

SUMMARY
Biotin biosynthesis is essential for survival and persistence of Mycobacterium tuberculosis (Mtb) in vivo. The aminotransferase BioA, which catalyzes the antepenultimate step in the biotin pathway, has been established as a promising target due to its vulnerability to chemical inhibition. We performed high-throughput screening (HTS) employing a fluorescence displacement assay and identified a diverse set of potent inhibitors including many diversity-oriented synthesis (DOS) scaffolds. To efficiently select only hits targeting biotin biosynthesis, we then deployed a whole-cell counter-screen in either biotin-free and biotin-containing medium against wild-type Mtb and in parallel with isogenic bioA Mtb strains that possess differential levels of BioA expression. This counter-screen proved crucial to filter out compounds whose whole-cell activity was off-target as well as identify hits with weak, but measurable whole-cell activity in BioA-depleted strains. Several of the most promising hits were co-crystallized with BioA to provide a framework for future structure-based drug design efforts.
INTRODUCTION
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis (Mtb) and related species that is most commonly observed as a chronic pulmonary infection (WHO, 2013). TB, once the leading cause of infectious disease mortality, was nearly eradicated from industrialized nations in the 20th century through a combination of public health measures and the introduction of antibiotics (CDC, 1999). However, the emergence of HIV that sensitizes latently TB-infected individuals and the inevitable development of drug-resistant TB strains through use of the same antibiotics for more than 50 years has led to a dramatic worldwide rise in TB mortality, which prompted the World Health Organization (WHO) to declare TB a global public health emergency. Current efforts to bring TB under control are focused on development of new antibiotics, improved diagnostics, and vaccines (NIH, 2014). Ultimately, success in each area will be required to control TB.
The development of new antibiotics for TB has traditionally been performed by empirically screening compound collections and natural product extracts for in vitro antitubercular whole-cell activity without any a priori knowledge of their mechanism-of-action (MOA) (Aldrich, et al., 2010; Marriner, et al., 2011). Streptomycin, the very first antibiotic effective against Mtb discovered by Albert Schatz in Selman Waksman’s laboratory at Rutgers in 1943, and bedaquiline, the newest TB drug developed by Koen Andries’ team at Janssen Pharmaceuticals and approved by the FDA for multidrug resistant-TB in 2012, were discovered in this manner (Andries, et al., 2005; Schatz, et al., 1944). An inverse and potentially more intellectually appealing strategy for antibiotic discovery is to identify a target essential for growth (or survival?) of the pathogen by comparative genomics and metabolic pathway analysis and then search for an inhibitor.
Target-based approaches have been immensely successful for antiviral drug discovery; however, they have been much less effective in the antibacterial arena for many reasons (Gopal and Dick, 2014; Payne, et al., 2007; Silver, 2011). One of the most significant challenges has been the inability to translate potent biochemical activity into whole-cell antibacterial activity. Moreover, many compounds with cell-based activity that were identified with biochemical assays, may be found later to not act through inhibition of the intended target. To overcome these limitations, target-based whole cell screening has been developed that combines the specificity of biochemical target-based approaches with the practical advantages of whole-cell phenotypic screens to identify cell-permeable target directed compounds (DeVito, et al., 2002; Forsyth, et al., 2002; Young, et al., 2006). In this approach, a target is differentially expressed in the bacterial cell, which potentially sensitizes the biochemical pathway to inhibition and counter-screening enables one to de-select compounds that retain activity presumably through alternate mechanisms. These strategies have been successfully used to identify new antibacterials for Gram positive bacteria (Phillips, et al., 2011; Wang, et al., 2006) and were recently applied for the first time to Mtb (Abrahams, et al., 2012).
We have genetically validated biotin biosynthesis as a promising pathway in Mtb that is essential for replication and persistence in vivo (Park, et al., 2011). The biotin pathway is absent in higher organisms, thus inhibitors of this pathway are expected to be intrinsically selective. BioA is a 5′-pyridoxal phosphate (PLP)-dependent aminotransferase that is responsible for the antepenultimate step of biotin biosynthesis (Figure 1) and catalyzes the reductive amination of 8-aminopelaragonic acid (KAPA, 5) to 7,8-diaminopelargonic acid (DAPA, 6) uniquely employing S-adenosyl-L-methionine (SAM) as the amino donor (Mann, et al., 2009; Mann and Ploux, 2006; Mann and Ploux, 2011). The natural product antibiotic amiclenomycin disrupts biotin metabolism in Mtb through inhibition of the BioA and possesses remarkably selective antimycobacterial activity, thereby providing chemical validation for this pathway (Kitahara, et al., 1975; Sandmark, et al., 2002). However, the chemical instability and highly polar nature of this compound precludes its use in vivo (Shi, et al., 2011). The chemical precedence provided by amiclenomycin in conjunction with our successful fragment-screening campaign supports the vulnerability of Mtb to chemical inhibition of BioA (Dai, et al., 2014; Edfeldt, et al., 2011).
Figure 1.

The biochemical pathway for biotin synthesis in analogy to the pathway in Escherichia coli is expected to proceed by methylation of malonyl-ACP 1 to the corresponding methyl ester 2 by BioC, which allows it to enter the fatty acid synthesis pathway (FAS-II) where it undergo two rounds of extension to pimeloyl-ACP methyl ester 3 (Lin, et al., 2010; Shapiro, et al., 2012). Hydrolysis of the methyl ester 3 by BioH is expected to furnish pimeloyl-ACP 4. Next BioF catalyzes the decarboxylative condensation of pimeloyl-ACP 4 with alanine to furnish 8-aminopelaragonic acid (KAPA, 5). Reductive amination of KAPA to 7,8-diaminopelargonic acid (DAPA, 6) is performed by the PLP-dependent aminotransferase BioA. Carboxylation of DAPA 6 to dethiobiotin 7 mediated by BioD followed by C-H activation and sulfur insertion by BioB affords biotin 8.
Herein we report the identification of potent BioA inhibitors by screening the Molecular Libraries Small Molecules Repository (MLSMR) compound collection of more than 350,000 compounds employing an innovative screening approach. A major challenge in hit discovery programs with biochemical assays is selecting compounds for further development since a typical hit rate of 0.1–0.3% on a library of this size can provide hundreds to thousands of confirmed hits with low micromolar activity. In order to rapidly identify compounds that operate through the desired mechanism of action we used a whole cell counter-screen with wild-type Mtb in biotin-free and biotin-containing medium as well as BioA under- and over-expressing Mtb strains. Integration of the resulting whole-cell activity profiles enabled rapid selection of compounds with BioA specific whole-cell activity. Moreover, the susceptible BioA-depleted Mtb strain allowed identification of compounds with modest, on-target whole-cell activity that would have been missed by enlisting only a wild-type Mtb strain. Several of the most promising scaffolds were co-crystallized with BioA and provide a foundation for future structure-based drug design studies.
