

Summary
The cell envelope of Gram-negative bacteria serves a critical role in maintenance of cellular homeostasis, resistance to external stress, and host-pathogen interactions. Envelope protein composition is influenced by the physiological and environmental demands placed on the bacterium. In this study, we report a comprehensive compilation of cell envelope proteins from the periodontal and systemic pathogen Aggregatibacter actinomycetemcomitans VT1169, an afimbriated serotype b strain. The urea-extracted membrane proteins were identified by mass spectrometry-based shotgun proteomics. The membrane proteome, isolated from actively growing bacteria under normal laboratory conditions, included 648 proteins representing 28% of the predicted ORFs in the genome. Bioinformatic analyses were used to annotate and predict the cellular location and function of the proteins. Surface adhesins, porins, lipoproteins, numerous influx and efflux pumps, multiple sugar, amino acid and iron transporters, and components of the type I, II and V secretion systems were identified. Periplasmic space and cytoplasmic proteins with chaperone function were also identified. 107 proteins with unknown function were associated with the cell envelope. Orthologs of a subset of these uncharacterized proteins are present in other bacterial genomes, while others are found exclusively in A. actinomycetemcomitans. This knowledge will contribute to elucidating the role of cell envelope proteins in bacterial growth and survival in the oral cavity.
Keywords: Periodontal disease, Secretion systems, Membrane proteins, Bioinformatics
Introduction
Periodontal disease is a common inflammatory condition that affects the tissue surrounding the tooth resulting in reduced bone levels and ultimately tooth loss (Darveau 2010). Aggregatibacter actinomycetemcomitans is a pathogenic bacterium of the Pasteurellaceae family that colonizes the oral cavity of humans and old-world primates (Zambon et al. 1983). The presence of A. actinomycetemcomitans in the subgingival plaque of teeth is strongly implicated in the development of chronic adult periodontal disease and an acute form of the disease, localized aggressive periodontal disease (LAP), that mainly affects children and young adults (Zambon et al. 1983). A. actinomycetemcomitans is also capable of disseminating to distant tissues via the bloodstream and causes infective endocarditis, the inflammation of heart valves (Paturel et al. 2004, Tang et al. 2008). Respiratory, soft tissue, and brain infections are also associated with this bacterium (Kaplan et al. 1989, Scannapieco 1999, Rahamat-Langendoen et al. 2011).
The survival of A. actinomycetemcomitans in these disparate environments is dependent on the protein composition of the cell envelope. The cell envelope of this and other Gram-negative bacteria is comprised of three layers: an inner membrane adjacent to the cytoplasm, a periplasmic space containing the cell wall, and an outer membrane separating the periplasm from the extracellular environment. Both membranes are composed of a phospholipid bilayer containing peripheral and integral membrane proteins with the outer membrane forming an asymmetric bilayer due to the incorporation of lipopolysaccharide (LPS) in the outer leaflet (Silhavy et al. 2010).
The cell envelope, as a whole, is a critical structure involved in maintaining cellular homeostasis (Silhavy et al. 2010). This equilibrium is maintained by the dynamic interaction between the compartments of the cell. These interactions are mediated by individual proteins or protein complexes that allow for the transport of macromolecules between the cytoplasm and the external milieu or to be incorporated into the envelope itself. Therefore, the cell envelope can be viewed as a single unit inclusive of proteins found in both membranes, the periplasmic space, and peripherally associated with the inner membrane.
The nature of the proteins that comprise the cell envelope will be dependent on the cellular environment. However, a proportion of proteins or protein orthologs will be present in the envelope of most, if not all, Gram-negative bacteria. Despite the importance of these proteins, few envelope proteins have been characterized in A. actinomycetemcomitans. In this work, we used a gel-free mass spectrometry approach to detect proteins present in whole membrane fractions of VT1169, an afimbriated serotype b strain of A. actinomycetemcomitans. A total of 648 unique proteins were consistently identified. Bioinformatic analyses indicate that these proteins have diverse functions including virulence determinants, secretion, transport, metabolism, and energy generation. In addition, 107 proteins in the dataset were identified which have not been characterized.
Materials and Methods
Bacterial strains and growth conditions
VT1169, a well characterized laboratory-adapted strain generated by Mintz and Fives-Taylor (1994), was used in this study. The genome of this afimbriated, serotype b strain of A. actinomycetemcomitans is homologous to the fimbriated, serotype b reference strain, HK1651. Bacteria were grown using TSBYE medium (3% tryptic soy broth, 0.6% YE; Becton Dickinson, Franklin Lakes, NJ) statically at 37°C in a humidified 10% CO2 atmosphere. For membrane preparations bacteria were first grown on solid media (TSBYE containing 1.5% agar) and 2-3 colonies were inoculated into 6 mL TSBYE broth. 250 mL of pre-warmed TSBYE was inoculated with 5 ml of a 16 hour liquid culture and grown to mid-logarithmic phase (OD495 = 0.3).
Cell envelope preparation
Bacterial cell envelopes were prepared by differential centrifugation based on previously described methods (Tang et al. 2012). Cells were harvested by centrifugation (8,000 x g for 10 minutes) and the resulting pellet was washed once with Dulbecco's phosphate buffered saline (PBS, 136.9 mM NaCl, 8.1 mM Na2HPO4, 2.68 mM KCl, 1.46 mM KH2PO4, 0.46 mM MgCl2, pH 7.4, Sigma Aldrich, St. Louis, MO). The washed cells were suspended in PBS containing protease inhibitors and 1 mM phenylmethylsulfonyl fluoride (Roche, Basel, Switzerland) and lysed using a French pressure cell press (Thermo Scientific, Waltham, MA) at 18,000 psi. Cell debris was removed by centrifugation at 10,000 x g for 30 min. Membrane and membrane-associated proteins were separated from cytoplasmic proteins by ultracentrifugation at 100,000 x g for 30 min (Optima TL Ultracentrifuge, Beckman, Brea, CA). The pellet was suspended in PBS and centrifuged as previously stated. The process was repeated and the final membrane pellet was stored frozen at -80°C.
