

Abstract
The mechanistic link of ketosis to neuroprotection under certain pathological conditions continues to be explored. We investigated whether chronic ketosis induced by ketogenic diet results in the partitioning of ketone bodies towards oxidative metabolism in brain. We hypothesized that diet-induced ketosis results in increased shunting of ketone bodies towards citric acid cycle (CAC) and amino acids with decreased carbon shunting from glucose. Rats were fed standard (STD) or ketogenic (KG) diets for 3.5 weeks and then infused with [U-13C]glucose or [U-13C]acetoacetate tracers. Concentrations and 13C-labeling pattern of CAC intermediates and amino acids were analyzed from brain homogenates using stable isotopomer mass spectrometry analysis. The contribution of [U-13C]glucose to acetyl-CoA and amino acids decreased by ~30% in the KG group vs STD, whereas [U-13C]acetoacetate contributions were more than 2-fold higher. The concentration of GABA remained constant across all groups; however, the 13C-labeling of GABA was markedly increased in the KG group infused with [U-13C]acetoacetate compared to STD. This study reveals that there is a significant contribution of ketone bodies to oxidative metabolism and GABA in diet-induced ketosis. We propose that this represents a fundamental mechanism of neuroprotection under pathological conditions.
Keywords: Ketone bodies, glucose, citric acid cycle, oxidative metabolism, GABA, brain amino acids
Introduction
The ketogenic diet has long been associated with neuroprotection but the underlying mechanisms remain elusive. The ketogenic diet has been primarily used as a clinical dietary treatment in children with drug-resistant epilepsy which has shown efficacy by significant reductions in frequency, duration of seizures, or becoming seizure free (Lennox 1928; Freeman et al. 1998;Wilder 1921;Neal et al. 2008; Taub et al. 2014). In animal studies, the ketogenic diet has been reported to be protective against epilepsy, ischemia-induced reperfusion injuries, traumatic brain injuries, as well as hypoxic insults (Bough and Eagles 1999;Masuda et al. 2005;Prins and Hovda 2009;Suzuki et al. 2002). Ketosis is a metabolic state resulting from hepatic production of ketone bodies (acetoacetate and β-hydroxybutyrate) via increased mitochondrial β-oxidation of fats. Overproduction of ketone bodies by the liver results in elevated blood levels that are metabolized by extra-hepatic tissues such as the brain (Hawkins et al. 1971;Williamson et al. 1971).
Researchers have speculated on mechanisms that link ketosis to neuroprotection (Freeman et al. 1998; Nebeling et al. 1995; Prins 2008; Puchowicz et al. 2008; Schwartzkroin 1999; Stafstrom and Rho 2012; Swink et al. 1997; Tallian et al. 1998; Uhlemann and Neims 1972; Veech 2004; Xu et al. 2010; Noh et al. 2008). Ketosis and neuroprotection are linked through metabolic regulation via four mechanisms: i) the “glucose sparing” effect which suggests that a decrease in glucose utilization and oxidation may be beneficial for brain function during recovery from neurological damage (LaManna et al. 2009; Zhang et al. 2013b), ii) the presence of brain ketone bodies in the reduction of glutamate neurotoxicity and promotion of GABA synthesis (Noh et al. 2006), iii) brain adaptation to chronic ketosis by induction of molecular regulatory proteins, such as monocarboxylate transporters (MCT) (Leino et al. 2001;Vannucci and Simpson 2003) and hypoxia-inducible factor (HIF1-α) that accounts for angiogenesis (Puchowicz et al. 2008), and iv) the reduction of reactive oxygen species (ROS) and subsequent oxidative stress in mitochondria (Bough and Eagles 1999; Maalouf et al. 2009; Sullivan et al. 2004).
The mechanism associated with the partitioning of fuels (as with glucose sparing) occurs in the cell during oxidative metabolism when there is an abundance of acetyl-CoA as a result of metabolic demand and regulation of glucose vs. ketone body oxidation. The percent contribution of fuels to oxidative metabolism can be defined as the amount of substrate entering the citric acid cycle (CAC) and amino acid pools. The mechanism associated with GABA (an amino acid and major inhibitory neurotransmitter) synthesis is through the oxidation of glutamate (via α-ketoglutarate) and is thought to explain seizure control in epilepsy (Melo et al. 2006; Waagepetersen et al. 1999; Yudkoff et al. 2008). The cellular compartmentalization of glutamate plays an important role in the balance between homeostasis and cytotoxicity. The production of GABA from neuronal glutamate in GABAergic neurons must maintain a metabolic balance with glial glutamine (Cerdan et al. 1990; Kunnecke et al. 1993). A classic study showed that GABA synthesis results in significant increases in glutamine labeling following infusions of 13C labeled acetate tracers (a glial-specific substrate) in ketotic mice (Yudkoff et al. 2005). The rationale was that both acetate and ketone bodies readily enter the CAC, bypassing pyruvate dehydrogenase (PDH, E.C number: 1.2.4.1) and pyruvate carboxylase (PC, E.C number: 6.4.1.1). It was proposed that ketosis may enhance glial glutamine synthesis through a ‘buffering process” allowing GABA synthesis in neurons via glial glutamine (Yudkoff et al. 2008; Greene et al. 2003;Maalouf et al. 2009). In short, the neuroprotective mechanisms of ketosis could be explained by the metabolic regulation of ketone body oxidation through the bypassing of PDH, as with glucose oxidation, and/or through stabilized GABA synthesis. What has remained unclear is the metabolic regulation of glucose in brain during chronic ketotic conditions. This is of particular interest to investigators studying defects in glucose metabolism associated with neuropathology and disease. To our knowledge, there is a lack of a quantitative data that directly measure the utilization of glucose vs. ketone bodies following metabolic adaptation to diet-induced ketosis. It also remains unclear how the partitioning of glucose and ketone bodies changes the capacity of the ‘buffering’ process of glial glutamine towards GABA synthesis.
Using Positron Emission Tomography (PET) and 2-18Fluoro-2-deoxy-D-glucose (FDG) tracer, we have recently shown that diet-induced ketosis suppresses cerebral metabolic rate of glucose (CMRglc) in rats (Zhang et al. 2013b). The basis of the study was founded on the concept that glucose oxidative metabolism is ‘spared’ while cerebral oxygen metabolic rate (CMRO2) was assumed to stays constant during ketosis (Dahlquist and Persson. 1976). A reduction in CMRglc with ketosis was shown to correlate with levels of ketosis. We also considered that CMRglc by PET analysis reflected the steady state phosphorylation rate of glucose (glucose conversion to glucose-6-phosphate; G6P), which only accounts for the first steps in glycolysis. The complete oxidation of glucose also includes the downstream metabolism from G6P towards production of pyruvate and entry into the CAC at the level of acetyl-CoA, as well as the partitioning of glucose away from the CAC, possibly towards brain amino acid synthesis.
In the present study we investigated whether diet-induced ketosis spares glucose oxidation by reducing glucose-derived carbon entry into brain CAC through the partitioning of substrates. A stable in vivo chronic ketotic rat model and sensitive analytical methods were applied to measure the fractional contribution of glucose and ketone bodies to CAC intermediates and amino acids in brain. This approach involved intravenous infusions of stable isotopes, [13C]glucose and [13C]acetoacetate, and mass spectrometry in lightly anesthetized rats. We presumed that the overall energy metabolism (CMRO2) remains constant in ketotic brain (Zhang et al. 2013b, Dahlquist and Persson. 1976). We hypothesized that there would be an increase in ketone body contribution to CAC and GABA through glutamate (Hasselbalch et al. 1994;Hawkins et al. 1971;Prins and Hovda 2009;Zhang et al. 2013a;Zhang et al. 2013b;Jiang et al. 2011) and a sparing of glucose oxidation with ketosis. We theorized that the enhancement of GABA synthesis under ketosis might explain the antiepileptic effects of the ketogenic diet.
