

Abstract
Epstein–Barr virus (EBV) and Kaposi's sarcoma‐associated herpesvirus (KSHV) are two human gammaherpesviruses associated with a broad spectrum of B‐cell lymphomas, most acutely in immuno‐compromised populations. However, there are no drugs which specifically target KSHV or EBV‐associated lymphomas. To identify small molecules which selectively inhibit the growth of EBV or KSHV‐associated B‐cell lines, we performed a fluorescence based high‐throughput screen on multiple stable GFP expressing virus‐infected or uninfected B‐cell lines. We identified 40 initial compounds with selective growth inhibition and subsequently determined the 50% growth inhibitory concentrations (GI50) for each drug. We further examined compounds with higher specificity to explore the underlying molecular mechanisms using transcription factor analysis, as well as a shRNA based knockdown strategy. Our data identified ten compounds with relatively high efficacy for growth inhibition. Two novel small molecules, NSC#10010 and NSC#65381 were potent growth inhibitors for gammaherpesvirus‐associated B‐lymphomas through activation of both the NF‐κB and c‐Myc‐mediated signaling pathways. These drugs can serve as potential lead compounds to expand the current therapeutic window against EBV or KSHV‐associated human B‐cell malignancies.
Keywords: EBV, KSHV, High throughput drug screening, Lymphoma
Highlights
Screening identified compounds lethal to EBV and KSHV‐infected lymphoma B‐cells.
Specific and effective compounds trigger an increase in NF‐κB and c‐Myc signaling.
In many cases, NF‐κB and c‐Myc knockdown sensitizes B‐cells to chemical apoptosis.
The identified lead compounds may be effective against KSHV and EBV malignancies.
1. Introduction
The human γ‐herpesviruses Epstein–Barr virus (EBV) and Kaposi's sarcoma‐associated herpesvirus (KSHV) are large enveloped DNA viruses which infect and establish lifelong persistence in B‐lymphocytes (Damania, 2007). Nearly ubiquitous in human populations, EBV is the causative agent of infectious mononucleosis and is associated with a variety of B‐cell malignancies; including Hodgkin's lymphoma, Burkitt's lymphoma (BL) and post‐transplant lymphoproliferative diseases (Saha et al., 2010).
KSHV is less widespread but its prevalence can reach over 50% depending on risk factors. KSHV is the causative agent of Kaposi's sarcoma (KS) and is associated with two major B‐cell lymphomas in AIDS patients: primary effusion lymphomas (PEL) and multicentric Castleman's disease (Hayward and Whitby, 2009). Solid organ and hematopoietic stem cell transplant recipients and HIV‐infected patients exhibit elevated risk for KSHV and EBV‐associated malignancies (Engels et al., 2011).
EBV and KSHV have evolved strategies to promote cell survival while protecting infected cells from apoptosis. Infections induce widespread genetic and proteomic responses in the host cell (Verma et al., 2009). While certain cancers have strict viral associations, most clinical therapies used to treat these malignancies do not specifically target oncogenic viral mechanisms.
Most chemotherapeutic drugs are mitotic inhibitors which target fundamental cellular structural and metabolic processes such as microtubule assembly, protein synthesis, or DNA synthesis. Therefore, therapeutic doses can non‐specifically inhibit cellular growth, causing significant side effects (Castellino et al., 2011; Herold and Hieke, 2002; Wilson et al., 2002). Several drugs such as the anti‐CD20 antibody Rituximab are specific to B‐cells. However, they are not effective enough to be used as single agents (Thomas et al., 2010). Considering the high prevalence of both EBV and KSHV‐associated B‐cell lymphomas, small molecules which can specifically target latent viral infections will improve current treatments. Virus‐specific treatments would be less likely to target basic metabolic mechanisms, and thus more efficiently kill virus infected cells with fewer potential side effects.
Our study has identified and characterized potential ‘lead compounds’ against latently infected EBV and KSHV positive B‐cell lymphomas, which can be rapidly translated into clinically viable drugs. We describe a high‐throughput fluorescence‐based screen (HTS) to identify small molecule drug candidates that are preferentially lethal to EBV or KSHV‐mediated B‐cell lymphomas. Further, we then identified transcription factor pathways affected by these small molecules, and showed that inhibiting those pathways increases the lethality of these small molecules. Our results yield new insights on targeted drug discovery through modulation of cellular pathways with potential therapeutic value in clinical trials against human oncogenic γ‐herpesvirus associated lymphomas.
2. Materials & methods
2.1. Materials
The ‘Mechanistic Set’ and the ‘Diversity Set II’ were obtained from the Developmental Therapeutics Program of the US National Cancer Institute (http://www.dtp.nci.nih.gov). The Mechanistic Set contains 879 growth inhibitory small molecules, while the Diversity Set II contains 1364 small molecules covering a range of chemical structures. 23 molecules are shared between both sets to total 2220 unique small molecules.
2.2. Cell lines and generation of stable GFP lines
BC3 and BCBL1 are KSHV‐positive pleural effusion lymphoma (PEL) B‐cell lines (Renne et al., 1996). LCL1 and LCL2 are in vitro EBV transformed B‐cell lines generated in our lab (Murakami et al., 2005). BJAB, Louckes, and Ramos are virus negative BL B‐cells obtained from Elliott Kieff (Harvard Medical School, Boston, MA) (Murakami et al., 2005). Cell lines were cultured as described previously (Murakami et al., 2013, 2013, 2005).
The lentiviral vector HIV‐CS‐CG contains a GFP cassette under the CMV promoter (Miyoshi et al., 1998), and Lentiviral packaging vector pCMV‐VSV‐G, pRSV‐Rev and pMDLg/pRRE were obtained from Addgene (Cambridge, MA). Lentivirus production and stable transduction of B‐cells for expression of GFP was done as previously described (Lu et al., 2009).
2.3. Small molecule HTS
40,000 cells of each GFP‐expressing B‐cell line in RPMI‐1640 were plated in 96‐well plate. Cells were incubated with 10 μM of each small molecule. Plates were imaged for GFP with an IVIS Lumina II (Caliper Life Sciences, Hopkinton, MA) at the University of Pennsylvania Small Animal Imaging Facility (SAIF) and quantified using the Odyssey software v3.0 (LI‐Cor Biosciences, Lincoln, NE). Growth inhibition was confirmed by light microscopy.
2.4. Determination of 50% growth inhibitory (GI50) concentrations
Similar to the HTS, small molecules were added to each well at 100 μM, 50 μM, 10 μM, 5 μM, 1 μM, 0.5 μM, 0.01 μM, and 0.05 μM. Plates were incubated for 5 days, and imaged for GFP signal with a Typhoon 9410 (GE Healthcare, Piscataway, NJ), and quantified using the Odyssey software v3.0 (LI‐Cor Biosciences, Lincoln, NE). No obvious difference was seen when the plates were scanned at 3 days or 5 days.