Liquid chromatography/Mass spectrometry (LC/MS)
Four biological replicates of A. actinomycetemcomitans cell envelope preparations were analyzed. The protein concentration of the preparations were quantified and 0.5 mg were suspended in 0.5 ml of solublization buffer (6 M urea, 2 M thiourea, 20 mM HEPES, pH = 8.0). Suspensions were sonicated on ice (3 bursts, 20 seconds each with 1 minute intervals) (Kontes 50W Ultrasonicator, Kontes, Vineland, NJ). Extracted proteins were reduced by addition of dithiothreitol (4.5 mM final concentration at 55°C for 30 min) and alkylated by addition of iodoacetamide (10 mM final concentration at room temperature in the dark for 15 min). The solution was diluted 4-fold with 20 mM HEPES, pH 8.0, and incubated with a 1:50 enzyme to substrate ratio with sequencing grade modified trypsin (Promega, Madison, WI) at 37°C overnight. The reaction was terminated by the addition of trifluoroacetic acid (TFA) to a final concentration of 1%. Resulting peptides were isolated (Sep-Pak tC18 cartridge; Waters, Milford, MA) and lyophilized. Peptides were reconstituted in 100 uL 1M HEPES, pH 7.5 and subjected to dimethyl labeling as previously described (Hsu et al. 2003, Boersema et al. 2009). Following labeling, TFA was added to a final concentration of 7%, peptides isolated, and lyophilized. Peptides were suspended in 150 μL of 10 mM potassium phosphate, pH 2.8, containing 25% CH3CN (Buffer A). The peptide solution (50μL) was fractionated using a strong cation exchange (SCX; PolySULFOETHYL A) solid phase extraction (SPE) TopTip (10-200 μL, PolyLC, Columbia, MD) and step elution (100 μL) with increasing concentrations of KCl added to Buffer A: 0 mM; 20-95 mM in 5 mM increments; 100 mM; 200 mM; and 350 mM. The fractions were dried under vacuum, desalted using Ziptips (Pierce, Rockford, IL) and dried again. Peptides were stored at -80°C prior to liquid chromatography/mass spectrometry (LC/MS) analysis.
Peptides from each of the 20 fractions were reconstituted in 2.5% CH3CN containing 2.5% formic acid and analyzed by nano-scale LC/MS on an LTQ-Orbitrap Discovery coupled to a Surveyor MS Pump Plus (Thermo Fisher Scientific, Waltham, MA). Half of the digest was loaded directly onto a 100 μm x 120 mm capillary column packed with MAGIC C18 (5 μm particle size, 20 nm pore size, Michrom Bioresources, Auburn, CA) at a flow rate of 500 nL/min, and peptides were separated by a gradient of CH3CN in 0.1 % formic acid (2.5-5% in 5 min, 5-35% in 100 min, 35-100% in 5 min, 100% in 10 min, followed by an immediate return to 2.5% and a hold at 2.5% for 15 min). Peptides were introduced into the linear ion trap via a nanospray ionization source and a laser pulled ∼3 μm orifice with a spray voltage of 1.8 kV. Mass spectrometry data was acquired in a data-dependent “Top 5” acquisition mode with lock mass function activated (protonated polydimethylcyclosiloxane (Si(CH3)2O)6; m/z 445.120011). An Orbitrap survey scan from m/z 360-1600 at 30,000 (FWHM) resolution was paralleled by 5 collision-induced dissociation (CID) MS/MS scans of the most abundant ions in the LTQ. MS/MS scans were acquired with the following parameters: isolation width: 2 m/z, normalized collision energy: 35%, Activation Q: 0.250 and activation time = 30 ms. Review mode for FTMS master scans was enabled. Dynamic exclusion was enabled (repeat count: 2; repeat duration: 30 sec; exclusion list size: 180; exclusion duration: 60 sec). The minimum threshold was 500. Singly charged ions were excluded for MS/MS.
Databases and data analysis
Product ion spectra were searched against the Los Alamos (Oralgen) annotation of the A. actinomycetemcomitans HK1651 genome. Sequences in forward and reverse orientations were analyzed using the SEQUEST search engine embedded in the Proteome Discoverer 1.4 software package (Thermo Fisher Scientific, Waltham, MA). Raw files from each fraction were processed as one contiguous input file and a single result file (.msf) was generated. Search parameters were as follows: full enzymatic activity and two missed cleavage sites allowed for trypsin; peptide MW of 350-5000 Da.; mass tolerance of 20 ppm and 0.8 Da for precursor and fragment ions, respectively; dynamic modifications on methionine (+15.9949 Da: oxidation) (4 maximum dynamic modifications allowed per peptide), static modification on cysteine (+57.0215 Da: carbamidomethylation) and on lysine and N-terminus (+28.0312984 Da). Cross-correlation (XCorr) values were applied to limit the false positive (FP) rates to less than 1% in each of the four data sets (with the Target/Decoy PSM Validator node). Proteins identified in three out of four biological replicates were included in the final analysis. All protein identification information from the result files (<1% FP; with protein grouping enabled) was exported to Excel spreadsheets, which are included as Supplementary Information.
