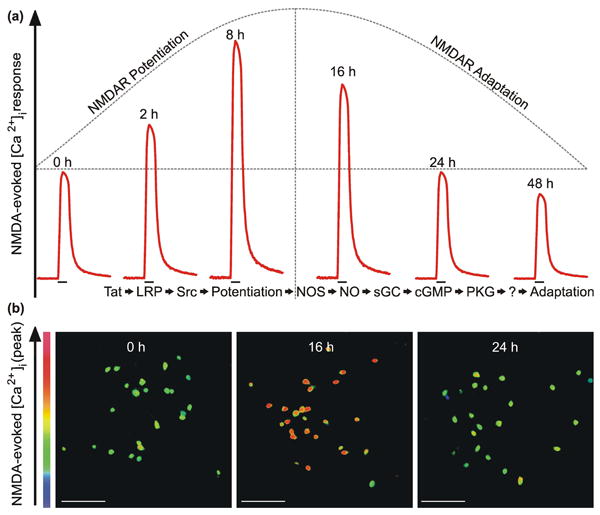
a, Stylized traces (
 ) depict time-dependent, biphasic changes in NMDA-evoked increases in [Ca2+]i following exposure to HIV-1 Tat for the time indicated above the trace. The previously reported (Krogh et al. 2014) sequence of events listed below the traces indicates that Tat activation of an LRP/Src kinase pathway resulted in NMDAR potentiation. Subsequently, NMDARs adapted following activation of the NOS/sGC/PKG signaling pathway. Identifying downstream effector(s) of PKG is the goal of this study. b, Representative pseudocolor images (scale bar = 100 μm) show peak response to NMDA (10μM, 30 s) in primary hippocampal neurons treated with Tat for the time indicated in the image panel. Images were scaled from 0 to 700 nM as indicated by the color bar on the y-axis.
) depict time-dependent, biphasic changes in NMDA-evoked increases in [Ca2+]i following exposure to HIV-1 Tat for the time indicated above the trace. The previously reported (Krogh et al. 2014) sequence of events listed below the traces indicates that Tat activation of an LRP/Src kinase pathway resulted in NMDAR potentiation. Subsequently, NMDARs adapted following activation of the NOS/sGC/PKG signaling pathway. Identifying downstream effector(s) of PKG is the goal of this study. b, Representative pseudocolor images (scale bar = 100 μm) show peak response to NMDA (10μM, 30 s) in primary hippocampal neurons treated with Tat for the time indicated in the image panel. Images were scaled from 0 to 700 nM as indicated by the color bar on the y-axis.
Materials and Experimental Methods
Drugs and Reagents
Materials were obtained from the following sources: HIV-1 Tat (Clade B, 1-86 amino acids) was acquired from Prospec Tany TechnoGene Ltd. (Rehovot, Israel); Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum, horse serum, fura-2-acetomethyl ester (Fura-2-AM), and glycine were obtained from Invitrogen (Carlsbad, CA). Phalloidin-Oleate, Y27632 (Trans-4-[(1R)-1-Aminoethyl]-N-4pyridinylcyclohexanecarboxamide dihydrochloride), and Cytochalasin D (zygosporium mansonii) were acquired from EMD Millipore (Billerica, MA); Rho Inhibitor I (exoenzyme C3 transferase) was obtained from Cytoskeleton, Inc. (Denver, CO); H1152 ((S)-(+)-2-Methyl-1-[(4-methyl-5-isoquinolinyl)sulfonyl]-hexahydro-1H-1,4-diazepine dihydrochloride), latrunculin A (4-[(1R,4Z,8E,10Z,12S,15R,17R)-17-Hydroxy-5,12-dimethyl-3-oxo-2,16-dioxabicyclo[13.3.1]nonadeca-4,8,10-trien-17-yl)-2-thiazolidinone), and jasplakinolide (Cyclo[(3R)-3-(4-hydroxyphenyl)-β-alanyl-(2S,4E,6R,8S)-8-hydroxy-2,4,6-trimethyl-4-nonenoyl-L-alanyl-2-bromo-N-methyl-D-tryptophyl]) were acquired from Tocris (Bristol, UK); ML-7 (1-(5-Iodonaphthalene-1-sulfonyl)-1H-hexahydro-1,4-diazepine hydrochloride) and NMDA were obtained from Sigma-Aldrich (St. Louis, MO).
DNA Constructs
Dominant Negative (DN)-RhoA (plasmid 15901) and constitutively active (CA)-RhoA (plasmid 15900) (Nobes & Hall 1999) were obtained from Addgene (Cambridge, MA). pTagRFP-N was acquired from Evrogen (Moscow, Russia). pEGFP-Actin was obtained from Clontech (Mountain View, CA).
Cell Culture
In accordance with the University of Minnesota's Institutional Animal Care and Use Committee and the NIH guide for the care and use of laboratory animals, maternal rats (Harlan Sprague Dawley) were euthanized by CO2 inhalation and fetuses were removed on embryonic day 17. Rat hippocampal neurons were grown in primary culture as described previously (Waataja et al. 2008) Hippocampi were dissected and placed in Ca2+- and Mg2+-free HEPES-buffered Hanks' salt solution (HHSS), pH 7.45. HHSS contained the following (in mM): HEPES 20, NaCl 137, CaCl2 1.3, MgSO4 0.4, MgCl2 0.5, KCl 5.0, KH2PO4 0.4, Na2HPO4 0.6, NaHCO3 3.0, and glucose 5.6. Cells were dissociated by triturating through a 5 mL pipette and a flame-narrowed Pasteur pipette and then re-suspended in DMEM without glutamine, supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/mL and 100 mg/mL, respectively). Dissociated cells were then plated at a density of 50,000 to 60,000 cells/dish onto a 25-mm-round cover glass (#1) pre-coated with matrigel (150 μL, 0.2 mg/mL). Neurons were grown in a humidified atmosphere of 10% CO2 (pH 7.4) at 37°C, and fed on days 1 and 7 by exchange of 75% of the media with DMEM supplemented with 10% horse serum and penicillin/streptomycin. Cells used in these experiments were cultured without mitotic inhibitors resulting in a mixed glial-neuronal culture consisting of 18 ± 2% neurons, 70 ± 3% astrocytes, and 9 ± 3% microglia as indicated by immunocytochemistry (Kim et al. 2011). The cultures used for experiments were 12 to 15 days in vitro (DIV).
[Ca2+]i imaging
Intracellular Ca2+ concentration ([Ca2+]i) was recorded as previously described (Li et al. 2013) with minor modifications. Cells were loaded with indicator by incubation with 5 μM fura-2 AM in 0.04% pluronic acid in HHSS for 30 min at 37°C followed by washing in the absence of indicator for 10 min. HIV-1 Tat and respective drugs were present during fura-2-AM loading, but were absent during the wash. Coverslips containing fura-2-AM loaded cells were transferred to a recording chamber, placed on the stage of an Olympus IX71 microscope (Melville, NY), and viewed through a 20X objective. Excitation wavelength was selected with a galvanometer-driven monochromator (8-nm slit width) coupled to a 75-W xenon arc lamp (Optoscan; Cairn Research). [Ca2+]i was monitored using sequential excitation of fura-2 at 340 and 380 nm; image pairs were collected every 1 s. All experimental recordings were conducted at room temperature (21°C). For experimental recordings, cells were superfused at a rate of 1-2 mL/min with HHSS for 1 min followed by 30 s perfusion of Mg2+-Free HHSS that contained 200 μM glycine and 10 μM NMDA. Fluorescence images (510/40 nm) were projected onto a cooled charge-coupled device camera (Cascade 512B; Roper Scientific) controlled by MetaFluor software (Molecular Devices). After background subtraction, the 340- and 380-nm image pairs were converted to [Ca2+]i using the formula [Ca2+]i = Kdβ(R − Rmin)/(Rmax − R) (Grynkiewicz et al. 1985). The dissociation constant (Kd) for fura-2 is 145 nM and R is the 340 nm / 380 nm fluorescence intensity ratio. Rmin, Rmax, and β were determined in a series of calibration experiments on intact cells. Rmin and Rmax values were generated by applying 10 μM ionomycin in Ca2+-free HHSS supplemented with 1 mM EGTA and saturating (5 mM) Ca2+, respectively. β is the ratio of fluorescence intensity acquired with 380 nm excitation measured in Ca2+-free buffer and buffer containing saturating (5 mM) Ca2+. Values for Rmin, Rmax, and β were 0.37, 9.38, and 6.46, respectively. These calibration constants were applied to all experimental recordings. For time course experiments, coverslips from the same cell culture plating were treated in parallel and each coverslip imaged only once. To generate pseudocolor images, a binary mask was created by applying an intensity threshold to the 380 nm image and then applied to [Ca2+]i images with colors assigned as indicated by the calibration bars in the figures (Fig. 1 b). The neuronal cell body was selected as the region of interest for all recordings. All neuronal cells types within imaging fields were included in the analysis and no exclusions were made.
