

Abstract
LIG4/Dnl4 is the DNA ligase that (re)joins DNA double-strand breaks (DSBs) via nonhomologous end joining (NHEJ), an activity supported by binding of its tandem BRCT domains to the ligase accessory protein XRCC4/Lif1. We screened a panel of 88 distinct ligase mutants to explore the structure-function relationships of the yeast Dnl4 BRCT domains and inter-BRCT linker in NHEJ. Screen results suggested two distinct classes of BRCT mutations with differential effects on Lif1 interaction as compared to NHEJ completion. Validated constructs confirmed that D800K and GG(868:869)AA mutations, which target the Lif1 binding interface, showed a severely defective Dnl4-Lif1 interaction but a less consistent and often small decrease in NHEJ activity in some assays, as well as nearly normal levels of Dnl4 accumulation at DSBs. In contrast, mutants K742A and KTT(742:744)ATA, which target the β3-α2 region of the first BRCT domain, substantially decreased NHEJ function commensurate with a large defect in Dnl4 recruitment to DSBs, despite a comparatively greater preservation of the Lif1 interaction. Together, these separation-of-function mutants indicate that Dnl4 BRCT1 supports DSB recruitment and NHEJ in a manner distinct from Lif1 binding and reveal a complexity of Dnl4 BRCT domain functions in support of stable DSB association.
Keywords: double-strand break, nonhomologous end joining, DNA ligase, BRCT domain, separation of function
1. Introduction
Nonhomologous end joining (NHEJ) is a major pathway for repairing DNA double strand breaks (DSBs) that is active throughout the cell cycle. The key components of the NHEJ pathway in budding yeast are the Ku DSB end-binding complex (Yku70–Yku80), the multi-functional MRX complex (Mre11–Rad50–Xrs2) and DNA ligase IV (Dnl4–Lif1–Nej1) [1, 2]. DNA ligation is the final and critical step of NHEJ, carried out by the Dnl4 catalytic subunit (LIG4 in humans) in complex with Lif1 (XRCC4 in humans) and Nej1 (XLF in humans) [2-4].
LIG4/Dnl4 protein in both human and yeast uses its tandem BRCA1 C-terminal (BRCT) domains to bind the coiled-coil region of its structural scaffold protein XRCC4/Lif1 (Figure 1A). This interaction is mediated primarily by the linker between the LIG4/Dnl4 BRCT domains, which intimately wraps around the XRCC4/Lif1 coiled coil [5, 6]. Deletions that ablate this conserved interface abolish NHEJ [7] while complete loss of yeast Lif1 prevents Dnl4 recruitment to a DSB [8]. This Dnl4–Lif1 interaction is thought to be the main reason that Lif1 and the Dnl4 BRCT region are required for NHEJ. However, Dnl4 that retains only the inter-BRCT linker is NHEJ deficient despite interacting with Lif1 [8]. Structural studies of both human and Saccharomyces cerevisiae DNA ligase IV indicate that, in addition to the inter-BRCT linker, LIG4/Dnl4 BRCT residues make contacts with XRCC4/Lif1 [5, 6], providing one possible reason that the BRCT domains themselves are required for NHEJ.
Figure 1. Separation of function in the Dnl4 BRCT mutation screen.
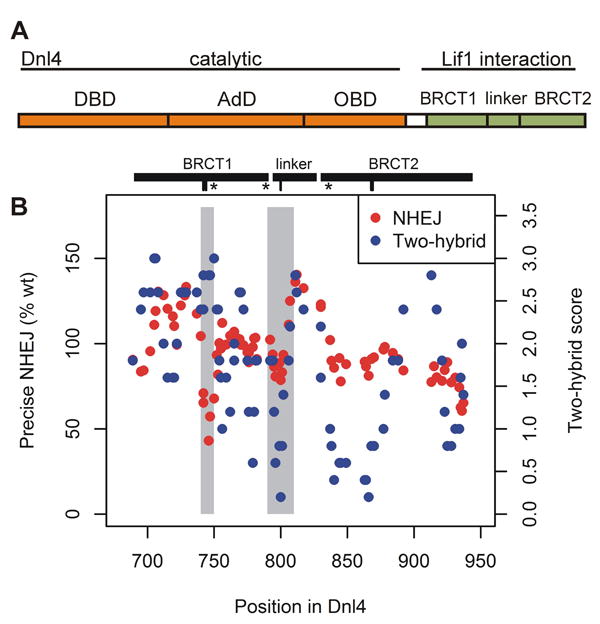
(A) Functional domains of Dnl4. DBD, DNA binding domain; AdD, adenylation domain; OBD, oligonucleotide binding domain; BRCT, BRCA1 C-terminal domain. (B) Panel showing precise NHEJ and Lif1 two-hybrid results for all point mutations tested in the Dnl4 BRCT mutation screen, plotted by their position in the Dnl4 protein. Each point represents a single mutant after application of a 5-point moving average over the position series to make regional patterns easier to visualize. Grey shading highlights two regions, 740-750 and 790-810, where different result patterns revealed a separation of function in the Dnl4 BRCT region. The spans of the two BRCT domains and inter-BRCT linker are denoted by black bars. Hash marks below denote the positions of mutations KTT(742:744)ATA, D800K, and GG(868:869)AA studied in further detail, and “*” indicates the position of an introduced stop codon.
Exactly how the LIG4/Dnl4–XRCC4/Lif1 interaction promotes DSB ligation is incompletely understood, but recent studies have suggested the importance of higher order complexes [9-14]. LIG4/Dnl4 is unstable without XRCC4/Lif1 [15] and accordingly is present predominantly in a constitutive LIG4–XRCC4 complex. XRCC4/Lif1 and XLF/Nej1 interact via their globular head domains [16, 17] and together form long super-helical multimer filaments in vitro [9, 10, 18, 19] and presumably also at DSBs in vivo. Such filaments might both bridge DSB ends for repair as well as deliver LIG4/Dnl4 to the site. In turn, evidence in humans suggests that binding of the LIG4 BRCT region might influence XRCC4–XLF multimerization, in part by preventing the XRCC4 C-terminus from interacting with the XRCC4–XLF interface [9, 12, 20, 21]. The human LIG4–XRCC4 interaction has also been shown to be required for proper nuclear localization and stability of XRCC4 [22].
Hypomorphic mutations in LIG4 lead to the ligase IV syndrome, characterized by microcephaly, growth retardation, developmental delay, radiosensitivity, immunodeficiency, bone marrow abnormalities and impaired end joining [23, 24], emphasizing the importance of understanding DNA ligase IV assembly and regulation. Recently, LIG4 mutation has also been reported for the related Dubowitz syndrome [25]. To further explore the consequences of LIG4/DNL4 mutations, we carried out mutational analysis of the Dnl4 BRCT region in S. cerevisiae, where we can assess the efficiency of multiple forms of NHEJ, Lif1 interaction, Dnl4 adenylation, and Dnl4–Lif1 recruitment to DSBs. We discovered a separation of function in the Dnl4 BRCT region in which destabilization of the Dnl4–Lif1 interaction by targeted mutations had a surprisingly small effect on NHEJ efficiency and Dnl4 recruitment in chromosomal DSB repair assays. In contrast, selected BRCT1 mutants showed a more severe defect in DSB recruitment and NHEJ despite having a relatively preserved Lif1 interaction, suggesting a previously unknown function of DNA ligase IV BRCT1 distinct from XRCC4/Lif1 binding.
2. Materials and methods
2.1. Dnl4 BRCT mutation screen
Detailed methods for the Dnl4 BRCT mutation screen are provided in Supplementary Materials and Methods.