Functional annotation of proteins
Protein sequences were given gene ontology terms (GO) (Ashburner et al. 2000) NCBI annotations, and KEGG pathway assignments via the BLAST tool (Altschul et al. 1990) using the Blast2GO software (http://www.blast2go.com/b2ghome) (Conesa et al. 2005). Sequences were also queried against the predicted proteomes of E. coli K-12 and A. actinomycetemcomitans ANH9381 with an e-value cutoff of 1-20 (www.ecogene.org, www.uniprot.org)(Jain et al. 2009, Zhou and Rudd 2013) to identify homologs of characterized proteins encoded in other genomes. Cluster of orthologous groups (COG) classes and protein family (PFAM) designations were assigned using the WebMGA server (http://weizhong-lab.ucsd.edu/metagenomic-analysis/)(Tatusov et al. 1997, Wu et al. 2011, Punta et al. 2012). COG classes that represented at least 10 sequences were manually divided into two broad categories (representing general cellular functions and transport) to construct pie charts. Transporters were identified by querying the transporter classification database (TCDB) using the BLAST algorithm with an e-value cutoff of 1-10 to identify significant hits (Saier et al. 2009). Secretion systems present in the dataset were manually identified by their annotation in Oralgen (www.oralgen.org), UniProt, the Human Oral Microbiome Database (HOMD) or BLAST searching against the HK1651 genome using known homologs (Chen et al. 2010). Curated protein annotations were assigned by manually synthesizing data from NCBI, Oralgen, EcoGene, HOMD, UniProt, and functional predictions from COG, PFAM, GO and TCDB.
Protein sequences without descriptive annotations were first analyzed for a predicted role in virulence or associated with other cellular processes using the VirulentPred server (http://203.92.44.117/virulent/index.html) (Garg and Gupta 2008) and querying the NCBI, HOMD, Oralgen, and PFAM databases. The PHMMER algorithm was utilized to predict the taxonomic distribution of these sequences (http://hmmer.janelia.org/)(Finn et al. 2011).
Protein localization prediction
Bioinformatic predictions were made based on a previously outlined approach (Emanuelsson et al. 2007). Localization to the cytoplasm, inner membrane, periplasm, outer membrane, or extracellular environment was predicted by combining data from the CELLO (http://cello.life.nctu.edu.tw/)(Yu et al. 2004) and PSORT algorithms (http://www.psort.org/psortb/index.html)(Yu et al. 2010). Predictions were refined by incorporating data from other bioinformatic tools: Sec signal peptides were predicted by SIGNALP (http://www.cbs.dtu.dk/services/SignalP/)(Petersen et al. 2011); lipoprotein signal peptides by LIPO (http://services.cbu.uib.no/tools/lipo)(Berven et al. 2006) and LIPOP (http://www.cbs.dtu.dk/services/LipoP/)(Juncker et al. 2003); twin arginine translocon signal peptides by TATP (http://www.cbs.dtu.dk/services/TatP/)(Bendtsen et al. 2005); transmembrane helices by TMHMM (http://www.cbs.dtu.dk/services/TMHMM-2.0/)(Krogh et al. 2001); and outer membrane localization by BOMP (http://services.cbu.uib.no/tools/bomp)(Berven et al. 2004). Annotation and GO data was utilized to resolve ambiguous predictions and to assign proteins to the nucleoid, ribosome, or as membrane associated. When possible, localization of characterized proteins was manually assigned.
Results
The proteomics approach utilized in this study was carefully optimized regarding SCX fractionation, nanoscale chromatography and mass spectrometry acquisition, to maximize the coverage of the membrane proteome. With the optimized approach, 666, 737 828, 764 proteins were identified in replicate 1 to 4, respectively (Supplementary Information). A total of 648 proteins were identified in 3 of the four biological replicates, 495 of which were found in all 4 replicates. The whole sequences of these proteins were queried against the A. actinomycetemcomitans HK1651 genome in the Oralgen and HOMD databases and separately against all genomes in NCBI to assign functional annotations. Homologs of all sequences were identified in the NCBI database, however >30% of these had ambiguous or conflicting annotation data. Annotations were improved by querying sequences against A. actinomycetemcomitans ANH9381 and E. coli K-12 predicted proteomes. This data was supplemented by manually incorporating information about conserved domains from the protein families (PFAM) and cluster of orthologous groups (COG) databases.
Sequences were queried against the PFAM database to identify function based on conserved domains. This database consists of defined groups of proteins organized into families based on related function or conservation among different organisms. PFAM data was available for 584 of the identified proteins. Clusters of Orthologous Groups (COG), which catalogs proteins based on conserved regions, was used as a complementary analysis to the PFAM data. The COG protein database compares predicted and known proteins in all completely sequenced microbial genomes to infer sets of orthologs. Each COG consists of any group of at least three proteins from distant genomes that are more similar to each other than they are to any other proteins from the same genomes (Tatusov et al. 1997). Each individual COG represents a specific motif and belongs to a larger group (COG class) with similar predicted function. The most common COG classes represented in our dataset were identified and are shown in Figure 1.
Figure 1. Functional predictions of identified proteins.
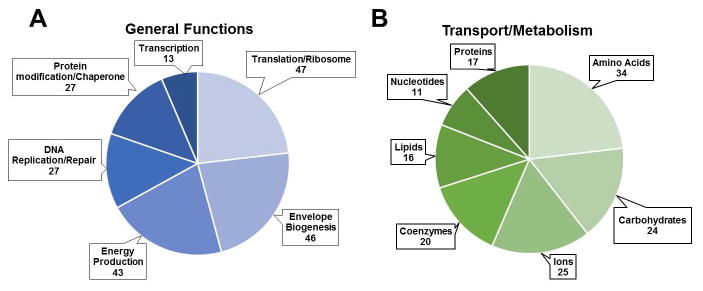
Proteins were assigned to functional groups (COG classes) utilizing the WebMGA server (http://weizhonglab.ucsd.edu/metagenomic-analysis/). Classes containing at least 10 sequences were utilized to generate pie charts of proteins involved in (A) general cellular functions and (B) transport and metabolic functions. COG class names were shortened for clarity. Classes represented are as follows for general cellular functions; Translation/ribosome: J, Envelope biogenesis: M, Energy production: C, DNA replication/repair: L, Protein modification/chaperone: O, Transcription: K, and for metabolism/transport of; Amino acids: E, Carbohydrates: G, Ions: P, Coenzymes: H, Lipids: I, Nucleotides: F, Proteins: U.
