

Abstract
Background
DNA polymerase epsilon (POLE) exonuclease domain mutations characterize a subtype of endometrial cancer (EC) with markedly increased somatic mutational burden. POLE mutant tumors were described as a molecular subtype with improved progression-free survival (PFS) by The Cancer Genome Atlas. This study investigates the frequency, spectrum, prognostic significance, and potential clinical application of POLE mutations in endometrioid EC patients.
Methods
PCR amplification and Sanger sequencing was used to test for POLE mutation in 544 tumors. Relationships between demographic, survival, clinicopathologic and molecular features were investigated. Statistical tests were two-sided.
Results
Thirty POLE mutations (5.6%) were identified. Mutations were associated with younger age (<60 years, P=.001). POLE mutations were detected in microsatellite stable (MSS) and unstable (MSI) tumors at similar frequencies (5.9 v 5.2%, respectively) and were most common in MSI tumors lacking MLH1 methylation (P<.001). There was no association with PFS (HR=0.22, P=.127).
Conclusions
Our discovery that mutations occur at equal frequency in MSS and MSI tumors and are most frequent in MSI tumors lacking MLH1 methylation has implications for Lynch syndrome screening and mutation testing. We show that POLE mutations are associated with somatic mutation in DNA mismatch repair genes in a subset of tumors. The absence of association between POLE mutation and PFS indicates POLE mutation status is unlikely to be a clinically useful prognostic marker. However, POLE testing in MSI ECs could serve as a marker of somatic origin of disease. As such, POLE tumor testing might be a valuable exclusionary criterion for Lynch syndrome gene testing.
Keywords: endometrial cancer, DNA mismatch repair, Lynch syndrome, mutation
Cancers have a mutator phenotype [1]. An elevated mutation rate is central to tumorigenesis in human malignancies and significantly contributes to the disruption of regulatory processes essential to genomic stability. Endometrial cancers (ECs) are frequently defective in DNA mismatch repair (MMR). Reduced post-replication surveillance and repair results in a 100-fold increase in somatic mutations in human tumor cell lines [2]. Recently, loss of DNA proofreading function in the DNA polymerase ε (POLE) has similarly been shown to be important in tumorigenesis in EC. Approximately 7% of ECs harbor mutations in the exonuclease domain of POLE [3, 4].
POLE encodes the major catalytic and proofreading subunits of the Polε DNA polymerase enzyme complex [5]. The Polε enzyme complex synthesizes the leading strand [5-8]. The proofreading (exonuclease) function locates and replaces erroneous bases in the daughter strand through failed complementary pairing with the parental strand. High fidelity incorporation of bases by POLE, coupled with its exonuclease proofreading function ensures a low mutation rate. POLE exonuclease domain mutations (EDMs) have shown to increase spontaneous mutation rates contributing to tumorigenesis in yeast and mouse models [9-14].
The Cancer Genome Atlas (TCGA) reported a POLE mutant subtype of EC [3]. Tumors with POLE EDMs are referred to as “POLE ultra-mutated”. ECs in this molecularly defined group are of endometrioid histology, predominantly have normal DNA MMR (microsatellite stable (MSS)) and have thousands of somatic mutations. Clinically, patients in the POLE ultra-mutated group were reported to have improved progression-free survival (PFS) [3].
We undertook an analysis of POLE mutations in a large cohort of endometrioid ECs to better understand clinicopathologic significance of POLE EDMs.
Methods
Study Population
Matched EC and normal tissues were prospectively collected at time of hysterectomy by the Division of Gynecology Oncology at Washington University School of Medicine. All research subjects consented to molecular analyses and follow-up (Washington University HRPO protocols 91-507 and 93-0828). The analyses performed at The Ohio State University in Columbus, OH were undertaken with IRB approval (2012C0117).
High molecular weight genomic DNA for 544 surgically staged endometrioid ECs was analyzed for POLE mutation. The tumor neoplastic cellularity and results of MSI and MLH1 methylation analyses have been previously described for the majority of cases [15, 16]. Microsatellite analysis was performed using 5 NCI consensus microsatellite markers (BAT25, BAT26, D2S123, D5S346 and D17S250) [17]. The COBRA method [18] evaluated methylation of the MLH1 promoter. PCR primers and conditions have been previously published [19]. Extensive data are available for all cases. The cohort has been previously described [15, 16, 20].
Mutation testing
The exonuclease domain of POLE (residues 268-471) was assessed for mutations using PCR amplification (AmpliTaq Gold® DNA Polymerase Applied Biosystems) and Sanger sequencing. Primers and conditions are provided (Supplementary Table 1). PCR products (AmpliTaq Gold® DNA Polymerase, Applied Biosystems®) were treated with ExoSAP-IT® (Affymetrix, Santa Clara, CA) and sequenced (ABI Prism BigDye Terminator Cycle Sequencing Kit version 3.1, Applied Biosystems®) at the Nucleic Acid Shared Resource laboratory at the Ohio State University in Columbus, OH (http://cancer.osu.edu/research/cancerresearch/sharedresources/na/services/dna_sequencing/pages/index.aspx). Sequences were analyzed in Sequencher (GeneCodes, Ann Arbor, MI) and all variants were tested in matched normal DNA to determine if they were somatic or germline alterations.
