

Abstract
Objectives
Vascular endothelial growth factor receptor 3 (VEGFR3) plays important roles both in lymphangiogenesis and angiogenesis. Upon stimulation by its ligand VEGF-C, VEGFR3 is able to form both homodimers as well as heterodimers with VEGFR2 and activates several downstream signal pathways including ERK1/2 and AKT. Despite certain similarities with VEGFR2, molecular features of VEGFR3 signaling are still largely unknown.
Approach and Results
Human dermal lymphatic endothelial cells (HDLECs) were used to examine VEGF-C-driven activation of signaling. Compared to VEGF-A activation of VEGFR2, VEGF-C-induced VEGFR3 activation led to a more extensive AKT activation while activation of ERK1/2 displayed a distinctly different kinetics. Furthermore, VEGF-C, but not VEGF-A, induced formation of VEGFR3/VEGFR2 complexes. Silencing VEGFR2 or its partner neuropilin-1 (NRP1) specifically abolished VEGF-C-induced AKT but not ERK activation, while silencing of NRP2 had little effect on either signaling pathway. Finally, suppression of vascular endothelial phosphotyrosine phosphatase (VE-PTP) but not other PTPs enhanced VEGF-C-induced activation of both ERK and AKT pathways. Functionally, both ERK and AKT pathways are important for LECs migration.
Conclusions
VEGF-C activates AKT signaling via formation of VEGFR3/VEGFR2 complex while ERK is activated by VEGFR3 homodimer. NRP1 and VE-PTP are involved in regulation of VEGFR3 signaling.
Keywords: VEGF-C, VEGFR3, VEGFR2, NRP1, NRP2, VE-PTP
INTRODUCTION
The vascular endothelial growth factor (VEGF) family of vascular growth factors in mammals is composed of five proteins (VEGFs A–D) and of a closely related placenta growth factor. VEGFs act by binding to three closely related receptor tyrosine kinases (VEGFR1-3) and two non-kinase receptors, neuropilin 1 (NRP1) and 2 (NRP2)1. Specificity of the biological activity of various VEGFs is determined, in part, by preferential binding of certain VEGFs to specific VEGFRs. Thus, VEGF-A and -B and placenta growth factor bind to VEGFR1, VEGF-A and -C to VEGFR2, and VEGF-C and -D predominantly bind to VEGFR32. Specificity is further determined by differential expression of VEGF receptors with VEGFR 1 and R2 being the predominant isoforms in blood endothelial cells. VEGFR3 is initially expressed during early embryonic development by both blood and lymphatic endothelial cells (LECs) later on becoming largely restricted to LECs3. However, its expression can be re-induced in blood endothelial cells during angiogenesis4 and it may play an important role in retinal blood vasculature formation5. VEGFR3 is also found in non-endothelial cells including neuronal progenitors, macrophages and osteoblasts2.
VEGFR2 signaling has been extensively studied as a prototype VEGF receptor. Recent advances include the appreciation of its interaction with VE-cadherin6, NRP17, various PTPs including density-enhanced phosphatase-1 (DEP1)8, vascular endothelial phosphotyrosine phosphatase (VE-PTP)9 and PTP1B)7, 10 as well as the role of its endocytosis in the extracellular signal-regulated kinases (ERK) cascade activation6, 11. In contrast, relatively little is known about molecular controls of VEGFR3 signaling. Although some details of VEGFR2 phosphorylation in blood endothelial cells have been reported12.
Previous studies demonstrated that when bound by their ligands, both VEGFR2 and VEGFR3 form homodimers and undergo autophosphorylation of cytoplasmic tyrosine residues leading to activation of their kinase activity. In addition to forming homodimers, upon VEGF-C or VEGF-A stimulation VEGFR3 can also form complexes with VEGFR213, 14. Interestingly, VEGF-C-induced complexes concentrate on the leading edge of LECs suggesting their involvement in cell migration. Yet the relative contribution of VEGFR3 homodimers vs. VEGFR3/R2 complexes to activation of various intracellular signaling pathways and LECs biology has not been established.
VEGFR signaling also involves co-receptor proteins NRP1 and NRP215. These transmembrane proteins are able to bind both VEGFs and semaphorins and regulate vascular and neural development16. In the vascular system, NRP1 is largely expressed in the arterial endothelium and lymphatic vessel valves whereas NRP2 is the predominant neuropilin in venous and lymphatic ECs17. During lymphatic vessel development, NRP1 acts as a semaphorin receptor in the valve endothelium17 while NRP2 is involved in VEGF-C-driven lymphatic vessel growth18. Despite these phenotypic observations, specific contributions of NRP1 and NRP2 to VEGF-C/VEGFR3 signaling have not been defined.
Recent studies have also highlighted the importance of PTPs in regulating VEGF receptors signaling. In the case of VEGFR2 these involve PTP1B10, 11, VE-PTP9, 19 and CD148/DEP18. In contrast, PTPs involvement in VEGFR3 signaling has not been defined. A PTP non-receptor type 14 (PTPN14) has recently been shown to be involved in lymphatic development but its role in VEGFR3 signaling is not established20.
In the present study, we set out to study VEGFR3 signaling in human dermal lymphatic endothelial cells (HDLECs). To this end, we examined VEGF-C vs. VEGF-A dependent activation of ERK1/2 and protein kinase B (AKT) signaling and examined the contribution of key signaling proteins including VEGFR2 and R3, NRP1 and NRP2, and various PTPs. We find that VEGFR2 and NRP1 are required for VEGF-C-induced AKT activation. Moreover, we found that VE-PTP, but not other phosphatases, is able to modulate VEGFR3 activation. Lastly we determined that VEGF-C stimulation of LEC migration requires activation of both ERK and AKT pathways.
MATERIALS AND METHODS
Materials and Methods are available in the online-only Data Supplement.