2.5. Luciferase reporter pathway assays
12 million Louckes, BC‐3 and LCL1 cells were transiently transfected with 20 μg of each Cignal Reporter plasmid from the Stress and Toxicity 10‐Pathway Reporter Assay (SA Biosciences, Frederick, MD) by electroporation with a Bio‐Rad GenePulser Xcell (Bio‐Rad, Hercules, CA) at 210 V and 20 ms pulse duration. After 2 days, small molecules were added at GI50 concentrations and incubated for an additional 2 days. Cells were washed with 1× phosphate buffered saline (PBS), lysed in Luciferase assay buffer (MBL International Corp., Woburn, MA) and assayed using a LMaxII384 Luminometer (Molecular Devices, LLC, Sunnyvale, CA) using the Luciferase substrates as recommended by the manufacturer.
2.6. Apoptosis assay
1 million cells were treated with drugs for 3 days, spun down and resuspended in 10 mg/mL of Acridine Orange and Ethidium Bromide. An aliquot of each sample was prepared on cover slips and apoptosis observed using an Olympus IX71 microscope at 200× magnification. Cell apoptosis and necrosis were evaluated based on established parameters (Ribble et al., 2005).
2.7. Generation of shRNA expressing constructs and viral transduction
Short‐hairpin oligonucleotides directed against c‐Myc and NF‐κB expressions were designed with siRNA Designer (Promega, Madison, WI). The sense strand of the c‐Myc‐shRNA sequence is 5′tcgagtgctgttgacagtgagcgaGAGGATATCTGGAAGAAATTCtagtgaagccacagatgtaga ATTTCTTCCAGATATCCTCgtgcctactgcctcggaa3′. The sense strand of the NF‐κB‐shRNA sequence is 5′tcgagtgctgttgacagtgagcgaGCTTCCCACACTATGGATTTCtagtgaagccacagatgtagaAATCCATAGTGTGGGAAGCgtgcctactgcctcggaa3′. shRNA expression constructs were previously described (Saha et al., 2011). Lentivirus production and transduction of B‐cells was as previously described (Lu et al., 2009). Cells were selected with 0.2 μg/mL puromycin (Sigma Aldrich, St. Louis, MO) for 3 weeks, confirmed by GFP visualization. Knockdown of expression was confirmed by Western blot using the monoclonal antibody anti‐c‐Myc (9E10) prepared from hybridoma cultures and rabbit polyclonal anti‐NF‐κB p50 (NLS) (Santa Cruz Inc., Santa Cruz, CA).
2.8. Western blotting
10 million cells were treated with DMSO and 10 selected compound for 24 h followed by Western blotting as described earlier (Lu et al., 2014). In brief, cell lysates subjected to SDS‐PAGE followed by western‐blot analysis. The nylon membrane was blocked with 5% skim milk in PBS for 1 h, followed by incubation with appropriate dilution of primary antibodies (in 1XPBS) for overnight at 4 °C. Secondary antibodies with IR‐dye tagged were used to detect the binding of primary antibodies. The membrane was scanned using an Odyssey imager LiCor Inc. (Lincoln, NE). Densitometry for the quantitation of the protein bands was carried out using Odyssey scanning software.
2.9. Electrophoretic mobility shift assay
Oligonucleotides containing the NF‐κB and Myc containing site were synthesized and annealed to the respective antisense strand by gradient cooling. All procedures were followed as described earlier (Verma et al., 2006). The dsDNA probe was packed in with γ‐32P‐labeled dCTP, dATP, dGTP, and dTTP using the Klenow fragment (New England Biolabs, Beverly, MA). Further the labeled probes were purified on a NucTrap probe purification column (Stratagene Inc., La Jolla, CA). The total incorporation of [γ‐32P]dCTP was measured, and approximately 1 million cpm of probe was used per reaction. Nuclear extracts prepared from BC3 and LCL1 cells were used as a source of Myc and NF‐κB transcription factor. Anti‐NF‐κB and anti‐Myc(9E10) antibody (Santa Cruz Biotechnology) was used for supershift analysis. Nuclear extract (5.0 μg of total protein) was used for binding to the indicated probes in electrophoretic mobility shift assay reactions. Unlabeled competitor (100‐fold) was added 5 min prior to the addition of radio labeled probes; 1.0 μg of mouse anti‐NF‐κB and anti‐9E10 was used to supershift the complex. The protein–DNA binding reaction mixture was incubated at 25 °C for 15 min. The bound complexes were loaded onto a 6.5% polyacrylamide gel containing 0.5 TBE (0.045 M Tris–borate, pH 8.2, 1 mM EDTA). The gel was resolved in TBE for 4 h at 150 V, dried, and autoradiographed using a PhosphorImager plate (Molecular Dynamics, Inc.).
3. Results
3.1. High‐throughput screen (HTS) of small molecules against human B‐lymphoma cell lines
To identify small molecule growth inhibitors of latently infected EBV and KSHV positive human B‐cells, we generated six GFP expressing B‐cell lines: two EBV‐mediated in vitro transformed lymphoblastoid cell lines LCL1 and LCL2, two KSHV‐positive pleural effusion lymphoma (PEL) B‐cell lines BC‐3 and BCBL1, and two virus negative Burkitt's Lymphoma (BL) B‐cell lines BJAB and Louckes (Figure 1A). We screened a collection of 2220 small molecule drugs from the Developmental Therapeutics Program (DTP) of the National Cancer Institute (NCI) of the United States.
Figure 1.
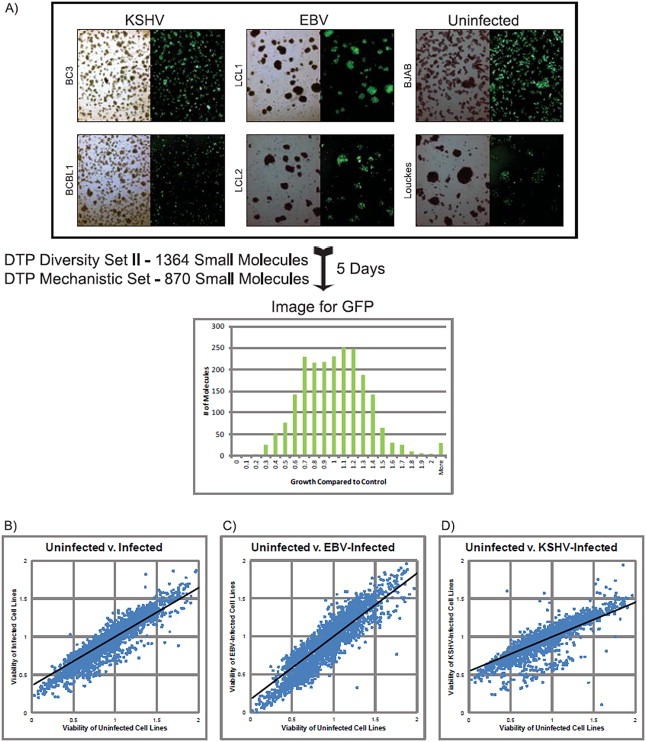
A) Schematic representation of high‐throughput small (HTS) molecule screening strategy and scatter plot representation. KSHV‐infected (BC3 and BCBL1), in vitro EBV‐transformed (LCL1 and LCL2), and uninfected B‐cell lines (BJAB, Louckes and Ramos) were infected with a lentiviral vector (HIV‐CSCG), which results in cell lines constitutively expressing GFP. A histogram showing the range of GFP signals is shown for 5 days post‐treatment. Growth is shown as ratios of signal compared to control growth. Growth inhibition was confirmed by light microscopy. B–D) Scatter plot representation of HTS data. Quantified data from HTS (as described in 1A) for each cell line were normalized and collectively averaged based on viral state in B‐cell lines. Data from 0% to 200% range was plotted in all three cases. Best fit lines were calculated and plotted. The clustering of the points indicates a similarity in the drug response between uninfected cell lines and KSHV‐infected cell lines and the lower slope of the LCL1 best fit line indicates that they are less responsive to cell growth inhibition compared to uninfected cell lines.