2.2. Protein structural analysis
Dnl4 and Lif1 protein structural analyses were performed using PyMol (http://www.pymol.org/) and the available crystal structure of the Dnl4–Lif1 interaction interface, PDB 1Z56 [6].
2.3. Yeast strains, growth and manipulation
For confirmation and exploration of phenotypes beyond the BRCT mutation screen, dnl4 mutations were recreated in the native chromosomal DNL4 gene. Yeast strains were isogenic derivatives of BY4741 [26] as previously described [27, 28]. Gene disruptions and modified alleles were made using a PCR-mediated technique [26] or a URA3 pop-in/pop-out method [29]. Truncations were created as stop codons after the indicated Dnl4 residues. Mutant alleles were confirmed by PCR and sequencing. Resulting strain genotypes are listed in Table S1. Yeast were grown at 30 °C in either rich medium containing 1% yeast extract, 2% peptone, 2% dextrose or 3% glycerol, and 40 μg/ml adenine, or synthetic defined medium with either 2% glucose or galactose.
2.4. Yeast two-hybrid assay
Detailed descriptions of the yeast two-hybrid assay have been provided previously [28, 30, 31]. Briefly, haploid yeast strains bearing Dnl4 bait (DNA binding domain) and Lif1 prey (transcriptional activating domain) plasmids were mated and resulting diploids spotted to indicator plates lacking histidine or adenine to score activation of the HIS3 and ADE2 interaction reporter alleles, respectively. Control plates lacking both leucine and tryptophan verified the health of the strains.
2.5. Immunoprecipitation and adenylation
Detailed descriptions of the methods and reagents used for detection of Lif1 interaction and Dnl4 adenylation status by immunoprecipitation have been provided previously [28]. Briefly, 13Myc-tagged Dnl4 was immunoprecipitated from yeast whole cell extracts and subjected to immunoblotting for detection of co-immunoprecipitated Lif1. Alternatively, immunoprecipitates were exposed to α-32P-ATP, with or without pre-treatment with sodium pyrophosphate to remove pre-formed Dnl4-AMP adducts, followed by phosphorimager exposure to detect newly formed Dnl4-AMP adducts with normalization to Dnl4 protein amounts determined by immunoblotting.
2.6. Chromosomal suicide deletion NHEJ assay
Detailed descriptions of the different versions of the suicide deletion assay have been provided previously [28, 31-33]. Briefly, the frequency of NHEJ repair of closely spaced DSBs in the ADE2 gene was measured as the ratio of colonies formed on DSB-inducing galactose plates compared to glucose plates. The nature of NHEJ events was revealed by scoring colonies as Ade+ or Ade-, by introducing fresh endonuclease into colonies to determine whether the DSB site could be recleaved, and finally by Sanger sequencing.
2.7. Plasmid recircularization NHEJ assay
Detailed descriptions of the plasmid recircularization assay have been provided previously [27, 28]. Briefly, the NcoI-digested URA3-marked pRS316 was transformed into yeast and colony yields normalized to transformations with undigested HIS3-marked pRS314, with the Ura+/His+ colony ratio serving as an indicator of NHEJ efficiency.
2.8. Chromatin immunoprecipitation
Detailed descriptions of the GAL1 promoter cut-site (GAL1-cs) and associated chromatin immunoprecipitation (ChIP) assays have been provided previously [28, 34]. Briefly, an HOendonuclease-mediated DSB was induced at the GAL1-cs by addition of galactose and stopped by addition of glucose. Repair of the break was monitored by flanking PCR as compared to an uncut control allele contained within the strains. Recruitment of chromosomal-13Myc-tagged proteins to the DSB by ChIP used quantitative PCR with one primer set on the same side of the GAL1-cs and a second set directed at the ACT1 control locus to determine fold enrichment at the DSB site.
3. Results
3.1. Dnl4 BRCT mutation screen reveals a separation of function
We constructed a screening panel of 88 different single and combined point mutations affecting 121 amino acids between Dnl4 residues 686 and 941, encompassing the tandem BRCT domains and inter-BRCT linker (Table S2). Targeted residues were chosen based on fungal primary sequence conservation (Figure S1A), supported by examination of yeast Dnl4 and human LIG4 crystal structures (Figure S1B) [5, 6]. Most mutations were alanine replacements, but a subset changed the charge of highly conserved residues. Controls included three silent mutations expected to act as wild-type and three premature stop codons that truncated from the middle of BRCT1 (L750*), just before the inter-BRCT linker (A789*), and at the beginning of the BRCT2 (F836*) (Figure 1B). Mutant constructs were introduced into yeast (Figure S2) in a manner that allowed us to score the Dnl4(651-944)–Lif1(137-265) BRCT–coiled-coil interaction by two-hybrid [16, 31] and both HO precise and +CA imprecise NHEJ using the two-DSB suicide deletion assay [31, 32, 35]. Table S2 and Figure S3 show the screen results for all constructs.
As expected, a wide range of Dnl4 screen phenotypes, ranging from wild-type to severely defective, was observed in both the Lif1 interaction and NHEJ assays. Precise and imprecise NHEJ were correlated (Figure S4, Pearson coefficient = 0.54), with no mutant showing a strong specific loss of only imprecise NHEJ as might have been expected if the Dnl4 BRCT domains had a central role in recruiting the Pol4 DNA polymerase required for +CA joint formation [29, 36]. In contrast, a separation of function in specific protein locations and a surprisingly poor correlation were observed for Lif1 interaction as compared to NHEJ activity (Figure 1B, Spearman coefficient = 0.15). Mutants in the inter-BRCT linker and the first half of BRCT2 showed severe defects in Lif1 interaction, as predicted by the position of Dnl4-Lif1 contacts in the reported co-crystal structure (Figure S5A-B) [5, 6]. However, despite their substantial Lif1 interaction defect, mutants in these regions showed nearly normal NHEJ in the screening assays (Figure 1B). Truncation mutation F836*, which lacks BRCT2, supported this result because it showed a substantial Lif1 interaction defect but normal precise NHEJ activity (Figure S3). In contrast, mutations in the Dnl4 region that includes β3 and α2 in BRCT1 (residues 742-753, Figure S5C) unexpectedly caused a substantial NHEJ defect despite a largely preserved Lif1 interaction (Figure 1B).
NHEJ results above were from screening assays with plasmid-expressed Dnl4 and uncloned mutant pools. For validation studies below, we focused on specific screen mutations deemed most likely to disrupt the Dnl4-Lif1 interface, D800K and GG(868:869)AA, and one in the β3-α2 region of BRCT1, KTT(742:744)ATA (Figure S5). D800K mutation alters the charge of one half of a Dnl4-Lif1 salt bridge at one of the few residues in the LIG4/Dnl4 BRCT region that is highly conserved region from yeast to mammals (Figure S1B). Residues GG(868:869) are less conserved but pack tightly against the XRCC4/Lif1 coiled coil in both yeast and humans (Figure S5B) [5, 6]. Residues KTT(742:744) only show sequence conservation among yeasts but reside in an interesting area of BRCT1 (see Discussion). Mutations at these sites were sequenced and recreated in the native DNL4 gene so that the apparent separation of function could be explored further.