COG class data was available for 349 proteins of the cell envelope. A total of 203 proteins were predicted to be involved in general cell functions such as DNA transcription, replication, protein translation, and energy generation. Within this group, a subset of 27 proteins were classified to be involved in protein modification and 46 involved in cell envelope biogenesis (Figure 1A). Metabolic and transport functions were associated with 146 proteins (Figure 1B). Specific metabolic enzymes were identified by incorporating annotation data and predictions from the KEGG database. These included enzymes for the synthesis or breakdown of amino acids (alanine, cysteine, glycine, tryptophan, tyrosine, and others), carbohydrates (glucose, fructose, amino sugars), and nucleotides (purines and pyrimidines).
Specific protein secretion systems were identified by analysis of combined annotation data. Three types of secretion systems (Type I, II, and V) were identified in the dataset. In addition, we also identified the general secretory machinery (Sec) and twin-arginine translocon (TAT) complexes (Table 1). The Sec translocon transports unfolded polypeptides across the inner membrane or integrates membrane proteins. The TAT actively transports folded proteins across the inner membrane (Lee et al. 2006). All components of the Type I secretion required for leukotoxin secretion (LtxBD and TolC) as well as the Type Va (Aae) and Vc autotransporter proteins (ApiA and EmaA) were present. Proteins encoded by the tight adherence (Tad) locus, classified as a subtype of the Type II secretion system, were identified. The fimbriae secretion apparatus (RcpC-TadG) was present in the preparations from this afimbriated strain. However, the structural subunit of fimbriae (Flp-1) and TadV, a processing enzyme of Flp-1, were not identified. Proteins associated with other characterized secretion systems (III, IV, VI-IX) were not present in genome and thus not in the cell envelope.
Table 1. Classical secretion pathways identified in the A. actinomycetemcomitans cell envelope.
| Secretion system | Protein(s) | Function | Sequence ID(s) |
|---|---|---|---|
| Type I (Leukotoxin secretion) | LtxB | Leukotoxin secretion ATPase | AA02805 |
| LtxD | Leukotoxin secretion channel | AA02803 | |
| TolC | Outer membrane pore | AA02077 | |
| Sec translocon | SecA | Sec translocon ATPase | AA01083 |
| SecB | Sec translocon chaperone | AA01506 | |
| SecYG | SecYEG complex channel | AA01237, AA00786 | |
| SecDFYajC | SecDFYajC inner membrane complex | AA02780-1, AA02779 | |
| LepB | Leader peptidase | AA03004 | |
| Twin-arginine translocon | TatA | Twin arginine translocon protein A | AA01154 |
| TatB | Twin arginine translocon protein B | AA01153 | |
| Type II (Fimbrial secretion) | RcpC-B | Rough colony protein C-B | AA00870-2 |
| TadZ-G | Tight adherence protein Z-G | AA00873-80 | |
| Type V (Autotransporters) | EmaA | Collagen adhesin | AA00234 |
| Aae | Epithelial cell adhesin | AA02347 | |
| ApiA | Epithelial cell adhesin | AA02485 |
We identified multiple accessory complexes responsible for envelope biogenesis (Table 2). Outer membrane proteins contain repeating alternate hydrophobic amino acids at the carboxyl terminus forming structural elements termed β-barrels (Koebnik et al. 2000). The β-barrel assembly proteins (Bam), which form complexes to assemble these outer membrane proteins (Rigel and Silhavy 2012), were identified in the A. actinomycetemcomitans cell envelope. Homologs of the lipoprotein transport system (Lol) of E. coli were present in the A. actinomycetemcomitans preparation. Lipoproteins comprise a subset of membrane proteins with a lipid-modified cysteine residue at the amino termini, which are used to anchor the protein to the membrane. Lipoproteins are localized to either the inner or the outer membrane in Gram-negative bacteria (Tokuda and Matsuyama 2004). The LPS transport systems responsible for translocation of both the lipid and sugar components across the cell envelope were also identified in our preparations. LPS is composed of a lipid-anchored carbohydrate moiety and exists only in the outer leaflet of the outer membrane. Chaperones are an important subset of proteins required for maintaining protein structure and degrading unfolded proteins during secretion. Eleven proteins were annotated as chaperones (Table 3). Overall, our annotation scheme allowed the assignment of informative annotations to 84% of protein sequences. The curated annotations for each identified protein in this study can be found in the Supplemental data.
Table 2. Outer membrane protein, LPS, and lipoprotein transport complexes.
| Transport system | Protein | Function | Sequence ID |
|---|---|---|---|
| Beta-barrel assembly module | BamA | Bam complex pore | AA00608 |
| BamCDE | Bam complex accessory lipoprotein | AA02350, AA01356, AA00998 | |
| LPS export system | MsbA | Transport of LPS/Lipid A across the inner membrane | AA01961 |
| LptBCFG | ABC transporter of LPS | AA02320, AA02323, AA01777, AA01776 | |
| LptADE | Periplasmic LPS transporter | AA02321, AA00919, AA01088 | |
| Lipoprotein releasing system | LolCDE | ABC transporter of lipoproteins to the periplasm | AA01615-6, AA01618 |
| LolB | Outer membrane protein for lipoprotein insertion | AA02743 |
Table 3. Chaperone proteins.
| Chaperone | Sequence ID |
|---|---|
| ClpB | AA01211 |
| ClpX | AA00120 |
| DegQ | AA01869 |
| DegS | AA01789 |
| DnaJ | AA00659 |
| DnaK | AA00657 |
| GrpE | AA00766 |
| GroEL | AA01284 |
| Lon | AA02395 |
| RseP | AA00606 |
| YidC | AA03011 |
The remaining 16% or 102 proteins were uncharacterized. In an attempt at annotation, these proteins were analyzed using the VirulentPred algorithm, a method to predict putative novel virulence factors, and taxonomic conservation using PHMMER. A combination of these programs identified 11 proteins that contained sequences similar to known virulence determinants and conserved across species (Table 4). The function of some of these proteins cannot be determined but are predicted to be associated with virulence. Taxonomic conservation of sequence is also considered to be of significance in terms of protein function. Analysis revealed conservation of these unknown proteins in many genera. Select proteins with interesting taxonomic distribution in terms of the diversity within families of prokaryotes are listed in Table 5.