Statistical Analyses
All analyses were based on available clinical and molecular data (as of 2/1/2014). SAS Version 9.2 (SAS Institute Inc., Cary, NC) and STATASE 10 (StataCorp, College Station, TX) were used for statistical analyses; P values were two-sided. Demographic and clinicopathologic features were compared between POLE mutation and wildtype using Chi-square test or Fisher's Exact test for categorical or dichotomized variables, or a two sample T-test for continuous variables. Due to numerous tests performed and to control the type I error, we considered a P value ≤0.01 significant.
Median time to death and recurrence were calculated using Kaplan-Meier estimates. The Kaplan-Meier curves were compared using a log-rank test. Overall survival (OS) was defined as time from surgery to death from any cause. Patients were censored if alive (with or without disease) at the time of last follow-up, had a peri-operative death, or no outcome data was available. PFS was defined as time from surgery to first recurrence or death from disease. For the PFS analysis, patients were censored if they were alive without disease at the time of last follow-up, were disease free and died of causes unrelated to their EC, had a peri-operative death, or no outcome data was available.
Multivariable Cox proportional hazard models were used to estimate survival hazard ratios (HRs) according to tumor POLE mutational status and progression HRs for all other clinicopathologic features. For OS, we used a stepwise modeling procedure starting with POLE mutation in the model and all significant univariate predictors at the 0.1 level. For PFS, we included all significant univariate predictors at the 0.1 level. For both outcomes, predictors with the highest P values were systematically removed from the model until the final model with all significant P values remained. All removed variables were added back in to verify whether they should be in the model.
Results
POLE EDMs in endometrioid EC
Mutations were identified in 30 of 535 (5.6%) successfully analyzed endometrioid tumors. Of the eight different mutations identified, six have previously been described (Table 1). Representative examples of the somatic mutations are shown (Figure 1). The p.Pro286Arg and p.Val411Leu mutations (Figure 1A,1B) were each present in 10 tumors. Two novel mutations, p.Ala426Val and p.His342Arg, were each seen once (Figure 1C,1D). The p.Ala426Val variant was reported as a rare SNP (rs374920539), but is clearly absent from the patient normal DNA. The p.Pro436Arg mutation (previously reported in colon cancer) was seen in one patient (Figure 1E). Six germline polymorphisms were seen in 17 cases (Table 1). Overall, POLE mutations were more common than polymorphisms (5.6% and 3.2%, respectively). Germline variants observed are rare (minor allele frequencies ≤0.01) and the three germline missense changes are predicted to have a deleterious impact on protein function (Table 1).
Table 1. POLE exonuclease domain variants identified in endometrioid endometrial cancers.
| Variants | Number observations | Predicted functional impact | ||
|---|---|---|---|---|
| SIFT Score/impact | MASS PIFS | PPH v2 score/impact | ||
| Missense mutations | ||||
| P286R c.857C>G | 10 | 0/Damaging | Medium | 1/Probably damaging |
| V411L* c.1231G>C | 10 | 0/Damaging | Medium | 1/Probably damaging |
| S297F* c.890C>T | 3 | 0/Damaging | Medium | 1/Probably damaging |
| A456P c.1366G>C | 3 | 0/Damaging | High | 1/Probably damaging |
| P436R c.1307C>G | 1 | 0.01/Damaging | High | 1/Probably damaging |
| A465V c.1394C>T | 1 | 0/Damaging | High | 1/Probably damaging |
| A426V† c.1277C>T | 1 | 0/Damaging | Medium | 1/Probably damaging |
| H342R† c.1025A>G | 1 | 0.26/Tolerated | Low | 0.04/Benign |
| Polymorphisms | ||||
| rs139075637 D287E | 2 | 0/Damaging | Medium | 0.997/Probably damaging |
| rs5744760 N336S | 2 | 0/Damaging | Medium | 1/Probably damaging |
| rs200403177 R446W | 1 | 0/Damaging | Medium | 0.998/Probably damaging |
| rs5744777 D490D | 8 | - | - | - |
| rs75135381 (intronic) | 2 | - | - | - |
| I300I (unassigned) | 2 | - | - | - |
Among the 10 known Lynch syndrome mutation carries in 535 cases, none had POLE mutations.
One case each with V411L and S297F mutation harbor two somatic MSH6 mutations and lack germline mutations (ref).
Novel mutations.
Figure 1. POLE exonuclease domain mutations in endometrioid endometrial cancer cases.