3.2. Dnl4-Lif1 interaction and Dnl4 stability
We first explored the Dnl4-Lif1 interaction phenotype using variations of the two-hybrid experiment. Results validated the screen observations that mutant KTT(742:744)ATA, in contrast to D800K or GG(868:869)AA, showed only a small decrease in Dnl4-Lif1 interaction when scored using constructs containing just the Dnl4 BRCT (residues 651-944) and Lif1 coiled coil regions (137-265) (Figure 2A and S6). We also made K742A and T744A single point mutants and observed that, similar to KTT(742:744)ATA from which they derive, each showed fully normal two-hybrid signals (Figure 2A), as expected from the fact that these residues are distant from the Lif1 interface (Figure S5C). Interestingly, switching to full-length Lif1 (1-421) markedly improved the D800K and GG(868:869)AA interactions (Figure 2A). This effect did not depend on Nej1 protein expressed in the two-hybrid host as it was also observed in a nej1Δ strain (Figure 2B). The Lif1 globular head domain (residues 1-136) thus stabilizes the Dnl4-Lif1 interaction in poorly understood ways, perhaps through secondary interaction interfaces or, most likely, by promoting Lif1 dimeric coiled-coil assembly. To explore the notion that impairing multiple contacts in the overall complex leads to more severe phenotypes, we made a D800K,GG(868:869)AA compound mutation and indeed observed a complete loss of two-hybrid signal even with full-length proteins (Figure 2A). Combining either D800K or GG(868:869)AA with KTT(742:744)ATA also decreased the interaction, especially for the GG(868:869)AA, KTT(742:744)ATA compound mutant (Figure 2C). These results again expose a small Dnl4-Lif1 interaction defect with KTT(742:744)ATA, but the effect of this β3-α2 region mutation on its own was less significant than for D800K and GG(868:869)AA.
Figure 2. Dnl4–Lif1 interaction, two-hybrid analysis.
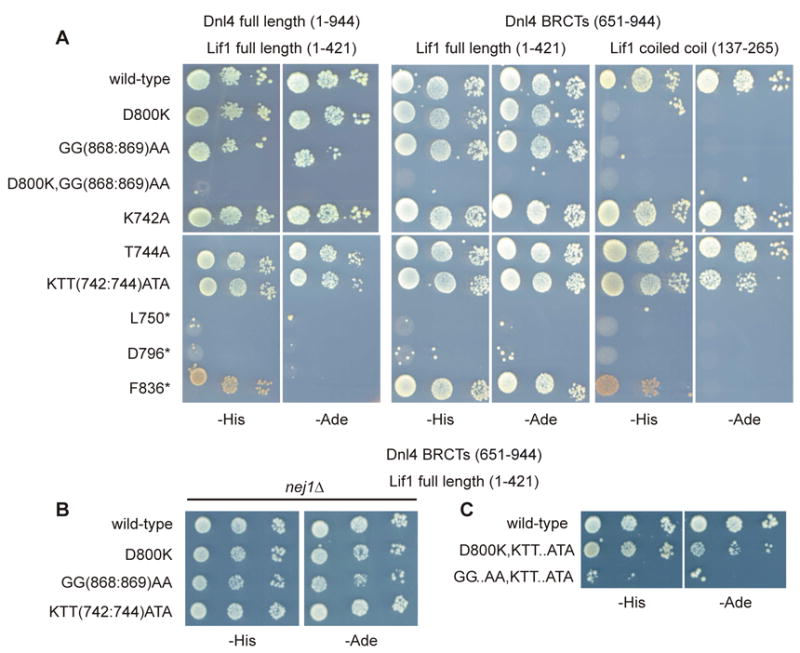
(A) Haploid strains expressing the indicated Dnl4 two-hybrid bait constructs, either as full-length Dnl4 (residues 1-944) or just its BRCT domains (651-944), were mated with prey strains expressing either full-length Lif1 (1-421) or just its coiled-coil region (137-265). Dnl4–Lif1 interaction was scored by spotting 10-fold serial dilutions to plates lacking either histidine (-His) or adenine (-Ade). Control plates are shown in Figure S6. Truncations that remove the inter-BRCT linker (L750* and D796*) are provided as negative controls, while a truncation that includes the linker but lacks BRCT2 (F836*) showed measurable interaction. The combination used in the BRCT mutation screen was Dnl4(651-944)–Lif1(137-265), which reveals a much greater Lif1 interaction defect for Dnl4 D800K and GG(868:869)AA mutants as compared to KTT(742:744)ATA. (B) Several of the analyses in (A) were repeated in a nej1Δ strain, which revealed that the Dnl4–Lif1 interaction was not dependent on Nej1 protein. (C) Additional compound point mutations were tested as an extension of panel (A) between the D800K, GG(868:869)AA (GG..AA), and KTT(742:744)ATA (KTT..ATA) mutations.
To explore the Dnl4-Lif1 interaction in a more physiological manner, 13Myc-tagged Dnl4 protein levels were assessed by immunoblotting, given that Dnl4 protein is known to be unstable if it is unable to interact with Lif1 [15, 16, 28]. Consistent with the two-hybrid results, either D800K or GG(868:869)AA mutations alone destabilized Dnl4 to a similar extent (Figure 3A). The D800K,GG(868:869)AA compound mutant was expressed at a similar level to both D800K and GG(868:869)AA, as well as to Dnl4 in the complete absence of Lif1 (lif1Δ; Figure 3B), indicating that the Dnl4-stabilizing effect of the Dnl4-Lif1 interaction was lost equivalently in each of these mutants. In contrast, KTT(742:744)ATA mutant protein was expressed similarly to wild-type (Figure 3A), indicating a sufficiently preserved Dnl4-Lif1 interaction to support Dnl4 stability. Unlike Dnl4 expression, 13Myc-Lif1 expression was not dependent on the Dnl4-Lif1 interaction, being normal even in dnl4Δ strains (Figure 3C).
Figure 3. Dnl4 stability, interaction with Lif1, and in vitro adenylation.
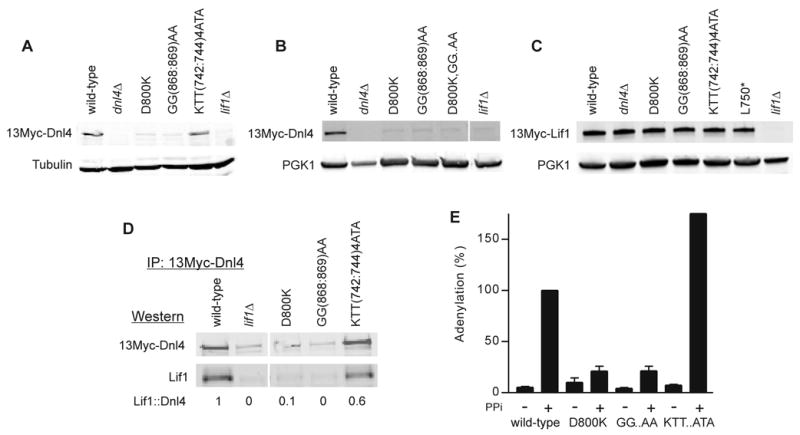
(A) Whole cell extracts from yeast expressing 13Myc-tagged Dnl4 were immunoblotted using anti-Myc and anti-tubulin antibodies, revealing an expression defect of Dnl4 D800K and GG(868:869)AA mutants as compared to KTT(742:744)ATA that indicates a Lif1 interaction defect of the former. (B) Similar to (A), now with the D800K,GG(868:869)AA compound mutant (D800K,GG..AA) and using anti-PGK1 as a loading control. (C) Similar to (A) and (B), now immunoblotting for 13Myc-tagged Lif1 expressed in strains bearing Dnl4 mutations. (D) 13Myc-Dnl4 protein was immunoprecipitated from yeast whole cell extracts. The amount of Lif1 that co-immunoprecipitated was assessed by immunoblotting. Numbers are the Lif1::Dnl4 ratio normalized to wild-type Dnl4, after correcting for the background band that migrates near the Lif1 position. Mutant KTT(742:744)ATA again showed substantial Lif1 interaction. (E) Immunoprecipitated 13Myc-Dnl4 was incubated with α-32P-ATP to form a radiolabeled enzymeadenylate complex followed by SDS-PAGE and immunoblotting. When indicated, protein was pre-treated with sodium pyrophosphate (PPi) to remove pre-existent covalently bound AMP. Blots were exposed to a phosphorimager to reveal in vitro adenylation and subsequently immunoblotted to reveal Dnl4 protein amounts. The phosphorimager signal was normalized to the Western signal and wild-type plus PPi set to 100%. GG..AA and KTT..ATA refer to GG(868:869)AA and KTT(742:744)ATA mutants, respectively. Results are the mean ± standard deviation of at least two independent experiments.