Table 4. Uncharacterized virulence-related proteins.
| Sequence ID | Predicted Localization | Predicted Function | Distribution | Conserved Domains | Domain Description |
|---|---|---|---|---|---|
| AA00179 | Outer Membrane | Unknown | Alphaproteobacteria | PF04575 | Unknown function |
| Betaproteobacteria | |||||
| Gammaproteobacteria | |||||
| AA00180 | Outer Membrane | Transferrin binding | Pasteurellaceae | PF01298 | Transferrin binding |
| Moraxellaceae | |||||
| Neisseriaceae | |||||
| AA00422 | Inner Membrane | Virulence protein secretion | Bacteria | PF04393 | Expression/localization of virulence factors |
| AA00702 | Outer Membrane | Unknown | Pasteurellaceae | None | N/A |
| AA00781 | Outer Membrane | Unknown | A. actinomycetemcomitans | None | N/A |
| AA01040 | Outer Membrane | Lipoprotein | Gammaproteobacteria | PF03923 | Lipoprotein |
| AA01688 | Outer Membrane | Unknown | Bacteria | PF07703 | Alpha-2-macroglobulin domain |
| AA02554 | Periplasmic | Unknown | Gammaproteobacteria | PF09829 | Unknown function |
| AA02614 | Outer Membrane | Unknown | Aggregatibacter | None | N/A |
| Pasteurella | |||||
| Gallibacterium | |||||
| AA02748 | Outer Membrane | Lipoprotein | Aggregatibacter | None | N/A |
| Haemophilus | |||||
| AA02811 | Outer Membrane | Lipoprotein | Pasteurellaceae | None | N/A |
Table 5. Selected uncharacterized proteins with taxonomic conservation.
| Sequence ID | Predicted Localization | Predicted Function | Distribution | Conserved Domains | Domain Description |
|---|---|---|---|---|---|
| AA00300 | Periplasmic | Unknown | Pasteurellaceae | PF07509 | Unknown function |
| Pseudomonadaceae | |||||
| Campylobacteraceae | |||||
| Helicobacteraceae | |||||
| AA01162 | Inner Membrane | Unknown | Proteobacteria | PF04224 | Quinol oxidase like |
| Actinobacteria | |||||
| Bacteroidetes | |||||
| AA01577 | Inner Membrane | Unknown | Proteobacteria | PF05128 | Unknown function |
| Cyanobacteria | |||||
| AA02643 | Inner Membrane | ABC Transporter | Bacteria | PF01061 | ABC transporter |
Cellular localization of the annotated proteins was assigned using a combination of general prediction algorithms (PSORT and CELLO), and manual combination with more specialized localization programs such as TMHMM, LipoP, and SignalP (Figure 2A). Where possible, these predictions were manually curated based on functional annotation data and localization of known proteins. Approximately 66% of all identified proteins were predicted to be associated with the cell envelope. There were 57 predicted outer membrane proteins represented in the dataset. These proteins contain an amino terminal signal sequence for transport across the inner membrane. Both integral and peripheral membrane proteins, including lipoproteins, are included in this compartment. We identified 60 periplasmic proteins in the envelope preparations. These proteins are also translocated across the inner membrane, but are predicted to lack large domains of hydrophobic amino acids and are considered to be soluble and not associated with the membrane. Inner membrane proteins composed the largest subset (170) of the membrane proteins. These proteins were assigned to the inner membrane based on the prediction of the presence of at least one amphipathic helix that can span the membrane. The majority of the proteins (80) are predicted to contain a single helical domain. However, diversity exists in the number of transmembrane domains and some of the proteins contain up to 16 transmembrane domains (Figure 2B). Many (38%) of these proteins had 5 or more transmembrane domains, indicating our ability to identify highly hydrophobic sequences with our mass spectrometry technique. Two proteins, leukotoxin and cytolethal distending toxin, which are considered extracellular, were identified but are expected to be present in the membrane during secretion to the extracellular milieu.
Figure 2. Predicted localization of identified proteins.
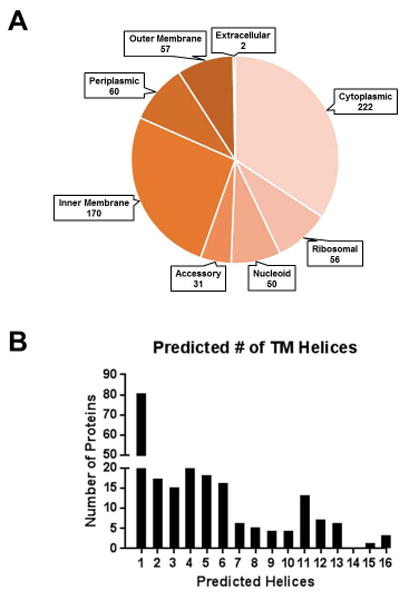
(A) The 665 identified protein sequences were analyzed using the CELLO and PSORT algorithms. Data from SIGNALP, LIPO, LIPOP, TATP, TMHMM, BOMP, and functional annotations were incorporated into the analysis to refine the initial predictions. Proteins were then assigned to one of eight localizations based on bioinformatic predictions combined with manual curation. (B) The proteins containing at least one predicted transmembrane helix by TMHMM arranged by number of predicted helices.
The localization of proteins that may interact directly with the polar head groups of lipids or directly with integral membrane proteins may not be correctly assigned using localization algorithms. Using the protein annotation data, proteins were designated membrane-associated if associated with cytoplasmic domains of transporters, ribosomal if associated with translation, or nucleoid if associated with DNA binding or replication. The remaining proteins were classified as cytoplasmic. These proteins are predicted to be soluble, contain no signal sequence, and are not predicted to be found as interacting partners with membrane proteins. Localization and annotation predictions for each protein can be found in the Supplemental data.