RESULTS
Molecular differences in VEGFR3 vs. VEGFR2 signaling in the lymphatic endothelium
To examine the differences in VEGFR3 vs. VEGFR2-induced activation of key endothelial signaling pathways such as MAPK and AKT, we compared the effect of VEGF-A- vs. VEGF-C stimulation of HDLECs that express both VEGFR2 and VEGFR3. Stimulation with VEGF-A resulted in a strong and rapid (within 5 min) activation of ERK1/2 pathway and a relatively mild activation of phosphoinositide 3-kinase (PI3K) signaling as determined by AKT phosphorylation (Fig. 1A and B). In contrast, when stimulated with VEGF-C, both ERK and AKT were activated with a much slower kinetics but the extent of AKT activation was far greater than seen with VEGF-A (Fig. 1A and B). These findings are in agreement with a previous in vitro study of VEGF-C signaling21.
Figure 1. Comparison of VEGFR2 and VEGFR3 signaling.
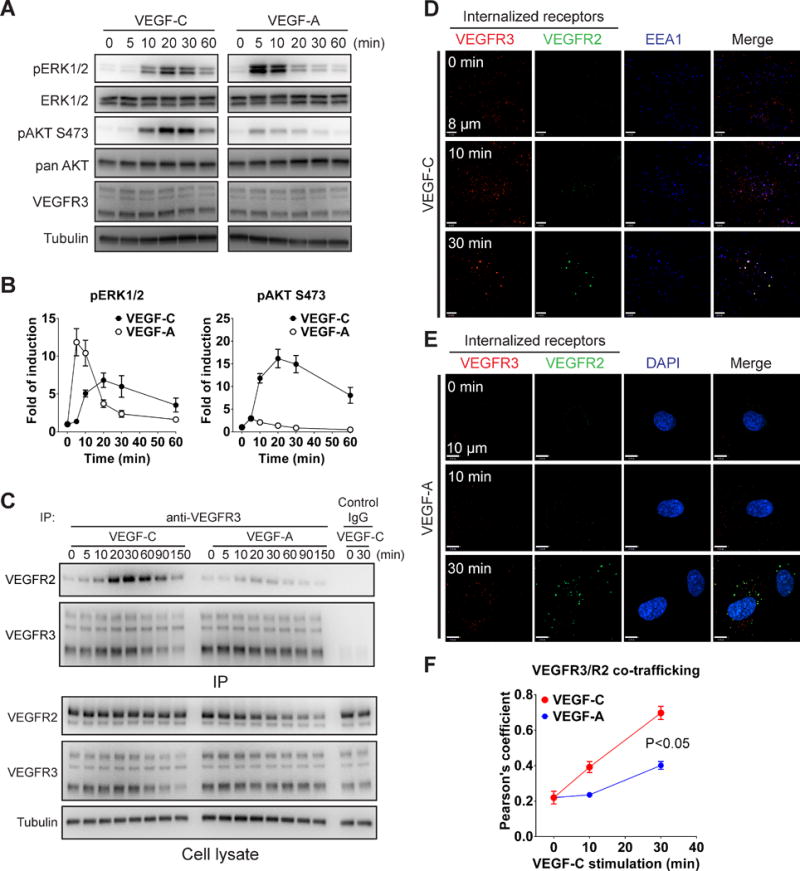
(A) Serum-starved HDLECs were stimulated with VEGF-C (100 ng/ml) or VEGF-A (50 ng/ml) and activation of ERK1/2 and AKT was determined by Western blotting. (B) Quantitative analyses of ERK and AKT phosphorylation in panel A normalized to total ERK and AKT levels, respectively. Data represent mean±SEM of three independent experiments. (C) Serum-starved HDLECs were stimulated with 100 ng/ml VEGF-C or 50 ng/ml VEGF-A for the indicated length of time. VEGFR3 immunoprecipitate was then probed with anti-VEGFR2 antibody (upper panel). Lower panel: Western blot analysis of total cell lysates corresponding to the upper panel. (D–E) Serum-starved HDLECs were incubated with anti-VEGFR3 and VEGFR2 antibodies and then stimulated with 100 ng/ml VEGF-C (D) or 50 ng/ml VEGF-A (E) for the indicated length of time. Internalized VEGFR3 (red) and VEGFR2 (green) were visualized by confocal microscopy. Early endosome was labeled with anti- early endosome antigen 1 (EEA1) (D, blue) and nuclear with DAPI (E, blue). (F) Co-localization of VEGFR3 with VEGFR2 was quantified using Pearson’s statistics. At least six fields of more than twenty cells were used for quantification. Statistical analysis was performed using two way’s ANOVA.
We next asked whether VEGFR3 signaling shares some of the key molecular elements involved in regulation of VEGFR2 signaling such as synectin (an adaptor protein which is critical to VEGFR2 trafficking and VEGF-A-induced ERK activation), and PTP1B, (an intracellular protein tyrosine phosphatase that has been shown to co-localize to VEGFR2 early endosomes and regulate its activation of ERK1/2)22. A knockdown of either synectin or PTP1b had no effect on VEGF-C-induced ERK or AKT activation in HDLEC (Fig. SI A,B). Moreover, VEGF-C failed to induce PTP1B co-localization with internalized VEGFR3 (Fig. SI C).