Materials and Methods
Chemicals
All chemicals were purchased from Sigma-Aldrich. [U13C]glucose (99.8 %) was purchased from Sigma-Isotec (St. Louis, MO, USA, Cat#389374); A 38.7 mM [U13C]glucose solution was prepared in 0.9 % NaCl. [U13C]acetoacetate ([U13C]AcAc) was derived from [U13C]ethylacetoacetate, (purchased from Sigma-Aldrich, St. Louis, MO, USA, Cat# CX1474), and prepared in 137 mM. [2H9]pentanoyl-CoA was prepared as previously described (Harris et al. 2013).
Biological experiments
Young adult male Wistar rats (Charles River, Wilmington MA, USA) weighing 150–200 grams at 40 days old were used in the study, with all procedures performed in strict accordance with the National Institutes of Health Guide for Care and were approved by Institutional Animal Care and Use Committee (IACUC) of Case Western Reserve University. Body weights were measured upon arrival and on the experimental day (Table 1). Littermates were housed in the Case Western Reserve University Animal Resource Center with 12h–12h light-dark cycle. All rats were allowed to acclimate for 1 week prior to initiating dietary protocols. Standard rodent (STD) diet was fed to all rats during the acclimation period (Cincinnati Lab Supply, Cincinnati, OH, USA, Labdiet Prolab RMH3000 5ANE). The ketogenic diet contains ~89% energy by fat, 10.4% energy by protein (Table S1). One week after their arrival, all rats were fasted overnight for 16 hours to deplete the liver glycogen storage and initiate ketosis. Rats were then randomly assigned to two diets, standard chow (STD) or ketogenic (ketogenic, KG; Research Diet, New Brunswick, NJ, USA, D12369b) and fed for 3–4 weeks ad libitum until the experiment day (Puchowicz et al. 2007;Zhang et al. 2013b). The STD (n=13) and KG (n=13) rats were further randomly assigned to receive infusions of either [U13C]glucose or [U13C]AcAc tracers. A total of 4 groups of rats were assigned.
Table 1.
Physiologic parameters. Rats were divided into four study groups: standard chow (STD) and ketogenic (KG), constantly infused with either [U13C]glucose or [U13C]AcAc (acetoacetate) for 50 minutes.
| Study 1: [U13C]glucose | Study 2: [U13C]AcAc | |||
|---|---|---|---|---|
|
| ||||
| Diet groups | STD (n=6) | KG (n=7) | STD (n=7) | KG (n=6) |
| Age (days) | 81 ± 12 | 69 ± 7 | 68 ± 6 | 77 ± 3* |
| Weight (g) | 363 ± 52 | 340 ± 32 | 340 ± 27 | 376 ± 29* |
| Arterial pH | 7.35 ± 0.02 | 7.33 ± 0.04 | 7.38 ± 0.07 | 7.37 ± 0.03 |
| Breath Rate (/min) | 62 ± 3 | 67 ± 5 | 68 ± 4 | 68 ± 4 |
| Hematocrit (%) | 45 ± 1 | 44 ± 2 | 42 ± 2 | 46 ± 2* |
| Plasma Parameters (t=50 min) | ||||
| BHB (mM) | 0.29 ± 0.17 | 2.28 ± 0.54* | 0.25 ± 0.10 | 3.91 ± 2.14* |
| AcAc (mM) | 0.17 ± 0.06 | 0.43 ± 0.07* | 0.26 ± 0.12 | 1.02 ± 0.52*† |
| BHB+AcAc (mM) | 0.45 ± 0.23 | 2.53 ± 0.35* | 0.51 ± 0.22 | 4.94 ± 2.60*† |
| BHB/AcAc ratio | 1.64 ± 0.34 | 4.97 ± 0.74* | 1.03 ± 0.19† | 3.82 ± 0.85*† |
| L-Lactate (mM) | 1.15 ± 0.31 | 0.68 ± 0.12* | 1.22 ± 0.22 | 0.75 ± 0.16* |
| D-Glucose (mM) | 10.1 ± 0.8 | 10.8 ± 1.3 | 9.3 ± 1.3 | 10.3 ± 1.6 |
| Brain Tissue Ketone Body Concentrations | ||||
| BHB (μmol/mg) | 34.10 ± 5.11 | 206.85 ± 57.67* | 36.21 ± 7.57 | 333.41 ± 162.80* |
| AcAc (μmol/mg) | 5.75 ± 2.35 | 29.71 ± 8.41* | 6.80 ± 1.89 | 45.62 ± 22.43* |
All data presented are mean ± SD.
p<0.05, KG compared to STD group with the same 13C- tracer infusion group;
p<0.05 when comparing the same diet but different 13C-tracer infusion group.
On the experimental day (after 3.5 weeks of diet) rats were fasted in the morning for 6 hours prior to infusion of [U13C] tracers. Rats were then anesthetized with vaporized 1.5% isoflurane balanced with pure oxygen delivered through a nose cone during the surgical placement of arterial and venous catheters: right jugular catheter (MRE, 0.035 mm ID and 0.084mm OD, Braintree Scientific Inc, Braintree, MA, USA) was advanced towards the atrium for isotope tracer infusion and the tail artery was cannulated with the same type of catheter for blood sampling during the experiment period. [U13C]tracers were infused at a constant rate via the jugular vein catheter (Harvard Apparatus syringe pump-11 Plus) for 50 min while maintained on isoflurane (1–2%); oxygen flow rate (0.05–0.2 liters per minute) and air flow rate (0.5–0.6 liters per minute) were adjusted to achieve a consistent physiological status across animals. Establishment of consistent, stable physiological status is essential as the anesthesia levels are known to affect substrate oxidation (Chowdhury et al. 2014). Absence of hind-leg pinch reflex was monitored throughout the experiment to ensure depth of anesthesia. Heart rate, respiratory rate (breaths/min), plethysmography and arterial oxygen saturation (%) were monitored via hind leg senor and recorded throughout the experiment using a pulse oximeter system (MouseOx, Starr life sciences, Oakmont, PA, USA) (Table 1). To maintain breath rates (~70 per minute) and normal blood gases throughout the 50 minutes infusion process, isoflurane was adjusted, as well as the oxygen percentage and flow rates (20–30%). The breath and heart rates were also recorded throughout the experimental process and were used as indicators for physiological status. Arterial blood pH were measured at t=0, 45 min (ABL5 Radiometer, Copenhagen, Denmark) to ensure the absence of respiratory acidosis.
Tracers were infused intravenously at constant rates: [U13C]glucose at 0.5mmol/kg/hr or [U13C]acetoacetate ([U13C]AcAc) at 0.5–1.0 mmol/kg/hr. The infusion rates were based on the estimated whole body substrate turnover for glucose and ketone bodies; the target plasma enrichments for [13C]glucose (M+6) was aimed at 10% and [13C]acetoacetate (M+4) was aimed at 20% (Zhang et al. 2013a).