RESULTS
Continuous Coupled Assay
To identify BioA inhibitors we used a coupled assay under initial velocity conditions as shown in Figure 2 wherein the BioA product DAPA is converted into dethiobiotin by BioD (Mann, et al., 2013; Wilson, et al., 2011). The BioA substrates KAPA and SAM, were held at 3 μM and 0.75 mM, respectively, which is near their KM values to provide balanced assay conditions (Copeland, 2000). Additionally, we employed 1 mM DTT to prevent false-positives caused by thiol reactive molecules and 0.0025% Igepal CA630, a non-ionic detergent to impede non-specific inhibitor aggregation (McGovern, et al., 2003). The use of BioD drives the overall reaction forward since the aminotransferase reaction is reversible. Detection of dethiobiotin is achieved with streptavidin and a fluorescently-labeled tracer that we developed, whose fluorescence quenching is relieved upon displacement by dethiobiotin (Wilson, et al., 2011). Termination of the assay is accomplished by addition of a 500 mM EDTA solution, which chelates the Mg2+ cofactor required for BioD activity.
Figure 2.

Overview of the coupled fluorescent displacement assay to measure BioA activity. BioA catalyzes the reversible transamination of KAPA 5 to DAPA 6, which is converted by BioD to dethiobiotin 7. Displacement of the fluorescently labeled dethiobiotin tracer 9 from streptavidin by the generated dethiobiotin 7 relieves the fluorescent quenching of 9 due to the tryptophan-rich environment of streptavidin (W79, W92, W108, and W120). The depicted binding of streptavidin to 7 and 9 does not accurately portray the molecular interactions. Streptavidin can bind four molecules of dethiobiotin 7 in each of the four equivalent biotin binding sites of the homotetramer while the dethiobiotin and the fluorescein moities of 9 are predicted to each occupy one of the four equivalent biotin binding sites (Wilson, et al., 2011).
Assay Screen
The assay was miniaturized to a 7.5 μL 1536-well format where it performed with a calculated Z′ factor of 0.825 ± 0.103, which is a measure of assay robustness (Zhang, et al., 1999). Screening was performed in duplicate at a single concentration of 10 μM against the Molecular Libraries Small Molecules Repository (MLSMR) collection of 356,486 diverse compounds (PubChem AID#602481). Notably, this library includes 83,000 compounds from the diversity-oriented synthesis (DOS) compound collection at the Broad Institute. We defined a hit as a compound showing greater or equal to 40% inhibition and 342 compounds were active at 10 μM by this criteria, representing a nominal hit rate of 0.095%. Hits were triaged to remove most pan-assay interference compounds (PAINS) (Baell and Holloway, 2010) yielding 327 hits of which 312 were available for re-testing. Re-testing was performed in an 8-point concentration-response format in triplicate and 298 of the 312 hits possessed IC50 values ≤ 20 μM with 55 compounds exceeding the activity threshold of IC50 ≤ 1 μM giving a confirmation rate of 96% (PubChem AID#651683) (See Supplemental Figure S1 for Assay Tree).
Secondary Assays
Next, all 298 hits were counter-screened in an 8-point dose response format against BioD using the substrate DAPA along with streptavidin and the fluorescent dethiobiotin tracer to identify compounds that cause fluorescence interference or inhibit BioD (PubChem AID#651679). None of the hits showed activity in the BioD counter-screen (IC50 > 20 μM). Since BioA is a PLP-dependent aminotransferase and because previous BioA inhibitors have been shown to covalently bind the PLP cofactor, we evaluated all compounds against aspartate transaminase (AST), a ubiquitous and functionally related PLP-dependent enzyme, in order to assess potential enzyme selectivity (AID#743184; Dai, et al., 2014; Sandmark, et al., 2002; Shi, et al., 2011; Zlitni, et al., 2013). All of the hits were inactive against AST suggesting they possess a useful level of selectivity. To evaluate the potential for mammalian cytotoxicity every hit was then screened against HepG2 human liver cells, HEK293 human kidney cells, and NIH3T3 murine fibroblast cells and none of the 298 hits demonstrated any cytotoxicity at 20 μM, the maximum compound concentration evaluated (PubChem AID#651898, AID#651899, and AID#651900) and were carried forward for further study.
Scaffold Analysis
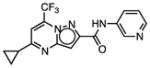
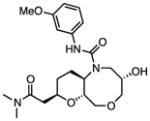
Cheminformatic analysis followed by manual culling of the 298 confirmed hits suggested chemical clustering into 65 groups of which 34 groups were defined by multiple examples sharing a common scaffold (Table S1) (Mulrooney, et al., 2013). Tetrahydroisoquinoline 10, from a DOS library and shown in Table 1, is the most abundant scaffold (36 analogs) representing nearly 12% of all hits and including the most potent hit identified with an IC50 value of 75 nM. Another interesting DOS scaffold (6 analogs) is the oxazocane typified by 17 (Gerard, et al., 2013). Several other DOS scaffolds registered as hits (Table S1) including benzooxathiazocines (Gerard, et al., 2011), monocyclic azetidine nitriles (Lowe, et al., 2012), and tricyclic pyridones (Marcaurelle, et al., 2009).
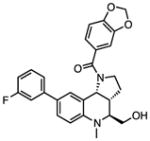
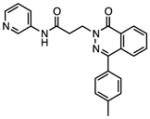
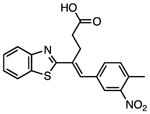
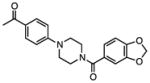
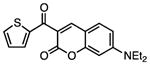
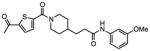
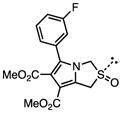
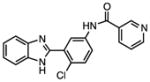
Several other chemotypes were identified with promising biological activity, among these 5,6-fused bicyclic heteroaromatics were the third most common scaffold (23 analogs) and pyrazolo[1,5-a]pyrimidine 11 is shown as an example. The piperidines (20 analogs) typified by 16, constitute the fourth most populous scaffold. The N-aryl piperazines, such as 14, represent the fifth most abundant scaffold (16 analogs). The coumarins, exemplified by 7-diethylaminocoumarin 15, were the sixth most abundant scaffold. Additionally, several other small clusters of 2–3 compounds demonstrated promising BioA inhibitory activity with pyrrolothiazolidine-S-oxide 18 and benzimidazole 19 provided as representative molecules. Among the compounds with no clear chemical analogs in the screening libraries were examples like phthalazinone 12 and 4-(benzothiazol-2-yl)pentenoic acid 13
Whole-Cell Evaluation
Representative members of each scaffold were evaluated against M. tuberculosis H37Rv in biotin-free and biotin-containing medium to identify compounds with biotin-dependent Mtb growth inhibitory activity. Additionally, compounds were tested against an Mtb strain that expresses approximately 20% of BioA relative to wild-type (WT) Mtb and is therefore sensitive to BioA inhibition. This strain was originally described as BioA TetON-5 (Park, et al., 2011) and is referred to here as BioA-underexpressor or BioA-UE. Select compounds were then screened against an Mtb strain that expresses approximately 1200% of BioA relative to WT Mtb and should therefore be more resistant to putative BioA inhibitors. The overexpressor was described as Bio TetON-1 (Park, et al., 2011) and is referred to here as BioA-OE. The results from these whole-cell screens allow prioritization of hits based on on-target whole-cell activity.