To identify anti‐proliferative compounds, we carried out HTS using GFP‐expressing B‐cell lines incubated with 10 μM of each molecule in a 96‐well plate (Figure 1A). Cells were titrated to be in exponential growth during HTS. In addition, DMSO‐treated cells were included as controls and analyzed 5 days post‐treatment. Plates were imaged and quantified for GFP expression as a marker of cell‐growth (Figure 1A) and inhibition was verified by microscopy.
3.2. KSHV infected B‐cells are more susceptible to drug treatment
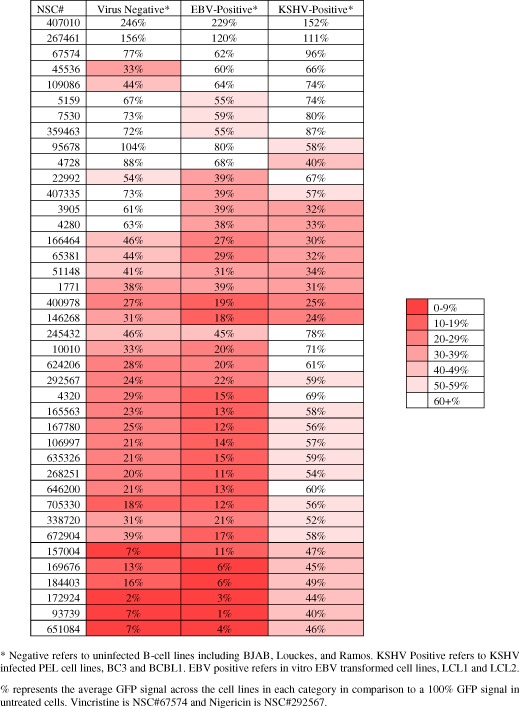
HTS data from each cell line was normalized and the data presented as scatter‐plots (Figure 1B). Based on the best fit line in the scatter‐plots of the primary screening data, KSHV‐infected lymphoma cell lines were more susceptible to drug‐induced cell death compared to other cell lines. This was even more evident when compared to the two EBV‐transformed Lymphoblastoid cell lines (LCLs) (Figure 1D). This is expected because EBV exhibits greater transformation capability compared to KSHV. The tighter clustering of values within the cell viability data demonstrated that the KSHV infected PEL cell lines and uninfected BL cell lines had a more similar response to the small molecules (Figure 1, compare B & D). EBV transformed LCLs were not as clustered, suggesting a wider distribution of responses to the compounds (Figure 1, compare B and C). Within the set of growth inhibitory drugs, we further selected 40 drugs based on three criteria: – drugs toxic to all cell lines, and drugs preferentially toxic to EBV, or those with preferential toxicity to KSHV‐infected cell lines. Cell viability data for each of the 40 compounds are shown by heat map in Table 1. We also selected Vincristine (NSC#67574), a known mitotic inhibitor currently used in chemotherapy regimens against multiple leukemia and lymphomas including EBV‐associated B‐cell lymphomas (Coiffier et al., 2002; Diehl et al., 2003; Kantarjian et al., 2004). Vincristine (NSC# 67574) showed greater than 50% viability against all cell lines, suggesting that it was not efficiently killing B‐lymphoma cells irrespective of viral status (Table 1). Nigericin (NSC#292567), a molecule previously identified to be selective against epithelial cancer stem cells, was also identified in our screen as selective against our KSHV‐infected B‐cell lines (Table 1) (Gupta et al., 2009). From this initial screen, more than 50% of the compounds showed efficient killing with less than 20% survival for the KSHV positive cell lines. Interestingly, when compared to the virus negative cells, these compounds also showed efficient killing of greater than 60%. The EBV positive cells were more resistant but showed sensitivity as shown by greater than 60% for at least 9 compounds shown (Table 1).
Table 1.
40 selected lethal small molecules based on high‐throughput screen (HTS) data.
3.3. Identification of compounds with preferential killing of viral positive and negative cell lines by GI50
To determine 50% growth inhibition (GI50) concentrations, we exposed GFP‐expressing LCL1, BC3, Louckes and Ramos cell lines to drugs from 0.05 to 100 μM for 5 days. GFP signal intensity and growth inhibition was corroborated by fluorescence and verified by light microscopy (Figure 2A).
Figure 2.
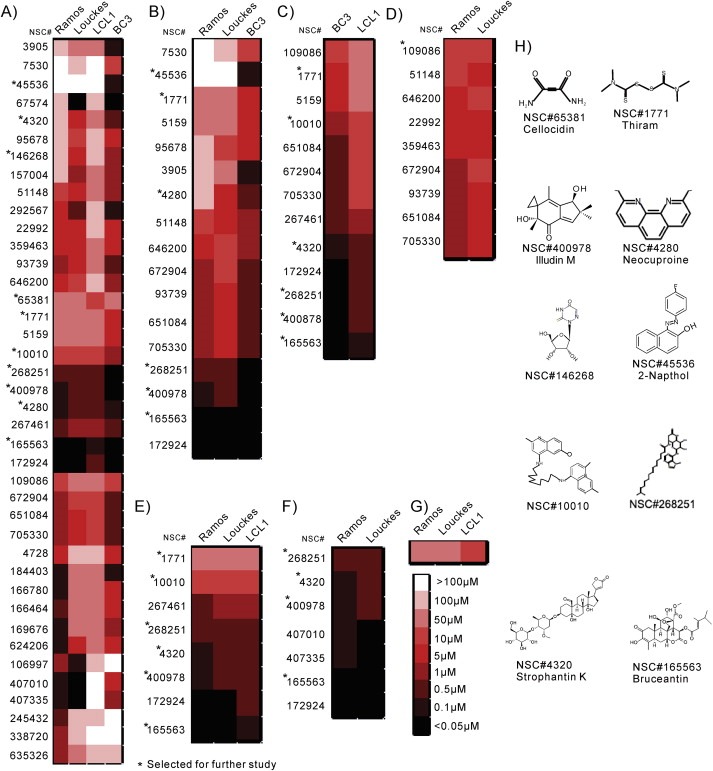
Determination of 50% growth inhibitory concentrations (GI 50 ) of 40 selected small molecules and chemical structures for 10 selected small molecules. GFP‐expressing BC3, LCL1, Louckes and Ramos cells were treated in a gradient of 8 drug concentrations from 100 μM to 0.05 μM in half log steps. Compounds which showed GI50 at 0.05 μM for a particular cell line is shown in highest intense color and as expected the cell lines showing a GI50 at 100 μM had the least intense color. Results were clustered by hierarchical clustering complete linkage. A) Results were ordered by hierarchical clustering based on lethality pattern. The molecules selected for further study show significant levels of specificity or overall lethality. B) Molecules specific to BC3. C) Molecules specific to KSHV and EBV‐infected cell lines. D) Molecules showing similar responses in Burkitt's lymphoma cell lines. E) Most generally lethal molecules, F) Molecules most lethal in Burkitt's lymphoma cell lines. G) Molecules specifically lethal to the EBV‐infected cell line. H) Structures and names of 10 molecules selected from growth inhibition data as specifically lethal to either EBV or KSHV‐infected cells or lethal to cell lines in our screen.