Transfection
Rat hippocampal neurons were transfected between 11-12 days in vitro using a modification of a calcium phosphate protocol described previously (Li et al. 2012). Briefly, hippocampal cultures were incubated for 30 min in DMEM supplemented with 1 mM kynurenic acid, 10 mM MgCl2, and 5 mM HEPES. A DNA/calcium phosphate precipitate containing 1 μg plasmid DNA per well was prepared, allowed to form for 30 min at 21°C then added to the culture. Following a 90 min incubation, cells were washed once with DMEM supplemented with MgCl2 and HEPES and then returned to conditioned media.
Confocal microscopy of EGFP-Actin
Glass-bottom petri dishes containing neurons transfected with EGFP-Actin and Tag-RFP were sealed with Parafilm, transferred to the stage of an inverted confocal microscope (Olympus Fluoview 300, Melville, NY) and viewed through a 60X oilimmersion objective (NA=1.4). EGFP-Actin was excited at 488 nm with an argon ion laser and emission collected at 530 nm (10 nm band pass). Tag-RFP was excited at 543 nm with a green HeNe laser and emission collected at >605 nm. 1 μm optical sections spanning 8 μm in the z-dimension were collected and these optical sections were combined through the z-axis into a compressed z-stack. To enable repeated imaging of the same cell, the location of the cell was recorded using micrometers attached to the stage of the microscope. The cell culture dish was returned to the CO2 incubator between image collections. Images were processed using MetaMorph 7.7 image processing software. Compressed z-stacks were created from the Tag-RFP and EGFP-Actin image stacks and then overlaid.
Statistical analysis
For [Ca2+]i imaging studies, an individual experiment (n=1) was defined as the change in NMDA-evoked [Ca2+]i response from a single neuron on a single coverslip. Changes in NMDA-evoked [Ca2+]i are presented as mean ± SEM. Each experiment was replicated using at least 4 separate coverslips from at least 2 separate cultures. Significant differences were determined by one-way ANOVA with Tukey's post hoc test for multiple comparisons (OriginPro v8.5)
Results
HIV Tat-induced potentiation of NMDAR function adapts via activation of RhoA
Our previous study showed that treating rat hippocampal neurons in culture with 50 ng/mL of the HIV Tat protein potentiated NMDA-evoked Ca2+ responses following 2 h exposure, which reached maximum response amplitudes by 8 h, and then NMDAR function adapted back to baseline by 24 h (Krogh et al. 2014). Adaptation of NMDAR function occurred after activation of a NOS/sGC/PKG signaling cascade, however effectors downstream of PKG had not yet been identified (Fig. 1). Because cytoskeletal changes regulate NMDAR function (Rosenmund & Westbrook 1993, Furukawa et al. 1995) and RhoA is a downstream effector of PKG that affects the actin cytoskeleton (Sauzeau et al. 2003, Rolli-Derkinderen et al. 2005, Sunico et al. 2010), we examined the possibility that adaptation of NMDARs required RhoA. We hypothesized that RhoA activation was necessary for the reduction in NMDA-evoked [Ca2+]i responses after Tat-induced potentiation. To test this hypothesis, we used pharmacological and genetic approaches. NMDAR function was assessed in rat hippocampal neurons (DIV12-15) using fura-2-based digital imaging to record [Ca2+]i increases evoked by superfusing NMDA (10 μM) and glycine (200 μM) in Mg2+-free HHSS onto the cells for 30 s. Cultures were pretreated with a cell-permeable inhibitor of RhoA, exoenzyme C3 transferase (ExoC3, 2.5 μg/mL), 1 h prior to and during exposure to Tat for 16 or 24 h. ExoC3 did not affect Tat-induced NMDAR potentiation, but did prevent adaptation of the NMDAevoked responses by 24 h (Fig. 2 a, b).
Fig. 2. Adaptation of NMDARs following Tat-induced potentiation requires RhoA.
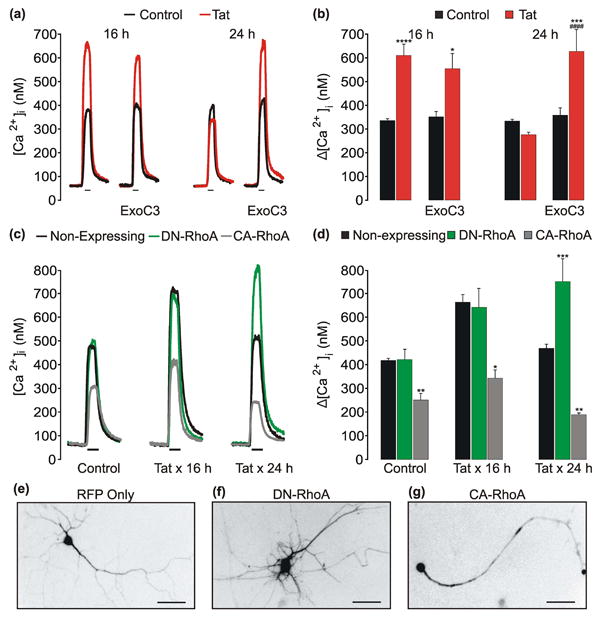
a, Representative traces show NMDA-evoked [Ca2+]i increases from control (
 ) neurons or neurons treated with 50 ng/mLTat (
) for 16 h or 24 h. Cells were pretreated with 2.5 μg/mL Exoenzyme C3 Transferase (ExoC3) 1 h prior to the addition of Tat. NMDA (10 μM, 30 s) was applied by superfusion at the times indicated by the horizontal bars. b, Bar graph shows net [Ca2+]i increase evoked by 10 μM NMDA in control (
) neurons or neurons treated with 50 ng/mLTat (
) for 16 h or 24 h. Cells were pretreated with 2.5 μg/mL Exoenzyme C3 Transferase (ExoC3) 1 h prior to the addition of Tat. NMDA (10 μM, 30 s) was applied by superfusion at the times indicated by the horizontal bars. b, Bar graph shows net [Ca2+]i increase evoked by 10 μM NMDA in control (
 ) cells or cells treated with Tat (
) cells or cells treated with Tat (
 ) for 16 h or 24 h. *p<0.05; ***p<0.001; ****p<0.0001 relative to respective control; ####p<0.0001 relative to 24 h Tat-treated neurons as determined by separate, one-way ANOVAs with 4 levels per treatment time followed by Tukey's post-test for multiple comparisons. c, representative traces show NMDA-evoked [Ca2+]i increases from non-expressing (
) neurons or neurons expressing DN-RhoA (
) for 16 h or 24 h. *p<0.05; ***p<0.001; ****p<0.0001 relative to respective control; ####p<0.0001 relative to 24 h Tat-treated neurons as determined by separate, one-way ANOVAs with 4 levels per treatment time followed by Tukey's post-test for multiple comparisons. c, representative traces show NMDA-evoked [Ca2+]i increases from non-expressing (
) neurons or neurons expressing DN-RhoA (
 ) or CA-RhoA (
) or CA-RhoA (
 ) that were left untreated (control) or treated with Tat for 16 h or 24 h. d, Bar graph shows net [Ca2+]i increase evoked by 10 μM NMDA in nonexpressing (
) neurons or neurons expressing DN-RhoA (
) that were left untreated (control) or treated with Tat for 16 h or 24 h. d, Bar graph shows net [Ca2+]i increase evoked by 10 μM NMDA in nonexpressing (
) neurons or neurons expressing DN-RhoA (
 ) or CA-RhoA (
) or CA-RhoA (
 ). Neurons were left untreated (control) or treated with Tat for 16 h or 24 h as indicated. *p<0.05; **p<0.01; ***p<0.001 relative to non-expressing neurons within respective treatment group as determined by separate, one-way ANOVAs with 3 levels per treatment time followed by Tukey's post-test for multiple comparisons. e-f, Representative images show neurons expressing RFP only (e) or RFP and DN-RhoA (f) or CA-RhoA (g) (scale bar = 50 μm). Images were inverted to enhance contrast between the neuron and background.