Co-immunoprecipitation of Lif1 with 13Myc-Dnl4 reinforced these observations. Here, D800K or GG(868:869)AA mutants were difficult to interpret due to the low recovery of Dnl4 itself, but Lif1 co-immunoprecipitated at nearly normal levels with the stable KTT(742:744)ATA mutant protein (Figure 3D). We further examined immunoprecipitated Dnl4 for the dependence of catalytic function on a stable Lif1 interaction. Interestingly, both D800K and GG(868:869)AA mutants showed substantially reduced in vitro adenylation, even after normalization for the reduced amount of Dnl4 that could be immunoprecipitated (Figure 3E). Given that the coimmunoprecipitation in Figure 3D was performed under the same conditions as adenylation in Figure 3E, we attribute this difference to a lack of Lif1 in the D800K and GG(868:869)AA immunoprecipitates and infer that Lif1 supports normal catalytic function, at least in vitro. Again consistent with its more preserved Lif1 interaction, KTT(742:744)ATA showed normal adenylation in vitro (Figure 3E).
3.3. NHEJ function of dnl4 mutants
The above results confirmed a substantially more severe Lif1 interaction defect with D800K or GG(868:869)AA Dnl4 mutations than with KTT(742:744)ATA. To further explore the ability of these mutants to complete NHEJ, we extended the HO suicide deletion screen results to a different suicide deletion assay based on the I-SceI endonuclease [28, 33]. In this system, it was again true that the KTT(742:744)ATA mutant had a more substantial NHEJ defect than D800K or GG(868:869)AA (Figure 4A). The more focused mutations in K742A and T744A suggested that K742 is a more important residue in the cluster than T744, although the T744A mutant was still partially NHEJ deficient (Figure 4A). The partially penetrant phenotype of D800K and GG(868:869)AA mutants was reinforced by the observation that NHEJ activity with the D800K,GG(868:869)AA compound mutant was undetectable and equivalent to dnl4Δ and the C-terminal truncations that eliminated the inter-BRCT linker (e.g. L750*, Figure 4A).
Figure 4. NHEJ efficiency in vivo.
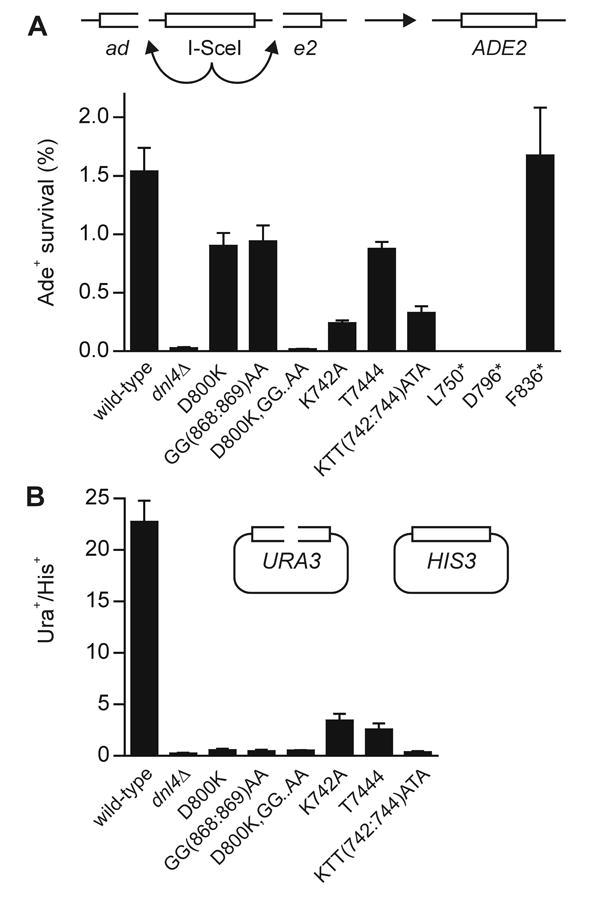
(A) NHEJ efficiency of yeast strains bearing the indicated Dnl4 mutations in the I-SceI chromosomal suicide deletion assay (ade2::SD2+::STE3-MET15), confirming the DSB repair defect of the KTT(742:744)ATA mutant. (B) NHEJ efficiency in the plasmid recircularization assay. Results are expressed as the ratio of the colony yield from NcoI-digested URA3-marked pRS316 [26] relative to undigested HIS3-marked pRS314 [26]. All results are the mean ± standard deviation of at least three independent experiments.
To reveal the nature of suicide deletion repair events we re-expressed I-SceI in them to identify recleavable precise simple-religation NHEJ (Table 1) and sequenced a subset of the remaining alleles to verify that they corresponded to imprecise NHEJ as expected (Figure S7). D800K and GG(868:869)AA mutants showed a slight bias toward imprecise joining (Table 1). In contrast, KTT(742:744)ATA showed a substantial loss of precise and, even more so, imprecise joining (Table 1). These different patterns are important because we recently demonstrated that catalytically inactive Dnl4 that accumulated at a DSB conferred a preferential loss of precise NHEJ and a relative preservation of imprecise NHEJ [28]. The fact that KTT(742:744)ATA mutation did not reproduce this phenotype provided a first indication that its defect is not in catalysis, consistent with Figure 3E, but that it instead does not accumulate normally at DSBs. Paradoxically, the Lif1 interface mutants showed a pattern more like the catalytic mutants [28].
Table 1. Joining precision in the suicide deletion assay.
For each Dnl4 allele, I-SceI+/total indicates the fraction of Ade+ suicide deletion survivors that could be re-cleaved by I-SceI and had thus used precise NHEJ. Subsequent columns indicate the percentage of Ade+ colonies that had survived by precise and imprecise NHEJ relative to all cells plated (colony survival) and the efficiency of precise and imprecise NHEJ for mutant strains relative to wild-type Dnl4 (relative efficiency). Data for wild-type and dnl4Δ have been reported previously [28] and are reproduced here for comparison as controls.
| Joint analysis (%) | Colony Survival (%) | Relative Efficiency (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| DNL4 genotype | I-SceI+/total | precise | Imprecise | total | precise | imprecise | total NHEJ | precise | imprecise |
| wild-type | 112/120 | 93 | 7 | 2.2 | 2.0 | 0.1 | 100 | 100 | 100 |
| dnl4Δ | 120/120 | 100 | 0 | 0.002 | 0.002 | 0 | 0.1 | 0.1 | 0 |
| D800K | 108/119 | 91 | 9 | 1.6 | 1.5 | 0.1 | 74 | 72 | 102 |
| GG(868:869)AA | 103/120 | 86 | 14 | 1.8 | 1.5 | 0.3 | 82 | 76 | 174 |
| KTT(742:744)ATA | 119/120 | 99 | 1 | 0.2 | 0.2 | 0.002 | 10 | 11 | 1 |
| D796* | 24/24 | 100 | 0 | 0.002 | 0.002 | 0 | 0.1 | 0.1 | 0 |
| F836* | 14/24 | 58 | 42 | 1.7 | 1.0 | 0.7 | 77 | 48 | 481 |
We also examined the ability of our Dnl4 BRCT mutants to complete NHEJ of transformed NcoI-cut linear plasmids. Consistent with prior observations that plasmid recircularization is more sensitive to DNA ligase IV impairment than is suicide deletion [27, 28, 37], even D800K and GG(868:869)AA mutants proved to be as deficient as dnl4Δ in the plasmid assay (Figure 4B). Plasmid recircularization was thus poorly informative with respect to the BRCT separation of function but did confirm an extensive NHEJ defect of KTT(742:744)ATA (Figure 4B).