Seven compounds were identified that effectively killed Louckes and Ramos at concentrations of 0.5 μM or lower, as well as 9 compounds that were similarly effective in killing these cells (Figure 2F and 2D, respectively).
Comparison of compounds with effective killing between EBV‐ and KSHV‐positive cell lines showed a range of effects. Five compounds were highly effective below 5 μM, 5 were effective at concentrations between 5 and 10 μM and 3 were effective between 10 and 100 μM. BC3 was consistently more sensitive towards these compounds at 5–10 μM concentrations compared to EBV‐positive transformed LCLs (Figure 2C).
In the HTS, the KSHV‐positive PEL cell lines were more sensitive to drug treatment, and EBV‐positive cell lines were more tolerant to treatment (Figure 1D and 1C). Similarly, 17 compounds showed effective killing of BC3 cells (Figure 2B), while only one drug, NSC#65381, showed an enhanced preference for killing LCL1 (Figure 2G).
NSC#1771, NSC#3905, NSC#4280, NSC#10010, and NSC#268251 exhibited greater lethality against BC3 compared to other cell lines. NSC#45536 was the most lethal to BC3 (Figure 2B).
Due to greater tolerance, drugs which killed LCL1 cells had an enhanced tendency to be similarly lethal against all B‐cells (Figure 2E). In particular, NSC#165563, NSC#172924, NSC#268251, and NSC#400978 showed lethality at 0.5 μM or less in all cell lines (Figure 2A).
Ten small molecules showing preferential or general growth inhibition were selected for follow up analysis. NSC#1771 and NSC#65381 are similar in structure and highly effective in killing KSHV and EBV infected cells. NSC#4280, NSC#10010, NSC#45536 and NSC#146268 have similar cyclic structures which may intercalate in DNA grooves or protein, inhibiting major cellular processes such as DNA replication and signaling activities. Structural similarities between NSC#4320, NSC#165563 and NSC#400978 suggest that they may affect similar mechanisms. Similarities in structure may be a predictor in generating analogs with less toxicity and greater preference for lethality against virally infected and/or negative B‐cell lymphomas (Figure 2H).
3.4. c‐Myc and NF‐κB mediated pathways were specifically activated by small molecule drugs in γ‐herpesvirus infected B‐cells
Human diseases are often linked to dysfunctional signal transduction networks. Small molecule drug candidates can modulate downstream transcription factors to disrupt or restore these signaling pathways (Wang et al., 2006). To determine which pathways these compounds were affecting, we used the Cignal™ transcription reporter assay for ten pathways: Myc/Max, SMAD2/3/4, NFAT, AP1, ELK1/SRF, E2F/DP1, Glucocorticoid Receptor (GR), NF‐κB, HIF‐1α, and p53. These reporters would provide direct evidence of the involvement of these pathways targeted by the small molecules.
Important signaling pathways were altered by these small molecules across all three different cell types (non‐infected, EBV infected and KSHV infected). NSC#1771 and NSC#4280 activated all the examined pathways in EBV transformed LCLs (Figure 3A). NSC#10010 and NSC#65381, each were highly preferential to BC3 and LCL1, respectively, and both activated NF‐κB and Myc/Max mediated pathways (Figure 3A). NSC#1771, a growth inhibitor of BC3, induced the broadest pathway activity in all three B‐cell lines. Transcription factors Myc/Max, SMAD2/3/4, ELK1/SRF and GR were shown to be activated in BC3 cells by NSC#1771 (Figure 3A).
Figure 3.
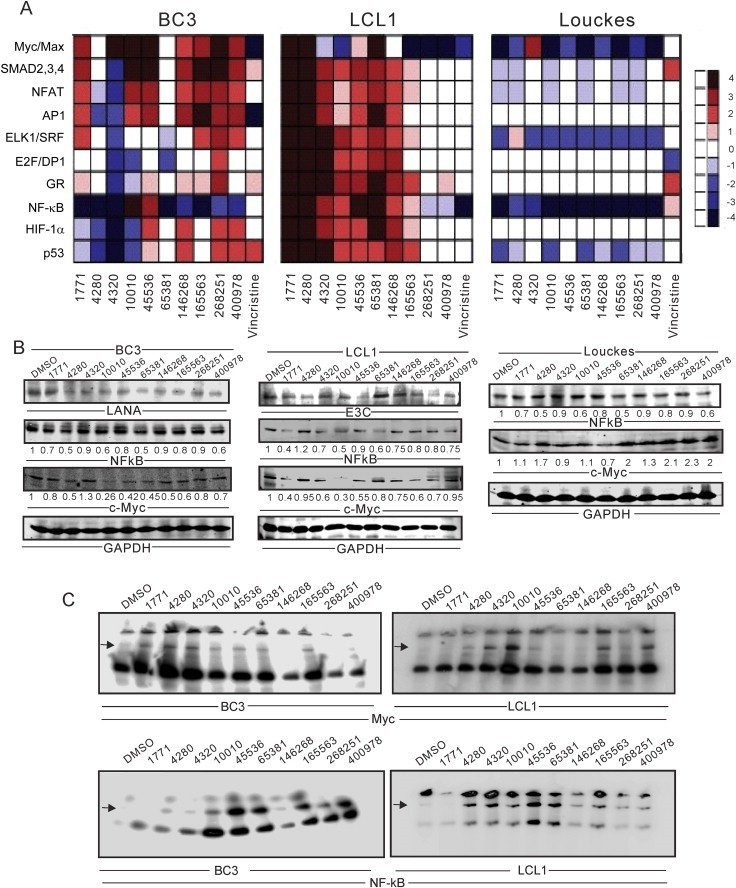
Myc/Max and NF‐κB mediated pathways are extensively modulated by drug treatment. (A) BC3, LCL1 and Louckes were transiently transfected with reporter plasmids and treated with 10 selected small molecules at their GI50. Two days post‐treatment, cells were lysed and assayed for Luciferase activity. The DMSO drug free control was used as a negative control. Transfection controls were normalized using the Renilla Luciferase, and the signaling intensities of virus positive cell lines were then compared with the values from the DMSO controls. (B) Western blot were performed in BC3, LCL1 and Louckes treated DMSO and 10 selected small molecules compounds. NF‐κB, c‐Myc, GAPDH, LANA and EBNA3C antibodies were applied. (C) Myc probe and antibody in BC3 and LCL1 and NF‐κB probe and antibody in BC3 and LCL1 were used in this study. EMSA was performed essentially as described in “Material and Methods” section. Super‐shift complexes were denoted by arrow on the line.