). Neurons were left untreated (control) or treated with Tat for 16 h or 24 h as indicated. *p<0.05; **p<0.01; ***p<0.001 relative to non-expressing neurons within respective treatment group as determined by separate, one-way ANOVAs with 3 levels per treatment time followed by Tukey's post-test for multiple comparisons. e-f, Representative images show neurons expressing RFP only (e) or RFP and DN-RhoA (f) or CA-RhoA (g) (scale bar = 50 μm). Images were inverted to enhance contrast between the neuron and background.
Next, we wanted to confirm the role of RhoA in reducing NMDA-evoked [Ca2+]i responses by co-expressing plasmids encoding either dominant negative (DN)-RhoA (T19N) or constitutively active (CA)-RhoA (Q63L) with a red-fluorescent protein (RFP) and imaging these cells in parallel. Neurons expressing the constructs of interest were compared to non-expressing neurons in the same imaging field for an internal comparison. Cells were treated with Tat for 16 or 24 h, loaded with fura-2, and then imaged (Fig. 2 c, d). In non-expressing control (untreated) cells, NMDA-evoked [Ca2+]i responses were similar to responses in neurons expressing DN-RhoA. However, expression of CA-RhoA inhibited NMDA-evoked responses by approximately 50%. Exposure to Tat for 16 h potentiated NMDAR function in non-expressing cells and in neurons expressing DN-RhoA. However, cells expressing CA-RhoA were markedly inhibited relative to non-expressing and DN-RhoA-expressing neurons with the same treatment time. After 24 h exposure to Tat, NMDAR function adapted in non-expressing cells, but cells expressing DN-RhoA remained potentiated. Additionally, neurons expressing DN-RhoA (Fig. 2 f) were morphologically distinct and exhibited increased dendritic branching relative to neurons expressing RFP only (Fig. 2 e), while expression of CA-RhoA (Fig. 2 g) caused dendritic simplification consistent with previous reports (Da Silva et al. 2003, Chen & Firestein 2007). Taken together, these data indicate that adaptation of NMDAR function following Tat-induced potentiation requires activation RhoA.
Tat-induced potentiation of NMDAR function adapts via activation of ROCK
The principal downstream effector of RhoA is ROCK (Matsui et al. 1996) and activation of ROCK results in rundown of NMDA-evoked currents via an actindependent mechanism (Beazely et al. 2008). Therefore, we hypothesized that ROCK activation was necessary for adaptation of NMDAR function after Tat-induced potentiation. To test this hypothesis, cultures were pretreated with an inhibitor of ROCK 1 h prior to and during Tat treatment for 16 or 24 h (Fig. 3). Inhibition of ROCK with Y27632 (10 μM) or H1152 (10 μM) did not affect Tat-induced NMDAR potentiation after 16 h treatment. However, the ROCK inhibitors prevented adaptation of NMDA-evoked [Ca2+]i after exposure to Tat for 24 h. These data suggest that adaptation of NMDAR function following Tat-induced potentiation requires activation ROCK.
Fig. 3. ROCK mediates adaptation of NMDA-evoked [Ca2+]i responses following Tat-induced potentiation.
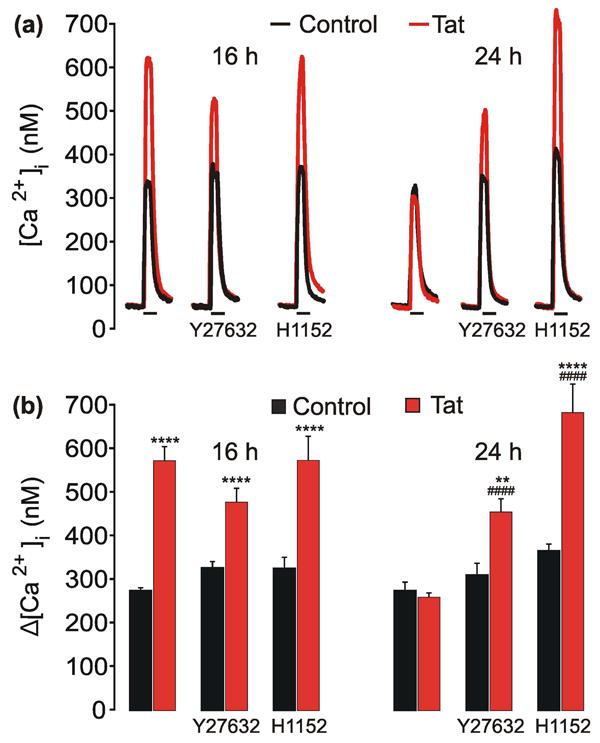
a, representative traces show NMDA-evoked [Ca2+]i increases from control (
) neurons or neurons treated with 50 ng/mLTat (
) for 16 h or 24 h. Cells were pretreated with 10 μM Y27632 or 10μM H1152 for 1 h prior to the addition of Tat. NMDA (10 μM, 30 s) was applied by superfusion at the times indicated by the horizontal bars. b, Bar graph shows net [Ca2+]i increase evoked by 10 μM NMDA in control (
) cells or cells treated with Tat (
) for 16 h or 24 h. **p<0.01, ****p<0.0001 relative to respective control; ####p<0.0001 relative to 24 h Tat-treated neurons as determined by separate, one-way ANOVAs with 6 levels per treatment time followed by Tukey's post-test for multiple comparisons.
Actin reorganization reduces NMDAR function
RhoA/ROCK signaling activates several targets that affect the actin cytoskeleton including LIM-kinase and cofilin (Maekawa et al. 1999), profilin (Da Silva et al. 2003), as well as myosin light chain phosphatase and myosin light chain kinase (MLCK) (Totsukawa et al. 2000). MLCK regulates NMDAR activity by increasing actomyosin contractility (Lei et al. 2001), thus we hypothesized that MLCK activation is necessary for the reduction in NMDA-evoked responses after Tat-induced potentiation. To test this hypothesis, cultures were pretreated with an inhibitor of MLCK, ML-7 (10 μM), for 1 h prior to and during Tat treatment for 16 or 24 h. ML-7 did not affect Tat-induced NMDAR potentiation by 16 h and, unexpectedly, did not block adaptation of NMDAR function by 24 h (Fig 4. a, b). Consistent with previous reports (Lei et al. 2001), ML-7 inhibited NMDA-evoked responses by approximately 40% and 25% following 16 h and 24 h treatment, respectively. Neurons exposed to ML-7 had vacuole-like structures present within the neuronal cell body similar to previous reports (Sulzer et al. 2008). These data do not support the hypothesized role for MLCK and suggest that adaptation of NMDAR function occurs via another effector downstream from ROCK.
Fig. 4. Adaptation of NMDA-evoked [Ca2+]i responses following Tat-induced potentiation requires remodeling of the actin cytoskeleton.