3.4. Dnl4 recruitment to DSBs is impaired by BRCT1 mutations
The above studies validated the screen finding that specific Dnl4 BRCT1 mutations can impair NHEJ function in a manner that does not correlate with Dnl4-Lif1 interaction. To explore the basis of this phenomenon, we employed the GAL1-cs system in which a single DSB is introduced into the chromosomal GAL1 promoter by induction of HO endonuclease [34]. DSBs are formed during a 1-hour induction period, after which the accumulation and disappearance of 13Myc-tagged Dnl4 (13Myc-Dnl4) at the DSB is measured by chromatin immunoprecipitation (ChIP) and any associated NHEJ is measured using flanking PCR. KTT(742:744)ATA and K742 mutants showed a very small degree of repair of the GAL1-cs DSB between 60 and 240 min relative to wild-type (Figure 5A). T744A showed an intermediate phenotype (Figure 5A). D800K and GG(868:869)AA mutants once again showed notably more NHEJ than KTT(742:744)ATA (compare Figure 5B to Figure 5A), while the compound mutant D800K,GG(868:869)AA with the complete Lif1 interaction defect showed no repair (Figure 5B). Overall, these repair efficiencies mirror those observed with suicide deletion. It is unclear if the apparent difference in repair kinetics with the partially functional T744A, D800K and GG(868:869)AA mutants are meaningful.
Figure 5. Dnl4 recruitment to a chromosomal DSB.
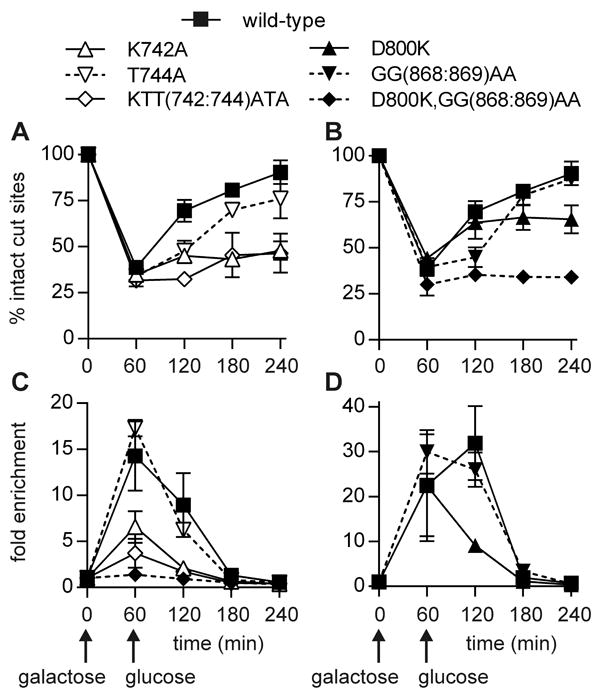
Yeast strains bearing the indicated 13Myc-Dnl4 mutations and the GAL1-cs allele were grown in galactose medium for 60 min to induce HO expression and DSB formation. Cells were then transferred to glucose to terminate HO expression and allow repair by NHEJ. (A) and (B) show the fraction of intact GAL1-cs HO cut sites, and thus DSB formation and repair over time, as determined by flanking PCR. (C) and (D) show the corresponding enrichment of 13Myc-Dnl4 at the DSB, relative to the ACT1 control gene, as determined by ChIP and quantitative PCR. Panels (C) and (D) were performed separately, where different lots of antibodies and other variables can lead to different apparent degrees of DSB enrichment, but all samples within each of panels (A) to (D) were handled together as part of the same set of experimental replicates. Combined results are the mean ± standard deviation of at least two independent experiments and demonstrate that the KTT(742:744)ATA mutation impairs NHEJ by reducing Dnl4 recruitment to DSBs even more than the reduction caused by direct disruption of the Dnl4-Lif1 interface.
Most importantly, the NHEJ repair efficiencies were predicted by the degree of Dnl4 recruitment to the GAL1-cs DSB. As suggested by the suicide deletion joint pattern (Table 1), the KTT(742:744)ATA mutant showed very poor accumulation at the DSB, as did K742A (Figure 5C). In contrast, D800K and GG(868:869)AA showed more normal recruitment, at least at the 60 min time point (Figure 5D), despite the fact that these were the mutations that targeted the Dnl4-Lif1 interface. Only when the interface was completely ablated in the compound mutant D800K,GG(868:869)AA did Dnl4 fail to appear at the DSB (Figure 5C). Importantly, we previously observed hyper-accumulation of catalytically inactive Dnl4 at unrepaired DSBs [28], which documents that failed NHEJ need not be synonymous with failed ligase recruitment. We thus conclude that the primary defect caused by KTT(742:744)ATA mutation is in stable DSB association.
Finally, we determined the effect of Dnl4 BRCT mutations on Lif1 recruitment to the GAL1-cs DSB. Repair profiles in 13Myc-Lif1 strains showed the same pattern as 13Myc-Dnl4 strains for KTT(742:744)ATA, D800K and GG(868:869)AA Dnl4 mutants (Figure 6A). Here, we additionally monitored dnl4Δ and L750* strains and observed, as expected from other assays, a complete GAL1-cs NHEJ deficiency (Figure 6A). Like Dnl4, Lif1 appeared at the GAL1-cs DSB at one and two hours after DSB induction, although the level of Lif1 enrichment was slightly lower (Figure 6B). Lif1 recruitment was not observed in the absence of Dnl4 or its BRCT domains and was reduced in each of the KTT(742:744)ATA, D800K and GG(868:869)AA Dnl4 mutants (Figure 6B). Relative to Dnl4 ChIP (Figure 5), the main difference in this pattern is that Lif1 recruitment was more impaired in D800K and GG(868:869)AA Dnl4 mutants than was Dnl4 recruitment, presumably reflecting their Lif1 interaction defect. Reduced Lif1-DSB association with KTT(742:744)ATA can be explained by the poor recruitment of Dnl4 itself.
Figure 6. Lif1 recruitment to a chromosomal DSB.
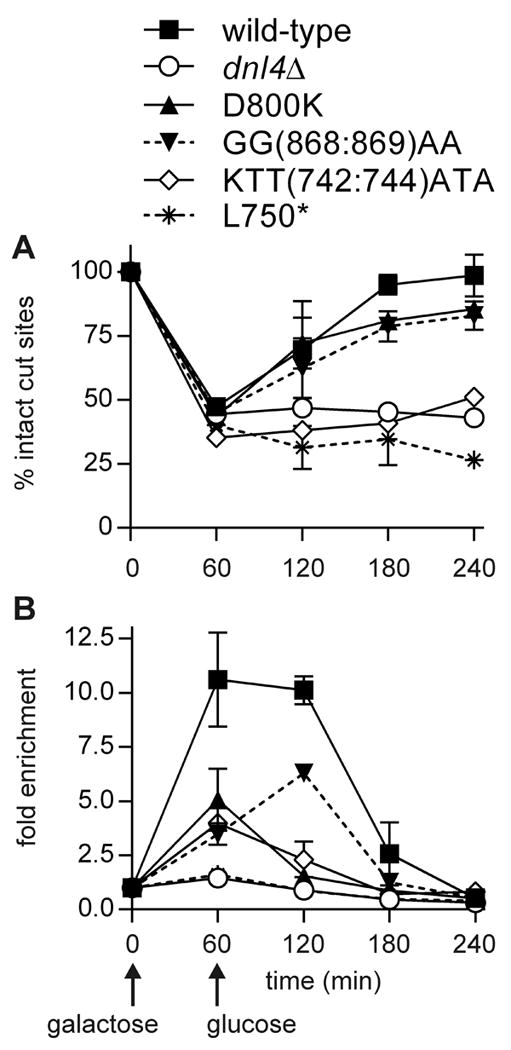
Similar to Figure 5, except using 13Myc-Lif1 strains. Note that Lif1 was otherwise wild-type. Mutant designations refer to the untagged dnl4 allele carried in the strains. (A) DSB formation and repair assessed by flanking PCR. (B) Enrichment of 13Myc-Lif1 at the GAL1-cs DSB by ChIP. Results are the mean ± standard deviation of two independent experiments. Like Dnl4, Lif1 recruitment to a DSB is impaired by Dnl4 KTT(742:744)ATA mutation.