The utility of this approach is illustrated with N-aryl piperazine 14 (Figure 4A), which shows modest activity against wild-type Mtb with a minimum inhibitory concentration that inhibits 90% of growth (MIC) at 26 μM in biotin-free medium. Addition of 1 μM biotin to the medium blocks the activity of 14 and chemically rescues the bacteria. Depletion of BioA in Mtb BioA-UE increases susceptibility to 14 yielding an MIC of 9 μM while BioA over-expression in Mtb BioA-OE confers resistance to 14 shifting the MIC to 99 μM. Collectively, these results are consistent with the desired mechanism of action (MOA) and present N-aryl piperazine 14 as a validated hit, which we will report on further in the future.
Figure 4.
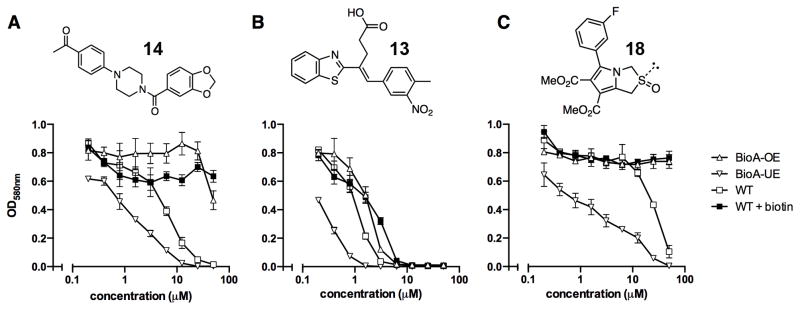
Whole-cell screening. Whole-cell screening was performed with 120 compounds in WT Mtb in biotin-free and biotin containing medium and BioA over- and under-expression strains, BioA-OE and BioA-UE, respectively. Examples of whole-cell screening with N-aryl piperazine 14 (A), 4-(benzothiazol-2-yl)pentenoic acid 13 (B), and pyrrolothiazolidine-S-oxide 18 (C) are shown. Error bars represent standard deviation. Full data is available in PubChem AID#743070 and AID#743350 for WT Mtb in biotin-free medium, AID#743071 and AID#743349 for WT Mtb in biotin-containing medium, AID#743073 and AID#743351 for BioA-OE, and AID#743072 and AID#743352 for BioA-UE.
In a similar manner this screening approach can be used to invalidate hits even though they may otherwise appear promising based on their biochemical and whole-cell activity. This is illustrated with 4-(benzothiazol-2-yl)pentenoic acid 13, which was the most potent whole-cell active hit identified with an MIC against wild-type Mtb of 2.5 μM in the absence of biotin. However, the MIC was only modestly shifted approximately 3-fold to 8.7 μM upon addition of biotin, suggesting the activity was largely biotin-independent. While assay of 13 using strains that under- or over-express BioA provided MICs of 0.7 and 4.7 μM, respectively. These MICs were relatively insensitive to BioA levels, which vary 60-fold between strains BioA-UE and BioA-OE (Park, et al., 2011). Thus, while 4-(benzothiazol-2-yl)pentenoic acid 13 had promising biochemical activity (IC50 = 153 nM), the observed whole-cell anti-mycobacterial activity showed lesser dependence on exogenous biotin or BioA protein levels suggesting that the observed whole-cell activity was primarily due to off-target effects. Consequently, hit 13 was de-prioritized for further development.
A third set of compounds was also identified from this screening method as exemplified by pyrrolothiazolidine-S-oxide 18, whose MIC against wild-type Mtb in biotin-free medium was greater than 50 μM. Compound 18 showed no observable growth inhibition in biotin-containing medium or against the BioA overexpression strain. In most screening work-flows this compound would be discarded. However, activity was revealed when this compound was evaluated against the sensitive strain BioA-UE, wherein BioA is depleted relative to WT, providing an MIC value of 35 μM which warrants further investigation. Several other hits also demonstrate weak whole-cell activity against BioA-UE, but are inactive against the other Mtb strains and growth conditions including pyrazolo[1,5-a]pyrimidine 11, piperidine 16, and DOS oxazocane 17 (see Table S1 for all active compounds).
The majority of the 255 hits showed no activity against any of the Mtb strains, which is in agreement with prior observations of biochemical screening campaigns (Payne, et al., 2007; Silver, 2011), and these hits can be rapidly de-prioritized without expending chemistry-intensive resources.
Structural Characterization
To facilitate future structure-based drug design seven of the most promising hits were subjected to co-crystallization with BioA. Three complex structures were obtained including 7-diethylaminocoumarin 15, pyrrolothiazolidine-S-oxide 18, and an N-aryl piperazine 14-Cl, which is otherwise identical to hit N-aryl piperazine 14 except the dioxolane ring is replaced with a meta-chloro substituent. Unambiguous electron density affirms that each inhibitor binds in the hydrophobic site adjacent to the PLP co-factor where substrate KAPA also binds as shown in Figure 5 (Dai, et al., 2014). This site exists at the interface of monomers in the BioA heterodimer; inhibitors are in contact with structural components of both monomers, distinguished by color in Figure 5 (panels A-C). Each inhibitor induces shifts in the conformation of sidechains of Tyr25 and Trp64 to accommodate the longer, flatter molecules that lie sandwiched between repositioned aromatic rings of the tyrosine below and the tryptophan above.
Figure 5.

Crystal structures with bound inhibitors. Panels A–C show 3σ omit density (Fo-Fc) from co-crystal structures of pyrrolothiazolidine-S-oxide 18 (A), 7-diethylaminocoumarin 15 (B) and N-aryl piperazine 14-Cl (C) (cyan), and the molecular environment surrounding the binding site near the PLP (Green). Comparable binding is observed in both active sites of the BioA heterodimer, but only one is shown. Residues from one monomer are identified with primes; those from the other are not. Panels D–F show ligand interaction maps. Hydrogen bonds are shown as green dashed lines, and hydrophobic contacts (closer than 3.9 Å) are identified by thin dashed red lines.