NF‐κB and c‐Myc mediated pathways were broadly affected by almost all the compounds tested in this assay. NF‐κB and c‐Myc broadly affects cellular functions, including cell proliferation and cell death (Kaileh and Sen, 2012). A NF‐κB promoter site lies upstream of c‐Myc, linking the expression of NF‐κB expression and c‐Myc in normal cells (Duyao et al., 1990) and in BL cell lines (Ji et al., 1994). NF‐κB and c‐Myc are typically expressed in response to a variety of drug treatments (Bourgarel‐Rey et al., 2001; Park et al., 2002). This may be expected since both KSHV and EBV have transcriptional activation strategies for usurping NF‐κB and c‐Myc activities (Ahmad et al., 2010; Faumont et al., 2009; Graham et al., 2013). Further we validated our results with Western blotting and EMSA binding assays. Interestingly, we observed that the small molecules were more effective in BC3 and LCL1 cells compared to the control Louckes cell line (Figure 3B). These results were followed by EMSAs where super‐shifted complexes were expected in the presence of specific binding (Verma et al., 2006). In support of our earlier results, EMSAs showed preferential interactions of the selected small molecules when compared to DMSO using nuclear lysates from BC3 and LCL1 cells (Figure 3C). We also determined the efficacy of probe binding with nuclear lysates in the presence of specific antibody and cold competitor (data not shown). Overall, the small molecules used in our studies induced NF‐κB and c‐Myc binding activity to a greater level when compared to the control (Figure 3A–C).
Our assays showed that these compounds affected multiple other cellular pathways, some more than others. In BC3, the SMAD2,3,4 and AP1 pathways were affected, whereas the Hif‐1α and p53 pathways were less affected. In contrast, the ELK1/SRF and p53 pathways were activated by a large number of compounds in the EBV negative B‐cell line Louckes (Figure 3A). However, we cannot rule out the involvement of other molecular pathways not explored in this study.
3.5. NSC#10010 and NSC#65381 induced necrotic cell death in γ‐herpesvirus infected B‐cells
To determine the mechanism of drug‐induced cell death in γ‐herpesvirus infected B‐cells, we counted the viable, apoptotic and necrotic cells after 3 days of drug treatment at GI50 concentrations using a modified Ethidium Bromide/Acridine Orange staining method (Ribble et al., 2005).
NSC#1771 induced apoptotic cell death across all four cell lines, and promoted necrotic cell death in EBV transformed LCL1 cells (Figure 4). NSC#10010 and NSC#65381 also displayed necrosis‐mediated cell death in EBV‐transformed LCL1 and BC3, respectively. When exposed to NSC#45536, the virus negative BL line, Ramos showed 40–50% apoptosis and necrosis in contrast to the other virus negative BL line, Louckes (Figure 4). These drugs induced a significant level of apoptosis, suggesting that NSC#10010 and NSC#65381 can enhance NF‐κB and c‐Myc‐regulated apoptosis (Figure 4).
Figure 4.
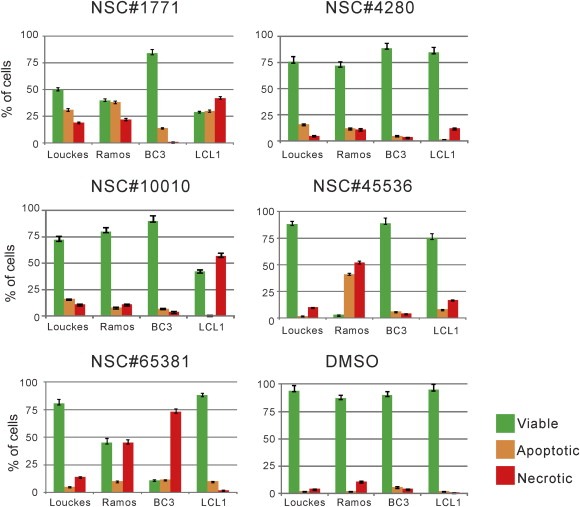
NSC#65381 and NSC#10010 induce specific cell death in both γ‐herpesvirus infected B‐cells. To delineate the underlying mechanism of growth inhibition by drug treatment, cells were treated with 10 selected small molecules at their GI50. Two days post‐treatment, cells were stained with Acridine Orange/Ethidium Bromide and subsequently visualized using Olympus IX71 microscope. The live (green), apoptotic (orange), or necrotic (red) state of the cells were then evaluated using at least 200 cells per sample (p < 0.05).
3.6. c‐Myc and NF‐κB knockdown γ‐herpesvirus infected B‐cells are more sensitive to drug induced cell death
In our study, the results suggested that the drugs that we selected can induce NF‐κB and c‐Myc activity as a response to stress. Considering that both of these pathways are associated with development or proliferation, shutdown of the NF‐κB and Myc pathways should handicap the normal cellular response, resulting in cell death. To characterize the influence of c‐Myc‐ and NF‐κB‐mediated pathways on drug treatment, we generated stable knockdowns for either c‐Myc or the NF‐κB p50 subunit, and repeated the apoptosis/necrosis assay. We further confirmed that these genes were knockdown by Western blot using specific antibodies against c‐Myc and NF‐κB p50.
As expected the knockdown of c‐Myc and NF‐κB expression did not significantly change the effect of drug treatment on apoptosis or necrosis in the Louckes as these were less affected as seen above (Figure 5A). However, the virus positive cell lines showed a more dramatic effect. Strikingly, knocking down either c‐Myc or NF‐κB dropped the viability of BC3 by greater than 50% with a corresponding increase in necrotic cell death after treatment with NSC#1771 (Figure 5B). In contrast, the lethality of NSC#10010 in BC3 increased necrosis in the NF‐κB knockdown cells but not in c‐Myc knockdown cells, indicating that while compounds may exert similar effects, they most likely do so by different mechanisms. NSC#10010 also caused a dramatic increase in apoptotic and necrotic cell death in LCL1 as a result of NF‐κB and c‐Myc knockdown (Figure 5C). Furthermore, NSC#65381 treatment induced substantial apoptotic cell death in BC3 when NF‐κB expression was reduced (Figure 5B).
Figure 5.