VEGFR3 has previously been reported to form complexes with VEGFR2 upon VEGF-C stimulation in cultured LEC and tip cells in vivo.13, 14 However, whether such complex formation affects VEGFR3 endocytosis and signaling has not been defined. To determine the role of VEGFR2 in these processes, we examined formation of VEGFR2/R3 complexes after VEGF-C and VEGF-A stimulation. In agreement with previous studies, VEGFR2 co-immunoprecipitated with VEGFR3 in HDLECs that were stimulated by VEGF-C while only marginal VEGFR2/R3 co-immunoprecipitation was observed after VEGF-A treatment (Fig. 1C). This was confirmed using confocal microscopy analysis of endocytosed receptors after VEGF-A or VEGF-C stimulation. Treatment of HDLEC with VEGF-C induced both VEGFR3 and VEGFR2 internalization and a significant degree of co-localization between the two receptors (Fig. 1D, F). In contrast, VEGF-A induced strong VEGFR2 but only marginal VEGFR3 internalization and a much lesser extent of co-localization of the two receptors was observed (Fig. 1E, F).
VEGFR2 is involved in VEGFR3 endocytosis and AKT activation
To examine the contribution of VEGFR2 to VEGFR3 signaling, we used VEGFR2 siRNA to knockdown its expression in HDLEC. Reduction in VEGFR2 expression significantly impaired VEGF-C-induced AKT activation but surprisingly had no effect on ERK activation (Fig. 2A, B). It also did not affect VEGF-C-induced VEGFR3 internalization (Fig. 2C).
Figure 2. VEGFR2 is involved in VEGF-C-induced AKT but not ERK activation.
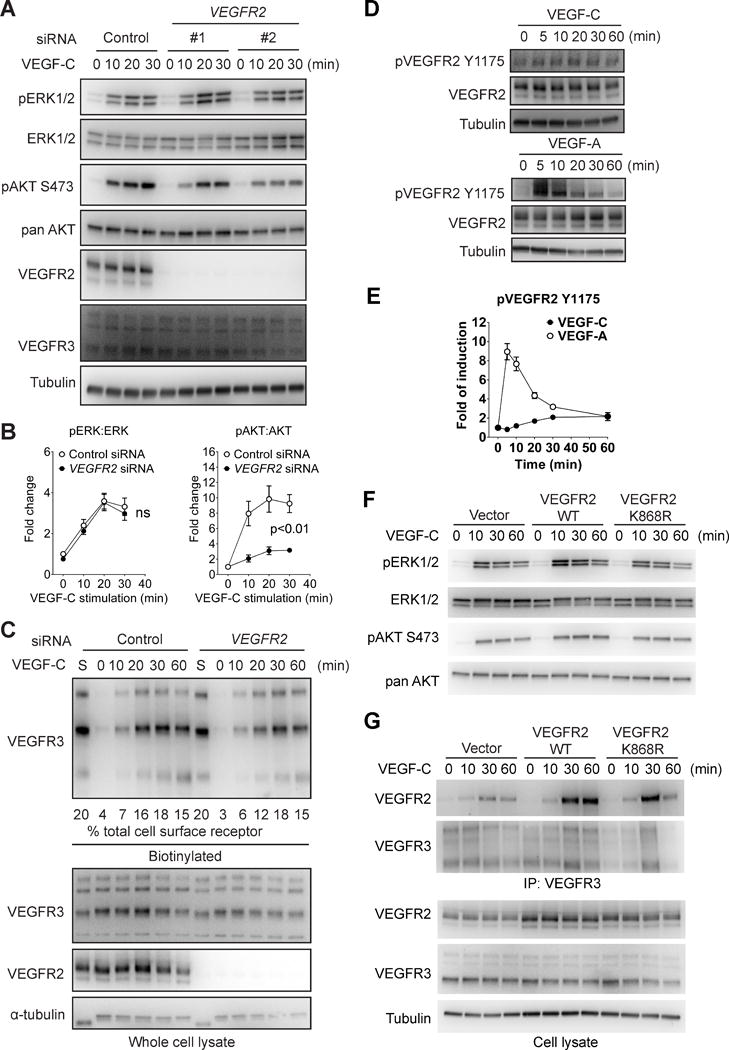
(A) HDLECs were transfected with two different siRNAs targeting human VEGFR2 or a negative control siRNA. The cells were then serum-starved and stimulated with 100 ng/ml VEGF-C. Activation of ERK1/2 and AKT was examined using Western blotting. (B) Quantitative analyses of ERK and AKT phosphorylation. Data represents Mean±SEM of three independent experiments with three distinct VEGFR2 siRNA sequences. Statistical analysis was performed using two way’s ANOVA. (C) HDLECs were biotin labeled as described in the Methods section and VEGF-C-induced VEGFR3 internalization in HDLECs treated with VEGFR2 or control siRNAs was determined by Western blotting of cell lysates at indicated time points (upper panel). Quantification was accomplished by deriving the percentage of internalized VEGFR3 to the amount of VEGFR3 present on the cell surface prior to VEGF-C treatment (lane “S”). Note that only 20% of lane S sample is loaded in the blot shown. Numbers under the blot refer to the % fraction of VEGFR3 internalized at various time points after VEGF-C stimulation. Note the absence of significant effect of VEGFR2 knockdown on VEGFR3 internalization. (D) Serum-starved HDLECs were stimulated with 100 ng/mL VEGF-C and VEGFR2 Y1175 phosphorylation was determined as shown. (E) Quantitative analyses of VEGFR2 Y1175 phosphorylation normalized to total VEGFR2 protein level. Data represents Mean±SEM of three independent experiments. (F) HDLECs infected with lentiviruses expressing either wild type VEGFR2, kinase-dead VEGFR2 mutant (K868R) or an empty control virus were stimulated with 100 ng/ml VEGF-C and activation of ERK and AKT was examined by Western blotting as indicated. (G) Upper panel: Western blot analysis of VEGFR3 antibody immunoprecipitate from HDLECs transduced with wild type or kinase-dead (K868R) VEGFR2 constructs or an empty virus control. Lower panel: Western blot analysis of total cell lysates corresponding to the upper panel.