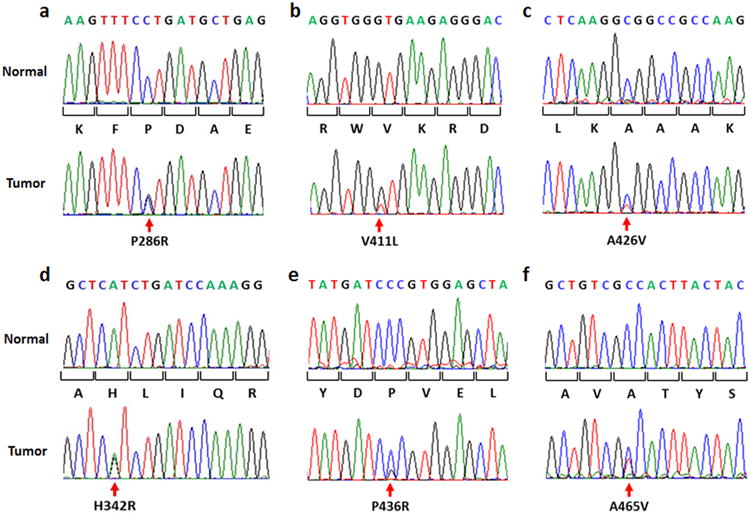
A) and B) are hotspot POLE mutations, C) and D) novel mutations, E) mutation previously seen in single colon cancer and F) an infrequent but known mutation.
The predicted functional impact of POLE EDM was assessed using mutation assessment prediction programs (Table 1). The majority of mutations were reported as having a damaging effect [21, 22], and a medium or high impact score [23]. The novel mutation p.H342R was predicted to have a tolerated impact, whereas the other novel mutation, p.A426V, was predicted to have a damaging impact on function (Table 1).
POLE mutations are similarly distributed between MSI and MSS tumors and are most common in MSI tumors lacking MLH1 methylation
Eighteen of 306 (5.9%) MSS and twelve of 229 (5.2%) MSI tumors harbored POLE EDMs. It is noteworthy that among MSI tumors, mutations were significantly more frequent in cases lacking MLH1 methylation (18% vs 2.4%, <0.001, Fisher's Exact test). Women whose tumors have MSI but lack MLH1 methylation are considered high risk for germline mutation in DNA MMR genes (Lynch syndrome (LS)). Tissues for immunohistochemical (IHC) analysis were available for three of the eight MSI, MLH1 unmethylated tumors with POLE EDMs. IHC for MSH2, MSH6, MLH1, and PMS2 revealed one tumor with normal expression of all markers, one did not express MSH6, and one tumor did not express MSH2 or MSH6. MMR proficiency in one case was somewhat unexpected, but could reflect an epitope stable mutation in one of the MMR genes. Loss of MSH6 alone, or MSH6 and MSH2, are characteristic of MSH6 and MSH2 mutations, respectively [24].
POLE EDMs are associated with younger age in women with EC
POLE mutation was associated with younger age at EC diagnosis. Seventy percent of women whose tumors harbored POLE EDMs were <60 years at diagnosis compared to 30% whose tumor had no mutation (P=0.001, Chi-square test). There were no statistically significant relationships between mutation and the other clinicopathologic factors assessed (Table 2). Although patients with mutations tended to present at an earlier stage (I and II vs III and IV) and have higher grade tumors (2 or 3), these associations did not reach the P value set for significance.
Table 2. Demographic and clinicopathologic features by POLE mutation status.
| Clinicopathologic Factor | POLE Mutant | POLE Wild-type | P value* |
|---|---|---|---|
| N (%) | N (%) | ||
| Age | |||
| <60 years | 21 (70) | 200 (39.6) | 0.001 |
| ≥60 years | 9 (30) | 305 (60.4) | |
| Stage | |||
| Early (I&II) | 29 (96.7) | 408 (81) | 0.027 |
| Advanced (III&IV) | 1 (3.3) | 96 (19) | |
| Grade | |||
| G1 | 9 (30) | 258 (51.2) | 0.024 |
| G2-3 | 21 (70) | 246 (48.8) | |
| LVSI | |||
| Present | 9 (30) | 172 (34.8) | 0.59 |
| Absent | 21 (70) | 322 (65.2) | |
| Depth of Invasion | |||
| ≥50% | 9 (33.3) | 148 (31.8) | 0.865 |
| <50% | 18 (66.7) | 318 (68.2) | |
| Adjuvant therapy | |||
| Any adjuvant therapy | 23 (76.7) | 347 (69.1) | 0.383 |
| No further treatment | 7 (23.3) | 155 (30.9) | |
| BMI | |||
| <30 mg/m2 | 12 (48) | 159 (35.3) | 0.199 |
| ≥30 mg/m2 | 13 (52) | 291 (64.7) | |
| Race | |||
| White | 28 (93.3) | 440 (87.5) | 0.609 |
| African American | 2 (6.7) | 61 (12.1) | |
| Other (Asian, Native American) | 0 (0) | 2 (0.4) |
Missing data includes: grade for 1 patient, stage for 1 patient, LVSI for 11 patients, invasion depth for 39 patients, adjuvant therapy for 3 patients, BMI for 55 patients, and race for 2 patients.
A P value of ≤0.01 was considered significant. Chi square test or Fisher's Exact test was used for categorical variables.