To verify that the infusions of the tracers of glucose or ketone bodies were at steady-state conditions, blood samples (100 μl) were taken from the tail artery at time point 0 (pre-infusion), and at 15, 30, 40, 50 minutes, and then immediately centrifuged. The plasma were frozen for GC-MS analysis for [U13C]tracer enrichments and concentrations of glucose and acetoacetate (AcAc), as well as beta-hydroxybutyrate (BHB). In addition, plasma D-glucose and L-lactate were also measured by YSI 2700 Biochemistry Analyzer (YSI Inc., Yellow Springs, OH, USA) at 40 minutes of infusions. The total amount of blood drawn from each animal during the experimental time was less than 1.5ml.
At the end of infusions, the rats were decapitated, brains were dissected immediately, frozen in liquid nitrogen within 30 seconds of removal and stored at −80 °C. Cortical sections (200–300 mg tissue) were then dissected under frozen conditions in dry ice (−78.5°C). For the isolation of metabolic intermediates the frozen tissue samples were then homogenized using an organic solvent mixture containing 5% acetic acid and methanol (1:1; methanol to water) (Harris et al. 2013). Briefly, samples were spiked with internal standards: [2H9]pentanoyl-CoA (0.4 nmol); [2H6]BHB (0.057 μmol); [13C4]succinate (0.083 μmol); [13C5]glutamate (0.167 μmol) and (RS)-3-hydroxy[2H4]glutarate (0.049 μmol) and then homogenized with 3 ml of cold methanol-H20 solvent mixture (1:1, v/v) using a Polytron homogenizer and then centrifuged for 30 minutes at 2500 RCF. The supernatant fractions were collected and stored at −80 °C freezer for analytical measurements.
Analytical methods
To probe brain oxidative metabolism in vivo, we utilized mass spectrometry and 13C stable isotope tracer technology to analyze the effects of ketosis on the fractional contributions to the CAC and amino acids. 13C labeled intermediates of the CAC, GABA, glutamate, glutamine and aspartate were measured in brain homogenates. This approach enables isotopically labeled metabolites to be measured with a high degree of sensitivity, while infusing 13C tracers substrates at low amounts.
GC-MS assays
Following homogenization and centrifugation, the supernatant fractions were decanted and reserved for measurements of concentrations and [13C]label of acetyl-CoA enrichment (Zhang et al. 2013a). The tissue pellets were further extracted using a mixture of acetonitrile and 2-propanol (3:1), centrifuged and then analyzed for CAC, amino acids and related intermediates (Kombu et al. 2011;Yang et al. 2008). Extracts were then dried by nitrogen gas for 1–2 hours and chemically derivatized using MTBSTFA + 1% TBDMCS reagent (N-methyl-N-(tert-butyldimethylsilyl) trifluoroacetamide + 1% tert-butyldimetheylchlorosilane, Regis Technologies, Inc. Morton Grove, IL, USA) at reacted at 70 °C for 30 minutes. The derivatized products were measured under Agilent 6890 Gas-Chromatography and Agilent 5973 Mass Spectrometry (GC-MS). A DB-17 MS capillary column (30m × 0.25mm × 0.25 μm) was used in all analysis. The starting oven temperature was set to 80 °C, the pressure was 14.82 psi, and the flow velocity was 45cm/sec. Temperature was then increased to linearly to 220 °C and held for 1 min. The mass spectrometer was in electron-impact (EI), sim mode. CAC and related intermediates, such as BHB (m/z=159), AcAc (m/z=160), succinate (m/z= 289), fumarate (m/z =287), malate (m/z =419), citrate (m/z=459) were measured. Other intermediates and amino acids, including 3-hydroxyglutarate (m/z= 433), aspartate (m/z =418), glutamate (m/z =432), glutamine (m/z =431) and GABA (m/z =274) were also measured.
The plasma glucose enrichment was also measured using GC-MS (Zhang et al. 2013a). Briefly, the plasma glucose were dried and incubated with 100 μl of pyridine:acetic anhydride (2:3) at 75 °C for 30 minutes. An Agilent 6890 GC system were used to determine the selective ion (Ammonia chemical ionization; CI) m/z 408–414.
LC-MS assays
Acetyl-CoA in brain was measured by LC-MS methods, as previously described, with slight modifications (Harris et al. 2013;Zhang et al. 2013a). Briefly, the supernatant fraction was loaded onto a Supelco solid-phase extraction cartridge ([2-(pyridyl)-ethyl functionalized silica gel] preconditioned with 3 ml of methanol and then added 3 ml of buffer A (1:1 methanol-H2O with 2% acetic acid). The cartridge was then washed with 3 ml of buffer A to elute impurities, followed sequentially by 3 ml of buffer B (1:1 methanol-H2O with 50 mM ammonium formate), 3 ml of buffer C (3:1 methanol-H2O with 50 mM ammonium formate), and 3 ml of methanol to elute the acyl-CoAs. The eluent was evaporated under nitrogen.
The LC was coupled with an API4000 Qtrap Mass Spectormeter (Applied Biosystems, Foster City, CA) operated under positive ionization mode (Harris et al. 2013). Acetyl-CoA was measured at the 14.2 minute elution time and the ions 810/303 – 812/305 were monitored.
Calculations
Fractional contributions of [U-13C]glucose or [U-13C]AcAc to intermediary metabolism
The fractional isotopic enrichments (via 13C label incorporation) for each of the intermediates were defined as molar percent enrichment (MPE), after background correction using a matrix method (Fernandez et al. 1996). Briefly, the MPEs (unit: percent) is calculated as
Where MPE(M+n) is the n th 13C labeled mass isotopomer enrichment of the metabolite. A is the abundance (arbitrary unit) measured by GC-MS. The variable i varied from zero to n, while n is no more than 6. A(0) is the abundance non-labeled metabolite, and A(i) is the abundance for mass isotopomer labeled with i 13C, regardless of position.
The M+2 MPE of each of the intermediates reflected the contribution of [U-13C]glucose or [U-13C]AcAc to the intermediate pool. For [U-13C]glucose the assumption was that the first turn of the CAC produced most of the M+2 [13C]labeled intermediates from [1,2-13C]acetyl-CoA, i.e, PDH was playing the major role instead of PC (Lapidot and Gopher 1994;Mason et al. 1992). For the fate of [U13C]AcAc, its entry into CAC in the first turn also produced [1,2-13C]acetyl-CoA. Thus, the first turn of the CAC (as measured by M+2 for each intermediates) reflected oxidative metabolism (Figure 1). Although not a focus of this study, different [13C]labeling patterns (isotopomers) of the intermediates such as, M+1 and M+3 could be expected. M+1 occurs with recycling of pyruvate with malic enzyme activities after the first turn of CAC (Cerdan et al. 1990; Melo et al. 2006). The metabolism of [U13C]glucose to [U13C]pyruvate could also generate M+3 malate via PC, which labels other intermediates of the CAC as M+3.
Figure 1.