4. Discussion
Our screening panel identified two functional classes of Dnl4 BRCT mutations that were subsequently validated by detailed study of fully characterized mutants. The first class, exemplified by D800K and GG(868:869)AA mutations, directly interfered with the Dnl4–Lif1 interface but had a surprisingly small effect on NHEJ efficiency in two chromosomal DSB repair assays. These mutants did confer a complete loss of NHEJ of transformed plasmids, consistent with the higher sensitivity of this assay for smaller degrees of impaired NHEJ function [27, 28, 37]. The second class of mutations, exemplified by KTT(742:744)ATA, had a smaller effect on Dnl4–Lif1 interaction but a larger effect on NHEJ efficiency in chromosomal assays. This pattern of results is not consistent with a model in which the sole function of the Dnl4 BRCT region is to bind Lif1 and indicates that BRCT1 in particular supplies a distinct NHEJ function.
4.1. Role of the LIG4/Dnl4 BRCT–Lif1 interaction
Critically, results reported here do not indicate that Dnl4–Lif1 interaction is not an important function of the Dnl4 BRCT region or that the interaction is not essential for NHEJ. Indeed, a clear importance of the Dnl4–Lif1 interface was documented by the complete NHEJ deficiency and failed recruitment of Dnl4 to a DSB with the D800K,GG(868:869)AA compound mutation (Figures 4-5), which interfered with the interface at two separate sites (Figure S5). In turn, it can be inferred that the weakened Lif1 binding seen with D800K and GG(868:869)AA single-site mutants (Figure 2) was sufficient to promote the required Dnl4–Lif1 interaction in chromosomal, although not plasmid, DSB repair (Figures 4-6). Nevertheless, the marked destabilization of Dnl4 protein caused by the single-site mutations indicated that they had a major impact on basal Dnl4–Lif1 interaction in the cell (Figure 3), consistent with the predictions of structural biology (Figure S5) [5, 6]. Such observations are also consistent with competitive displacement studies demonstrating that disruption of a single helix within the human LIG4 inter-BRCT linker was sufficient to destabilize the LIG4/XRCC4 interaction [22].
One implication of this result pattern is that maintaining a normal Dnl4 protein level isn’t the most critical function of Dnl4–Lif1 interaction. The system was able to compensate for the reduced protein levels and accumulate Dnl4 at DSBs regardless (note that the same 13Myc-Dnl4 strains were used for Western blotting and ChIP in Figures 3 and 5, respectively). Further, it is apparent that the NHEJ protein assembly at chromosomal DSBs is more able to overcome a partial impairment of the Dnl4–Lif1 interface than are the free proteins. A higher order complex is one way to explain this behaviour (see more below). Finally, it was interesting that Lif1 recruitment was more affected by D800K and GG(868:869)AA mutations than was Dnl4 recruitment (Figure 5 and 6). It is tempting to interpret that Dnl4 is the lead protein that subsequently recruits Lif1. However, that overly simplistic idea fails to account for the complete absence at DSBs of Dnl4 forms that cannot interact with Lif1 (Figure 5), the dependence of Dnl4 recruitment on Lif1 [8], and the fact that Ku and MRX make redundant recruiting contacts to Dnl4 and Lif1, respectively [31]. A more consistent view is that Dnl4 and Lif1 are co-dependent in their appearance at DSBs, supported by the Dnl4–Lif1 interface.
Dnl4 Lif1-interaction mutants D800K and GG(868:869)AA also significantly impaired in vitro auto-adenylation of Dnl4 (Figure 3E), an effect most likely due to the relative absence of Lif1 in the immunoprecipitates used in these experiments (Figure 3D). This suggests that binding of Lif1 to Dnl4 stimulates Dnl4 catalytic function. However, the importance of this effect appears exaggerated by the in vitro assay, given the results of chromosomal NHEJ assays with D800K and GG(868:869)AA mutant yeast (Figures 4-6).
4.2. Role of LIG4/Dnl4 BRCT1 in NHEJ
The Dnl4 D800K and GG(868:869)AA Lif1-interaction mutants further established by comparison that the second class of BRCT1 mutations could not be exerting their effect exclusively through the Dnl4–Lif1 interaction. Not surprisingly, KTT(742:744)ATA and K742A mutants, which affected residues distant from Lif1 in the complex (Figure S5), showed a greater ability to interact with Lif1 than the Lif1-interface mutants, as revealed by two hybrid, Dnl4 protein stability, and co-immunoprecipitation (Figures 2-3). They did have a small interaction defect that was especially exposed in the GG(868:869)AA,KTT(742:744)ATA compound mutant (Figure 2). In contrast, KTT(742:744)ATA mutants showed a greater NHEJ defect in chromosomal DSB assays (Figures 4-6). Such a separation of function dictates that BRCT1 must be doing something other than promoting Lif1 interaction via the inter-BRCT clamp. This function is not primarily catalytic, to the extent that our limited in vitro assays could reveal (Figure 3E), but instead promotes assembly of Dnl4–Lif1 at a chromosomal DSB (Figures 5-6, supported by Table 1). Thus, stable binding of Dnl4 to a DSB critically depends on its tandem BRCT region, but this function is more complex than the singular Dnl4–Lif1 interface depicted in Figure S5.
A series of recent studies provide a unifying model for understanding the potential roles of LIG4/Dnl4 BRCT1 in the NHEJ assembly. A combination of small angle X ray scattering, crystallography, electron microscopy, and atomic modelling have revealed that LIG4/Dnl4, XRCC4/Lif1 and XLF/Nej1 form DNA-influenced multimers in vitro via head domain interactions of XRCC4/Lif1 and XLF/Nej1 [9-14]. Lessons to be learned from these higher-order structures include the potential for a multiplicity of contacts, some of which may be partially or wholly redundant, with even large impairment of a single binary interaction overcome by high local protein concentrations. These ideas provide a possible explanation for the surprisingly small effect of single-site Lif1 interface mutants discussed above. Further, some NHEJ protein interactions might only be realized in the context of the multimer assembled at a DSB. Building from these insights, we suggest that Dnl4 BRCT1 makes such a contact that contributes to the assembly of the Dnl4-Lif1-Nej1 multimer.
Importantly, the hypothesized contact made by BRCT1 could be to either protein or DNA. In this regard, it is striking that the relevant Dnl4 mutations are in the β3-α2 region of BRCT1. This region is precisely where other tandem-BRCT proteins make contacts to phosphorylated residues in bound phosphopeptides [38-41], which might suggest a similar function in Dnl4. However, in vitro studies of BRCA1, XRCC1, TopBP1 and E. coli DNA ligase BRCT domains separately document their ability to bind DNA [42-45]. One idea is that the same site that recognizes phosphopeptides in some BRCT domains might recognize DNA phosphates in others [38, 39, 41, 46]. Interestingly, there is evidence that LIG4 makes DNA contacts outside of its catalytic domain, specifically via a patch of basic residues upstream of BRCT1 that is argued to help align the ligase on DNA relative to the XRCC4 DNA-binding channel [20]. Further work is required to determine whether Dnl4 BRCT1 facilitates such a ligase-DNA interaction or contributes to NHEJ chromatin assembly in a different manner, for example by mediating a protein-protein interaction. It is finally noteworthy that α2 of BRCT1 is the specific location in human LIG4 that is required for its targeted degradation during adenovirus infection, although at present is not clear how this fact might be linked to LIG4 function in NHEJ [47].