Discussion
In this study, we have adopted an alternative protocol for improved identification of the proteins associated with the cell envelope of A. actinomycetemcomitans. The protocol used in this study improves upon prior methodologies by eliminating limitations associated with 2D gel electrophoresis (2D-GE). Techniques based on 2D-GE are restricted amount of protein that can be loaded, which affects the available protein for extraction and subsequent identification by MS, leading to false negatives. This phenomenon is especially significant when low abundance proteins are of interest (Gygi et al. 2000). Furthermore, specific nonionic or zwitterionic detergents are required for isoelectric focusing (IEF) and highly hydrophobic proteins are relatively insoluble in these detergents (Braun et al. 2007). There is also poor resolution of highly hydrophobic proteins by pH gradients and precipitation of these proteins at pH values close to their isoelectric points during IEF (Rabilloud 2009).
We have eliminated these potential pitfalls by using gel–free mass spectrometry-based shotgun sequencing (multidimensional protein identification technique, MudPIT)(Liu et al. 2002). SCX chromatography is routinely used as the first dimension of separation in MudPIT (Washburn 2004). In this study, SCX separation was performed on a SPE column, which in conjunction with LC/MS analysis serves as a robust method for characterizing complex proteomes, as previously reported (Dephoure and Gygi 2011). To yield the maximum proteome coverage, we carefully optimized the number of SCX fractions (20 fractions) as well as the elution concentrations so that peptides were equally distributed over the 20 fractions for subsequent LC/MS shotgun sequencing. Ion exchange chromatography of urea extracted membrane preparations and analysis of individual fractions by LC/MS increased the identification of envelope proteins ∼4-fold in this study from previous studies of the A. actinomycetemcomitans proteome (Rylev et al. 2011, Zijnge et al. 2012).
Functional annotation and cellular localization of the identified proteins were assigned using a combination of bioinformatic algorithms due to inherent weaknesses associated with the individual programs. To overcome this problem, we used programs in parallel and compared results looking for inconsistencies. Disagreements were resolved by incorporating results from other algorithms. For example: The localization assigned by PSORT is highly accurate. However, the program does not assign cellular localization to all queries. Assignments can be deduced using the CELLO program, which is less accurate but provides greater coverage (Gardy and Brinkman 2006). Disagreements between programs can then be mediated by the outcome of an independent tool, e.g. TMHMM, which is optimized for identification of specific motifs (Krogh et al. 2001). Using multiple algorithms takes advantage of the strengths while mitigating the weaknesses of the individual programs. This ultimately leads to more accurate bioinformatic predictions for the proteins associated with the cell envelope (Solis and Cordwell 2011).
The cell envelope of Gram-negative bacteria serve as the locus for many facets of bacterial physiology: transport of nutrients; metabolism; replication; and colonization. A. actinomycetemcomitans is saccharolytic and utilizes a multitude of sugars as the primary carbon source (Olsen et al. 1999). The media used in this study contains glucose as the major sugar. Therefore, the presence of a glucose transporter (AA01876) was expected. We also found transporters specific for other sugars such as mannose (AA01465-AA01467) and fructose (AA00332, AA00335). These sugars are not presumed to be present in the medium based on the manufacturer's stated composition. This indicates that these transporters are either upregulated, constitutively expressed, or contaminants are present in the medium suggesting transporter regulation is exquisitely sensitive to the respective substrates. However, the observation of Brown and Whiteley (2007) suggests that transcripts corresponding to a subset of sugar transporters in A. actinomycetemcomitans are not changed in abundance by carbon source availability (Brown and Whiteley 2007).
Potential carbon sources for A. actinomycetemcomitans are free amino acids, which are found in abundance in vivo and in laboratory media (Syrjanen et al. 1990). However, except for the characterization of a single amino acid transporter (Jorth and Whiteley 2010), little is known about amino acid utilization in A. actinomycetemcomitans. Amino acid transporters are broadly categorized as being specific for polar or nonpolar amino acids, with individual transporters having high specificity for a single amino acid (Saier 2000). Six specific amino acid transporters were identified when grown under the culture conditions used in this study: arginine (AA00855, AA00858); cysteine/cystine (AA01509, AA01510, AA01524, AA01525) glutamic acid (AA00994); methionine (AA0415-AA0417) proline (AA00680); serine (AA00092). Transporters for oligopeptides (AA02893, AA02897-AA02799) and dipeptides (AA01568, AA01573) were identified, as well as for related amine containing compounds e.g. spermidine (AA02718-AA02721) and glycine betaine (AA02352). Interestingly, RNAs encoding amino acid transporters and metabolic proteins have been identified in an in vivo model of infection, implying a potential role for amino acid metabolism in disease (Jorth et al. 2013).
Cysteine is suggested to be an integral amino acid for the growth of A. actinomycetemcomitans. Substitution of yeast extract with cystine in TSBYE does not alter the growth kinetics of the bacterium (Sreenivasan et al. 1993). Cysteine can be acquired by the bacterium from the degradation of polypeptides or de novo synthesis from serine. Notably, metabolic enzymes required for the synthesis of cysteine (AA02412, AA01502) were present. This suggests that even in the presence of a protein rich media, cysteine synthesis is still maintained. The biosynthesis of cysteine may be required in vivo due to the limitation of cysteine in gingival crevicular fluid of individuals with periodontal disease (Syrjanen et al. 1990). Taken together, the identification of multiple transport and metabolic systems imply that the metabolism of A. actinomycetemcomitans is more diverse than previously appreciated.
The composition of a subset of transporters include both membrane and membrane associated proteins. Both ATP binding cassette (ABC) and phosphoenolpyruvate transferase system (PTS) transporter families require integral membrane components that associate with soluble proteins on the inner leaflet of the inner membrane for function (Postma et al. 1993, Davidson and Chen 2004). The soluble components of these transporters are easily overlooked by de novo bioinformatic tools as they are indistinguishable from cytoplasmic proteins. Our annotation based approach allowed for manual curation of these proteins based on their predicted function as components of transporters. The majority of sugar transporters identified in this study belong to the PTS family. The remaining transporters are categorized as members of the major facilitator superfamily, which do not contain large cytoplasmic domains (Pao et al. 1998). The majority of amino acid transporters expressed in A. actinomycetemcomitans belong to the ABC transporter family. Both of these transporter classes appear to be important in A. actinomycetemcomitans physiology.