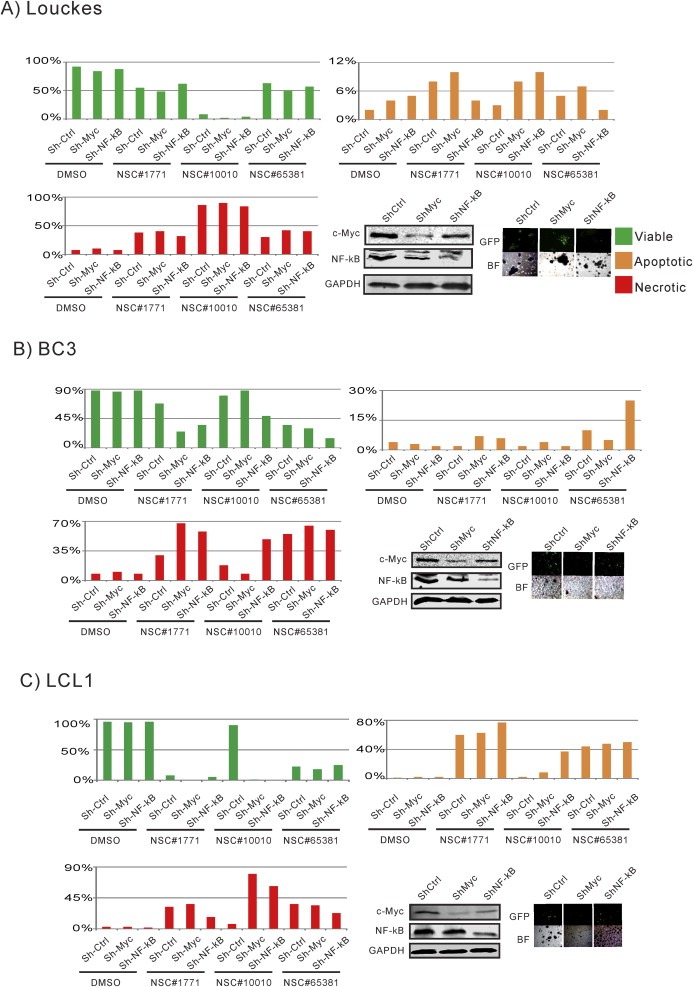
Down‐regulation of c‐Myc and NF‐κB resulted in significantly increased drug sensitivity. A) Louckes, B) BC3, and C) LCL1 cells were transduced with 3 specific lentiviral constructs producing shRNA sequences for control, c‐Myc and NF‐κB knockdown. Knockdown was confirmed by Western blot. Selected cells were then treated with different small molecule drugs at GI50 for 2 days. Post‐treatment, the extent of live (green), apoptosis (orange) and necrotic (red), cells were stained with Acridine Orange/Ethidium Bromide (10 mg/mL). At least 200 cells (p < 0.05) were counted per sample and visualized at 200× optical amplification using Olympus IX71 microscope.
Overall, our results strongly suggest that γ‐herpesvirus‐infected B‐cells are more sensitive to drug treatment when NF‐κB and c‐Myc are reduced. Thus, in the context of γ‐herpesvirus infected B‐lymphoma cells, at expression levels above a certain threshold, NF‐κB and c‐Myc may potentially function as tumor suppressors rather than oncoproteins (Figure 5).
4. Discussion
Novel anti‐tumor drugs with specific mechanisms are needed to treat cancer, particularly those refractory to standard therapies. There are no chemotherapeutic regimens which specifically target virally‐associated B‐cell lymphomas (Little and Yarchoan, 2003). We have identified drug candidates and characterized their modes of action against γ‐herpesvirus infected B‐cell lines. After high‐throughput screening, a panel of 40 potential lethal candidate drugs was selected and 50% growth inhibitory concentrations (GI50) were determined for each cell line. Analysis of these drugs led to selection of two novel drugs, NSC#10010 and NSC#65381, which displayed preferential growth inhibition towards γ‐herpesvirus infected B‐lymphoma cells through the modulation of both NF‐κB and c‐Myc‐mediated signaling.
NSC#10010[N,N′‐bis(6‐methoxy‐2‐methylquinolin‐4‐yl)nonane‐1,9‐diamine] showed consistent growth inhibition against the KSHV‐infected PEL cell lines. NSC#65381(2‐Butynediamide or Cellocidin) demonstrated anti‐proliferative activities against EBV‐transformed cell lines. Interestingly, NSC#10010 and NSC#65381 are structurally different but resulted in similar pathway activation.
We found that both NSC#10010 and NSC#65381 enhanced Myc/Max‐mediated transcriptional activities, preferentially killing γ‐herpesvirus infected B‐cells. Knockdown of c‐Myc expression in LCLs resulted in a significant increase of apoptotic cell death when treated with NSC#10010, possibly through the p53 regulated pathway. This indicated that treatment involving interference with the c‐Myc‐mediated pathway may be a promising strategy in combination with conventional anti‐tumor therapy.
Rationale drug design targeting central cellular regulators of cell‐propagation and apoptosis is a major focus in the development of novel cancer therapies (Bianco et al., 2006; Kasibhatla and Tseng, 2003; Mandal et al., 2009). Future treatments could be improved by adding these compounds to traditional regimens (Bianco et al., 2006). c‐Myc sensitizes cancer cells to apoptosis induced by insufficient mitogenic stimuli, treatment with cytotoxic drugs, as well as radiotherapy (Hoffman and Liebermann, 2008). Also, c‐Myc may be an important consideration in lower dosage chemotherapy strategies designed to overcome drug resistance, toxicity and side effects (Prochownik, 2004). Preclinical studies showed that patients with epithelial ovarian cancer treated with platinum compounds had higher c‐Myc levels compared to non‐responders, and higher survival rates (Iba et al., 2004; von Bueren et al.). Apoptotic response towards many chemotherapeutic drugs such as etoposide, doxorubicin, Camptothecin, and Taxol are enhanced by the presence of c‐Myc (Albihn et al., 2007; von Bueren et al.). Reduction in c‐Myc levels can also improve the effect of many cytotoxic drugs as well as increased sensitivity to the treatment with γ‐radiation by activating p53‐independent apoptotic pathway (Hoffman and Liebermann, 2008; Meyer et al., 2006).
There is significant enthusiasm from both in vitro as well as in vivo animal experiments for the use of NF‐κB inhibitors as a new anti‐cancer therapy (Baud and Karin, 2009; Madonna et al., 2012; Yamamoto and Gaynor, 2001). However, the role of NF‐κB subunits was shown to be diverse in a variety of human cancers (Dolcet et al., 2005). Thus, current NF‐κB inhibitors suffer from unintended effects and non‐specificity (Aggarwal, 2004). Constitutive NF‐κB is one of the core survival mechanisms of B‐lymphoma cells infected with EBV or KSHV (de Oliveira et al., 2010), and inhibition of NF‐κB activity results in spontaneous apoptosis in these virus‐associated B‐lymphomas (Keller et al., 2006). Therapeutically targeting NF‐κB signaling would rely on an understanding of its role in B‐cells. We showed that NSC#10010 and NSC#65381 resulted in activation of the NF‐κB and c‐Myc pathways inducing cell death in these γ‐herpesvirus infected B‐cells. This suggests that strategies aimed at temporal activation of c‐Myc and NF‐κB to boost cell death may be effective for viral‐associated hematological cancers.
The Developmental Therapeutics Program of the US National Cancer Institute has tested NSC#65381 against the L1210 and P388 leukemia mouse models (http://dtp.nci.nih.gov/docs/dtp_search.html). In these experiments, NSC#65381 did not reach therapeutic cut‐offs for further study. However, both of these animal models are leukemia models with known weaknesses as a primary screen for chemotherapeutic agents (Waud, 2004). Also, neither the L1210 nor the P388 cell lines are known to carry murine herpesviruses, so these animal models would not share the virus‐associated vulnerabilities that we are targeting.
We believe that the identified small molecules are promising leads as treatments against EBV and KSHV‐associated B‐cell lymphomas through preferential targeting of the NF‐κB and c‐Myc pathways.
Funding
This work was supported by public health service grants by the National Cancer Institute at the National Institutes of Health (grant numbers CA137894, CA171979, CA174439, CA177423, P30‐DK‐050306 and P01‐CA‐174439 to E.S.R.). Support was also obtained from The Abramson Cancer Center Director's Fund.
Dzeng Richard K., Jha Hem Chandra, Lu Jie, Saha Abhik, Banerjee Sagarika, Robertson Erle S., (2015), Small molecule growth inhibitors of human oncogenic gammaherpesvirus infected B-cells, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.09.006.