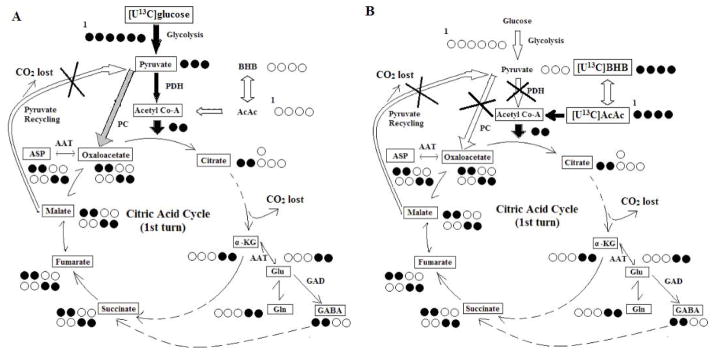
Schemes of metabolite 13C labeling patterns as a result of [U13C]glucose (Panel A) or [U13C]AcAc (Panel B) infusions. Schemes do not account for compartmentation of glutamine-glutamate cycling between glial cells and neurons and pyruvate recycling. All positional carbons are noted from left to right (C1–C6), and 13C are noted by filled black circles. The major entrance pathways of labeled 13C to citrate acid cycle (CAC) are noted in filled black arrows; the minor pathways are note by gray arrows. Pathways that do not contributes to first turn of CAC 13C labeling were marked with cross. Panel A: labeling patterns of brain metabolite isomers (M+2) from [U13C]glucose, only considering pyruvate dehydrogenase (PDH) pathway and first turn of CAC from citrate to oxaloacetate. Panel B: labeling patterns of brain metabolite isomers (M+2) from [U13C]AcAc as a result of the first turn of the CAC. ASP: aspartate; AAT, aspartate aminotransferase; α-KG, α –ketoglutarate; BHB:β-hydroxybutyrate; GAD :Glutamate acid decarboxylase; Glu : glutamate ; Gln : glutamine; PC, pyruvate carboxylase.
Concentrations of intermediates in brain
The concentrations of the endogenous tissue metabolites were determined by standard curves for each metabolite and the known amount internal standard added to the sample. For GABA and fumarate, the concentrations were calculated using the internal standard of [13C4] succinate; 2-hydroxyglutrate, aspartate, malate and citrate concentrations were calculated using (RS)-3-hydroxy-[2H4]glutarate; glutamine and glutamate concentrations were calculated using [13C5]glutamate; BHB and AcAc concentrations were calculated using [13C6]BHB and the acetyl-CoA concentration was calculated using [2H9]pentanoyl-CoA.
Statistical analysis
All values were presented as mean ± SD. The comparisons between two diet groups or between two infusion groups was analyzed with 2-tailed t tests. Statistical significance was considered at the level of p<0.05.
Results
Plasma 13C tracer enrichments (MPE)
Infusions of [U13C]glucose resulted in ~10% MPE M+6 of plasma glucose in both diet groups, similar with what we previously reported (Zhang et al. 2013a). These values were 9.1 ± 0.6 % (Mean ± SD) for the STD rats and 10.1 ± 1.3% for the KG rats and were not statistically different. Infusions of [U13C]AcAc resulted in 17–26% MPE M+4 of AcAc. The plasma MPE M+4 of BHB in STD and KG groups were not different (11.5 ± 1.4 % for STD and 11.0 ± 3.5 % for KG). The plasma MPE M+4 of AcAc in STD group was higher than the KG group (26.3 ± 4.2 % for STD and 17.0 ± 6.5 % for KG, p=0.01).
Physiologic parameters
The rats in all four groups were of similar age, weights and hematocrits (Table 1). Breath rates were monitored throughout the experiment to ensure consistency across groups. There were no signs of hypoglycemia or hyperglycemia during anesthesia and tracer infusions, as there were no significant changes in arterial blood glucose concentrations with KG diet compared to STD. Ketosis was evident by the significant increases in plasma and tissue ketone body concentrations. The range of ketosis (plasma total ketone body concentrations) reached in the KG diet groups was 1.7–3.9 mM. Plasma L-lactate levels were significantly lower in the KG groups compared to STD groups. Within the same dietary condition, tracer infusions ([U13C]glucose or [U13C]AcAc) did not significantly alter plasma BHB, glucose and lactate concentrations. Plasma BHB/AcAc ratios were significantly elevated in the ketogenic diet groups compared to STD, but remained within physiological range (Veech 2004).
Partitioning of glucose and ketone body oxidative metabolism with ketosis
The percentage of 13C labeling of CAC intermediates and amino acids was considered a result of the utilization from either [U13C]glucose or [U13C]AcAc. As indicated in Figure 1, the first turn of the CAC shows the 13C substrates label the two carbons of acetyl-CoA and downstream intermediates of CAC. Thus, the M+2 labeling pattern for CAC intermediates and amino acids was used to evaluate intermediary metabolism from the oxidation of [U13C]glucose or [U13C]AcAc. When [U-13C]glucose was administered, acetyl-CoA M+2 was 33% lower with ketosis compared to the STD group (Fig. 2A). Mass spectra analysis also revealed ~ 30% decreases in succinate M+2; similar M+2 trends were also observed with glutamate, glutamine and GABA.
Figure 2.

Brain metabolite M+2 percent 13C enrichments as a result of first turn of CAC. Panel A: metabolite 13C labeling patterns show generally decreased M+2 with ketosis in the group infused with [U13C]glucose. Panel B : metabolite 13C labeling patterns show increased M+2 with ketosis in the group infused with [U13C]AcAc. All data are presented as mean ± SD. *p<0.05 for ketogenic diet (KG) compared to standard (STD) diet. N.D., not detected. AcAc: acetoacetate.
In the [U13C]AcAc infusion study, acetyl-CoA M+2 increased ~2 fold with KG compared to STD diet (Fig. 2B). Analysis also revealed substantial increases in M+2 of all of the measured CAC intermediates from citrate through malate, as well as with glutamate, glutamine and GABA. Similar with acetyl-CoA, the M+2 enrichments increased greater than 2.5 fold with ketosis, with citrate reaching ~8 fold compared to STD. In the STD group no M+2 enrichment was detected in fumarate and GABA (Fig. 2 B). However, M+2 fumarate and GABA was detected with ketosis. These data show that with diet-induced ketosis, there was an increased contribution of ketone bodies to CAC intermediates and amino acids.
Concentrations of intermediates in ketotic brain
The metabolite concentrations (μmol/g) for each of the diet and infusion groups are shown in Figure 3. In the [U13C]glucose infusion study, malate concentrations were significantly increased, as well as glutamate and glutamine, by ~40% in the KG group compared to STD (Fig. 3A). Whereas in the groups infused with [U13C]AcAc, glutamate and glutamine concentrations significantly decreased ~35% in the KG group compared to STD. The concentrations of CAC intermediates followed similar trends as with the [U13C]glucose groups, with no significant differences between KG and STD groups, except for malate (Fig. 3B). GABA concentrations were not significantly different irrespective of dietary or infusion conditions (Fig 3A, B).
Figure 3.

Brain metabolite concentrations (sums of non-labeled metabolites plus 13C-labeled metabolites). Panel A : metabolite concentrations in the standard diet (STD) group vs ketogenic diet (KG) group, both infused with [U13C]glucose. Panel B : metabolite concentrations in STD and KG, both groups infused with [U13C]AcAc. All data are presented as mean ± SD. *p<0.05 compared to standard (STD) diet. AcAc :Acetoacetate.
Acetyl-coA concentrations in brain were not changed with dietary or infusion conditions ([U13C]glucose STD diet group 5.36±1.31 nmol/g and KG 6.87±2.02 nmol/g; for [U13C]AcAc infusion, STD was 6.65±2.24 nmol/g and KG was 6.44±2.19 nmol/g).