Interactions between these inhibitors and BioA are primarily hydrophobic and arise from two subsites. A subsite on the right (orientation defined by Figure 5) is formed by the juxtaposition of Pro24 and Trp 64 with residues 91′-93′ and 316′-318′ (primes denote residues of the other BioA monomer). The m-fluoro-phenyl group of 18, the thienyl ketone of 15, and the acetophenone group of 14-Cl all lie in this subsite with the aromatic groups in planar coincidence. The fluorine of 18 is inserted most deeply into this subsite, where it is nestled between Gly′93 and Met′91. The acetocarbonyl of 14-Cl accepts an H-bond from the amide NH of Gly′93, the only H-bond donor or acceptor present in this otherwise hydrophobic pocket. A subsite on the left exists between the sidechain of Tyr25, Tyr157 and the loop formed by Gly172-Met174. The subsite is capped by Arg403, although none of these inhibitors extend far enough to achieve direct contact with it. The chlorobenzyl of 14-Cl extends to the limit of this subsite, with the aromatic ring stacked in what could be a π-π interaction along the flat face of the peptide bond joining Gly173 and Gly173. Inhibitor 18 is too short to extend into this subsite. One branch of the diethylamine of 15 lies in a similar position, while the other branch extends upward toward a niche between Tyr25 and Met174 not occupied by other inhibitors.
Inhibitor 18 is too short to contact the left subsite, but it is the only inhibitor that extends down to make direct contact with the PLP. The S-oxide of the pyrrolothiazolidine is positioned to donate an H-bond to one oxygen of the PLP phosphate and accept one from the hydroxyl group of Tyr157.
Isothermal Titration Calorimetry
To futher confirm the activity of N-aryl piperazine 14, we independently synthesized it (see Supplemental Information) and measured binding to BioA by isothermal titration calorimetry (Figure 6). Consistent with our kinetic assay, 14 binds BioA tightly with a KD of 110 ± 9 nM in a 1:1 stoichiometry. The affinity is enthalpically driven (ΔH = 11.8 ± 0.8 kcal/mol) with a small unfavorable entropic compenent (−TΔS = +2.8 ± 0.8 kcal/mol).
Figure 6.

Isothermal titration calorimetry profile of BioA (10 μM) with 14 (103 μM). Experiments were performed as described in the Experimental Procedures. The top panel shows data obtained from automatic titrations of 200 μL of 14. The bottom panel shows the integrated curve showing experimental points (➄) and the best fit (–). A fit of the data to a one-set-of-sites model produced the following values for the binding of 14 to BioA (average from duplicate experiments): n = 0.73 ± 0.11, ΔH = −11.8 ± 0.8 kcal mmol−1, −TΔS = 2.3 ± 0.8 kcal mol−1; and KA = (9.23 ± 0.89) × 106 M−1.
DISCUSSION
Target-based Strategies
Reductionist target-based approaches have gained considerable notoriety in antibacterial drug discovery because of their limited success (Payne, et al., 2007; Silver, 2011). There are several potential reasons for the poor outcome of target-based strategies for antibacterial development including the challenge of converting a biochemical inhibitor into a compound that leads to bacterial accumulation, rapid evolution of resistance to single targets, and the difficulty of developing broad-spectrum compounds. With respect to TB, narrow spectrum agents that do not affect the commensal microbiota are in fact preferred, thus removing a major constraint typically faced by antibiotic discovery programs. The other major concern that has plagued antibiotic development is the high resistance frequencies of single-target agents, but this is less important for diseases like TB and HIV since combination drug therapy is always employed. For intracellular cytosolic targets in Mtb, which represent the majority of novel targets, the lack of efficacy of potent biochemical hits has generally been ascribed to lack of penetration through the lipophilic cell envelope; however, drug efflux is increasingly being recognized as a major contributor to intrinsic resistance to antibiotics in Mtb. As recently demonstrated by Lee and co-workers, efflux pump mediated resistance may be successfully overcome through chemical modification of the parent antibiotic scaffold (Lee, et al., 2014). Based on the unique requirements for TB chemotherapeutic agents as noted above combined with the potential ability to overcome efflux-mediated resistance, target-based strategies could be well-suited for TB drug development.
Target-based Whole Cell Screening (TB-WCS)
Rudimentary target-based whole-cell screening was first performed in the 1960s using pathway mutants in cell-wall biosynthesis (Silver, 2013). Modern TB-WCS approaches employing antisense and siRNA target knockdown were reported by several groups (DeVito, et al., 2002; Forsyth, et al., 2002; Yin, et al., 2004) and this led to the discovery of the antibiotics platensimycin and kibdelomycin (Phillips, et al., 2011; Wang, et al., 2006; Young, et al., 2006). Mizrahi and co-workers were the first to apply TB-WCS to Mtb and used a more sophisticated method with conditional mutants that underexpress essential genes for inhibitor screening since antisense and siRNA methods are largely ineffective in Mtb (Abrahams, et al., 2012). Our results extend these prior findings and highlight the utility of whole-cell studies with genetically modified strains for mode-of-action determination. Remarkably, the workflow described here identified selective biotin-dependent on-target compounds that inhibit growth of Mtb, even though BioA requires more than 95% depletion to affect growth (Park, et al., 2011).
While TB-WCS is gaining momentum, it is important to recognize the limitations of this approach as sensitivity to antibiotics does not always monotonically increase with target expression levels (Palmer and Kishony, 2014). Thus, respective target under-expression in E. coli paradoxically reduces susceptibility to fluoroquinolines when the target DNA gyrase is underexpressed and has no effect on the sulfonamide antibacterials when the target dihydropteroate synthase (DHPS) is underexpressed. As astutely noted by Palmer and Kishony, deviations from the expected monotonic relationship between sensitivity and target expression levels suggests more complex mechanisms of action (Palmer and Kishony, 2014).
Promising Scaffolds
We identified and structurally characterized several inhibitors with novel chemotypes that potently inhibit BioA and possess on-target whole cell activity. The N-aryl piperazine scaffold of 14 is the most promising compound identified in this HTS campaign based on its chemical tractability for further development, potent biochemical activity with an IC50 of 155 nM, and on-target biotin-dependent whole-cell activity. The co-crystal structure of this scaffold with BioA reveals several opportunities to enhance potency and selectivity. Additionally, several other meritorious scaffolds were identified using the sensitive BioA knockdown Mtb strain including pyrazolopyrimidine 11, piperidine 16, and pyrrolothiazolidine-S-oxide 18. Coumarin 15 and benzimidazole 19 possess attractive biotin- and BioA-dependent whole-cell activity and lack cytotoxicity. However, the activity profile indicates partial off-target activity as the activity is not eliminated upon BioA overexpression or addition of exogenous biotin. These compounds therefore warrant further study given their favorable therapeutic index and promising activity, but do not represent useful molecular scaffolds for preparation of a probe for in vivo chemical validation of biotin synthesis in Mtb.