POLE mutation is not associated with survival
TCGA reported improved PFS for patients in the POLE, ultra-mutated subgroup [3]. Univariate analysis for our cohort revealed advanced stage (stages III/IV), higher grade (G2 vs 1 and G3 vs 1), presence of LVSI, deep myometrial invasion and adjuvant therapy had significantly higher PFS HRs (Table 3A). POLE mutation, however, was not associated with PFS (Table 3A). Kaplan-Meier curves similarly demonstrated no difference (P=0.093) (Figure 2A). There was one recurrence among the 30 patients (3.4%) whose tumors had a POLE EDM (median follow-up time of 68.4 months). The recurrence rate of wild-type patients was 17% (median follow-up time 70.6 months). A multivariable model that included six factors with P<0.10 in univariate analysis (POLE and BMI excluded), revealed stage, grade and presence of LVSI were significant (Table 3B).
Table 3. A: Univariate analyses of progression-free and overall survival in endometrioid EC.
| Variable | Progression-free survival | Overall Survival | ||
|---|---|---|---|---|
| Hazard ratio (95% CI) | P value | Hazard ratio (95% CI) | P value* | |
| Advanced age (≥60 y) | 1.53 (0.93-2.52) | .09 (NS) | 2.32 (1.64-3.26) | <.001 |
| Advanced stage (I/II vs III/IV) | 4.98 (3.11-7.97) | <.001 | 3.03 (2.18-4.23) | <.001 |
| Grade 1 vs 2 | 2.81 (1.52-5.18) | 0.001 | 1.75 (1.24-2.46) | .001 |
| Grade 1 vs 3 | 7.49 (4.01-13.98) | <.001 | 4.17 (2.84-6.12) | <.001 |
| Presence of LVSI | 3.94 (2.42-6.42) | <.001 | 2.61 (1.94-3.53) | <.001 |
| Deep myometrial invasion (≥50%) | 3.41 (2.09-5.55) | <.001 | 2.01 (1.48-2.72) | <.001 |
| Adjuvant therapy (any kind) | 3.13 (1.95-5.01) | <.001 | 1.68 (1.25-2.27) | .001 |
| BMI ≥30 mg/m2 | 0.99 (0.97-1.02) | .574 (NS) | 0.98 (0.97-1.00) | .085 (NS) |
| POLE mutation | 0.22 (0.03-1.55) | .127 (NS) | 0.27 (0.08-0.83) | .023 (NS) |
|
| ||||
| *In order to correct for multiple comparisons, a P value ≤0.01 was considered significant. | ||||
| B: Multivariable analysis of progression-free and overall survival in endometrioid EC | ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Variable | Progression-free survival | Overall Survival | ||||||
| Hazard ratio (95% CI) | Std. Error | z | P value | Hazard ratio (95% CI) | Std. Error | z | P value | |
| Advanced age (>60 y) | 1.21 (0.72-2.06) | .326 | 0.72 | .472 | 2.02 (1.35-3.01) | .413 | 3.42 | .001 |
| Advanced stage (I/II vs III/IV) | 2.65 (1.39-5.07) | .876 | 2.96 | .003 | 2.95 (1.74-5.00) | .795 | 4.01 | <.001 |
| High grade (1 vs 2/3) | 2.51 (1.36-4.62) | .782 | 2.96 | .003 | 1.51 (1.06-2.16) | .275 | 2.28 | .023 |
| Presence of LVSI | 1.80 (1.02-3.18) | .523 | 2.03 | .042 | 1.60 (1.10-2.33) | .308 | 2.45 | .014 |
| Deep myometrial invasion (≥50%) | 1.59 (0.88-2.87) | .478 | 1.55 | .122 | 1.38 (0.92-2.07) | .285 | 1.57 | .117 |
| Adjuvant therapy (any kind) | 0.99 (0.52-1.88) | .324 | -0.05 | .964 | 0.65 (0.40-1.05) | .16 | -1.76 | .079 |
| BMI ≥30 mg/m2 | - | - | - | - | 1.00 (0.98-1.02) | .01 | -0.17 | .863 |
| POLE mutation | - | - | - | - | 0.37 (0.09-1.54) | .27 | -1.37 | .172 |
Variables with a p value <0.1 in univariate analysis were included in the multivariable model. Std. error: standard error.
Figure 2. Kaplan-Meier estimates according to POLE mutational status.
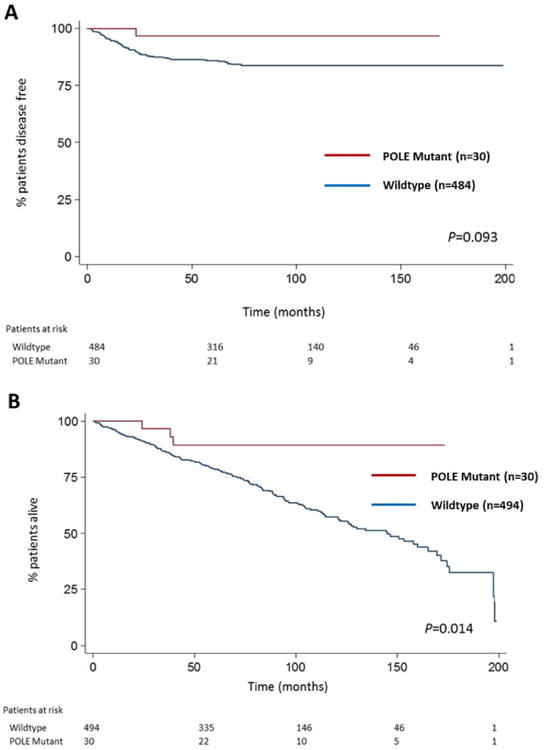
A) Progression-free survival. B) Overall survival. P values calculated using log-rank test (two-sided).