Partitioning of glucose and ketone bodies with ketosis: contribution of ketones to GABA
The effects of ketosis on the partitioning of substrates glucose and acetoacetate to GABA and glutamate are shown in Figure 4. The relative percent of carbon that contributed to GABA from a pool of glutamate enriched with [13C] label is presented as the ratios of GABA/glutamate. A large fraction of the carbon that contributed to 13C GABA from a pool of 13C glutamate was shown to be derived from [13C]glucose tracer irrespective of diet conditions (Figure 4A).
Figure 4.
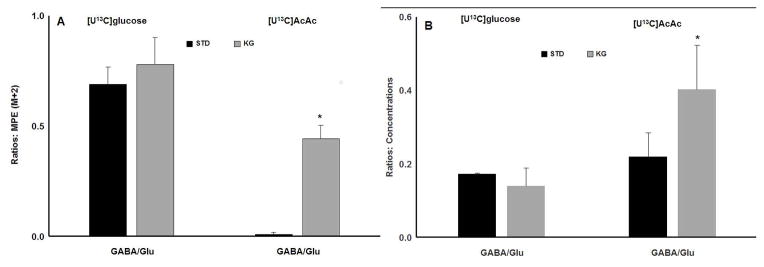
Brain amino acid ratios during ketosis. Panel A : GABA vs. Glutamate M+2 enrichment ratios for standard diet (STD) vs. ketogenic diet (KG) conditions, with either 13C tracer infusions. The M+2 enrichment indicates the contributions of the 13C tracer towards amino acids in the first turn of citrate acid cycle. Panel B: total brain GABA vs glutamate concentration (sum of 13C labeled metabolite plus non-labeled metabolite) ratios for STD vs. KG, with either 13C tracer infusions. All values are shown as mean ± SD. * p<0.05 when comparing the same infusion of [U13C] tracers but different diet groups. Glu: glutamate. Gln: glutamine. AcAc: acetoacetate.
The most dramatic effect of the KG diet on carbon entry into GABA and glutamate was with the STD rats infused with [13C]AcAc. In STD condition, there was a trace amount of 13C GABA carbon detected that comes from 13C glutamate (Figure 4A). The percentage of 13C labeled glutamate was significantly less in the STD group, compared to KG (see Figure 2B). In contrast, the KG group infused with [13C]AcAc tracer showed that the 13C labeled GABA/glutamate ratio was significantly increased compared to STD (Figure 4A), which was consistent with the large increase in GABA enrichment. Additionally, the glutamine/glutamate 13C ratios were not statistically different in the KG groups irrespective of [13C] tracer infused (ratios not graphed).
We also investigated whether ketosis and substrate partitioning affected pool sizes (μmol/g tissue) of GABA and glutamate (Figure 4B). For this purpose, the 13C labeled and non-labeled GABA/glutamate and glutamine/glutamate total concentration ratios were calculated for each of the groups. Consistent with the 13C label incorporation ratios (Figure 4A), there were no significant changes in the GABA/glutamate and glutamine/glutamate concentration ratios (ratio not graphed) in the dietary groups infused with [U13C]glucose (Figure 4B). Although there were no significant changes in total GABA concentrations with diet or infusion conditions (Figure 3), there were significant increases in the GABA/glutamate concentration ratio in the KG diet group infused with [U13C]AcAc tracer, compared to STD (Figure 4B). This can be attributed to the 3-fold decrease in glutamate pool (both in ketotic group) compared to the group infused with [13C]glucose tracer (see Figure 3A vs 3B). The glutamine/glutamate concentration ratio was modestly increased in the KG group infused with [U13C]AcAc tracer compared to STD (0.62±0.05 in STD and 0.71±0.03 in KG, p=0.01). The physiological relevance may be attributed to a greater decrease in glutamate pool size vs glutamine in ketosis, compared to STD (Figure 3B).
Discussion
Our diet-induced ketotic rat model includes metabolic adaptation and stabilization of ketosis which we consider necessary for neuroprotection (Puchowicz et al. 2007;Puchowicz et al. 2008). Adaptation to chronic ketosis includes up-regulation of monocarboxylate transporters (MCT) at the blood brain barrier (Gjedde and Crone 1975;Leino et al. 2001;Vannucci and Simpson 2003) and moderate to high blood levels of ketosis (2–6 mM) (Leino et al. 2001;Prins and Hovda 2009). However, the extent at which the brain switches from glucose to ketone oxidation with short term fasting or acute ketone body administrations has not been well defined. In the current study, we used 13C stable isotope analysis and an established chronic diet-induced ketotic rat model and demonstrated that the glucose and ketone bodies partitioned differently in both brain CAC and amino acids.
Contribution of ketone bodies to oxidative metabolism
13C stable isotopomer analysis revealed that brain metabolism of [U13C]glucose and [U13C]AcAc were distinctive in ketotic brain compared to non-ketotic brain. The 13C label incorporation from [U13C]AcAc into acetyl-CoA, CAC intermediates and related amino acids were significantly increased in KG as compared to STD. In contrast, 13C label incorporation from [U13C]glucose into acetyl-CoA, CAC intermediates and related amino acids were significantly decreased in KG compared to STD. These findings are consistent with previous reports that ketosis spares glucose utilization through decreased phosphorylation rates (LaManna et al. 2009;Zhang et al. 2013b;Hasselbalch et al. 1994;Redies et al. 1989). In support of our data, it has been recently described that ketone body oxidation can account up to 62% of neuronal substrate oxidation (Chowdhury et al. 2014). The oxidation of ketone bodies result in increased production of acetyl-CoA which has been described to inhibit glycolysis at the level of PDH (Melo et al. 2006; Yudkoff et al. 2005). In this study, the use of stable isotopomer analysis enables us to determine the effect of ketosis on glucose oxidation downstream of PDH at the level of CAC and amino acids. The measurements of 13C labeled intermediates revealed decreased glucose entry downstream of PDH with chronic ketosis. Moreover, there were no significant changes in CAC and acetyl-CoA concentrations with diet or infusion conditions, and the brain amino acids concentration values were similar to what has been previously reported (Yudkoff et al. 2001; Jiang et al. 2011), indicating that chronic ketosis resulted in a metabolic partitioning of substrates without altering pool sizes of the intermediates.
Additionally, in the [U13C]glucose infusion study, the contribution of 13C labeled glucose to GABA, glutamate and glutamine decreased proportionately (approximately 35%) in KG group compared to STD. These data advocate that glucose is the preferred substrate for amino acid synthesis under STD diet conditions. Unexpectedly, there was a significant increase in glutamate and glutamine concentrations with KG diet and [U13C]glucose infusion. We interpret these findings as a result of the combination of chronic ketosis and infusions of [U13C]glucose tracer, irrespective of the lack of change in plasma glucose concentrations or the low amount of tracer infused. Another explanation could be that increased glucose partitioning towards pyruvate carboxylation with ketosis occurred (Melo et al. 2006). Whereas in the [U13C]AcAc tracer group, a 30% decrease in glutamate and glutamine concentrations in the KG group, as well as a greater than 4-fold increase in 13C labeling compared to the STD were observed.