Prior structural studies with Mtb BioA have clearly shown that the active site is highly adaptable in the presence of different bound ligands (Dai, et al., 2014; Dey, et al., 2010; Shi, et al., 2011). This plasticity is not unexpected, given the need for the enzyme to recognize, bind, and catalyze chemical transformations of such diverse substrates as KAPA and SAM. Nevertheless, specific conformational changes that support ligand binding are not easily predicted a priori. Each of the high resolution inhibitor complexes described here represents a novel induced protein conformation that constitutes a possible ground-state for use in modeling or design of analogs derived from these scaffolds.
SIGNIFICANCE
Biotin biosynthesis is a genetically validated pathway in Mtb for antibiotic development. The chemical precedence and narrow spectrum activity afforded by the antibiotic amiclenomycin that inhibits biotin biosynthesis at BioA, highlights the unique vulnerability of this pathway in Mtb. Here we describe the identification of potent on-target BioA- and biotin-dependent Mtb whole-cell active compounds through high-throughput screening followed by phenotypic screening employing isogenic Mtb strains that differentially express BioA. The use of Mtb strains that allow modulation of BioA protein levels was crucial to distinguish among hits with non-specific versus on-target activity. More importantly, the use of BioA depleted Mtb strains revealed hits with on-target whole-cell activity that would be overlooked by traditional antibiotic discovery workflows. The structural characterization of three different scaffolds bound in the BioA active site directly adjacent to the PLP-cofactor demonstrate the remarkable plasticity of BioA to accommodate structurally diverse ligands, which may account for the unique vulnerability of this target to chemical inhibition by a wide array of diverse chemical types.
EXPERIMENTAL PROCEDURES
Materials
BioA and BioD were expressed and purified as described (Geders, et al., 2012; Wilson, et al., 2011). The aspartate transaminase assay kit was purchased from BioAssay Systems (Hayward, CA, USA). The fluorescent tracer N1-{3-[2-(2-{3-[(Fluorescein-5-yl)carbonyl]aminopropoxy}ethoxy)ethoxy]propyl}dethiobiotinamide 9 (Wilson, et al., 2011), 7-keto-8-aminopelargonic acid (KAPA, 5) (Wilson, et al., 2011) and 7,8-diaminopelargonic acid (Vasantkumar, 2007) were synthesized as reported. 6-(2-Fluorophenyl)-1,3-dioxolo[4,5-g]quinolin-8(5H)-one (CHM-1) previously identified (Wilson, et al., 2011) as a BioA inhibitor from screening the LOPAC library (Sigma-Aldrich) was employed as a positive control and purchased from R&D Systems (Minneapolis, MN, USA). All other buffers, salts and reagents for the HTS assay (streptavidin from Streptomyces avidiniii as a salt-free lyophilized powder, S-(5′-adenosyl)-L-methionine p-toluenesulfonate salt (SAM), adenosine 5′-triphosphate disodium salt hydrate (ATP), dithiothreitol (DTT), pyridoxal 5′-phosphate hydrate (PLP), MgCl2, NaHCO3, bicine, 1% w/v Igepal CA630) were obtained from Sigma-Aldrich (St. Louis, MO, USA), EMD Millipore (Billerica, MA, USA) or Fisher Scientific (Pittsburgh, PA, USA). Mycobacterium tuberculosis H37Rv, Mycobacterium tuberculosis SD1 and Mycobacterium tuberculosis SD5 strains have been described (Park, et al., 2011). The reagents in Sauton’s medium (0.5 g of KH2PO4, 0.5 g of MgSO4·7H2O, 2.0 g of citric acid, 0.05 g of ferric ammonium citrate, 60 ml of glycerol, 4.0 g of asparagine, 0.1 ml of 1% ZnSO4 and 0.02% tyloxapol in one liter) for the Mtb whole-cell studies were obtained from Sigma-Aldrich. HEK293 (ATCC# CRL-1573), HepG2 (ATCC# HB-8065), and NIH3T3 (ATCC# CRL-1658) were obtained from the American Type Culture Collection (Manassas, VA, USA) and cell culture medium and reagents (DMEM medium, fetal bovine serum, 0.25% trypsin-EDTA, Pen/Strep/L-Glutamine, MG132) and were purchased from Gibco. Mitoxantrone dihydrochloride was obtained from Enzo Life Sciences (Farmingdale, NY, USA) and the CellTiterGlo assay kit was purchased from Promega (Madison, WI, USA).
HTS Assay protocol
Compounds in 10 mM dimethylsulfoxide (DMSO) stock solution (7.5 μL) were robotically dispensed into Aurora black 1536 well plates (cat#00019180BX) plates using a Echo 555 acoustic liquid handler. The first and last four columns contained positive (compound CHM-1, CID357860) and negative (DMSO only) controls. Next, 3.75 μL of reaction buffer (100 nM BioA, 640 nM BioD, 1.5 mM SAM, 20 nM Fluorescent-DTB tracer 9, 92.5 nM streptavidin, 2 mM DTT, 5 mM ATP, 50 mM NaHCO3, 1 mM MgCl2, 0.1 mM PLP, 0.0025% Igepal CA630, 100 mM Bicine, pH 8.6) containing all reaction components except KAPA was dispensed into all wells of the plate. The reaction was started by addition of 3.75 μL of freshly prepared KAPA initiation solution (6 μM KAPA, 50 mM NaHCO3, 1 mM MgCl2, 0.0025% Igepal CA630, 100 mM Bicine, pH 8.6) and incubated at 25 °C for 45 min. The reaction was terminated by addition of 1.5 μL of 500 mM EDTA into each well of the assay plate. After 5 min equilibration, the plate was read on a Viewlux microplate reader in fluorescence mode with a excitation of 485 nM, an emission of 530 nM, and a cutoff of 530 nM (FITC filters). The library was screened in duplicate and hits were procured as dry powders and purity and structural assignment was verified by LC-MS analysis. Primary HTS data were analyzed in Genedata Screener Assay Analyzer. All values were normalized against DMSO treated samples and the positive control. Z’ values were calculated for each plate from the positive and negative controls and plates were considered technically acceptable for Z′-factors ≥ 0.75. For the HTS, the average of two replicates was used to rank order activity and to choose compounds for retests. A hit was defined when the average value of both replicates resulted in greater than 40% inhibition. The BioD counter-screen was performed analogously, but BioA was excluded from the reaction buffer, BioD was reduced to 10 nM (final concentration), and the reaction was initiated with DAPA initiation solution (8 μM DAPA, 50 mM NaHCO3, 1 mM MgCl2, 0.0025% Igepal CA630, 100 mM Bicine, pH 8.6). For dose-response studies two-fold dilutions of compound stock solutions were prepared from 10–0.078 mM in DMSO and 7.5 nL of these stock solutions were transferred to 1536 well assay plates as described above to provide final compound concentrations in the well ranging from 10–0.078 μM. IC50 curves were generated using a 4-parameter Hill equation with the SmartFit algorithm in Genedata Screener version 7.0.3. The data was normalized to negative and positive controls as 0 and 100% inhibition, respectively, and the curves were then fit to the percent activities.