Phosphorylation of VEGFR2 at Y1175 is crucial for activation of both ERK1/2 and AKT by the receptor. Similar to VEGF-A, VEGF-C was also able to induce VEGFR2 Y1175 phosphorylation in HDLEC (Fig. 2D). However, the extent of VEGF-C induced phosphorylation was much weaker and the kinetics slower than that by VEGF-A (Fig. 2E). To determine whether VEGFR2 kinase activity is required for VEGFR3 signaling, we expressed either a control (wild type) or a dominant-negative “kinase-dead” VEGFR2 mutant (K868R) construct23 in HDLECs (Fig. SII). The expression of the mutant receptor had no effect on VEGF-C-induced ERK or AKT activation (Fig 2F) or formation of VEGFR2/R3 complexes (Fig 2G), indicating that VEGFR2 kinase activity is dispensable for VEGFR3 signaling.
NRP1 but not NRP2 is involved in VEGFR3 signaling in lymphatic endothelial cells
Besides VEGFR2, NRP2, highly expressed in lymphatic and venous endothelial cells, is thought to play a role in VEGFR3 signaling. In addition, NRP1, while predominantly found in arterial ECs, is also expressed in a portion of lymphatic ECs. Therefore, we next set out to examine the roles of NRP1 and NRP2 in VEGF-C signaling in HDLECs.
We first tested whether the two NRPs interact with VEGFR3 upon VEGF-C stimulation. NRP2 but not NRP1 could be immunoprecipitated with VEGFR3 following VEGF-C stimulation (Fig 3A, B). However, while a knockdown of NRP2 expression had no effect on VEGF-C-induced ERK or AKT activation (Fig 3C, D), NRP1 knockdown severely impaired VEGF-C-induced AKT but not ERK activation (Fig 3C, E), thus resembling the effect of VEGFR2 knock-down. Knocking down both NRP1 and NRP2 had a similar effect on VEGFR3 signaling to that observed with a single NRP1 knock-down indicating that NRP1 but not NRP2 plays a predominant role in regulating VEGF-C signaling (Fig 3C).
Figure 3. NRP1 but not NRP2 is required for VEGF-C signaling.
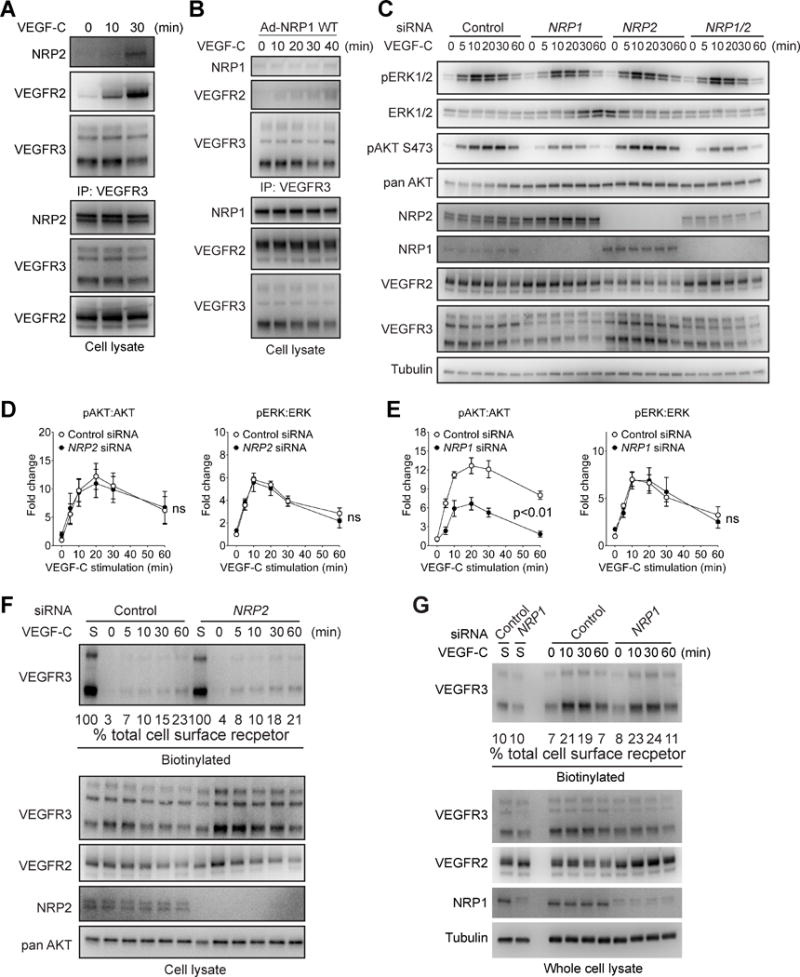
(A, B) Serum-starved HDLECs were stimulated with 100 ng/ml VEGF-C and then subjected to Western blotting with anti-NRP2 (A) or NRP1 (B) antibodies of VEGFR3 antibody immunoprecipitates (upper panels). Lower panels: Western blotting of whole cell lysates. (C) HDLECs transfected with NRP1, NRP2 or control siRNAs were serum-starved and stimulated with 100 ng/ml VEGF-C. Activation of VEGFR3 signaling was examined by Western blotting as indicated. (D–E) Quantification of ERK and AKT phosphorylation after NRP2 (D) or NRP1 (E) knockdowns. Data represents Mean±SEM of three independent experiments. Statistical analysis was performed using two way’s ANOVA. (F–G) VEGF-C-induced VEGFR3 internalization following NRP2 (F) or NRP1 (G) knocked down and control HDLECs was determined by serial Western blotting of cell lysates after cell surface biotinylation as described in Methods. siRNAs was determined by Western blotting of cell lysates at indicated time points (upper panel). Quantification was accomplished by deriving the percentage of internalized VEGFR3 to the amount of VEGFR3 present on the cell surface prior to VEGF-C treatment (lane “S”). Note that 100% of lane S sample was loaded in Panel F and 10% of lane S loaded in panel G. The numbers below the blots refer to percentage of internalized VEGFR3.