Contribution of ketone bodies to GABA
GABA is an inhibitory neurotransmitter and neurotrophin (Waagepetersen et al. 1999), as well as a metabolic intermediate. An unexpected finding in our study was there were no changes in GABA concentrations with KG diet (Yudkoff et al. 2001), even though partitioning of substrate was observed. We also observed very low 13C labeling from [U13C]AcAc to GABA in the STD condition and a remarkable increase in 13C labeling from [U13C]AcAc to GABA with ketosis. Additionally, there was a significant decrease in the [U13C]glucose contribution to GABA during ketosis. These data suggest that the KG diet results in increased turnover of GABA while maintaining a metabolic equilibrium of the CAC. The decreased contribution of glucose to GABA, also reflects that ketones are utilized in place of glucose through the partitioning towards GABA through metabolic adaptation. Explanation of the paradoxical partitioning of GABA in comparison to that of glutamate and glutamine necessitates further analysis of the neuron-glia models.
The glutamate-glutamine two-pool model is often used to describe the partitioning of brain amino acids synthesis in neurons and glia as explored by NMR studies (Jeffrey et al. 2013;Kunnecke et al. 1993;Lapidot and Gopher 1994;Lebon et al. 2002;Mason et al. 1992;McKenna 2007). Documentation of the two-pool model was presented from a study showing the metabolism of [U-13C]glucose resulted in labeling patterns of GABA (position C3, C4) that were similar to glutamate but different from glutamine (Lapidot and Gopher 1994). These reports suggest that the contribution of glucose to GABA is mostly through neuronal glutamate, at least in non-ketotic conditions. Although our study was limited to non-positional carbon tracing, we formulated our interpretations on two fundamental well-accepted assumptions: the glutamate pool is dominantly neuronal, whereas the glutamine pool is dominantly glial (Lebon et al. 2002).
With [U13C]AcAc infusions, the 13C labeling of glutamine and glutamate were significantly increased in KG, which highlights that the ultimate fate of ketone bodies is through the production of neuronal glutamate. The rationale is based on the scheme shown in Figure 5: the highly labeled neuronal glutamate pool comes from highly labeled glial glutamate and glutamine as a result of an enriched pool of acetyl-CoA (via oxidation of ketone bodies by glia). The lack of 13C labeling of GABA in STD diet conditions can be explained by low ketone body utilization of glia (as well as neurons) in non-ketotic state. In ketosis, utilization of ketone bodies by glia occurs through the ‘pulling’ of neuronal glutamate from glial glutamine towards GABA synthesis. Yudkoff et al. advocated that ketotic brain shunts ketone bodies towards glial glutamine pool, whereby favoring the “buffering” of synaptic glutamate (Yudkoff et al. 2008). This premise together with our findings supports that ketosis results in the partitioning of ketone bodies towards buffering of glial glutamine to synthesize GABA, a potential mechanism explaining neuroprotection by ketosis.
Figure 5.
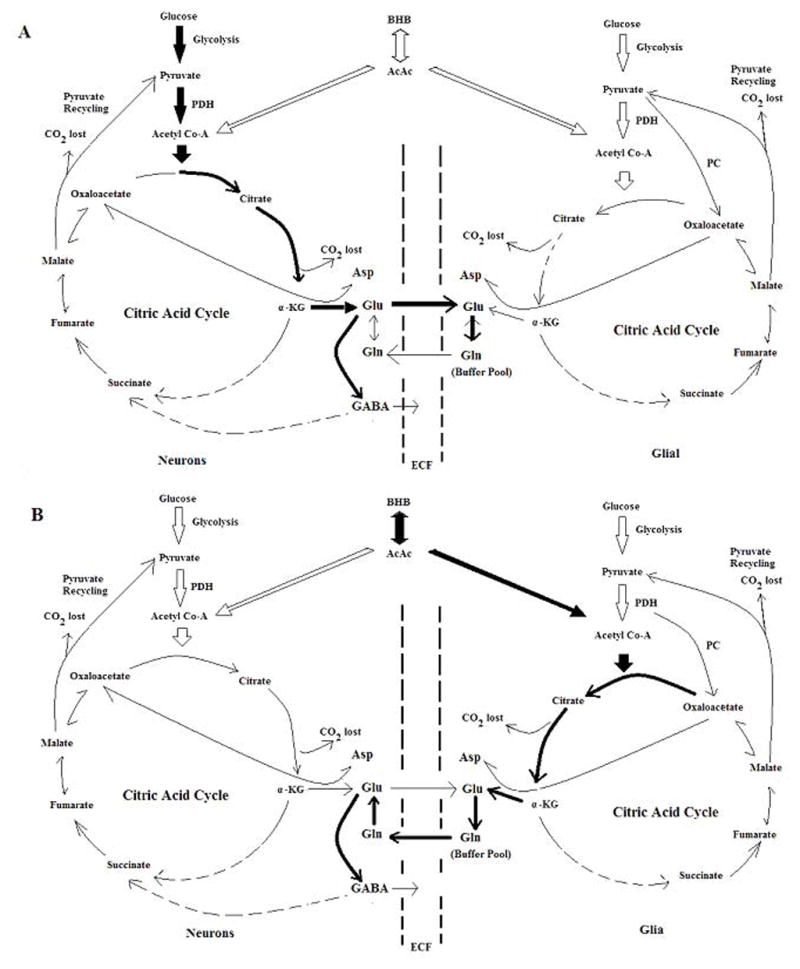
Scheme illustrating the effects of ketosis on the partitioning of glucose and ketone bodies towards oxidative metabolism in neurons and glial cells. Black-heavy arrows indicate the major metabolic pathways of the fate of glucose or ketone bodies in oxidative metabolism; hollow arrows indicate less dominant pathways. Panel A: Proposed partitioning of glucose and ketone bodies during non-ketotic, standard diet (STD) conditions. In this state, glucose is the dominant fuel in oxidative metabolism while ketone bodies were used insignificantly. In neurons, glucose is partitioned towards the synthesis of neuronal glutamate from which GABA is produced. Ketone bodies are also utilized by glial cells and are the primary substrates for glial synthesis of glutamine, where it is buffered in a large glial glutamine pool, and not highly contributing to neuronal glutamate and thus GABA. Panel B: Proposed partitioning of glucose and ketone bodies during ketotic (KG) conditions. During diet-induced ketosis where the brain is well adapted to this metabolic state, overall glucose utilization is decreased and ketone bodies utilization is greatly increased. In neurons, glucose contributions towards neuronal glutamate decreases (showing in hollow arrows), which creates a “pulling” of carbon supply for neuronal GABA synthesis via glial glutamine. Consequently, ketone bodies (black-heavy arrows) are partitioned towards the synthesis of glutamate (via glial glutamine), thus initiating GABA synthesis from ketone bodies in KG conditions. AcAc: Acetoacetate; α-KG, α –ketoglutarate; Asp: aspartate. BHB: β-hydroxybutarate; ECF: extracellular fluid; Gln, glutamine; Glu, glutamate; PC: pyruvate carboxylase; PDH, pyruvate dehydrogenase; OAA, oxaloacetate.
Summary
In this study we determined that diet-induced ketosis results in increased utilization of ketone bodies as an oxidative substrate with a concomitant decrease in glucose oxidation, thus reflecting the partitioning of substrates at the level of oxidative metabolism. We advocate that the metabolic compensation for decreased glucose contribution to neuronal glutamate and GABA is through increased ketone body contribution to glutamate via glial glutamine. Ketone body contribution to GABA requires the sequestering of carbons when glucose contribution to GABA in neurons is decreased. Chronic adaptation to ketosis results in increased ketone body contribution to GABA through glial metabolism, which indicates a neuroprotective mechanism of ketosis.