4.3. Implications for human DNA ligase IV
Human ligase IV syndrome results from hypomorphic LIG4 mutations. Disease-associated nonsense mutations that lead to truncation of the LIG4 BRCT domains (R580* and R814*) behave similarly to analogous Dnl4 mutants described here, with each showing impairment of the interaction with XRCC4/Lif1, reduced protein stability and impaired formation of preadenylated enzyme [48, 49]. The relevance of our findings to human LIG4 function is further supported by recent observations by Liu et al. that mutation of a core tryptophan residue in either of LIG4 BRCT1 or BRCT2 reduced chromatin association of LIG4 and XRCC4 and correspondingly increased sensitivity to resistance to ionizing radiation [50]. These same authors also observed that the tandem BRCT domains alone could bind to chromatin. These results attest to an importance of the LIG4 BRCT domains themselves in NHEJ repair that closely parallels our observations with Dnl4 β3-α2 region mutations. Altogether, results here contribute to an understanding of key residues in the LIG4/Dnl4 BRCT domains that confer two related but separable functions: interaction with XRCC4/Lif1 and association with chromosomal DSBs. Improved understanding of these intertwined functions has the potential to lead to novel anticancer therapeutics that alter NHEJ capacity by targeting the LIG4 BRCT domains [51].
Supplementary Material
Highlights (for review).
A mutational screen of the yeast DNA ligase IV BRCT domains is reported.
NHEJ efficiency and the Dnl4-Lif1 interaction (LIG4-XRCC4 in humans) were examined.
A separation of function suggesting multiple roles of the BRCT domains was observed.
Dnl4 BRCT1 supported NHEJ in a manner distinct from Lif1 interaction.
Acknowledgments
We thank Alan Tomkinson for the kind gift of the Lif1 antibody.
Funding: This work was supported by the National Institutes of Health [CA102563 to T.E.W.]. Funding for open access charge: National Institutes of Health.
Abbreviations
- BRCT
BRCA1 C-terminal domain
- ChIP
chromatin immunoprecipitation
- DSB
double-strand break
- GAL1-cs
GAL1 promoter cut-site
- MRX
Mre11–Rad50–Xrs2 complex
- NHEJ
nonhomologous end-joining
- PCR
polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Daley JM, Palmbos PL, Wu D, Wilson TE. Nonhomologous end joining in yeast. Annual review of genetics. 2005;39:431–451. doi: 10.1146/annurev.genet.39.073003.113340. [DOI] [PubMed] [Google Scholar]
- 2.Chiruvella KK, Liang Z, Wilson TE. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol. 2013;5:a012757. doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA endjoining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem. 2008;77:313–338. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu PY, Frit P, Meesala S, Dauvillier S, Modesti M, Andres SN, Huang Y, Sekiguchi J, Calsou P, Salles B, Junop MS. Structural and functional interaction between the human DNA repair proteins DNA ligase IV and XRCC4. Molecular and cellular biology. 2009;29:3163–3172. doi: 10.1128/MCB.01895-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dore AS, Furnham N, Davies OR, Sibanda BL, Chirgadze DY, Jackson SP, Pellegrini L, Blundell TL. Structure of an Xrcc4-DNA ligase IV yeast ortholog complex reveals a novel BRCT interaction mode. DNA repair. 2006;5:362–368. doi: 10.1016/j.dnarep.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Grawunder U, Zimmer D, Leiber MR. DNA ligase IV binds to XRCC4 via a motif located between rather than within its BRCT domains. Curr Biol. 1998;8:873–876. doi: 10.1016/s0960-9822(07)00349-1. [DOI] [PubMed] [Google Scholar]
- 8.Teo SH, Jackson SP. Lif1p targets the DNA ligase Lig4p to sites of DNA double-strand breaks. Curr Biol. 2000;10:165–168. doi: 10.1016/s0960-9822(00)00317-1. [DOI] [PubMed] [Google Scholar]
- 9.Hammel M, Yu Y, Fang S, Lees-Miller SP, Tainer JA. XLF regulates filament architecture of the XRCC4.ligase IV complex. Structure. 2010;18:1431–1442. doi: 10.1016/j.str.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ropars V, Drevet P, Legrand P, Baconnais S, Amram J, Faure G, Marquez JA, Pietrement O, Guerois R, Callebaut I, Le Cam E, Revy P, de Villartay JP, Charbonnier JB. Structural characterization of filaments formed by human Xrcc4-Cernunnos/XLF complex involved in nonhomologous DNA endjoining. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:12663–12668. doi: 10.1073/pnas.1100758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammel M, Rey M, Yu Y, Mani RS, Classen S, Liu M, Pique ME, Fang S, Mahaney BL, Weinfeld M, Schriemer DC, Lees-Miller SP, Tainer JA. XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J Biol Chem. 2011;286:32638–32650. doi: 10.1074/jbc.M111.272641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahaney BL, Hammel M, Meek K, Tainer JA, Lees-Miller SP. XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2013;91:31–41. doi: 10.1139/bcb-2012-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andres SN, Vergnes A, Ristic D, Wyman C, Modesti M, Junop M. A human XRCC4-XLF complex bridges DNA. Nucleic Acids Res. 2012;40:1868–1878. doi: 10.1093/nar/gks022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roy S, Andres SN, Vergnes A, Neal JA, Xu Y, Yu Y, Lees-Miller SP, Junop M, Modesti M, Meek K. XRCC4's interaction with XLF is required for coding (but not signal) end joining. Nucleic Acids Res. 2012;40:1684–1694. doi: 10.1093/nar/gkr1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bryans M, Valenzano MC, Stamato TD. Absence of DNA ligase IV protein in XR-1 cells: evidence for stabilization by XRCC4. Mutat Res. 1999;433:53–58. doi: 10.1016/s0921-8777(98)00063-9. [DOI] [PubMed] [Google Scholar]
- 16.Deshpande RA, Wilson TE. Modes of interaction among yeast Nej1, Lif1 and Dnl4 proteins and comparison to human XLF, XRCC4 and Lig4. DNA repair. 2007;6:1507–1516. doi: 10.1016/j.dnarep.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 18.Andres SN, Junop MS. Crystallization and preliminary X-ray diffraction analysis of the human XRCC4-XLF complex. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67:1399–1402. doi: 10.1107/S1744309111033549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Q, Ochi T, Matak-Vinkovic D, Robinson CV, Chirgadze DY, Blundell TL. Non-homologous end-joining partners in a helical dance: structural studies of XLF-XRCC4 interactions. Biochem Soc Trans. 2011;39(suppl 1382):1387–1392. doi: 10.1042/BST0391387. p following 1392. [DOI] [PubMed] [Google Scholar]
- 20.Ochi T, Wu Q, Chirgadze DY, Grossmann JG, Bolanos-Garcia VM, Blundell TL. Structural insights into the role of domain flexibility in human DNA ligase IV. Structure. 2012;20:1212–1222. doi: 10.1016/j.str.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ochi T, Wu Q, Blundell TL. The spatial organization of non-homologous end joining: From bridging to end joining. DNA Repair. 2014 doi: 10.1016/j.dnarep.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Francis DB, Kozlov M, Chavez J, Chu J, Malu S, Hanna M, Cortes P. DNA Ligase IV regulates XRCC4 nuclear localization. DNA Repair (Amst) 2014;21:36–42. doi: 10.1016/j.dnarep.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chistiakov DA. Ligase IV syndrome. Adv Exp Med Biol. 2010;685:175–185. doi: 10.1007/978-1-4419-6448-9_16. [DOI] [PubMed] [Google Scholar]