To determine if either NRP is involved in VEGF-C-induced VEGFR3 internalization, we followed the fate of surface biotinylated VEGFR3 following VEGF-C stimulation. A knockdown of neither NRP2 (Fig. 3F) nor NRP1 (Fig. 3G) had any significant effect on VEGF-C-induced VEGFR3 internalization. Both NRPs also had no significant role in formation of VEGF-C-induced VEGFR2/R3 complex (Fig. SIII A,B). A knockdown of VEGFR2 did not have any effect on VEGF-C induced VEGFR3/NRP2 interaction (Fig SIII C).
VE-PTP regulates VEGF-C/VEGFR3 signaling
To determine if a protein tyrosine phosphatase is involved in regulation of VEGFR3 signaling, we knocked down PTPs expressed in LEC and observed the effect of these knock-downs on VEGF-C-dependent activation of ERK and AKT activation. While knockdowns of PTP1B, PTPu, Src homology region 2 domain-containing phosphatase-1 (SHP1) and DEP-1 did not show any significant impact on VEGF-C-induced ERK and AKT activation, a VE-PTP knockdown significantly activated these pathways (Fig. SIV A,B and Fig 4A,B). In agreement with a previously established role of VE-PTP in regulation of VEGFR2 phosphorylation9, 19, the activation of this receptor, as judged by Y1175 phosphorylation, was also increased (Fig. 4A,B).
Figure 4. VE-PTP negatively regulates VEGFR3 signaling and endocytosis.
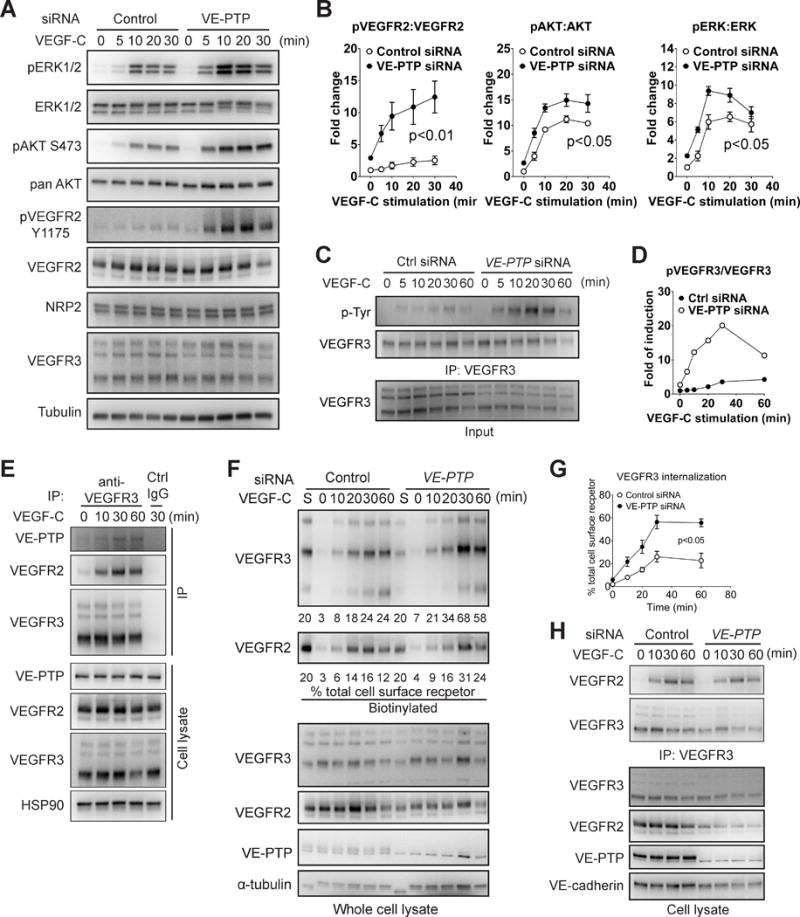
(A) HDLECs transfected with VE-PTP or control siRNAs were serum-starved and stimulated with 100 ng/ml VEGF-C. Activation of VEGFR3 signaling was examined by Western blotting as indicated. (B) Quantitative analyses of ERK, AKT and VEGFR2 phosphorylation shown in panel A. Data represents mean±SEM of three independent experiments. Statistical analysis was performed using two way’s ANOVA. (C) HDLECs transfected with VE-PTP or control siRNAs were serum-starved and stimulated with 100 ng/ml VEGF-C. VEGFR3 tyrosine phosphorylation was examined by blotting of VEGFR3 antibody immunoprecipitate with anti-tyrosine antibody. (D) Quantification of panel C. Data represents mean±SEM of three independent experiments. (E) Serum-starved HDLECs were stimulated with 100 ng/ml VEGF-C and then VEGFR3/VE-PTP interaction was examined by Western blotting with anti-VE-PTP antibody of VEGFR3 immunoprecipitate. Ctrl IgG: negative control. (F–G) Surface biotinylation assay analysis of VEGF-C-induced VEGFR3 internalization following HDLEC treatment with VE-PTP or control siRNAs. Upper panel: Western blot analysis of internalized VEGFR3. Lower panel: Western blots of total cell lysates corresponding to the upper panel. Quantification was performed as described in Fig 3F,G. Note that 20% of lane S sample was loaded. Data in G is a summary of the quantification and represented mean±SEM of at least three independent experiments. Statistical analysis was performed using two way’s ANOVA. (H) Serum-starved HDLECs treated with anti-VE-PTP or control siRNAs were stimulated with 100 ng/ml VEGF-C followed by Western blotting with anti-VEGFR2 antibody of VEGFR3 immunoprecipiates. VEGFR3/VEGFR2 interaction was determined by IP using antibodies against VEGFR3 and western blot.