POLE mutation trended towards a lower OS HR (P=0.023; Table 3A) and Kaplan-Meier curves showed a significant association with a lower OS HR (P=0.014) (Figure 2B). However, in multivariable analysis, there was no significant association between POLE mutation and OS (Table 3B).
Discussion
Our analysis of a large cohort of endometrioid ECs confirms prior reports that POLE EDMs are present in 5-8% of sporadic ECs [3, 4]. In the POLE ultra-mutated group described by TCGA, there were 17 endometrioid ECs with POLE EDMs reported (6.9% overall rate). One serous tumor (TCGA-AP-A1DQ) had a POLE EDM. Church and colleagues [4] described 13 POLE EDMs among 173 tumors tested (7.5% overall rate). Our observation that POLE EDMs are seen at equal frequencies in MSS and MSI tumors was unexpected. Previous studies (including TCGA) in colorectal and ECs have pointed to a POLE mutant, hypermutated state occurring predominantly in MSS tumors [3, 4, 25-28]. Our data clearly indicate POLE EDMs are seen in MSI ECs. Furthermore, POLE EDMs are more common in MSI tumors lacking MLH1 methylation compared to those methylated ones (epigenetic silencing of MLH1) (8 of 44 vs 4 of 167, P=0.001, Fisher's Exact test). Although POLE mutations have been reported to occur predominantly in MSS cases, detailed analysis of TCGA mutation data revealed 15 MSI, MLH1 unmethylated tumors, of which seven had POLE EDMs. Six of these seven cases were included in the 17 cases reported by TCGA (Table 4). Combining our data with TCGA data, we estimate that 25% of endometrioid tumors with MSI, but lacking MLH1 methylation, have POLE EDMs.
Table 4. POLE and somatic mismatch repair gene mutations in MSI, unmethylated tumors in TCGA dataset.
| Gene/Mutation type | ||||||
|---|---|---|---|---|---|---|
| Tumor ID | POLE | Cluster | MLH1 | MSH2 | MSH6 | PMS2 |
| TCGA-D1-A17Q | P286R | POLE | E34*/Nonsense | Q76H/Neutral | D390N/Neutral | |
| E908*/Nonsense | ||||||
| TCGA-BS-A0UV | P286R | POLE | K241N/Neutral | K871N/Deleterious | K1013N/Neutral | |
| N566H/Neutral | ||||||
| E483*/Nonsense | ||||||
| TCGA-AP-A059 | S297F | POLE | G529C/Neutral | |||
| E1234*/Nonsense | ||||||
| R178H/Neutral | ||||||
| TCGA-AX-A0J0 | P286R | POLE | G1299D/Deleterious | |||
| K1101N/Neutral | ||||||
| TCGA-D1-A16Y | V411L | POLE | ||||
| TCGA-AP-A056 | V411L | POLE | R929Q/ Neutral | R482Q/Deleterious | R628Q/Neutral | |
| N960T/Neutral | ||||||
| TCGA-B5-A11H | Q453R | MSI | S677fs/FS del | |||
| G67W/Deleterious | ||||||
| TCGA-A5-A0VP | - | MSI | N335fs/FS del | |||
| TCGA-D1-A174 | - | MSI | ||||
| TCGA-B5-A11G | - | MSI | A702D/Deleterious | |||
| TCGA-BS-A0UJ | - | MSI | Q288R/Deleterious | |||
| TCGA-B5-A11X | - | NA | ||||
| TCGA-B5-A11Y | - | CN LOW | ||||
| TCGA-BG-A187 | - | CN LOW | ||||
| TCGA-D1-A15Z | - | CN LOW | ||||
MSI: microsatellite unstable; NA: Not assigned to a cluster by TCGA; CN LOW: copy-number low cluster. Mutation prediction using Condel.