WCS protocol
Mtb WT, BioA-UE, and BioA-OE were grown in Sauton’s medium containing 1 μM biotin to an OD580 nm between 1.0 and 1.2, harvested by centrifugation, washed twice with biotin-free Sauton’s medium, and diluted in 96 well plates to an OD580 nm of 0.03. The compounds were added to final concentrations between 50 and 0.2 μM. Wells containing no compound were used as controls. 1 μM biotin was added when indicated. 200 ng/mL of anhydrotetracycline were added to the Mtb BioA-UE and BioA-OE to achieve the desired level of BioA expression. Plates were incubated at 37°C and optical density was measured after 14 days. All growth assays were performed in triplicate. MIC values were calculated using Prism (version 5.01).
Crystallization
BioA was co-crystallized with 14-Cl, 15, 16, and 18 by the vapor diffusion method in a hanging drop at 20 °C as previously described (Dai, et al., 2014). Protein solution containing 10 mg/mL BioA in 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5, 50 mM NaCl, 0.1 mM tris(2-carboxyethyl)phosphine (TCEP) was mixed with reservoir solution (9–14% PEG 8000, 100 mM HEPES pH 7.5, 100 mM MgCl2, and 500 μM compound) and a seed solution (a reservoir solution containing crushed BioA crystals) in a 4:3:1 ratio 2 μL BioA protein:1.5 μL reservoir solution: 0.5 μL crushed BioA seed solution). Crystals appear in the drop within 24 hours and grow to their full size in 72 hours. BioA-compound co-crystals were cryo-protected by transferring to a cryo solution (15% PEG400, 15% PEG 8000, 100 mM HEPES pH 7.5, 100 mM MgCl2, and 5 mM compound) using an appropriately sized fiber loop of a cryo pin from Hampton Research and then flash frozen in liquid nitrogen.
Data collection, processing and model building
The diffraction data for a co-crystal with 18 were collected at 100 K using Cu Kα radiation on a Rigaku HighFlux HomeLab rotating-anode system with a Saturn 944+ CDD detector in the Kahlert Structural Biology Laboratory at the University of Minnesota. Diffraction data for 15 and 14-Cl, co-crystals were collected on a NOIR-1 CCD at ALS beamline 4.2.2 (Berkeley, CA). The data were processed, integrated, and scaled with d*TREK (Pflugrath, 1999). The structures were solved by molecular replacement using Phaser (McCoy, et al., 2007) in the CCP4 package (Winn, et al., 2011) using atomic coordinates from PDB code 3TFT as a search model (Shi, et al., 2011). Refinement and model building were done using Refmac5 (Murshudov, et al., 2011), Phenix (Adams, et al., 2010) and coot (Emsley and Cowtan, 2004). Ligand geometry restraints were generated with PRODRG (Schuttelkopf and van Aalten, 2004). Figure 5 was prepared with PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.), and Ligplot+ (Laskowski and Swindells, 2011). A summary of crystallographic data and refinement statistics is provided as Supplemental Information Table S2. Atomic coordinates have been deposited in the PDB with accession codes 4W1V, 4W1W, and 4W1X, for complexes with 18, 15, and 14-Cl, respectively.
Isothermal Titration Calorimetry
The ITC experiments were conducted on an automated microcalorimeter (Malvern Instruments Ltd, UK). The experiments were performed at 25 °C in ITC buffer (25 mM HEPES (pH 7.5), 50 mM NaCl). BioA was exchanged (2 × 13 mL) into ITC buffer using an Amicon Ultra concentrator and the final filtrate was used to prepare a solution of 14. In the titration experiments, 14 was injected into a solution of the enzyme. Ligand and protein concentrations were 10.2 μM for BioA (determined by Bradford analysis using BSA as a standard) and 103 μM for 14 (determined by weighing sample on a ultramicrobalance accurate 0.001 mg). Titrations were carried with a stirring speed of 750 rpm and 150 s interval between 4 μL injections. The first injection was excluded from data fitting. Titrations were run past the point of enzyme saturation to determine and correct for heats of dilution. The experimental data were fit to a theoretical titration curve using the Origin software package (version 7.0) provided with the instrument to afford values of KA (the association constant in M−1), n (the number of binding sites per monomer) and ΔH (the binding enthalpy change in kilocalories per mole). The thermodynamic parameters ΔG and ΔS were calculated using equation 1:
| (1) |
where ΔG, ΔH and ΔS are the changes in free energy, enthalpy, and entropy of binding, respectively, R = 1.98 cal mol−1 K−1, and T is the absolute temperature. The affinity of the ligand for the protein is given as the dissociation constant (KD = 1/KA). ITC experiments were performed in duplicate and analyzed independently, and the thermodynamic values obtained were averaged.
Supplementary Material
Figure 3.

Summary of data from all >350,000 wells of the entire MLSMR library screen. (A) Replicate comparison plot of screened compounds. Activity of 0 represents the average of the DMSO neutral control wells, −100 the average of the positive control wells. Compounds with all replicates passing the −40% activity score threshold are considered active (blue), those with the mean of the replicates above −40% are considered inactive (red), and those with a mean replicate activity below −40% but with less than half the replicates passing the threshold are inconclusive (green). (B) Mean activities of all replicates for each compound. Coloring and activity calls are as in panel A. The −40% activity threshold is shown.
Table 1.