- 24.Woodbine L, Gennery AR, Jeggo PA. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair. 2014 doi: 10.1016/j.dnarep.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 25.Yue J, Lu H, Lan S, Liu J, Stein MN, Haffty BG, Shen Z. Identification of the DNA repair defects in a case of Dubowitz syndrome. PloS one. 2013;8:e54389. doi: 10.1371/journal.pone.0054389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 27.Palmbos PL, Wu D, Daley JM, Wilson TE. Recruitment of Saccharomyces cerevisiae Dnl4-Lif1 complex to a double-strand break requires interactions with Yku80 and the Xrs2 FHA domain. Genetics. 2008;180:1809–1819. doi: 10.1534/genetics.108.095539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiruvella KK, Liang Z, Birkeland SR, Basrur V, Wilson TE. Saccharomyces cerevisiae DNA ligase IV supports imprecise end joining independently of Its catalytic activity. PLoS Genet. 2013;9:e1003599. doi: 10.1371/journal.pgen.1003599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daley JM, Laan RL, Suresh A, Wilson TE. DNA joint dependence of pol X family polymerase action in nonhomologous end joining. J Biol Chem. 2005;280:29030–29037. doi: 10.1074/jbc.M505277200. [DOI] [PubMed] [Google Scholar]
- 30.Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, Qureshi-Emili A, Li Y, Godwin B, Conover D, Kalbfleisch T, Vijayadamodar G, Yang M, Johnston M, Fields S, Rothberg JM. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 31.Palmbos PL, Daley JM, Wilson TE. Mutations of the Yku80 C terminus and Xrs2 FHA domain specifically block yeast nonhomologous end joining. Molecular and cellular biology. 2005;25:10782–10790. doi: 10.1128/MCB.25.24.10782-10790.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Della M, Palmbos PL, Tseng HM, Tonkin LM, Daley JM, Topper LM, Pitcher RS, Tomkinson AE, Wilson TE, Doherty AJ. Mycobacterial Ku and ligase proteins constitute a two-component NHEJ repair machine. Science. 2004;306:683–685. doi: 10.1126/science.1099824. [DOI] [PubMed] [Google Scholar]
- 33.Karathanasis E, Wilson TE. Enhancement of Saccharomyces cerevisiae end-joining efficiency by cell growth stage but not by impairment of recombination. Genetics. 2002;161:1015–1027. doi: 10.1093/genetics/161.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu D, Topper LM, Wilson TE. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics. 2008;178:1237–1249. doi: 10.1534/genetics.107.083535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous endjoining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson TE, Lieber MR. Efficient processing of DNA ends during yeast nonhomologous end joining. Evidence for a DNA polymerase beta (Pol4)-dependent pathway. J Biol Chem. 1999;274:23599–23609. doi: 10.1074/jbc.274.33.23599. [DOI] [PubMed] [Google Scholar]
- 37.Wilson TE. A genomics-based screen for yeast mutants with an altered recombination/end-joining repair ratio. Genetics. 2002;162:677–688. doi: 10.1093/genetics/162.2.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clapperton JA, Manke IA, Lowery DM, Ho T, Haire LF, Yaffe MB, Smerdon SJ. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat Struct Mol Biol. 2004;11:512–518. doi: 10.1038/nsmb775. [DOI] [PubMed] [Google Scholar]
- 39.Kilkenny ML, Dore AS, Roe SM, Nestoras K, Ho JC, Watts FZ, Pearl LH. Structural and functional analysis of the Crb2-BRCT2 domain reveals distinct roles in checkpoint signaling and DNA damage repair. Genes Dev. 2008;22:2034–2047. doi: 10.1101/gad.472808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shiozaki EN, Gu L, Yan N, Shi Y. Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide: implications for signaling. Mol Cell. 2004;14:405–412. doi: 10.1016/s1097-2765(04)00238-2. [DOI] [PubMed] [Google Scholar]
- 41.Lee MS, Edwards RA, Thede GL, Glover JN. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J Biol Chem. 2005;280:32053–32056. doi: 10.1074/jbc.C500273200. [DOI] [PubMed] [Google Scholar]
- 42.Yamane K, Katayama E, Tsuruo T. The BRCT regions of tumor suppressor BRCA1 and of XRCC1 show DNA end binding activity with a multimerizing feature. Biochem Biophys Res Commun. 2000;279:678–684. doi: 10.1006/bbrc.2000.3983. [DOI] [PubMed] [Google Scholar]
- 43.Yamane K, Tsuruo T. Conserved BRCT regions of TopBP1 and of the tumor suppressor BRCA1 bind strand breaks and termini of DNA. Oncogene. 1999;18:5194–5203. doi: 10.1038/sj.onc.1202922. [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi M, Figaroa F, Meeuwenoord N, Jansen LE, Siegal G. Characterization of the DNA binding and structural properties of the BRCT region of human replication factor C p140 subunit. J Biol Chem. 2006;281:4308–4317. doi: 10.1074/jbc.M511090200. [DOI] [PubMed] [Google Scholar]
- 45.Wilkinson A, Smith A, Bullard D, Lavesa-Curto M, Sayer H, Bonner A, Hemmings A, Bowater R. Analysis of ligation and DNA binding by Escherichia coli DNA ligase (LigA) Biochim Biophys Acta. 2005;1749:113–122. doi: 10.1016/j.bbapap.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 46.Coquelle N, Green R, Glover JN. Impact of BRCA1 BRCT domain missense substitutions on phosphopeptide recognition. Biochemistry. 2011;50:4579–4589. doi: 10.1021/bi2003795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gilson T, Greer AE, Vindigni A, Ketner G, Hanakahi LA. The alpha2 helix in the DNA ligase IV BRCT-1 domain is required for targeted degradation of ligase IV during adenovirus infection. Virology. 2012;428:128–135. doi: 10.1016/j.virol.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, Hirsch B, Gennery A, Palmer SE, Seidel J, Gatti RA, Varon R, Oettinger MA, Neitzel H, Jeggo PA, Concannon P. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. 2001;8:1175–1185. doi: 10.1016/s1097-2765(01)00408-7. [DOI] [PubMed] [Google Scholar]
- 49.Girard PM, Kysela B, Harer CJ, Doherty AJ, Jeggo PA. Analysis of DNA ligase IV mutations found in LIG4 syndrome patients: the impact of two linked polymorphisms. Hum Mol Genet. 2004;13:2369–2376. doi: 10.1093/hmg/ddh274. [DOI] [PubMed] [Google Scholar]
- 50.Liu S, Liu X, Kamdar RP, Wanotayan R, Sharma MK, Adachi N, Matsumoto Y. C-Terminal region of DNA ligase IV drives XRCC4/DNA ligase IV complex to chromatin, Biochem. Biophys Res Commun. 2013;439:173–178. doi: 10.1016/j.bbrc.2013.08.068. [DOI] [PubMed] [Google Scholar]
- 51.Srivastava M, Nambiar M, Sharma S, Karki SS, Goldsmith G, Hegde M, Kumar S, Pandey M, Singh RK, Ray P, Natarajan R, Kelkar M, De A, Choudhary B, Raghavan SC. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell. 2012;151:1474–1487. doi: 10.1016/j.cell.2012.11.054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.