The high rate of somatic POLE EDM in ECs with defective DNA MMR has implications for LS screening and mutation testing. MMR IHC and/or MSI analysis of ECs is used to screen for patients at increased risk for LS (germline mutation in MLH1, MSH2, MSH6, and PMS2). Most tumors with defective DNA MMR are due to somatic methylation of the MLH1 promoter region and loss of MLH1 expression [29-31]. MLH1 methylation can be used to triage IHC results, and exclude patients from germline MMR gene mutation testing [32, 33]. Alternatively, gene testing is indicated for women with ECs that have MSI or defective DNA MMR but lack MLH1 methylation [34]. Our analysis and review of TCGA data suggest that 25% of MSI tumors lacking MLH1 methylation have POLE defects. Somatic POLE EDMs could phenocopy defective DNA MMR (by giving rise to strand slippage mutation and MSI) or lead to somatic inactivation of MMR genes (Figure 3). Of the three POLE mutant, MSI, MLH1 unmethylated tumors investigated for DNA MMR protein expression, two lacked one or more MMR proteins. Two tumors with a POLE EDM (1442 and 1269) have previously been shown to each have two somatic MSH6 mutations and lack germline mutations [35]. Somatic MSH6 mutations in these cases are likely secondary to the hypermutator state conferred by POLE EDM. Of the seven MSI, MLH1 unmethylated, POLE mutant cases in TCGA (1 of which was excluded in POLE cluster by TCGA and classified as MSI), four have clear loss-of-function somatic mutations in the DNA MMR genes (MLH1, MSH2 and MSH6 frameshift or nonsense mutations) and two additional cases have deleterious missense changes (Table 4) (cBio Portal for Cancer Genomics [36] http://www.cbioportal.org/). Among the ten cases with known LS in our cohort, none had POLE mutations. Combined tumor and germline MMR and somatic POLE mutation testing should shed light on whether MMR defects secondary to POLE mutation are common. POLE mutation testing in IHC abnormal/MSI/MLH1 unmethylated tumors may be important in clinical decision making for MMR gene mutation testing in EC patients.
Figure 3. Relationship POLE mutations, tumor microsatellite instability (MSI) and DNA mismatch repair (MMR) defects.
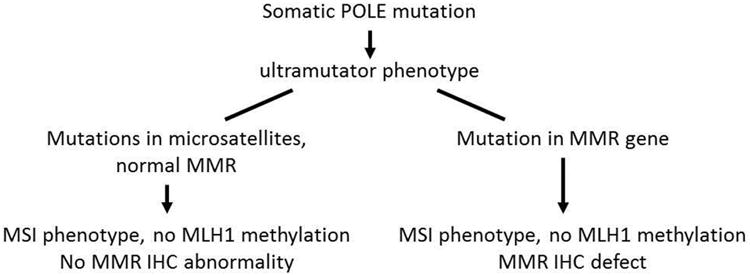
Somatic POLE exonuclease domain mutations could phenocopy defective DNA mismatch repair (normal MMR but strand slippage mutations) or could lead to somatic inactivation of MMR genes with associated MSI and/or immunohistochemical (IHC) defect in tumors lacking MLH1 promoter methylation.
POLE mutation and patient outcomes
POLE mutation was not associated with survival outcomes. This was unexpected given that TCGA reported a subtype of ECs with POLE EDMs with improved PFS. By focusing our outcome analyses on endometrioid tumors, the histologic subtype in which POLE defects are most common, we have provided an important clinical context for the POLE ultra-mutated subtype. A recent publication from Meng and colleagues [37] described improved PFS for patients with grade 3 endometrioid POLE mutant tumors in an analysis that combined TCGA data and findings from their own patient cohort. None of the 16 women with POLE mutations recurred (8 of which were from TCGA). The significance of POLE mutations in grade 3 tumors remains uncertain, particularly in light of the fact that one POLE mutant grade 3 patient in our cohort recurred.
The reported improved survival for POLE mutant patients from TCGA was based on comparison of four molecularly defined subgroups [3]. POLE (ultra-mutated), MSI (hypermutated), copy-number low (endometrioid) and copy-number high (serous like) subgroups were compared. The greatest difference in PFS was for copy-number high and POLE subgroups. Outcomes for women with serous ECs are worse than those with endometrioid tumors [38-40]. The poor outcome associated with serous histology, coupled with the fact POLE mutations are infrequent in serous cancers [3, 4], may explain differences in survival seen for the subgroups.
We recognize that our study is based on POLE mutation status and is not an integrated genomic analysis classification as was performed by TCGA. We do not have whole exome mutation burden that in part defines TCGA's POLE ultra-mutated subgroup. However, given that TCGA cases with POLE EDMs predicted to affect function were all endometrioid carcinomas, we believe our approach to assessing the clinical significance of POLE EDM is appropriately focused on endometrioid tumors.
Among the 30 cases with POLE mutations, there was one recurrence in a 55 year-old patient with stage IB, grade 3 endometrioid tumor who had a pelvic recurrence 23 months after surgery. In the TCGA series, there were no recurrences among the 17 POLE cases. Together our studies suggest an overall low rate of recurrence among endometrioid EC cases (1 in 47 combined). If any difference in outcome does exist, it is unlikely to be clinically useful in planning treatments for endometrioid tumors, given the traditionally impactful clinicopathologic features that would be considered.
POLE mutation status was associated with improved OS in univariate analysis in our cohort. POLE EDM cases had a HR of death of 0.27 (95% CI 0.08-0.83) (Table 3A, Figure 2B). In multivariable analysis however, POLE mutation was no longer statistically significant. This is not surprising given that POLE EDMs were more common in women diagnosed at a younger age (mean age 58.8 years vs 63.7 years for non-EDM cases), as well as trended towards being more common in early stage tumors (P=0.027)(Table 2). These factors are expected to contribute to improved OS. The univariate and multivariable analyses for these and other factors reveal the importance of advanced stage, higher grade and presence of LVSI in risk for recurrence and these coupled with advanced age for OS (Table 3A, B). The final multivariable models, following a step wise modeling procedure (see Methods) for PFS and OS, illustrate these conclusions with adjusted HRs (Supplementary Table 2).