Selected BioA and Mtb Hits
| Compound | structure | IC50, μMa | MIC90, μM BioA-UEb |
MIC90, μM WT -biotinc |
MIC90, μM WT+biotind |
MIC90, μM BioA-OEe |
|---|---|---|---|---|---|---|
| 10 |
|
0.075 | >100 | >100 | >100 | >100 |
| 11 |
|
0.144 | 15.1 | 75.1 | >100 | 57.8 |
| 12 |
|
0.148 | 79.3 | >100 | >100 | >100 |
| 13 |
|
0.153 | 0.7 | 2.5 | 8.7 | 4.7 |
| 14 |
|
0.155 | 9.3 | 26.3 | >100 | 98.6 |
| 15 |
|
0.195 | 5.6 | 20.7 | 65.0 | 77.9 |
| 16 |
|
0.508 | 35.0 | >100 | >100 | >100 |
| 17 |
|
0.596 | 98.7 | >100 | >100 | >100 |
| 18 |
|
0.659 | 34.9 | >100 | >100 | >100 |
| 19 |
|
0.788 | 14.5 | 46 | 72.9 | 39.5 |
Full data are available in PubChem (go to: http://pubchem.ncbi.nlm.nih.gov/# and search under the bioassay tab with the given six-digit AID number).
IC50 value against Mtb BioA in the described biochemical assay: AID#651683;
Minimum inhibitory concentration that results in 90% inhibition against the Mtb BioA under-expression strain: AID#743072 and AID#743352,
Minimum inhibitory concentration that results in 90% inhibition against Mtb H37Rv without supplemental biotin: AID#743070 and AID#743350,
Minimum inhibitory concentration that results in 90% inhibition against Mtb H37Rv with 1 μM supplemental biotin: AID#743071 and AID#743349,
Minimum inhibitory concentration that results in 90% inhibition against the Mtb BioA over-expression strain: AID#743073 and AID#743351.
Acknowledgments
This work was funded in part by grants from the National Institutes of Health (R03 MH096537 to C.C.A. and R01AI091790 to D.S.) and by the NIH-MLPCN program (1 U54 HG005032-1 awarded to S.L.S.). We also gratefully acknowledge support from the University of Minnesota Bighley Graduate fellowship (to R.D.) and resources from the University of Minnesota Supercomputing Institute.
Footnotes
Supplemental information includes 1 Figure, 4 Tables, and Supplemental Experimental Procedures and can be found with this article online at: doi10.1016/j.chem.biol.2014.xx.xxx
AUTHOR CONTRIBUTIONS
S.W.P. performed the whole cell studies with Mtb. D.C. performed the primary HTS and secondary biochemical and cytotoxicity assays.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahams GL, Kumar A, Savvi S, Hung AW, Wen S, Abell C, Barry CE, 3rd, Sherman DR, Boshoff HI, Mizrahi V. Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem Biol. 2012;19:844–854. doi: 10.1016/j.chembiol.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich CC, Boshoff HI, Remmel RP. Antitubercular Agents. In: Rotella D, Abraham DJ, editors. Burgers Medicinal Chemistry, Drug Discovery, and Development. 7. Vol. 7. Wiley; Hoboken, N. J: 2010. pp. 713–812. [Google Scholar]
- Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- CDC Centers for Disease Control and Prevention. Achievements in Public Health, 1900–1999: Control of Infectious Diseases. MMWR. 1999;48:621–629. [Google Scholar]
- Copeland RA. Enzymes: A practical introduction to structure, mechanism and data analysis. New York, NY: John Wiley & Sons Inc; 2000. [Google Scholar]
- Dai R, Wilson DJ, Geders TW, Aldrich CC, Finzel BC. Inhibition of Mycobacterium tuberculosis transaminase BioA by aryl hydrazines and hydrazides. Chembiochem. 2014;15:575–586. doi: 10.1002/cbic.201300748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVito JA, Mills JA, Liu VG, Agarwal A, Sizemore CF, Yao Z, Stoughton DM, Cappiello MG, Barbosa MD, Foster LA, et al. An array of target-specific screening strains for antibacterial discovery. Nat Biotechnol. 2002;20:478–483. doi: 10.1038/nbt0502-478. [DOI] [PubMed] [Google Scholar]
- Dey S, Lane JM, Lee RE, Rubin EJ, Sacchettini JC. Structural characterization of the Mycobacterium tuberculosis biotin biosynthesis enzymes 7,8-diaminopelargonic acid synthase and dethiobiotin synthetase. Biochemistry. 2010;49:6746–6760. doi: 10.1021/bi902097j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edfeldt FN, Folmer RH, Breeze AL. Fragment screening to predict druggability (ligandability) and lead discovery success. Drug Discov Today. 2011;16:284–287. doi: 10.1016/j.drudis.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, Trawick JD, Wall D, Wang L, Brown-Driver V, Froelich JM, et al. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol. 2002;43:1387–1400. doi: 10.1046/j.1365-2958.2002.02832.x. [DOI] [PubMed] [Google Scholar]
- Geders TW, Gustafson K, Finzel BC. Use of differential scanning fluorimetry to optimize the purification and crystallization of PLP-dependent enzymes. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68:596–600. doi: 10.1107/S1744309112012912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard B, Duvall JR, Lowe JT, Murillo T, Wei J, Akella LB, Marcaurelle LA. Synthesis of a stereochemically diverse library of medium-sized lactams and sultams via S(N)Ar cycloetherification. ACS Comb Sci. 2011;13:365–374. doi: 10.1021/co2000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard B, Lee MDt, Dandapani S, Duvall JR, Fitzgerald ME, Kesavan S, Lowe JT, Marie JC, Pandya BA, Suh BC, et al. Synthesis of stereochemically and skeletally diverse fused ring systems from functionalized C-glycosides. J Org Chem. 2013;78:5160–5171. doi: 10.1021/jo4000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard B, O’Shea MW, Donckele E, Kesavan S, Akella LB, Xu H, Jacobsen EN, Marcaurelle LA. Application of a catalytic asymmetric Povarov reaction using chiral ureas to the synthesis of a tetrahydroquinoline library. ACS Comb Sci. 2012;14:621–630. doi: 10.1021/co300098v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal P, Dick T. Reactive dirty fragments: implications for tuberculosis drug discovery. Curr Opin Microbiol. 2014;21C:7–12. doi: 10.1016/j.mib.2014.06.015. [DOI] [PubMed] [Google Scholar]