To find out if VE-PTP regulates VEGFR3 phosphorylation, we performed phosphotyrosine blotting of immunoprecipitated VEGFR3 following VEGF-C stimulation from HDLEC treated with VE-PTP or control siRNA sequences. VE-PTP knockdown resulted in a nearly 6 fold increase in VEGFR3 tyrosine phosphorylation compared to control knockdown (Fig. 4C, D). To further confirm the interaction between these two molecules, we next examined the presence of VE-PTP in VEGFR3 immunoprecipitate. While essentially no VE-PTP was present prior to VEGF-C treatment, there was a marked increase in its presence following VEGF-C exposure (Fig 4E).
The presence of VE-PTP in fact also has a significant effect on VEGFR3 internalization. Using a cell surface biotinylation assay, we observed a significant increase in VEGF-C-induced VEGFR3 internalization after siRNA VE-PTP knockdown (Fig 4F,G). However, VE-PTP is not involved in VEGFR2/VEGFR3 interaction as knocking down VE-PTP had no effect on the kinetics or extent of complex formation (Fig. 4H).
VEGFR2 and NRP1 are required for VEGF-C-induced cell migration
Having established roles played by NRPs, VE-PTP and VEGFR2 in VEGFR3 signaling we next examined the effect of these signaling changes on LEC migration using a wound healing assay. Knockdown of VEGFR2 and NRP1 abolished cell migration of HDLEC in response to VEGF-C while knocking down VE-PTP strikingly showed no effect (Fig. 5A). In agreement with the minor role played by NRP2 in VEGFR3-dependent ERK and AKT activation, its knockdown had only a marginal effect on VEGF-C induced migration (Fig 5A).
Figure 5. VEGFR2 and NRP1 but not VE-PTP or NRP2 are required for VEGF-C-induced cell migration.
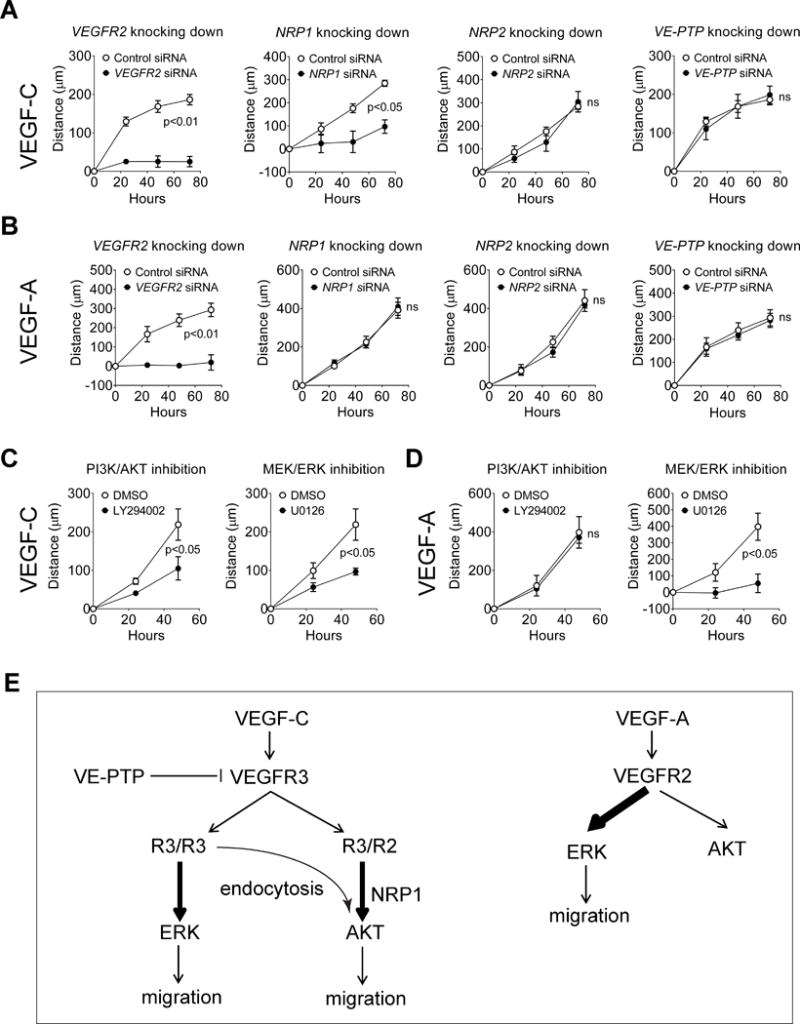
(A–B) Effect of knockdown of VEGFR2, NRP1, NRP2 and VE-PTP on VEGF-C- (A) and VEGF-A- (B) induced migration of HDLECs. (C–D) Effect of inhibition of MEK/ERK or PI3K/AKT pathways on VEGF-C- (C) and VEGF-A- (D) induced migration of HDLECs. In A-D, data represent the results of the distance with VEGF stimulation minus that without VEGF. Therefore they specifically show VEGF-induced migration. Data in A–D represent mean±SEM of at least four independent experiments. Statistical analyses were performed using two way’s ANOVA. (E) A schematic model of VEGF-C/VEGFR3 signaling.
To compare the role of the same proteins in VEGF-A signaling, we examined the effect of their knockdown on VEGF-A-induced HDLEC migration. As expected, knockdown of VEGFR2 completely abolished VEGF-A-induced cell migration (Fig. 5B). However, we did not observe any significant effect of NRP1 knockdown on VEGF-A-induced cell migration, indicating that NRP1 plays a distinct role in VEGF-C vs. VEGF-A-induced cell migration. Similar to our observation in VEGF-C-induced cell migration, knockdown of NRP2 and VE-PTP did not show any effect on the ability of HDLEC to migrate in response to VEGF-A, indicating that both of the two molecules are likely dispensable for these processes. Lastly, we investigated the importance of ERK and AKT activation in VEGF-C- vs. VEGF-A-induced cell migration. Inhibition of either ERK or AKT suppressed VEGF-C-induced cell migration while only ERK but not AKT inhibition suppressed VEGF-A-induced cell migration (Fig 5C,D).