References
- Aggarwal, B.B. , 2004. Nuclear factor-kappaB: the enemy within. Cancer Cell. 6, 203–208. [DOI] [PubMed] [Google Scholar]
- Ahmad, A. , Groshong, J.S. , Matta, H. , Schamus, S. , Punj, V. , Robinson, L.J. , Gill, P.S. , Chaudhary, P.M. , 2010. Kaposi sarcoma-associated herpesvirus-encoded viral FLICE inhibitory protein (vFLIP) K13 cooperates with Myc to promote lymphoma in mice. Cancer Biol. Ther. 10, 1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albihn, A. , Mo, H. , Yang, Y. , Henriksson, M. , 2007. Camptothecin-induced apoptosis is enhanced by Myc and involves PKCdelta signaling. Int. J. Cancer. 121, 1821–1829. [DOI] [PubMed] [Google Scholar]
- Baud, V. , Karin, M. , 2009. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 8, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco, R. , Melisi, D. , Ciardiello, F. , Tortora, G. , 2006. Key cancer cell signal transduction pathways as therapeutic targets. Eur. J. Cancer. 42, 290–294. [DOI] [PubMed] [Google Scholar]
- Bourgarel-Rey, V. , Vallee, S. , Rimet, O. , Champion, S. , Braguer, D. , Desobry, A. , Briand, C. , Barra, Y. , 2001. Involvement of nuclear factor kappaB in c-Myc induction by tubulin polymerization inhibitors. Mol. Pharmacol. 59, 1165–1170. [DOI] [PubMed] [Google Scholar]
- Castellino, S.M. , Geiger, A.M. , Mertens, A.C. , Leisenring, W.M. , Tooze, J.A. , Goodman, P. , Stovall, M. , Robison, L.L. , Hudson, M.M. , 2011. Morbidity and mortality in long-term survivors of Hodgkin lymphoma: a report from the Childhood Cancer Survivor Study. Blood. 117, 1806–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coiffier, B. , Lepage, E. , Briere, J. , Herbrecht, R. , Tilly, H. , Bouabdallah, R. , Morel, P. , Van Den Neste, E. , Salles, G. , Gaulard, P. , Reyes, F. , Lederlin, P. , Gisselbrecht, C. , 2002. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 346, 235–242. [DOI] [PubMed] [Google Scholar]
- Damania, B. , 2007. DNA tumor viruses and human cancer. Trends Microbiol. 15, 38–44. [DOI] [PubMed] [Google Scholar]
- de Oliveira, D.E. , Ballon, G. , Cesarman, E. , 2010. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol. 18, 248–257. [DOI] [PubMed] [Google Scholar]
- Diehl, V. , Franklin, J. , Pfreundschuh, M. , Lathan, B. , Paulus, U. , Hasenclever, D. , Tesch, H. , Herrmann, R. , Dorken, B. , Muller-Hermelink, H.K. , Duhmke, E. , Loeffler, M. , 2003. Standard and increased-dose BEACOPP chemotherapy compared with COPP-ABVD for advanced Hodgkin's disease. N. Engl. J. Med. 348, 2386–2395. [DOI] [PubMed] [Google Scholar]
- Dolcet, X. , Llobet, D. , Pallares, J. , Matias-Guiu, X. , 2005. NF-kB in development and progression of human cancer. Virchows Arch. 446, 475–482. [DOI] [PubMed] [Google Scholar]
- Duyao, M.P. , Buckler, A.J. , Sonenshein, G.E. , 1990. Interaction of an NF-kappa B-like factor with a site upstream of the c-myc promoter. Proc. Natl. Acad. Sci. U. S. A. 87, 4727–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels, E.A. , Pfeiffer, R.M. , Fraumeni, J.F. , Kasiske, B.L. , Israni, A.K. , Snyder, J.J. , Wolfe, R.A. , Goodrich, N.P. , Bayakly, A.R. , Clarke, C.A. , Copeland, G. , Finch, J.L. , Fleissner, M.L. , Goodman, M.T. , Kahn, A. , Koch, L. , Lynch, C.F. , Madeleine, M.M. , Pawlish, K. , Rao, C. , Williams, M.A. , Castenson, D. , Curry, M. , Parsons, R. , Fant, G. , Lin, M. , 2011. Spectrum of cancer risk among US solid organ transplant recipients. J. Am. Med. Assoc. 306, 1891–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faumont, N. , Durand-Panteix, S. , Schlee, M. , Gromminger, S. , Schuhmacher, M. , Holzel, M. , Laux, G. , Mailhammer, R. , Rosenwald, A. , Staudt, L.M. , Bornkamm, G.W. , Feuillard, J. , 2009. c-Myc and Rel/NF-kappaB are the two master transcriptional systems activated in the latency III program of Epstein-Barr virus-immortalized B cells. J. Virol. 83, 5014–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, C. , Matta, H. , Yang, Y. , Yi, H. , Suo, Y. , Tolani, B. , Chaudhary, P.M. , 2013. Kaposi's sarcoma-associated herpesvirus oncoprotein K13 protects against B cell receptor-induced growth arrest and apoptosis through NF-kappaB activation. J. Virol. 87, 2242–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, P.B. , Onder, T.T. , Jiang, G. , Tao, K. , Kuperwasser, C. , Weinberg, R.A. , Lander, E.S. , 2009. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 138, 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward, G. , Whitby, D. , 2009. KSHV epidemiology and subtype evolution. In Damania B., Pipas J.M.(Eds.), DNA Tumor Viruses. Springer Science + Business Media; New York: pp. xxvi, 794 pp., [794] p. of plates [Google Scholar]
- Herold, M. , Hieke, K. , 2002. Costs of toxicity during chemotherapy with CHOP, COP/CVP, and fludarabine. Eur. J. Health Econ. 3, 166–172. [DOI] [PubMed] [Google Scholar]
- Hoffman, B. , Liebermann, D.A. , 2008. Apoptotic signaling by c-MYC. Oncogene. 27, 6462–6472. [DOI] [PubMed] [Google Scholar]
- Iba, T. , Kigawa, J. , Kanamori, Y. , Itamochi, H. , Oishi, T. , Simada, M. , Uegaki, K. , Naniwa, J. , Terakawa, N. , 2004. Expression of the c-myc gene as a predictor of chemotherapy response and a prognostic factor in patients with ovarian cancer. Cancer Sci. 95, 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha, H.C. , Upadhyay, S.K. , A J Prasad, M. , Lu, J. , Cai, Q. , Saha, A. , Robertson, E.S. , 2013. H2AX phosphorylation is important for LANA-mediated Kaposi's sarcoma-associated herpesvirus episome persistence. J. Virol. 87, 5255–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha, H.C. , Lu, J. , Saha, A. , Cai, Q. , Banerjee, S. , Prasad, M.A. , Robertson, E.S. , 2013. EBNA3C-mediated regulation of aurora kinase B contributes to Epstein-Barr virus-induced B-cell proliferation through modulation of the activities of the retinoblastoma protein and apoptotic caspases. J. Virol. 87, 12121–12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, L. , Arcinas, M. , Boxer, L.M. , 1994. NF-kappa B sites function as positive regulators of expression of the translocated c-myc allele in Burkitt's lymphoma. Mol. Cell Biol. 14, 7967–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaileh, M. , Sen, R. , 2012. NF-kappaB function in B lymphocytes. Immunol. Rev. 246, 254–271. [DOI] [PubMed] [Google Scholar]