- Kitahara T, Hotta K, Yoshida M, Okami Y. Biological studies of amiclenomycin. J Antibiot (Tokyo) 1975;28:215–221. doi: 10.7164/antibiotics.28.215. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Lee RE, Hurdle JG, Liu J, Bruhn DF, Matt T, Scherman MS, Vaddady PK, Zheng Z, Qi J, Akbergenov R, et al. Spectinamides: a new class of semisynthetic antituberculosis agents that overcome native drug efflux. Nat Med. 2014;20:152–158. doi: 10.1038/nm.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Hanson RE, Cronan JE. Biotin synthesis begins by hijacking the fatty acid synthetic pathway. Nat Chem Biol. 2010;6:682–688. doi: 10.1038/nchembio.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe JT, Lee MDt, Akella LB, Davoine E, Donckele EJ, Durak L, Duvall JR, Gerard B, Holson EB, Joliton A, et al. Synthesis and profiling of a diverse collection of azetidine-based scaffolds for the development of CNS-focused lead-like libraries. J Org Chem. 2012;77:7187–7211. doi: 10.1021/jo300974j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann S, Colliandre L, Labesse G, Ploux O. Inhibition of 7,8-diaminopelargonic acid aminotransferase from Mycobacterium tuberculosis by chiral and achiral anologs of its substrate: biological implications. Biochimie. 2009;91:826–834. doi: 10.1016/j.biochi.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Mann S, Eveleigh L, Lequin O, Ploux O. A microplate fluorescence assay for DAPA aminotransferase by detection of the vicinal diamine 7,8-diaminopelargonic acid. Anal Biochem. 2013;432:90–96. doi: 10.1016/j.ab.2012.09.038. [DOI] [PubMed] [Google Scholar]
- Mann S, Ploux O. 7,8-Diaminoperlargonic acid aminotransferase from Mycobacterium tuberculosis, a potential therapeutic target. Characterization and inhibition studies. FEBS J. 2006;273:4778–4789. doi: 10.1111/j.1742-4658.2006.05479.x. [DOI] [PubMed] [Google Scholar]
- Mann S, Ploux O. Pyridoxal-5′-phosphate-dependent enzymes involved in biotin biosynthesis: structure, reaction mechanism and inhibition. Biochim Biophys Acta. 2011;1814:1459–1466. doi: 10.1016/j.bbapap.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Marcaurelle LA, Johannes C, Yohannes D, Tillotson BP, Mann D. Diversity-oriented synthesis of a cytisine-inspired pyridone library leading to the discovery of novel inhibitors of Bcl-2. Bioorg Med Chem Lett. 2009;19:2500–2503. doi: 10.1016/j.bmcl.2009.03.037. [DOI] [PubMed] [Google Scholar]
- Marriner GA, Nayyar A, Uh E, Wong SY, Mukherjee T, Via LE, Carroll M, Edwards RL, Gruber TD, Choi I, et al. The medicinal chemistry of tuberculosis chemotherapy. Top Med Chem. 2011;7:47–124. [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- Mulrooney CA, Lahr DL, Quintin MJ, Youngsaye W, Moccia D, Asiedu JK, Mulligan EL, Akella LB, Marcaurelle LA, Montgomery P, et al. An informatic pipeline for managing high-throughput screening experiments and analyzing data from stereochemically diverse libraries. J Comput Aided Mol Des. 2013;27:455–468. doi: 10.1007/s10822-013-9641-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH. NIH Fact Sheet: Tuberculosis. 2014 http://report.nih.gov/nihfactsheets/ViewFactSheet.aspx?csid=31.
- Palmer AC, Kishony R. Opposing effects of target overexpression reveal drug mechanisms. Nat Commun. 2014;5:article number 4296. doi: 10.1038/ncomms5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-W, Klotzsche M, Wilson DJ, Boshoff HI, Eoh H, Manjunatha U, Blumenthal A, Rhee K, Barry CE, 3rd, Aldrich CC, et al. Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLoS Pathog. 2011;7:e1002264. doi: 10.1371/journal.ppat.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- Phillips JW, Goetz MA, Smith SK, Zink DL, Polishook J, Onishi R, Salowe S, Wiltsie J, Allocco J, Sigmund J, et al. Discovery of kibdelomycin, a potent new class of bacterial type II topoisomerase inhibitor by chemical-genetic profiling in Staphylococcus aureus. Chem Biol. 2011;18:955–965. doi: 10.1016/j.chembiol.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Sandmark J, Mann S, Marquet A, Schneider G. Structural basis for the inhibition of the biosynthesis of biotin by the antibiotic amiclenomycin. J Biol Chem. 2002;277:43352–43358. doi: 10.1074/jbc.M207239200. [DOI] [PubMed] [Google Scholar]
- Schatz A, Bugie E, Waksman S. Streptomycin, a substance exhibiting antibiotic activity against Gram-positive and Gram-negative bacteria. Proc Soc Expt Biol Med. 1944;55:66–69. doi: 10.1097/01.blo.0000175887.98112.fe. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Shapiro MM, Chakravartty V, Cronan JE. Remarkable diversity in the enzymes catalyzing the last step in synthesis of the pimelate moiety of biotin. PloS one. 2012;7:e49440. doi: 10.1371/journal.pone.0049440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Geders TW, Park SW, Wilson DJ, Boshoff HI, Abayomi O, Barry CE, 3rd, Schnappinger D, Finzel BC, Aldrich CC. Mechanism-based inactivation by aromatization of the transaminase BioA involved in biotin biosynthesis in Mycobaterium tuberculosis. J Am Chem Soc. 2011;133:18194–18201. doi: 10.1021/ja204036t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LL. Viable screening targets related to the bacterial cell wall. Ann N Y Acad Sci. 2013;1277:29–53. doi: 10.1111/nyas.12006. [DOI] [PubMed] [Google Scholar]
- Vasantkumar GR. Hydrolysis of cyclic ureas under microwave irradiation: Synthesis and characterization of 7,8-diaminopelargonic Acid. Synth Commun. 2007;37:2633–2639. [Google Scholar]
- Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature. 2006;441:358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- WHO (World Health Organization) Global Tuberculosis Report 2013 2013 [Google Scholar]
- Wilson DJ, Shi C, Duckworth BP, Muretta JM, Manjunatha U, Sham YY, Thomas DD, Aldrich CC. A continuous fluorescence displacement assay for BioA: an enzyme involved in biotin biosynthesis. Anal Biochem. 2011;416:27–38. doi: 10.1016/j.ab.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin D, Fox B, Lonetto ML, Etherton MR, Payne DJ, Holmes DJ, Rosenberg M, Ji Y. Identification of antimicrobial targets using a comprehensive genomic approach. Pharmacogenomics. 2004;5:101–113. doi: 10.1517/phgs.5.1.101.25679. [DOI] [PubMed] [Google Scholar]
- Young K, Jayasuriya H, Ondeyka JG, Herath K, Zhang C, Kodali S, Galgoci A, Painter R, Brown-Driver V, Yamamoto R, et al. Discovery of FabH/FabF inhibitors from natural products. Antimicrob Agents Chemother. 2006;50:519–526. doi: 10.1128/AAC.50.2.519-526.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zlitni S, Ferruccio LF, Brown ED. Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat Chem Biol. 2013;9:796–804. doi: 10.1038/nchembio.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.