Clinical implications for POLE EDMs
Our data does not support survival advantages for POLE EDM in endometrioid EC patients, and as such, POLE mutation is unlikely to be a useful prognostic marker. We recognize that low prevalence of the mutation and low recurrence rate limit the power of our study. A much larger study could prove a survival advantage does exist for women with POLE mutant tumors. However, two factors make it unlikely that POLE mutation will impact therapeutic decision making, even where a survival advantage is demonstrated. First is the relatively low frequency of POLE EDMs (5-8%); the second relates to the importance of tumor stage and grade in use of adjuvant therapies for EC patients. Only with a validated and large effect on improvement in survival of POLE mutant patients (large effect size) would POLE mutation likely be part of a nomogram for EC patients.
Although POLE mutation is unlikely to serve as a prognostic marker, the vast resource of genomic information provided by TCGA will lead the way to discovery of other prognostic markers and molecular targets to serve as potential predictive factors. Markers that are clinically useful in determining treatment choices and improving outcomes require extensive validation [41], and should be universally available and cost-effective.
In summary, somatic POLE EDMs are common in endometrioid EC, are seen at equal frequencies in MSS and MSI tumors, and are not associated with survival. The majority of the MSI tumors with POLE EDMs lacked MLH1 methylation. POLE EDMs may provide an alternative pathway for MSI in these tumors, and combining our results with TCGA data, we estimate that up to 25% of MSI, unmethylated tumors will harbor a POLE EDM. Following future studies assessing somatic and germline defects in the MMR genes of POLE mutant tumors, a positive tumor POLE mutation may serve as a marker for somatic origin of disease, and act as an exclusionary criterion for LS testing in these patients.
Supplementary Material
Acknowledgments
We would like to the study team and patients treated at Washington University School of Medicine for making this work possible, and recognize Dr. Li Ding (The Genome Institute at Washington University) for her valuable input.
Funding: National Institutes of Health (P30 CA016058), additional funding from the James Comprehensive Cancer Center and Department of Obstetrics and Gynecology at the Ohio State University College of Medicine.
Footnotes
Disclosures: The authors have no financial disclosures to report.
References
- 1.Loeb LA. A mutator phenotype in cancer. Cancer Research. 2001;61(8):3230–3239. [PubMed] [Google Scholar]
- 2.Parsons R, Li GM, Longley MJ, et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75:1227–1236. doi: 10.1016/0092-8674(93)90331-j. [DOI] [PubMed] [Google Scholar]
- 3.Kandoth C, Schultz N, Cherniack AD, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Church DN, Briggs SE, Palles C, et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum Mol Genet. 2013;22(14):2820–8. doi: 10.1093/hmg/ddt131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pursell ZF, Isoz I, Lundstrom EB, et al. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317(5834):127–30. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larrea AA, Lujan SA, Nick McElhinny SA, et al. Genome-wide model for the normal eukaryotic DNA replication fork. Proc Natl Acad Sci U S A. 2010;107(41):17674–9. doi: 10.1073/pnas.1010178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyabe I, Kunkel TA, Carr AM. The major roles of DNA polymerases epsilon and delta at the eukaryotic replication fork are evolutionarily conserved. PLoS Genet. 2011;7(12):e1002407. doi: 10.1371/journal.pgen.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kunkel TA, Burgers PM. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 2008;18(11):521–7. doi: 10.1016/j.tcb.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simon M, Giot L, Faye G. The 3′ to 5′ exonuclease activity located in the DNA polymerase delta subunit of Saccharomyces cerevisiae is required for accurate replication. EMBO J. 1991;10(8):2165–70. doi: 10.1002/j.1460-2075.1991.tb07751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldsby RE, Lawrence NA, Hays LE, et al. Defective DNA polymerase-delta proofreading causes cancer susceptibility in mice. Nat Med. 2001;7(6):638–9. doi: 10.1038/88963. [DOI] [PubMed] [Google Scholar]
- 11.Goldsby RE, Hays LE, Chen X, et al. High incidence of epithelial cancers in mice deficient for DNA polymerase delta proofreading. Proc Natl Acad Sci U S A. 2002;99(24):15560–5. doi: 10.1073/pnas.232340999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albertson TM, Ogawa M, Bugni JM, et al. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A. 2009;106(40):17101–4. doi: 10.1073/pnas.0907147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrison A, Bell JB, Kunkel TA, et al. Eukaryotic DNA polymerase amino acid sequence required for 3′----5′ exonuclease activity. Proc Natl Acad Sci U S A. 1991;88(21):9473–7. doi: 10.1073/pnas.88.21.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrison A, Sugino A. The 3′-]5′ Exonucleases of Both DNA-Polymerases Delta and Epsilon Participate in Correcting Errors of DNA-Replication in Saccharomyces-Cerevisiae. Molecular & General Genetics. 1994;242(3):289–296. doi: 10.1007/BF00280418. [DOI] [PubMed] [Google Scholar]