Supplementary Material
Acknowledgments
We would thank the CASE Mouse Metabolic Phenotyping Center (MMPC; U24 DK76174), for assisting with GC-MS assays and Drs J.C. LaManna and K. Xu (Case Western Reserve University, Departments of Physiology & Biophysics and Neurology) for their scientific discussions and contributions to this project. This research has been supported by the National Institutes of Health, R01 HL092933-01, R21 NS062048-01. These studies were submitted as partial fulfillment of requirements for the degree of Doctor of Philosophy, School of Biomedical Engineering (Y. Zhang).
Abbreviations
- GCMS
Gas Chromatography Mass Spectrometry
- STD
standard (diet)
- KG
ketogenic
- LCMS
Liquid-Chromatography Mass Spectrometry
- AcAc
acetoacetate
- BHB
beta-hydroxybutyric acid
- CAC
citric acid cycle, GABA, gamma-aminobutyric acid
- GLU
glutamate
- GLN
glutamine
- PDH
pyruvate dehydrogenase
- PC
pyruvate carboxylase
- CMRO2
cerebral metabolic rate of oxygen
Footnotes
Disclosure/Conflicts of Interest: Authors have no duality of interest to declare.
Reference List
- Bough KJ, Eagles DA. A ketogenic diet increases the resistance to pentylenetetrazole-induced seizures in the rat. Epilepsia. 1999;40:138–143. doi: 10.1111/j.1528-1157.1999.tb02066.x. [DOI] [PubMed] [Google Scholar]
- Cerdan S, Kunnecke B, Seelig J. Cerebral metabolism of [1,2–13C2]acetate as detected by in vivo and in vitro 13C NMR. J Biol Chem. 1990;265:12916–12926. [PubMed] [Google Scholar]
- Chowdhury GM, Jiang L, Rothman DL, Behar KL. The contribution of ketone bodies to basal and activity-dependent neuronal oxidation in vivo. Journal of Cerebral Blood Flow & Metabolism. 2014;34:1233–1242. doi: 10.1038/jcbfm.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlquist G, Persson B. The rate of cerebral utilization of glucose, ketone bodies, and oxygen: a comparative in vivo study of infant and adult rats. Pediatric research. 1976;10(11):910–917. doi: 10.1203/00006450-197611000-00002. [DOI] [PubMed] [Google Scholar]
- Fernandez CA, Des Rosiers C, Previs SF, David F, Brunengraber H. Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J Mass Spectrom. 1996;31:255–262. doi: 10.1002/(SICI)1096-9888(199603)31:3<255::AID-JMS290>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Freeman JM, Vining EPG, Pillas DJ, Pyzik PL, Casey JC, Kelly MT. The efficacy of the ketogenic diet - 1998: a prospective evaluation of intervention in 150 children. Pediatrics. 1998;102:1358–1363. doi: 10.1542/peds.102.6.1358. [DOI] [PubMed] [Google Scholar]
- Gjedde A, Crone C. Induction processes in blood brain transfer of ketone bodies during starvation. A J Physiol. 1975;229:1165–1169. doi: 10.1152/ajplegacy.1975.229.5.1165. [DOI] [PubMed] [Google Scholar]
- Greene AE, Todorova MT, Seyfried TN. Perspectives on the metabolic management of epilepsy through dietary reduction of glucose and elevation of ketone bodies. J Neurochem. 2003;86:529–537. doi: 10.1046/j.1471-4159.2003.01862.x. [DOI] [PubMed] [Google Scholar]
- Harris SR, Zhang GF, Sadhukhan S, Wang H, Shi C, Puchowicz MA, Anderson VE, Salomon RG, Tochtrop GP, Brunengraber H. Metabolomics and mass isotopomer analysis as a strategy for pathway discovery: pyrrolyl and cyclopentenyl derivatives of the pro-drug of abuse, levulinate. Chem Res Toxicol. 2013;26:213–220. doi: 10.1021/tx3003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselbalch SG, Knudsen GM, Jakobsen J, Hageman LP, Holm S, Paulson OB. Brain metabolism during short-term starvation in humans. J Cereb Blood Flow Metab. 1994;14:125–131. doi: 10.1038/jcbfm.1994.17. [DOI] [PubMed] [Google Scholar]
- Hawkins RA, Williamson DH, Krebs HA. Ketone-body utilization by adult and suckling rat brain in vivo. Biochem J. 1971;122:13–18. doi: 10.1042/bj1220013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey FM, Marin-Valencia I, Good LB, Shestov AA, Henry PG, Pascual JM, Malloy CR. Modeling of brain metabolism and pyruvate compartmentation using (13)C NMR in vivo: caution required. J Cereb Blood Flow Metab. 2013;33:1160–1167. doi: 10.1038/jcbfm.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Mason GF, Rothman DL, de Graaf RA, Behar KL. Cortical substrate oxidation during hyperketonemia in the fasted anesthetized rat in vivo. J Cereb Blood Flow Metab. 2011;31:2313–2323. doi: 10.1038/jcbfm.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kombu RS, Brunengraber H, Puchowicz MA. Analysis of the citric acid cycle intermediates using gas chromatography-mass spectrometry. Methods Mol Biol. 2011;708:147–157. doi: 10.1007/978-1-61737-985-7_8. [DOI] [PubMed] [Google Scholar]
- Kunnecke B, Cerdan S, Seelig J. Cerebral metabolism of [1,2–13C2]glucose and [U-13C4]3-hydroxybutyrate in rat brain as detected by 13C NMR spectroscopy. NMR Biomed. 1993;6:264–277. doi: 10.1002/nbm.1940060406. [DOI] [PubMed] [Google Scholar]
- LaManna JC, Salem N, Puchowicz M, Erokwu B, Koppaka S, Flask C, Lee Z. Ketones suppress brain glucose consumption. Adv Exp Med Biol. 2009;645:301–306. doi: 10.1007/978-0-387-85998-9_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot A, Gopher A. Cerebral metabolic compartmentation. Estimation of glucose flux via pyruvate carboxylase/pyruvate dehydrogenase by 13C NMR isotopomer analysis of D-[U-13C]glucose metabolites. J Biol Chem. 1994;269:27198–27208. [PubMed] [Google Scholar]
- Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI, Rothman DL. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci. 2002;22:1523–1531. doi: 10.1523/JNEUROSCI.22-05-01523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leino RL, Gerhart DZ, Duelli R, Enerson BE, Drewes LR. Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem Int. 2001 May;38(6):519–27. doi: 10.1016/s0197-0186(00)00102-9. [DOI] [PubMed] [Google Scholar]
- Lennox WB. Ketogenic Diet in the treatment of epilepsy. New England Journal of Medicine. 1928;199:74–75. [Google Scholar]
- Maalouf M, Rho JM, Mattson MP. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev. 2009;59:293–315. doi: 10.1016/j.brainresrev.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason GF, Rothman DL, Behar KL, Shulman RG. NMR determination of the TCA cycle rate and alpha-ketoglutarate/glutamate exchange rate in rat brain. J Cereb Blood Flow Metab. 1992;12:434–447. doi: 10.1038/jcbfm.1992.61. [DOI] [PubMed] [Google Scholar]
- Masuda R, Monahan JW, Kashiwaya Y. D-beta-hydroxybutyrate is neuroprotective against hypoxia in serum-free hippocampal primary cultures. J Neurosci Res. 2005;80:501–509. doi: 10.1002/jnr.20464. [DOI] [PubMed] [Google Scholar]