DISCUSSION
The results of this study define relative contributions of various VEGFR3 partners to VEGF-C-induced signaling in LECs. In particular we find that while VEGFR3/VEGFR2 complex formation is critical to VEGF-C-induced AKT activation, ERK activation by this growth factor is primarily driven by VEGFR3 homodimer. Furthermore, whereas NRP1 plays an essential role in AKT activation, it is dispensable for ERK activation. At the same time, a plasma membrane protein VE-PTP acts as a VEGFR3 tyrosine phosphatase and modulates both ERK and AKT activation by VEGF-C. Functionally, both ERK and AKT pathways play an equally important role in regulating LEC migration (Fig 5E).
One unexpected finding is the role of VEGFR2/R3 complex in VEGF-C driven AKT activation. The role of VEGFR2 in this complex is apparently to bring in its partner NRP1. This is suggested by the fact that VEGFR2 kinase activity is not required for either the complex formation or AKT activation as well as by the observed decrease in AKT activation following NRP1 knockdown. The fact that Nrp1 was not detected on co-IP with VEGFR2 is likely attributable to very low levels of this protein in LECs. The function of NRP1 in this setting is distinctly different from the role it plays in VEGFR2-driven ERK activation where it is involved in intracellular trafficking of VEGFR27. How NRP1 facilitates AKT activation in the case of VEGR3/R2 complex is not known.
Taken together with the observation that knockdown of VEGFR2 and NRP1 did not affect VEGFR3 endocytosis and ERK activation but specifically abolished AKT activation, our data argues that VEGFR3-driven ERK but not AKT activation is dependent on receptor endocytosis. While this endocytosis dependence of VEGFR3/ERK signaling is similar to that of VEGFR2 activation of ERK22, in the distinction to the latter VEGFR3 intracellular signaling does not seem affected by its subsequent intracellular trafficking as neither an intracellular phosphatase PTP1b nor synectin, regulators of VEGFR2 trafficking, affected this pathway. These findings are further in agreement with recent in vivo demonstration of the role of VEGFR2 in lymphatic vessel morphogenesis24.
The other key finding is the role of VE-PTP in modulating of VEGFR3 signaling. VE-PTP knockdown enhanced both ERK and AKT activation. The effect of VE-PTP is likely attributable to a direct dephosphorylation of VEGF-C-activated VEGFR3 on the plasma cell membrane thus diminishing ERK and AKT activation. While VEGFR3/VE-PTP complexes have not been previously reported, we observed co-immunoprecipitation of the two proteins following, but not before, VEGF-C treatment of HDLECs. An unexpected finding is that VE-PTP knockdown does not increase HDLEC migration above baseline. This is likely due to the fact that migration is already fully activated by the normal VEGF signaling.
Another unexpected finding is the lack of significant contribution of NRP2 to VEGFR3 signaling despite of its role in lymphatic biology18. The two proteins do form a complex as suggested by immunoprecipitation experiments25, but there seems to be little impact from NRP2 knockdown on either VEGF-C-driven ERK and AKT activation or LEC migration. This may reflect intrinsic limitations of an in vitro study although it should be noted that there is no clear data supporting or refuting NRP2 contributions to lymphatics biology in vivo. In addition, AKT activation in this study was dispensable for VEGF-A-induced LEC migration though a previous report suggested its involvement in this process.24 The reason for this difference is not clear.
In summary, these studies have uncovered novel feature of VEGFR3 lymphatic endothelium signaling involving regulation of AKT activation via VEGFR3/VEGFR2/NRP1 complex, ERK via VEGFR3/R3 homodimer as well as regulatory roles of VE-PTP.
Supplementary Material
Significance.
Lymphatic endothelial cells play a key biological role in a number of disease processes. Yet our knowledge of how signal transduction takes place in this cell type is incomplete. The principle signaling receptors in the lymphatic endothelium are vascular endothelial growth factor receptor 2 and 3. In this study we investigated how these receptors transmit signal from VEGF-C, the growth factor responsible for much of the lymphatic biology. We find that VEGFR3 homodimer activates ERK-1/2 pathway while VEGFR2/R3 complex activates AKT signaling. Furthermore, VEGFR3-driven ERK but not AKT activation is dependent on the receptor endocytosis while activation of both pathways is modulated by a transmembrane phosphatase VE-PTP.
Acknowledgments
We would like to thank Dr. D. Vestweber, Max Planck Institute for Molecular Biomedicine for kindly providing anti-VE-PTP antibodies.
This work was supported in part by NIH grants HL084619, HL107205 (MS) and a Leducq Foundation Transatlantic Network grant (YD, MS).
Abbreviation/Acronyms
- VEGF
Vascular endothelial growth factor
- VEGFR
Vascular endothelial growth factor receptor
- VE-PTP
Vascular endothelial phosphotyrosine phosphatase
- NRP
Neuropilin
- ERK
Extracellular signal-regulated kinases
- AKT
Protein kinase B
- PI3K
Phosphoinositide 3-kinase
- PTP
Phosphotyrosine phosphatase
- DEP1
Density-enhanced phosphatase-1
- LEC
Lymphatic endothelial cell
- HDLEC
Human dermal lymphatic endothelial cell
- SHP1
Src homology region 2 domain-containing phosphatase-1
- EEA1
Early Endosome Antigen 1
Footnotes
Disclosure: The authors have declared that no conflict of interest exists.