- 15.Zighelboim I, Goodfellow PJ, Gao F, et al. Microsatellite instability and epigenetic inactivation of MLH1 and outcome of patients with endometrial carcinomas of the endometrioid type. J Clin Oncol. 2007;25(15):2042–8. doi: 10.1200/JCO.2006.08.2107. [DOI] [PubMed] [Google Scholar]
- 16.Zighelboim I, Schmidt AP, Gao F, et al. ATR mutation in endometrioid endometrial cancer is associated with poor clinical outcomes. J Clin Oncol. 2009;27(19):3091–6. doi: 10.1200/JCO.2008.19.9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boland CR, Thibodeau SN, Hamilton SR, et al. A national cancer institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Research. 1998;58(22):5248–5257. [PubMed] [Google Scholar]
- 18.Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25(12):2532–4. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitcomb BP, Mutch DG, Herzog TJ, et al. Frequent HOXA11 and THBS2 promoter methylation, and a methylator phenotype in endometrial adenocarcinoma. Clin Cancer Res. 2003;9(6):2277–87. [PubMed] [Google Scholar]
- 20.Novetsky AP, Zighelboim I, Thompson DM, Jr, et al. Frequent mutations in the RPL22 gene and its clinical and functional implications. Gynecol Oncol. 2013;128(3):470–4. doi: 10.1016/j.ygyno.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Research. 2011;39(17):e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baudhuin LM, Burgart LJ, Leontovich O, et al. Use of microsatellite instability and immunohistochemistry testing for the identification of individuals at risk for Lynch syndrome. Familial Cancer. 2005;4(3):255–265. doi: 10.1007/s10689-004-1447-6. [DOI] [PubMed] [Google Scholar]
- 25.Valle L, Hernandez-Illan E, Bellido F, et al. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu058. [DOI] [PubMed] [Google Scholar]
- 26.Briggs S, Tomlinson I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013;230(2):148–53. doi: 10.1002/path.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45(2):136–44. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muzny DM, Bainbridge MN, Chang K, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simpkins SB, Bocker T, Swisher EM, et al. MLH1 promoter methylation and gene silencing is the primary cause of microsatellite instability in sporadic endometrial cancers. Human Molecular Genetics. 1999;8:661–666. doi: 10.1093/hmg/8.4.661. [DOI] [PubMed] [Google Scholar]
- 30.Bettstetter M, Dechant S, Ruemmele P, et al. Distinction of hereditary nonpolyposis colorectal cancer and sporadic microsatellite-unstable colorectal cancer through quantification of MLH1 methylation by real-time PCR. Clin Cancer Res. 2007;13(11):3221–8. doi: 10.1158/1078-0432.CCR-06-3064. [DOI] [PubMed] [Google Scholar]
- 31.Esteller M, Levine R, Baylin SB, et al. MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene. 1998;17(18):2413–7. doi: 10.1038/sj.onc.1202178. [DOI] [PubMed] [Google Scholar]
- 32.Buchanan DD, Tan YY, Walsh MD, et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J Clin Oncol. 2014;32(2):90–100. doi: 10.1200/JCO.2013.51.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez-Carbonell L, Alenda C, Paya A, et al. Methylation analysis of MLH1 improves the selection of patients for genetic testing in Lynch syndrome. J Mol Diagn. 2010;12(4):498–504. doi: 10.2353/jmoldx.2010.090212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lancaster JM, Powell CB, Kauff ND, et al. Society of Gynecologic Oncologists Education Committee statement on risk assessment for inherited gynecologic cancer predispositions. Gynecol Oncol. 2007;107(2):159–62. doi: 10.1016/j.ygyno.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 35.Goodfellow PJ, Buttin BM, Herzog TJ, et al. Prevalence of defective DNA mismatch repair and MSH6 mutation in an unselected series of endometrial cancers. Proc Natl Acad Sci U S A. 2003;100(10):5908–13. doi: 10.1073/pnas.1030231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meng B, Hoang LN, McIntyre JB, et al. POLE exonuclease domain mutation predicts long progression-free survival in grade 3 endometrioid carcinoma of the endometrium. Gynecol Oncol. 2014 doi: 10.1016/j.ygyno.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 38.Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecologic Oncology. 1983;15:10–17. doi: 10.1016/0090-8258(83)90111-7. [DOI] [PubMed] [Google Scholar]
- 39.Felix AS, Weissfeld JL, Stone RA, et al. Factors associated with Type I and Type II endometrial cancer. Cancer Causes Control. 2010;21(11):1851–6. doi: 10.1007/s10552-010-9612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore KN, Fader AN. Uterine papillary serous carcinoma. Clin Obstet Gynecol. 2011;54(2):278–91. doi: 10.1097/GRF.0b013e318218c755. [DOI] [PubMed] [Google Scholar]
- 41.Simon RM, Paik S, Hayes DF. Use of Archived Specimens in Evaluation of Prognostic and Predictive Biomarkers. Journal of the National Cancer Institute. 2009;101(21):1446–1452. doi: 10.1093/jnci/djp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.