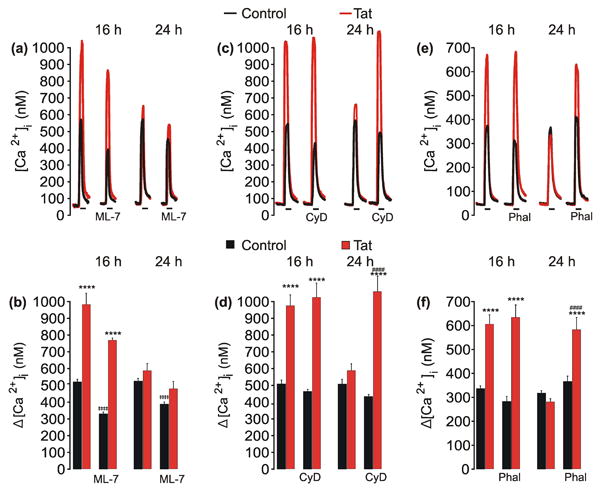
a, c, e, representative traces show NMDA-evoked [Ca2+]i increases from control (
) neurons or neurons treated with 50 ng/mLTat (
) for 16 h or 24 h. Cells were pretreated with ML-7 (10 μM), Cytochalasin D (CyD, 1 μM), or Phalloidin (Phal, 1 μM) for 1 h prior to the addition of Tat. NMDA (10 μM, 30 s) was applied by superfusion at the times indicated by the horizontal bars. b, d, f, bar graphs show net [Ca2+]i increase evoked by 10 μM NMDA in control (
) cells or cells treated with Tat (
) for 16 h or 24 h. Cells were pretreated with ML-7, CyD, or Phal as indicated. ****p<0.0001 relative to respective control; ‡‡‡‡p<0.0001 relative to untreated control with same treatment time; ####p<0.0001 relative to 24 h Tat-treated neurons as determined by separate, one-way ANOVAs with 4 levels per treatment time followed by Tukey's post-test for multiple comparisons.
We next examined the role of the actin cytoskeleton in Tat induced regulation of NMDAR function. Cultures were pretreated with cytochalasin D (1 μM), which binds to the barbed end of F-actin to prevent the addition of G-actin monomers (Cingolani & Goda 2008), for 1 hour prior to and during treatment with Tat. Cytochalasin D did not affect Tat-induced NMDAR potentiation by 16 h, but did prevent the reduction of NMDA-evoked [Ca2+]i responses observed after 24 h treatment with Tat (Fig. 4 c, d). Additionally, cultures were pretreated with phalloidin (1 μM), which binds to and stabilizes F-actin polymers (Cingolani & Goda 2008), for 1 hour prior to and during treatment with Tat. Phalloidin did not affect Tat-induced NMDAR potentiation by 16 h, but did prevent the reduction of NMDA-evoked [Ca2+]i responses after 24 h treatment with Tat (Fig. 4 e, f). These data suggest that Tat-induced reorganization of the actin cytoskeleton reduces NMDA-evoked [Ca2+]i responses.
Because phalloidin and cytochalasin D have opposing effects on the actin cytoskeleton, but similarly prevented adaptation, we further examined the effects of actin polymerization and depolymerization on the neuronal distribution of actin and NMDA-evoked responses. To induce changes in the actin cytoskeleton, cultures were treated for 1 h with jasplakinolide (10 μM), which promotes F-actin polymerization, or latrunculin A (5 μM), which sequesters G-actin to promote F-actin depolymerization (Cingolani & Goda 2008). Treatment for 1 h with jasplakinolide induced robust somatic and dendritic redistribution of the actin cytoskeleton and caused retraction of dendritic spines (Fig. 5 a). Latrunculin A dispersed the actin cytoskeleton, revealed fibers in the neuronal cell body, and caused a dramatic loss of dendritic spines (Fig. 5 b). To determine if changes in the actin cytoskeleton affect NMDAR function, cultures were treated for 1 h to 24 h with jasplakinolide (10 μM) or latrunculin A (5 μM). Treatment with jasplakinolide attenuated NMDA-evoked [Ca2+]i responses as early as 1 h (Fig. 5 c, d). Treatment with latrunculin A also reduced NMDA-evoked [Ca2+]i responses, but required 24 h for significant functional impairment to develop (Fig. 5 c, d). Such rapid and persistent changes to the actin cytoskeleton caused by jasplakinolide and latrunculin A are consistent with previous reports (Okamoto et al. 2004) and indicate that bidirectional reorganization of actin reduces NMDAR function.
Fig. 5. Polymerization or depolymerization of the actin cytoskeleton reduces NMDA-evoked [Ca2+]i responses.
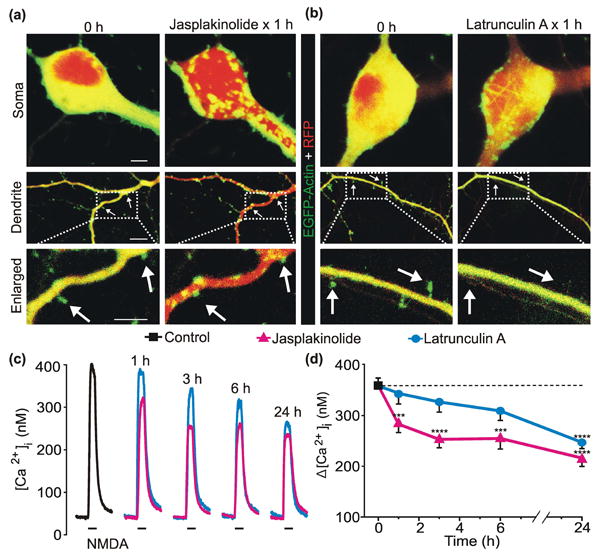
a, b, Representative confocal images (Scale bar = 10 μm) of neurons expressing EGFP-Actin and Tag-RFP before (0 h) and after 1 h treatment with 10μM Jasplakinolide (a) or 5 μM Latrunculin A (b). Compressed z-stacks of red (Tag-RFP) and green (EGFP-Actin) images were superimposed. c, representative traces show NMDA-evoked [Ca2+]i increases from control (
) neurons or neurons treated with 10 μM jasplakinolide (
 ) or 5 μM latrunculin A (
) or 5 μM latrunculin A (
 ) for the times indicated above the traces. NMDA (10 μM, 30 s) was applied by superfusion at the times indicated by the horizontal bars below the traces. d, plot summarizes time-dependent changes in NMDA-evoked [Ca2+]i increases from control (
) for the times indicated above the traces. NMDA (10 μM, 30 s) was applied by superfusion at the times indicated by the horizontal bars below the traces. d, plot summarizes time-dependent changes in NMDA-evoked [Ca2+]i increases from control (
 ) neurons and neurons treated with 10 μM jasplakinolide (
) neurons and neurons treated with 10 μM jasplakinolide (
 ) or 5 μM latrunculin A (
) or 5 μM latrunculin A (
 ).***p<0.001, ****p<0.0001 relative to control as determined by separate, one-way ANOVAs with 5 levels per treatment followed by Tukey's post-test for multiple comparisons.
).***p<0.001, ****p<0.0001 relative to control as determined by separate, one-way ANOVAs with 5 levels per treatment followed by Tukey's post-test for multiple comparisons.
Discussion
NMDAR dysfunction contributes to the neurotoxicity that underlies many neurodegenerative disorders including HAND (Danysz & Parsons 2003, Hallett & Standaert 2004, Spalloni et al. 2013, Rossi et al. 2013, Young et al. 1988). Although the neuropathogenesis of HAND likely involves a combination of factors, evidence suggests that the HIV protein Tat plays a prominent role by altering the activity of NMDARs (Haughey et al. 2001, Song et al. 2003, Chandra et al. 2005, Krogh et al. 2014) and impairing cognitive function (Bachani et al. 2013, Carey et al. 2012, Fitting et al. 2010). Here we used digital Ca2+ imaging to study the pathways that modulate NMDA-evoked [Ca2+]i responses during prolonged exposure to Tat. Previous work described a biphasic change in NMDA-evoked increases in [Ca2+]i during prolonged exposure to Tat (Popescu 2014). Initially, Tat potentiated NMDAR function via LRP-dependent activation of Src kinase. Subsequently, NMDAR function adapted after activation of the NOS/sGC/PKG pathway (Krogh et al. 2014).
The goal of this study was to identify effectors downstream of PKG responsible for adaptation of NMDA-evoked responses following Tat-induced NMDAR potentiation. We determined that a RhoA/ROCK-dependent remodeling of the actin cytoskeleton was an obligatory step in NMDAR adaptation (Fig. 6). Adaptation of NMDAR function may be a neuroprotective mechanism to prevent excessive NMDAR-mediated Ca2+ influx.