- Kantarjian, H. , Thomas, D. , O'Brien, S. , Cortes, J. , Giles, F. , Jeha, S. , Bueso-Ramos, C.E. , Pierce, S. , Shan, J. , Koller, C. , Beran, M. , Keating, M. , Freireich, E.J. , 2004. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer. 101, 2788–2801. [DOI] [PubMed] [Google Scholar]
- Kasibhatla, S. , Tseng, B. , 2003. Why target apoptosis in cancer treatment?. Mol. Cancer Ther. 2, 573–580. [PubMed] [Google Scholar]
- Keller, S.A. , Hernandez-Hopkins, D. , Vider, J. , Ponomarev, V. , Hyjek, E. , Schattner, E.J. , Cesarman, E. , 2006. NF-kappaB is essential for the progression of KSHV- and EBV-infected lymphomas in vivo. Blood. 107, 3295–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little, R.F. , Yarchoan, R. , 2003. Treatment of gammaherpesvirus-related neoplastic disorders in the immunosuppressed host. Semin. Hematol. 40, 163–171. [DOI] [PubMed] [Google Scholar]
- Lu, J. , Verma, S.C. , Murakami, M. , Cai, Q. , Kumar, P. , Xiao, B. , Robertson, E.S. , 2009. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus (KSHV) upregulates survivin expression in KSHV-associated B-lymphoma cells and contributes to their proliferation. J. Virol. 83, 7129–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J. , Jha, H.C. , Verma, S.C. , Sun, Z. , Banerjee, S. , Dzeng, R. , Robertson, E.S. , 2014. Kaposi's sarcoma-associated herpesvirus-encoded LANA contributes to viral latent replication by activating phosphorylation of survivin. J. Virol. 88, 4204–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madonna, G. , Ullman, C.D. , Gentilcore, G. , Palmieri, G. , Ascierto, P.A. , 2012. NF-kappaB as potential target in the treatment of melanoma. J. Transl. Med. 10, 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal, S. , Moudgil, M. , Mandal, S.K. , 2009. Rational drug design. Eur. J. Pharmacol. 625, 90–100. [DOI] [PubMed] [Google Scholar]
- Meyer, N. , Kim, S.S. , Penn, L.Z. , 2006. The Oscar-worthy role of Myc in apoptosis. Semin. Cancer Biol. 16, 275–287. [DOI] [PubMed] [Google Scholar]
- Miyoshi, H. , Blomer, U. , Takahashi, M. , Gage, F.H. , Verma, I.M. , 1998. Development of a self-inactivating lentivirus vector. J. Virol. 72, 8150–8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami, M. , Lan, K. , Subramanian, C. , Robertson, E.S. , 2005. Epstein-Barr virus nuclear antigen 1 interacts with Nm23-H1 in lymphoblastoid cell lines and inhibits its ability to suppress cell migration. J. Virol. 79, 1559–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J.K. , Chung, Y.M. , Kang, S. , Kim, J.U. , Kim, Y.T. , Kim, H.J. , Kim, Y.H. , Kim, J.S. , Yoo, Y.D. , 2002. c-Myc exerts a protective function through ornithine decarboxylase against cellular insults. Mol. Pharmacol. 62, 1400–1408. [DOI] [PubMed] [Google Scholar]
- Prochownik, E.V. , 2004. c-Myc as a therapeutic target in cancer. Expert Rev. Anticancer Ther. 4, 289–302. [DOI] [PubMed] [Google Scholar]
- Renne, R. , Zhong, W. , Herndier, B. , McGrath, M. , Abbey, N. , Kedes, D. , Ganem, D. , 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2, 342–346. [DOI] [PubMed] [Google Scholar]
- Ribble, D. , Goldstein, N.B. , Norris, D.A. , Shellman, Y.G. , 2005. A simple technique for quantifying apoptosis in 96-well plates. BMC Biotechnol. 5, 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha, A. , Halder, S. , Upadhyay, S.K. , Lu, J. , Kumar, P. , Murakami, M. , Cai, Q. , Robertson, E.S. , 2011. Epstein-Barr virus nuclear antigen 3C facilitates G1-S transition by stabilizing and enhancing the function of cyclin D1. PLoS Pathog. 7, e1001275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha, A. , Kaul, R. , Murakami, M. , Robertson, E.S. , 2010. Tumor viruses and cancer biology: modulating signaling pathways for therapeutic intervention. Cancer Biol. Ther. 10, 961–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, D.A. , O'Brien, S. , Faderl, S. , Garcia-Manero, G. , Ferrajoli, A. , Wierda, W. , Ravandi, F. , Verstovsek, S. , Jorgensen, J.L. , Bueso-Ramos, C. , Andreeff, M. , Pierce, S. , Garris, R. , Keating, M.J. , Cortes, J. , Kantarjian, H.M. , 2010. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J. Clin. Oncol. 28, 3880–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma, S.C. , Lan, K. , Choudhuri, T. , Robertson, E.S. , 2006. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen modulates K1 expression through its cis-acting elements within the terminal repeats. J. Virol. 80, 3445–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma, S.C. , Cai, Q. , Bajaj, B.G. , Robertson, E.S. , 2009. Overview of the large DNA tumor viruses. In Damania B., Pipas J.M.(Eds.), DNA Tumor Viruses. Springer Science + Business Media; New York: pp. xxvi, 794 pp., [794] p. of plates [Google Scholar]
- von Bueren, A.O., Oehler, C., Shalaby, T., von Hoff, K., Pruschy, M., Seifert, B., Gerber, N.U., Warmuth-Metz, M., Stearns, D., Eberhart, C.G., Kortmann, R.D., Rutkowski, S., Grotzer, M.A. c-MYC expression sensitizes medulloblastoma cells to radio- and chemotherapy and has no impact on response in medulloblastoma patients. BMC Cancer 11, 74. [DOI] [PMC free article] [PubMed]
- Wang, W. , Kim, S.H. , El-Deiry, W.S. , 2006. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc. Natl. Acad. Sci. U. S. A. 103, 11003–11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waud, W.R. , 2004. Murine L1210 and P388 leukemias. In Teicher B.A., Andrews P.A.(Eds.), Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval. second ed. Humana Press; Totowa, N.J: 79–97. [Google Scholar]
- Wilson, W.H. , Grossbard, M.L. , Pittaluga, S. , Cole, D. , Pearson, D. , Drbohlav, N. , Steinberg, S.M. , Little, R.F. , Janik, J. , Gutierrez, M. , Raffeld, M. , Staudt, L. , Cheson, B.D. , Longo, D.L. , Harris, N. , Jaffe, E.S. , Chabner, B.A. , Wittes, R. , Balis, F. , 2002. Dose-adjusted EPOCH chemotherapy for untreated large B-cell lymphomas: a pharmacodynamic approach with high efficacy. Blood. 99, 2685–2693. [DOI] [PubMed] [Google Scholar]
- Yamamoto, Y. , Gaynor, R.B. , 2001. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J. Clin. Invest. 107, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]