References
- 1.Shibuya M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. Journal of biochemistry. 2013;153:13–19. doi: 10.1093/jb/mvs136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437:169–183. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- 3.Alitalo K. The lymphatic vasculature in disease. Nat Med. 2011;17:1371–1380. doi: 10.1038/nm.2545. [DOI] [PubMed] [Google Scholar]
- 4.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 5.Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, et al. Notch-dependent vegfr3 upregulation allows angiogenesis without vegf-vegfr2 signalling. Nature. 2012;484:110–114. doi: 10.1038/nature10908. [DOI] [PubMed] [Google Scholar]
- 6.Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls vegfr-2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174:593–604. doi: 10.1083/jcb.200602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lanahan A, Zhang X, Fantin A, Zhuang Z, Rivera-Molina F, Speichinger K, et al. The neuropilin 1 cytoplasmic domain is required for vegf-a-dependent arteriogenesis. Developmental cell. 2013;25:156–168. doi: 10.1016/j.devcel.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lampugnani GM, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, et al. Contact inhibition of vegf-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase dep-1/cd148. J Cell Biol. 2003;161:793–804. doi: 10.1083/jcb.200209019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayashi M, Majumdar A, Li X, Adler J, Sun Z, Vertuani S, et al. Ve-ptp regulates vegfr2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nature communications. 2013;4:1672. doi: 10.1038/ncomms2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lanahan AA, Lech D, Dubrac A, Zhang J, Zhuang ZW, Eichmann A, et al. Ptp1b is a physiologic regulator of vegf signaling in endothelial cells. Circulation. 2014 doi: 10.1161/CIRCULATIONAHA.114.009683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, et al. Vegf receptor 2 endocytic trafficking regulates arterial morphogenesis. Developmental cell. 2010;18:713–724. doi: 10.1016/j.devcel.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salameh A, Galvagni F, Bardelli M, Bussolino F, Oliviero S. Direct recruitment of crk and grb2 to vegfr-3 induces proliferation, migration, and survival of endothelial cells through the activation of erk, akt, and jnk pathways. Blood. 2005;106:3423–3431. doi: 10.1182/blood-2005-04-1388. [DOI] [PubMed] [Google Scholar]
- 13.Dixelius J, Makinen T, Wirzenius M, Karkkainen MJ, Wernstedt C, Alitalo K, et al. Ligand-induced vascular endothelial growth factor receptor-3 (vegfr-3) heterodimerization with vegfr-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. The Journal of biological chemistry. 2003;278:40973–40979. doi: 10.1074/jbc.M304499200. [DOI] [PubMed] [Google Scholar]
- 14.Nilsson I, Bahram F, Li X, Gualandi L, Koch S, Jarvius M, et al. Vegf receptor 2/-3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. The EMBO journal. 2010;29:1377–1388. doi: 10.1038/emboj.2010.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koch S. Neuropilin signalling in angiogenesis. Biochem Soc Trans. 2012;40:20–25. doi: 10.1042/BST20110689. [DOI] [PubMed] [Google Scholar]
- 16.Raimondi C, Ruhrberg C. Neuropilin signalling in vessels, neurons and tumours. Seminars in cell & developmental biology. 2013;24:172–178. doi: 10.1016/j.semcdb.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Bouvree K, Brunet I, Del Toro R, Gordon E, Prahst C, Cristofaro B, et al. Semaphorin3a, neuropilin-1, and plexina1 are required for lymphatic valve formation. Circulation research. 2012 doi: 10.1161/CIRCRESAHA.112.269316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Y, Yuan L, Mak J, Pardanaud L, Caunt M, Kasman I, et al. Neuropilin-2 mediates vegf-c-induced lymphatic sprouting together with vegfr3. J Cell Biol. 2010;188:115–130. doi: 10.1083/jcb.200903137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mellberg S, Dimberg A, Bahram F, Hayashi M, Rennel E, Ameur A, et al. Transcriptional profiling reveals a critical role for tyrosine phosphatase ve-ptp in regulation of vegfr2 activity and endothelial cell morphogenesis. FASEB J. 2009;23:1490–1502. doi: 10.1096/fj.08-123810. [DOI] [PubMed] [Google Scholar]
- 20.Au AC, Hernandez PA, Lieber E, Nadroo AM, Shen YM, Kelley KA, et al. Protein tyrosine phosphatase ptpn14 is a regulator of lymphatic function and choanal development in humans. American journal of human genetics. 2010;87:436–444. doi: 10.1016/j.ajhg.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Makinen T, Veikkola T, Mustjoki S, Karpanen T, Catimel B, Nice EC, et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the vegf-c/d receptor vegfr-3. The EMBO journal. 2001;20:4762–4773. doi: 10.1093/emboj/20.17.4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simons M. An inside view: Vegf receptor trafficking and signaling. Physiology (Bethesda) 2012;27:213–222. doi: 10.1152/physiol.00016.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanes MG, Oubaha M, Rautureau Y, Gratton JP. Phosphorylation of tyrosine 801 of vascular endothelial growth factor receptor-2 is necessary for akt-dependent endothelial nitric-oxide synthase activation and nitric oxide release from endothelial cells. The Journal of biological chemistry. 2007;282:10660–10669. doi: 10.1074/jbc.M609048200. [DOI] [PubMed] [Google Scholar]
- 24.Dellinger MT, Meadows SM, Wynne K, Cleaver O, Brekken RA. Vascular endothelial growth factor receptor-2 promotes the development of the lymphatic vasculature. PloS one. 2013;8:e74686. doi: 10.1371/journal.pone.0074686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karpanen T, Heckman CA, Keskitalo S, Jeltsch M, Ollila H, Neufeld G, et al. Functional interaction of vegf-c and vegf-d with neuropilin receptors. FASEB J. 2006;20:1462–1472. doi: 10.1096/fj.05-5646com. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.