Fig. 6. Proposed mechanism of action.
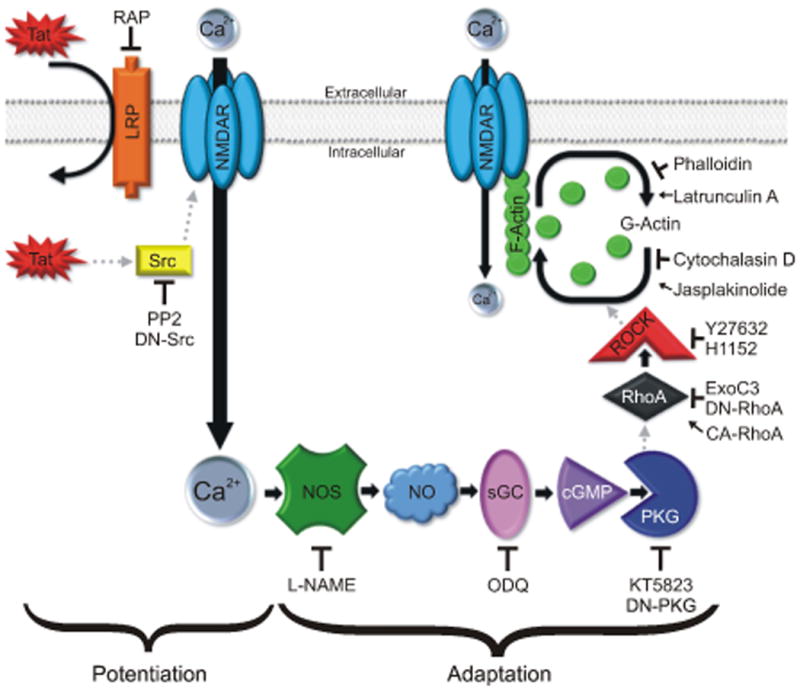
HIV-1 Tat interacts with LRP leading to Src-dependent potentiation of the NMDAR and activation of the NOS/sGC/PKG signaling pathway. PKG activates a RhoA/ROCK signaling pathway leading to remodeling of the actin cytoskeleton and subsequent reduction of NMDAR function.
The small GTPase RhoA is a well-known regulator of the actin cytoskeleton (Kaibuchi et al. 1999). The expression and function of RhoA is regulated by NO-mediated activation of PKG in vascular smooth muscle cells (Sauzeau et al. 2003, Rolli-Derkinderen et al. 2005) consistent with our hypothesis that Tat activates a NOS/sGC/PKG signaling cascade upstream from RhoA. Tat activates RhoA in endothelial cells (Urbinati et al. 2005, Zhong et al. 2010, Yuen et al. 2010, Xu et al. 2012), however much less is known about the effects of Tat on RhoA in neurons. Our data show that adaptation of NMDAR function following Tat-induced potentiation required activation of RhoA. Pharmacologic or genetic inhibition of RhoA prevented adaptation of NMDAR function following Tat-induced potentiation. Furthermore, constitutive activation of RhoA attenuated NMDA-evoked responses by approximately 50% relative to non-expressing neurons in the same imaging field, consistent with our hypothesis that RhoA activation reduces NMDAR function. Thus, Tat-induced activation of RhoA attenuates NMDAR function. Because inhibition of RhoA had no effect on NMDA-evoked responses until the cell had been treated with Tat for over 8 h, we speculate that the adaptive response is a mechanism to compensate for the excess Ca2+ influx resulting from potentiation of NMDARs.
Activation and inhibition of RhoA profoundly affected neuronal morphology. Consistent with previous reports (Da Silva et al. 2003, Chen & Firestein 2007), neurons expressing DN-RhoA exhibited increased dendritic branching while cells expressing CARhoA displayed simplified dendritic structures with minimal branching and no noticeable dendritic spines. Interestingly, activation of RhoA causes synapse loss in vitro (Sunico et al. 2010) and loss of dendritic spines resulting in cognitive impairment in vivo (Pozueta et al. 2013). Similarly, treatment with Tat causes synapse loss in vitro (Kim et al. 2008, Shin et al. 2012) and loss of dendritic spines resulting in cognitive impairment in vivo (Fitting et al. 2010, Carey et al. 2012). Whether RhoA activation is required for Tat-induced synaptodendritic changes in vivo remains unknown.
ROCK is the primary downstream target of RhoA and is a serine/threonine kinase that modifies the cytoskeleton to regulate cell migration and proliferation (Matsui et al. 1996, Amano et al. 2010). Tat activates ROCK in endothelial cells of the blood brain barrier (Zhong et al. 2012) and activation of ROCK in neurons induces rundown of NMDAR currents via an actin-dependent mechanism (Beazely et al. 2008). Based on these studies, we hypothesized that Tat-induced activation of ROCK would attenuate NMDA-evoked responses. Consistent with our hypothesis, we found that inhibition of ROCK prevented adaptation of NMDAR function following Tat-induced potentiation. These data indicate that Tat-induced activation of ROCK reduces NMDAR function. Abnormal activation of ROCK is observed in many models of neuronal disorders including Alzheimer's disease, spinal-cord injury, neuropathic pain, and excitotoxicity (Mueller et al. 2005). However, little is known about the role of ROCK in HAND.
RhoA activates ROCK to regulate the actin cytoskeleton by acting on multiple targets including MLCK (Maekawa et al. 1999). We found that adaptation of NMDA-evoked responses did not require MLCK. Inhibition of MLCK with ML-7 significantly inhibited NMDAR function, consistent with previous reports indicating that MLCK regulates NMDAR activity by contraction of actomyosin (Lei et al. 2001). The effector between ROCK and the actin cytoskeleton remains unknown.
Activation of ROCK increases F-actin formation (Da Silva et al. 2003). The status of F-actin polymerization affects NMDAR function. Indeed, depolymerization of the actin cytoskeleton reduces NMDAR-mediated currents (Rosenmund & Westbrook 1993), while polymerization of actin enhances glutamate receptor function (Furukawa et al. 1995). As anticipated, we found that F-actin depolymerization with latrunculin A reduced NMDAR function. Unexpectedly, increased F-actin polymerization with jasplakinolide also decreased NMDAR function. Both jasplakinolide (Bubb et al. 1994) and CA-RhoA (Ishizaki et al. 1997) increase the formation of F-actin and both caused approximately 50% reduction in NMDAR function, suggesting that increased actin polymerization reduces NMDAR function. Although the mechanism leading to impaired NMDAR function remains unclear, there is precedent for modulation of NMDAR function through tension exerted on the NMDAR via its physical connection with the actin cytoskeleton (Lei et al. 2001). Perhaps the robust somatic and dendritic redistribution of actin shown in figure 5 disrupts the connections between the NMDAR and the actin cytoskeleton resulting in attenuated NMDAR function.
Adaptation of NMDAR function following Tat-induced potentiation was blocked by cytochalasin D and phalloidin, agents that inhibit polymerization and depolymerization, respectively (Cingolani & Goda 2008). These results suggest that adaptation occurs in response to remodeling of the actin cytoskeleton, an effect that requires both depolymerization and polymerization of actin. Tat is known to affect the cytoskeleton; it depolymerizes actin in endothelial cells (Wu et al. 2004) and produces a loss of F-actin puncta in hippocampal neurons in vitro (Bertrand et al. 2014). Our data indicate that Tat activates a RhoA/ROCK pathway leading to remodeling of the actin cytoskeleton resulting in reduced NMDA-evoked responses.