The transport of proteins across cellular membranes is also important for cellular physiology. Conserved secretion systems were identified in the strain of A. actinomycetemcomitans utilized in this study. Type I, II, and V secretion systems were present in this bacterium. Proteomic and genomic analysis of other A. actinomycetemcomitans strains indicate the conservation of all these systems. Consistent with the study of Zijnge et al. (2012), we did not find type III or IV secretion systems in the envelope proteome consistent with the absence of these genes in the chromosome. Interestingly, a type VI secretion system is present in the genome of A. aphrophilus, a bacterium closely related to A. actinomycetemcomitans (Di Bonaventura et al. 2009). These secretion systems are typically involved in direct cell-to-cell interactions and delivery of effector molecules into the host (Tseng et al. 2009). We hypothesized a similar system may exist in A. actinomycetemcomitans, however, we found no evidence for a type VI secretion system in the genome based on BLAST searches against the NCBI database or annotation in the KEGG database.
The major virulence associated proteins of A. actinomycetemcomitans were identified in the studied strain. These included adhesins: EmaA, ApiA, and Aae (Asakawa et al. 2003, Rose et al. 2003, Mintz 2004); outer membrane proteins: Omp34, Omp18, Omp39, and Omp64 (Fives-Taylor et al. 1999, Komatsuzawa et al. 2002); and soluble toxins: leukotoxin and cytolethal distending toxin (Lally et al. 1989, Mayer et al. 1999). In addition to these factors, several poorly annotated proteins shared characteristics of virulence proteins based on bioinformatics analyses. These proteins may also be factors that contribute to the pathogenicity of this organism.
A. actinomycetemcomitans is typically isolated with fimbriae, which are considered an important virulence determinant. The fimbriae are composed of repeating subunits (Flp1) secreted through a subclass of the Type II secretion system consisting of 11 proteins encoded by the tight adherence (tad) locus (Tomich et al. 2007). The tad operon is suggested to be regulated by a single promoter (Kram et al. 2008). In the cell envelope of the afimbriated strain used in this study, the complete fimbrial secretion apparatus (RcpC-TadG) was present. However, the fimbriae subunit (Flp1) and the prepilin peptidase (TadV) were not detected. Typically, the loss of fimbriation is due to point mutations in the promoter region of the locus (Wang et al. 2005). However, studies in our laboratory indicate that the promoter is functional and nonsense mutations are present in flp1 (data not shown) of the studied strain. The absence of Flp1 in the data set is explained by the presence of these stop codons in the gene. However, the absence of TadV and presence of RcpC-TadG suggests that a second promoter is present in the tad locus. Studies are underway to determine if the secretion apparatus is functional and if a second promoter exists in this locus in this strain of A. actinomycetemcomitans.
In addition to being a platform for membrane proteins, the cell envelope also interacts with structures traditionally considered to be purely cytoplasmic. Electron micrographs have shown an intimate association between the chromosome and bacterial membrane (Ryter 1968). Furthermore, regions of DNA transcribing genes coding for membrane proteins are found in close proximity to the membrane (Libby et al. 2012). This is consistent with the observation that ribosomes directly interact with the Sec translocon through the signal recognition particle (SRP) while translating membrane proteins (Herskovits and Bibi 2000). As transcription and translation are linked, this provides an elegant means to protect membrane proteins from interacting with the cytoplasmic environment. These known interactions explain the presence of ribosome and nucleoid proteins in cell envelope preparations found in this and other proteomic studies (Huang et al. 2006, Marti et al. 2006).
Cell envelopes of Gram-negative bacteria are complex structures containing proteins critical for cell physiology and virulence. The proteome presented in this study includes both well characterized and hypothetical proteins predicted to exist based only on genomic sequencing. Some of these proteins share sequence identity with known proteins, whereas others remain functionally uncharacterized. This study may serve as a roadmap to study proteins required for colonization and survival in the oral cavity.
Supplementary Material
Acknowledgments
We thank James Vincent (Vermont Genetics Network Bioinformatics Core) for his help in generating sequence files. This study was supported by NIH grant RO1-DE018889 (K.P.M.). The Vermont Genetics Network Proteomics Facility is supported through NIH grant P20GM103449 from the INBRE Program of the National Institute of General Medical Sciences.