- McKenna MC. The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res. 2007;85:3347–3358. doi: 10.1002/jnr.21444. [DOI] [PubMed] [Google Scholar]
- Melo TM, Nehlig A, Sonnewald U. Neuronal-glial interactions in rats fed a ketogenic diet. Neurochem Int. 2006;48:498–507. doi: 10.1016/j.neuint.2005.12.037. [DOI] [PubMed] [Google Scholar]
- Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, Whitney A, Cross JH. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 2008;7:500–506. doi: 10.1016/S1474-4422(08)70092-9. [DOI] [PubMed] [Google Scholar]
- Nebeling LC, Miraldi F, Shurin SB, Lerner E. Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: two case reports. J Am Coll Nutr. 1995;14:202–208. doi: 10.1080/07315724.1995.10718495. [DOI] [PubMed] [Google Scholar]
- Noh HS, Hah YS, Nilufar R, Han J, Bong JH, Kang SS, Cho GJ, Choi WS. Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J Neurosci Res. 2006;83:702–709. doi: 10.1002/jnr.20736. [DOI] [PubMed] [Google Scholar]
- Noh HS, Kim YS, Choi WS. Neuroprotective effects of the ketogenic diet. Epilepsia. 2008;49(Suppl 8):120–123. doi: 10.1111/j.1528-1167.2008.01855.x. [DOI] [PubMed] [Google Scholar]
- Prins ML. Cerebral metabolic adaptation and ketone metabolism after brain injury. J Cereb Blood Flow Metab. 2008;28:1–16. doi: 10.1038/sj.jcbfm.9600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ML, Hovda DA. The effects of age and ketogenic diet on local cerebral metabolic rates of glucose after controlled cortical impact injury in rats. J Neurotrauma. 2009;26:1083–1093. doi: 10.1089/neu.2008.0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchowicz MA, Xu K, Sun X, Ivy A, Emancipator D, LaManna JC. Diet-induced ketosis increases capillary density without altered blood flow in rat brain. Am J Physiol Endocrinol Metab. 2007;292:E1607–E1615. doi: 10.1152/ajpendo.00512.2006. [DOI] [PubMed] [Google Scholar]
- Puchowicz MA, Zechel JL, Valerio J, Emancipator DS, Xu K, Pundik S, LaManna JC, Lust WD. Neuroprotection in diet-induced ketotic rat brain after focal ischemia. J Cereb Blood Flow Metab. 2008;28:1907–1916. doi: 10.1038/jcbfm.2008.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redies C, Hoffer LJ, Beil C, Marliss EB, Evans AC, Lariviere F, Marrett S, Meyer E, Diksic M, Gjedde A. Generalized decrease in brain glucose metabolism during fasting in humans studied by PET. Am J Physiol. 1989;256:E805–E810. doi: 10.1152/ajpendo.1989.256.6.E805. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA. Mechanisms underlying the anti-epileptic efficacy of the ketogenic diet. Epilepsy Res. 1999;37:171–180. doi: 10.1016/s0920-1211(99)00069-8. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Rho JM. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol. 2012;3:59. doi: 10.3389/fphar.2012.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Rippy NA, Dorenbos K, Concepcion RC, Agarwal AK, Rho JM. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann Neurol. 2004;55:576–580. doi: 10.1002/ana.20062. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Suzuki M, Kitamura Y, Mori S, Sato K, Dohi S, Sato T, Matsuura A, Hiraide A. Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn J Pharmacol. 2002;89:36–43. doi: 10.1254/jjp.89.36. [DOI] [PubMed] [Google Scholar]
- Swink TD, Vining EPG, Freeman JM. The Ketogenic Diet: 1997. Adv Pediatr. 1997;44:297–329. [PubMed] [Google Scholar]
- Tallian KB, Nahata MC, Tsao CY. Role of the Ketogenic Diet in Children with Intractable Seizures. Annals Pharmacotherapy. 1998;32:349–385. doi: 10.1345/aph.16245. [DOI] [PubMed] [Google Scholar]
- Taub KS, Kessler SK, Bergqvist AG. Risk of seizure recurrence after achieving initial seizure freedom on the ketogenic diet. Epilepsia. 2014;55(4):579–583. doi: 10.1111/epi.12583. [DOI] [PubMed] [Google Scholar]
- Uhlemann ER, Neims AH. Anticonvulsant properties of the ketogenic diet in mice. J Pharm Exper Ther. 1972;180:231–238. [PubMed] [Google Scholar]
- Vannucci SJ, Simpson IA. Developmental switch in brain nutrient transporter expression in the rat. Am J Physiol Endocrinol Metab. 2003;285:E1127–E1134. doi: 10.1152/ajpendo.00187.2003. [DOI] [PubMed] [Google Scholar]
- Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. 2004;70:309–319. doi: 10.1016/j.plefa.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Waagepetersen HS, Sonnewald U, Schousboe A. The GABA paradox: multiple roles as metabolite, neurotransmitter, and neurodifferentiative agent. J Neurochem. 1999;73:1335–1342. doi: 10.1046/j.1471-4159.1999.0731335.x. [DOI] [PubMed] [Google Scholar]
- Wilder RM. The effects of ketonemia on the course of epilepsy. Mayo Clinic Bulletin. 1921;2:304–314. [Google Scholar]
- Williamson DH, Bates MW, Page MA, Krebs HA. Activities of enzymes involved in acetoacetate utilization in adult mammalian tissues. Biochem J. 1971;121:41–47. doi: 10.1042/bj1210041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Sun X, Eroku BO, Tsipis CP, Puchowicz MA, LaManna JC. Diet-induced ketosis improves cognitive performance in aged rats. Adv Exp Med Biol. 2010;662:71–75. doi: 10.1007/978-1-4419-1241-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Kasumov T, Kombu RS, Zhu SH, Cendrowski AV, David F, Anderson VE, Kelleher JK, Brunengraber H. Metabolomic and mass isotopomer analysis of liver gluconeogenesis and citric acid cycle: II. Heterogeneity of metabolite labeling pattern. J Biol Chem. 2008;283:21988–21996. doi: 10.1074/jbc.M803455200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudkoff M, Daikhin Y, Horyn O, Nissim I, Nissim I. Ketosis and brain handling of glutamate, glutamine, and GABA. Epilepsia. 2008;49(Suppl 8):73–75. doi: 10.1111/j.1528-1167.2008.01841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudkoff M, Daikhin Y, Nissim I, Horyn O, Lazarow A, Luhovyy B, Wehrli S, Nissim I. Response of brain amino acid metabolism to ketosis. Neurochem Int. 2005;47:119–128. doi: 10.1016/j.neuint.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Yudkoff M, Daikhin Y, Nissim I, Lazarow A, Nissim I. Brain amino acid metabolism and ketosis. Journal of neuroscience research. 2001;66(2):272–281. doi: 10.1002/jnr.1221. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kuang Y, LaManna JC, Puchowicz MA. Contribution of brain glucose and ketone bodies to oxidative metabolism. Adv Exp Med Biol. 2013a;765:365–370. doi: 10.1007/978-1-4614-4989-8_51. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kuang Y, Xu K, Harris D, Lee Z, LaManna J, Puchowicz MA. Ketosis proportionately spares glucose utilization in brain. J Cereb Blood Flow Metab. 2013b;33:1307–1311. doi: 10.1038/jcbfm.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.