The primary cultures used for this study were composed of approximately 70% astrocytes, 20% neurons and 10% microglia (Kim et al. 2011), all of which can be affected by Tat and the pharmacological agents used in these experiments. Therefore, it is possible that the observed effects of Tat on NMDAR function may result from both direct effects on neurons and indirect effects on glia. Indeed, Tat promotes the release of glutamate (Eugenin et al. 2003), inflammatory cytokines (Chen et al. 1997), chemokines (Conant et al. 1998), and reactive oxygen species (Kruman et al. 1998) from glia and neurons. These substances all influence NMDAR function. However, studies in which dominant negative constructs were expressed selectively in neurons determined that potentiation of NMDAR function required activation of neuronal Src kinase and adaptation required activation of a neuronal NO signaling pathway (Krogh et al. 2014). Indeed, Tat-induced NO production was shown previously to require NMDAR-mediated Ca2+ influx in neurons (Eugenin et al. 2007). Furthermore, neuronal expression of dominant negative RhoA prevented NMDAR adaptation while constitutively active RhoA inhibited NMDAR function. Thus, potentiation and adaptation of NMDAR function produced by Tat require the activation of signaling pathways in neurons. Clearly microglia and astrocytes are capable of releasing factors following exposure to Tat that could subsequently act on neurons and would thus be acting upstream of the neuronal pathways that are the focus of this study.
Adaptation of Tat-induced NMDAR potentiation might improve neuronal survival. However, increasing evidence suggests that over compensation following neuronal insult can lead to excessive inhibitory tone (Hargus & Thayer 2013, Wu et al. 2014) and impaired connectivity due to lost excitatory synapses (Kim et al. 2008). Reduced NMDAR function during prolonged exposure to HIV neurotoxins may contribute to cognitive impairment analogous to the impairment seen in transgenic animals with reduced NMDAR expression (Tsien et al. 1996, Shimizu et al. 2000) or humans given NMDAR antagonists (Krystal et al. 1994, Malhotra et al. 1996). Thus, inhibiting ROCK might prevent cognitive decline. Indeed, RhoA/ROCK are promising targets for treating various neurological disorders. ROCK inhibition lowers brain levels of amyloid-β in a transgenic mouse model of Alzheimer's disease (Zhou et al. 2003) and improves cognitive function in aged rats (Huentelman et al. 2009). Inhibition of ROCK also improves neurological function and reduces infarct size in models of ischemic stroke (Satoh et al. 2001, Toshima et al. 2000) and accelerates functional recovery from spinal cord injuries (Tanaka et al. 2004, Sung et al. 2003, Fournier et al. 2003, Hara et al. 2000, Dergham et al. 2002). Thus, ROCK is a valuable drug target with neuroprotective and neuroregenerative potential. The ROCK inhibitor, fasudil, is a well-tolerated vasodilator that has been used clinically in Japan since 1995 for the treatment of subarachnoid hemorrhage (Shibuya et al. 1992) and improves physical performance in patients with angina pectoris (Shimokawa et al. 2002). Importantly, fasudil is neuroprotective in models of Alzheimer's disease (Song et al. 2013), amyotrophic lateral sclerosis (Takata et al. 2013), and Parkinson's disease (Tonges et al. 2012). Whether fasudil or other ROCK inhibitors are effective against HAND remains to be determined.
In summary, we show that adaptation of NMDAR function following Tat-induced potentiation results from RhoA/ROCK-dependent remodeling of the actin cytoskeleton. These findings suggest that different signaling pathways mediate the neurotoxicity induced by HIV Tat over the course of prolonged exposure. As such, drugs that modulate ROCK signaling may warrant exploration as neuroprotective agents for the treatment of HAND.
Acknowledgments
This work was supported by a grant from National Institutes of Health (DA07304) to SAT. A National Institute on Drug Abuse Training Grant (DA007097) supported KK and EL.
Footnotes
The authors declare no conflict of interest.
References
- Albright AV, Shieh JT, Itoh T, Lee B, Pleasure D, O'Connor MJ, Doms RW, Gonzalez-Scarano F. Microglia express CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human immunodeficiency virus type 1 dementia isolates. J Virol. 1999;73:205–213. doi: 10.1128/jvi.73.1.205-213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton. 2010;67:545–554. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An SF, Groves M, Gray F, Scaravilli F. Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J Neuropathol Exp Neurol. 1999;58:1156–1162. doi: 10.1097/00005072-199911000-00005. [DOI] [PubMed] [Google Scholar]
- Antinori A, Arendt G, Becker JT, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachani M, Sacktor N, McArthur JC, Nath A, Rumbaugh J. Detection of anti-tat antibodies in CSF of individuals with HIV-associated neurocognitive disorders. Journal of neurovirology. 2013;19:82–88. doi: 10.1007/s13365-012-0144-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beazely MA, Weerapura M, MacDonald JF. Abelson tyrosine kinase links PDGFbeta receptor activation to cytoskeletal regulation of NMDA receptors in CA1 hippocampal neurons. Mol Brain. 2008;1:20. doi: 10.1186/1756-6606-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand SJ, Mactutus CF, Aksenova MV, Espensen-Sturges TD, Booze RM. Synaptodendritic recovery following HIV Tat exposure: neurorestoration by phytoestrogens. J Neurochem. 2014;128:140–151. doi: 10.1111/jnc.12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- Carey AN, Sypek EI, Singh HD, Kaufman MJ, McLaughlin JP. Expression of HIV-Tat protein is associated with learning and memory deficits in the mouse. Behav Brain Res. 2012;229:48–56. doi: 10.1016/j.bbr.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra T, Maier W, Konig HG, Hirzel K, Kogel D, Schuler T, Chandra A, Demirhan I, Laube B. Molecular interactions of the type 1 human immunodeficiency virus transregulatory protein Tat with N-methyl-d-aspartate receptor subunits. Neuroscience. 2005;134:145–153. doi: 10.1016/j.neuroscience.2005.02.049. [DOI] [PubMed] [Google Scholar]
- Chang HC, Samaniego F, Nair BC, Buonaguro L, Ensoli B. HIV-1 Tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. AIDS. 1997;11:1421–1431. doi: 10.1097/00002030-199712000-00006. [DOI] [PubMed] [Google Scholar]
- Chen H, Firestein BL. RhoA regulates dendrite branching in hippocampal neurons by decreasing cypin protein levels. J Neurosci. 2007;27:8378–8386. doi: 10.1523/JNEUROSCI.0872-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Mayne M, Power C, Nath A. The Tat protein of HIV-1 induces tumor necrosis factor-alpha production. Implications for HIV-1-associated neurological diseases. J Biol Chem. 1997;272:22385–22388. doi: 10.1074/jbc.272.36.22385. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3117–3121. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cysique LA, Maruff P, Brew BJ. Prevalence and pattern of neuropsychological impairment in human immunodeficiency virus–infected/acquired immunodeficiency syndrome (HIV/AIDS) patients across pre- and post-highly active antiretroviral therapy eras: A combined study of two cohorts. Journal of neurovirology. 2004;10:350–357. doi: 10.1080/13550280490521078. [DOI] [PubMed] [Google Scholar]
- Da Silva JS, Medina M, Zuliani C, Di Nardo A, Witke W, Dotti CG. RhoA/ROCK regulation of neuritogenesis via profilin IIa-mediated control of actin stability. J Cell Biol. 2003;162:1267–1279. doi: 10.1083/jcb.200304021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danysz W, Parsons CG. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer's disease: preclinical evidence. International journal of geriatric psychiatry. 2003;18:S23–32. doi: 10.1002/gps.938. [DOI] [PubMed] [Google Scholar]
- Davis LE, Hjelle BL, Miller VE, et al. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology. 1992;42:1736–1739. doi: 10.1212/wnl.42.9.1736. [DOI] [PubMed] [Google Scholar]
- Del Valle L, Croul S, Morgello S, Amini S, Rappaport J, Khalili K. Detection of HIV-1 Tat and JCV capsid protein, VP1, in AIDS brain with progressive multifocal leukoencephalopathy. Journal of neurovirology. 2000;6:221–228. doi: 10.3109/13550280009015824. [DOI] [PubMed] [Google Scholar]
- Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570–6577. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, D'Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tatinduced apoptosis. J Neurochem. 2003;85:1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW. HIV-tat induces formation of an LRP-PSD-95- NMDARnNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A. 2007;104:3438–3443. doi: 10.1073/pnas.0611699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Ignatowska-Jankowska BM, Bull C, et al. Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry. 2013;73:443–453. doi: 10.1016/j.biopsych.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Xu R, Bull C, Buch S, El-Hage N, Nath A, Knapp PE, Hauser KF. Interactive Comorbidity between Opioid Drug Abuse and HIV-1 Tat: Chronic Exposure Augments Spine Loss and Sublethal Dendritic Pathology in Striatal Neurons. Am J Pathol. 2010;177:1397–1410. doi: 10.2353/ajpath.2010.090945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, Takizawa BT, Strittmatter SM. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci. 2003;23:1416–1423. doi: 10.1523/JNEUROSCI.23-04-01416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Smithswintosky VL, Mattson MP. Evidence that actin depolymerization protects hippocampal neurons against excitotoxicity by stabilizing [ca2+](i) Experimental Neurology. 1995;133:153–163. doi: 10.1006/exnr.1995.1018. [DOI] [PubMed] [Google Scholar]
- Genis P, Jett M, Bernton EW, et al. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–1718. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, Standaert DG. Rationale for and use of NMDA receptor antagonists in Parkinson's disease. Pharmacol Ther. 2004;102:155–174. doi: 10.1016/j.pharmthera.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Hara M, Takayasu M, Watanabe K, Noda A, Takagi T, Suzuki Y, Yoshida J. Protein kinase inhibition by fasudil hydrochloride promotes neurological recovery after spinal cord injury in rats. J Neurosurg. 2000;93:94–101. doi: 10.3171/spi.2000.93.1.0094. [DOI] [PubMed] [Google Scholar]
- Hargus NJ, Thayer SA. Human Immunodeficiency Virus-1 Tat Protein Increases the Number of Inhibitory Synapses between Hippocampal Neurons in Culture. J Neurosci. 2013;33:17908–17920. doi: 10.1523/JNEUROSCI.1312-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. Journal of Neurochemistry. 2001;78:457–467. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Clifford DB, Franklin DR, Jr, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75:2087–2096. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton RK, Franklin DR, Ellis RJ, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. Journal of neurovirology. 2011;17:3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton RK, Marcotte TD, Mindt MR, Sadek J, Moore DJ, Bentley H, McCutchan JA, Reicks C, Grant I. The impact of HIV-associated neuropsychological impairment on everyday functioning. J Int Neuropsychol Soc. 2004;10:317–331. doi: 10.1017/S1355617704102130. [DOI] [PubMed] [Google Scholar]
- Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, Male D, Everall I. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. Journal of neurovirology. 2000;6:145–155. doi: 10.3109/13550280009013158. [DOI] [PubMed] [Google Scholar]
- Huentelman MJ, Stephan DA, Talboom J, et al. Peripheral delivery of a ROCK inhibitor improves learning and working memory. Behavioral neuroscience. 2009;123:218–223. doi: 10.1037/a0014260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki T, Naito M, Fujisawa K, Maekawa M, Watanabe N, Saito Y, Narumiya S. p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Lett. 1997;404:118–124. doi: 10.1016/s0014-5793(97)00107-5. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Piggee C, Heyes MP, Murphy C, Quearry B, Bauer M, Zheng J, Gendelman HE, Markey SP. Glutamate is a mediator of neurotoxicity in secretions of activated HIV-1-infected macrophages. Journal of Neuroimmunology. 2001;117:97–107. doi: 10.1016/s0165-5728(01)00315-0. [DOI] [PubMed] [Google Scholar]
- Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem. 1999;68:459–486. doi: 10.1146/annurev.biochem.68.1.459. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Martemyanov KA, Thayer SA. Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci. 2008;28:12604–12613. doi: 10.1523/JNEUROSCI.2958-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Shin AH, Thayer SA. Activation of Cannabinoid Type 2 Receptors Inhibits HIV-1 Envelope Glycoprotein gp120-Induced Synapse Loss. Mol Pharmacol. 2011;80:357–366. doi: 10.1124/mol.111.071647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh KA, Wydeven N, Wickman K, Thayer SA. HIV-1 protein Tat produces biphasic changes in NMDA-evoked increases in intracellular Ca concentration via activation of Src kinase and nitric oxide signaling pathways. J Neurochem. 2014 doi: 10.1111/jnc.12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Experimental Neurology. 1998;154:276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Lei S, Czerwinska E, Czerwinski W, Walsh MP, MacDonald JF. Regulation of NMDA receptor activity by F-actin and myosin light chain kinase. J Neurosci. 2001;21:8464–8472. doi: 10.1523/JNEUROSCI.21-21-08464.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Li G, Steiner J, Nath A. Role of Tat Protein in HIV Neuropathogenesis. Neurotox Res. 2009;16:205–220. doi: 10.1007/s12640-009-9047-8. [DOI] [PubMed] [Google Scholar]
- Li Y, Krogh KA, Thayer SA. Epileptic stimulus increases Homer 1a expression to modulate endocannabinoid signaling in cultured hippocampal neurons. Neuropharmacology. 2012;63:1140–1149. doi: 10.1016/j.neuropharm.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Popko J, Krogh KA, Thayer SA. Epileptiform stimulus increases Homer 1a expression to modulate synapse number and activity in hippocampal cultures. J Neurophysiol. 2013;109:1494–1504. doi: 10.1152/jn.00580.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D, Breier A. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology. 1996;14:301–307. doi: 10.1016/0893-133X(95)00137-3. [DOI] [PubMed] [Google Scholar]
- Matsui T, Amano M, Yamamoto T, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov. 2005;4:387–398. doi: 10.1038/nrd1719. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Popescu GK. Dynamic plasticity of NMDA receptor-mediated calcium entry in neurons exposed to HIV-tat. J Neurochem. 2014 doi: 10.1111/jnc.12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, Ribe EM, Troy CM, Arancio O, Shelanski M. Caspase-2 is required for dendritic spine and behavioural alterations in J20 APP transgenic mice. Nat Commun. 2013;4:1939. doi: 10.1038/ncomms2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick L, Berger JR, Shapshak P, Tourtellotte WW. Early penetration of the blood-brain-barrier by HIV. Neurology. 1988;38:9–14. doi: 10.1212/wnl.38.1.9. [DOI] [PubMed] [Google Scholar]
- Rolli-Derkinderen M, Sauzeau V, Boyer L, Lemichez E, Baron C, Henrion D, Loirand G, Pacaud P. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ Res. 2005;96:1152–1160. doi: 10.1161/01.RES.0000170084.88780.ea. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Westbrook GL. Calcium-Induced Actin Depolymerization Reduces NMDA Channel Activity. Neuron. 1993;10:805–814. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- Rossi S, Studer V, Moscatelli A, et al. Opposite roles of NMDA receptors in relapsing and primary progressive multiple sclerosis. PLoS One. 2013;8:e67357. doi: 10.1371/journal.pone.0067357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh S, Utsunomiya T, Tsurui K, Kobayashi T, Ikegaki I, Sasaki Y, Asano T. Pharmacological profile of hydroxy fasudil as a selective rho kinase inhibitor on ischemic brain damage. Life Sci. 2001;69:1441–1453. doi: 10.1016/s0024-3205(01)01229-2. [DOI] [PubMed] [Google Scholar]
- Sauzeau V, Rolli-Derkinderen M, Marionneau C, Loirand G, Pacaud P. RhoA expression is controlled by nitric oxide through cGMP-dependent protein kinase activation. J Biol Chem. 2003;278:9472–9480. doi: 10.1074/jbc.M212776200. [DOI] [PubMed] [Google Scholar]
- Schneider J, Kaaden O, Copeland TD, Oroszlan S, Hunsmann G. Shedding and interspecies type sero-reactivity of the envelope glycopolypeptide gp120 of the human immunodeficiency virus. Journal of General Virology. 1986;67:2533–2538. doi: 10.1099/0022-1317-67-11-2533. [DOI] [PubMed] [Google Scholar]
- Shibuya M, Suzuki Y, Sugita K, et al. Effect of AT877 on cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Results of a prospective placebo-controlled double-blind trial. J Neurosurg. 1992;76:571–577. doi: 10.3171/jns.1992.76.4.0571. [DOI] [PubMed] [Google Scholar]
- Shimizu E, Tang YP, Rampon C, Tsien JZ. NMDA receptor-dependent synaptic reinforcement as a crucial process for memory consolidation. Science. 2000;290:1170–1174. doi: 10.1126/science.290.5494.1170. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Hiramori K, Iinuma H, et al. Anti-anginal effect of fasudil, a Rho-kinase inhibitor, in patients with stable effort angina: a multicenter study. J Cardiovasc Pharmacol. 2002;40:751–761. doi: 10.1097/00005344-200211000-00013. [DOI] [PubMed] [Google Scholar]
- Shin AH, Kim HJ, Thayer SA. Subtype selective NMDA receptor antagonists induce recovery of synapses lost following exposure to HIV-1 Tat. Br J Pharmacol. 2012;166:1002–1017. doi: 10.1111/j.1476-5381.2011.01805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin AH, Thayer SA. Human immunodeficiency virus-1 protein Tat induces excitotoxic loss of presynaptic terminals in hippocampal cultures. Molecular and Cellular Neuroscience. 2013;54:22–29. doi: 10.1016/j.mcn.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodroski J, Patarca R, Rosen C, Wong-Staal F, Haseltine W. Location of the trans-activating region on the genome of human T-cell lymphotropic virus type III. Science. 1985a;229:74–77. doi: 10.1126/science.2990041. [DOI] [PubMed] [Google Scholar]
- Sodroski J, Rosen C, Wong-Staal F, Salahuddin SZ, Popovic M, Arya S, Gallo RC, Haseltine WA. Trans-acting transcriptional regulation of human T-cell leukemia virus type III long terminal repeat. Science. 1985b;227:171–173. doi: 10.1126/science.2981427. [DOI] [PubMed] [Google Scholar]
- Song L, Nath A, Geiger JD, Moore A, Hochman S. Human Immunodeficiency Virus Type 1 Tat Protein Directly Activates Neuronal N -methyl- D -aspartate Receptors at an Allosteric Zinc-Sensitive Site. Journal of neurovirology. 2003;9:399–403. doi: 10.1080/13550280390201704. [DOI] [PubMed] [Google Scholar]
- Song Y, Chen X, Wang LY, Gao W, Zhu MJ. Rho kinase inhibitor fasudil protects against beta-amyloid-induced hippocampal neurodegeneration in rats. CNS Neurosci Ther. 2013;19:603–610. doi: 10.1111/cns.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalloni A, Nutini M, Longone P. Role of the N-methyl-d-aspartate receptors complex in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2013;1832:312–322. doi: 10.1016/j.bbadis.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Mosharov E, Talloczy Z, Zucca FA, Simon JD, Zecca L. Neuronal pigmented autophagic vacuoles: lipofuscin, neuromelanin, and ceroid as macroautophagic responses during aging and disease. J Neurochem. 2008;106:24–36. doi: 10.1111/j.1471-4159.2008.05385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JK, Miao L, Calvert JW, Huang L, Louis Harkey H, Zhang JH. A possible role of RhoA/Rho-kinase in experimental spinal cord injury in rat. Brain Res. 2003;959:29–38. doi: 10.1016/s0006-8993(02)03717-4. [DOI] [PubMed] [Google Scholar]
- Sunico CR, Gonzalez-Forero D, Dominguez G, Garcia-Verdugo JM, Moreno-Lopez B. Nitric Oxide Induces Pathological Synapse Loss by a Protein Kinase G-, Rho Kinase-Dependent Mechanism Preceded by Myosin Light Chain Phosphorylation. J Neurosci. 2010;30:973–984. doi: 10.1523/JNEUROSCI.3911-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M, Tanaka H, Kimura M, et al. Fasudil, a rho kinase inhibitor, limits motor neuron loss in experimental models of amyotrophic lateral sclerosis. Br J Pharmacol. 2013;170:341–351. doi: 10.1111/bph.12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Yamashita T, Yachi K, Fujiwara T, Yoshikawa H, Tohyama M. Cytoplasmic p21(Cip1/WAF1) enhances axonal regeneration and functional recovery after spinal cord injury in rats. Neuroscience. 2004;127:155–164. doi: 10.1016/j.neuroscience.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Tonges L, Frank T, Tatenhorst L, Saal KA, Koch JC, Szego EM, Bahr M, Weishaupt JH, Lingor P. Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson's disease. Brain. 2012;135:3355–3370. doi: 10.1093/brain/aws254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima Y, Satoh S, Ikegaki I, Asano T. A new model of cerebral microthrombosis in rats and the neuroprotective effect of a Rho-kinase inhibitor. Stroke. 2000;31:2245–2250. doi: 10.1161/01.str.31.9.2245. [DOI] [PubMed] [Google Scholar]
- Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol. 2000;150:797–806. doi: 10.1083/jcb.150.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozzi V, Balestra P, Lorenzini P, et al. Prevalence and risk factors for human immunodeficiency virus–associated neurocognitive impairment, 1996 to 2002: Results from an urban observational cohort. Journal of neurovirology. 2005;11:265–273. doi: 10.1080/13550280590952790. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The Essential Role of Hippocampal Ca1 Nmda Receptor-Dependent Synaptic Plasticity in Spatial Memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Urbinati C, Bugatti A, Giacca M, Schlaepfer D, Presta M, Rusnati M. alpha(v)beta3-integrin-dependent activation of focal adhesion kinase mediates NF-kappaB activation and motogenic activity by HIV-1 Tat in endothelial cells. J Cell Sci. 2005;118:3949–3958. doi: 10.1242/jcs.02518. [DOI] [PubMed] [Google Scholar]
- Uthman OA, Abdulmalik JO. Adjunctive therapies for AIDS dementia complex. Cochrane database of systematic reviews. 2008:CD006496. doi: 10.1002/14651858.CD006496.pub2. Online. [DOI] [PubMed] [Google Scholar]
- Waataja JJ, Kim HJ, Roloff AM, Thayer SA. Excitotoxic loss of postsynaptic sites is distinct temporally and mechanistically from neuronal death. J Neurochem. 2008;104:364–375. doi: 10.1111/j.1471-4159.2007.04973.x. [DOI] [PubMed] [Google Scholar]
- Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, Alvarez X, Lackner AA. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med. 2001;193:905–915. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RF, Gu Y, Xu YC, Mitola S, Bussolino F, Terada LS. Human immunodeficiency virus type 1 Tat regulates endothelial cell actin cytoskeletal dynamics through PAK1 activation and oxidant production. J Virol. 2004;78:779–789. doi: 10.1128/JVI.78.2.779-789.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Guo Z, Gearing M, Chen G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzhiemer's disease model. Nat Commun. 2014;5:4159. doi: 10.1038/ncomms5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Feng X, Xie X, Zhang J, Wu D, Xu L. HIV-1 Tat protein increases the permeability of brain endothelial cells by both inhibiting occludin expression and cleaving occludin via matrix metalloproteinase-9. Brain Res. 2012;1436:13–19. doi: 10.1016/j.brainres.2011.11.052. [DOI] [PubMed] [Google Scholar]
- Young AB, Greenamyre JT, Hollingsworth A, Albin R, D'Amato C, Shoulson I, Penney JB. NMDA receptor losses in putamen from patients with huntington's disease. Science. 1988;241:981–983. doi: 10.1126/science.2841762. [DOI] [PubMed] [Google Scholar]
- Yuen EY, Zhong P, Yan Z. Homeostatic regulation of glutamatergic transmission by dopamine D4 receptors. Proc Natl Acad Sci U S A. 2010;107:22308–22313. doi: 10.1073/pnas.1010025108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Hennig B, Toborek M. Intact lipid rafts regulate HIV-1 Tat proteininduced activation of the Rho signaling and upregulation of P-glycoprotein in brain endothelial cells. J Cereb Blood Flow Metab. 2010;30:522–533. doi: 10.1038/jcbfm.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Zhang B, Eum SY, Toborek M. HIV-1 Tat triggers nuclear localization of ZO-1 via Rho signaling and cAMP response element-binding protein activation. J Neurosci. 2012;32:143–150. doi: 10.1523/JNEUROSCI.4266-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Su Y, Li B, et al. Nonsteroidal Anti-Inflammatory Drugs Can Lower Amyloidogenic A{beta}42 by Inhibiting Rho. Science. 2003;302:1215–1217. doi: 10.1126/science.1090154. [DOI] [PubMed] [Google Scholar]