References Cited
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Asakawa R, Komatsuzawa H, Kawai T, Yamada S, Goncalves RB, Izumi S, Fujiwara T, Nakano Y, Suzuki N, Uchida Y, et al. Outer membrane protein 100, a versatile virulence factor of Actinobacillus actinomycetemcomitans. Mol Microbiol. 2003;50:1125–1139. doi: 10.1046/j.1365-2958.2003.03748.x. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S. Prediction of twin-arginine signal peptides. BMC Bioinformatics. 2005;6:167. doi: 10.1186/1471-2105-6-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berven FS, Flikka K, Jensen HB, Eidhammer I. BOMP: a program to predict integral beta-barrel outer membrane proteins encoded within genomes of Gram-negative bacteria. Nucleic Acids Res. 2004;32:W394–399. doi: 10.1093/nar/gkh351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berven FS, Karlsen OA, Straume AH, Flikka K, Murrell JC, Fjellbirkeland A, Lillehaug JR, Eidhammer I, Jensen HB. Analysing the outer membrane subproteome of Methylococcus capsulatus (Bath) using proteomics and novel biocomputing tools. Arch Microbiol. 2006;184:362–377. doi: 10.1007/s00203-005-0055-7. [DOI] [PubMed] [Google Scholar]
- Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc. 2009;4:484–494. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- Braun RJ, Kinkl N, Beer M, Ueffing M. Two-dimensional electrophoresis of membrane proteins. Anal Bioanal Chem. 2007;389:1033–1045. doi: 10.1007/s00216-007-1514-6. [DOI] [PubMed] [Google Scholar]
- Brown SA, Whiteley M. A novel exclusion mechanism for carbon resource partitioning in Aggregatibacter actinomycetemcomitans. J Bacteriol. 2007;189:6407–6414. doi: 10.1128/JB.00554-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Yu WH, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010;2010 doi: 10.1093/database/baq013. baq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- Davidson AL, Chen J. ATP-binding cassette transporters in bacteria. Annu Rev Biochem. 2004;73:241–268. doi: 10.1146/annurev.biochem.73.011303.073626. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Gygi SP. A solid phase extraction-based platform for rapid phosphoproteomic analysis. Methods. 2011;54:379–386. doi: 10.1016/j.ymeth.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bonaventura MP, DeSalle R, Pop M, Nagarajan N, Figurski DH, Fine DH, Kaplan JB, Planet PJ. Complete genome sequence of Aggregatibacter (Haemophilus) aphrophilus NJ8700. J Bacteriol. 2009;191:4693–4694. doi: 10.1128/JB.00447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fives-Taylor PM, Meyer DH, Mintz KP, Brissette C. Virulence factors of Actinobacillus actinomycetemcomitans. Periodontol 2000. 1999;20:136–167. doi: 10.1111/j.1600-0757.1999.tb00161.x. [DOI] [PubMed] [Google Scholar]
- Gardy JL, Brinkman FS. Methods for predicting bacterial protein subcellular localization. Nat Rev Microbiol. 2006;4:741–751. doi: 10.1038/nrmicro1494. [DOI] [PubMed] [Google Scholar]
- Garg A, Gupta D. VirulentPred: a SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinformatics. 2008;9:62. doi: 10.1186/1471-2105-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi SP, Corthals GL, Zhang Y, Rochon Y, Aebersold R. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc Natl Acad Sci U S A. 2000;97:9390–9395. doi: 10.1073/pnas.160270797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovits AA, Bibi E. Association of Escherichia coli ribosomes with the inner membrane requires the signal recognition particle receptor but is independent of the signal recognition particle. Proc Natl Acad Sci U S A. 2000;97:4621–4626. doi: 10.1073/pnas.080077197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JL, Huang SY, Chow NH, Chen SH. Stable-isotope dimethyl labeling for quantitative proteomics. Anal Chem. 2003;75:6843–6852. doi: 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- Huang CZ, Lin XM, Wu LN, Zhang DF, Liu D, Wang SY, Peng XX. Systematic identification of the subproteome of Escherichia coli cell envelope reveals the interaction network of membrane proteins and membrane-associated peripheral proteins. J Proteome Res. 2006;5:3268–3276. doi: 10.1021/pr060257h. [DOI] [PubMed] [Google Scholar]
- Jain E, Bairoch A, Duvaud S, Phan I, Redaschi N, Suzek BE, Martin MJ, McGarvey P, Gasteiger E. Infrastructure for the life sciences: design and implementation of the UniProt website. BMC Bioinformatics. 2009;10:136. doi: 10.1186/1471-2105-10-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorth P, Trivedi U, Rumbaugh K, Whiteley M. Probing bacterial metabolism during infection using high-resolution transcriptomics. J Bacteriol. 2013;195:4991–4998. doi: 10.1128/JB.00875-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorth P, Whiteley M. Characterization of a novel riboswitch-regulated lysine transporter in Aggregatibacter actinomycetemcomitans. J Bacteriol. 2010;192:6240–6250. doi: 10.1128/JB.00935-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003;12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan AH, Weber DJ, Oddone EZ, Perfect JR. Infection due to Actinobacillus actinomycetemcomitans: 15 cases and review. Rev Infect Dis. 1989;11:46–63. doi: 10.1093/clinids/11.1.46. [DOI] [PubMed] [Google Scholar]
- Koebnik R, Locher KP, Van Gelder P. Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol Microbiol. 2000;37:239–253. doi: 10.1046/j.1365-2958.2000.01983.x. [DOI] [PubMed] [Google Scholar]
- Komatsuzawa H, Asakawa R, Kawai T, Ochiai K, Fujiwara T, Taubman MA, Ohara M, Kurihara H, Sugai M. Identification of six major outer membrane proteins from Actinobacillus actinomycetemcomitans. Gene. 2002;288:195–201. doi: 10.1016/s0378-1119(02)00500-0. [DOI] [PubMed] [Google Scholar]
- Kram KE, Hovel-Miner GA, Tomich M, Figurski DH. Transcriptional regulation of the tad locus in Aggregatibacter actinomycetemcomitans: a termination cascade. J Bacteriol. 2008;190:3859–3868. doi: 10.1128/JB.00128-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Lally ET, Golub EE, Kieba IR, Taichman NS, Rosenbloom J, Rosenbloom JC, Gibson CW, Demuth DR. Analysis of the Actinobacillus actinomycetemcomitans leukotoxin gene. Delineation of unique features and comparison to homologous toxins. J Biol Chem. 1989;264:15451–15456. [PubMed] [Google Scholar]
- Lee PA, Tullman-Ercek D, Georgiou G. The bacterial twin-arginine translocation pathway. Annu Rev Microbiol. 2006;60:373–395. doi: 10.1146/annurev.micro.60.080805.142212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby EA, Roggiani M, Goulian M. Membrane protein expression triggers chromosomal locus repositioning in bacteria. Proc Natl Acad Sci U S A. 2012;109:7445–7450. doi: 10.1073/pnas.1109479109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Lin D, Yates JR., 3rd Multidimensional separations for protein/peptide analysis in the post-genomic era. Biotechniques. 2002;32:898, 900, 902. doi: 10.2144/02324pt01. passim. [DOI] [PubMed] [Google Scholar]
- Marti S, Sanchez-Cespedes J, Oliveira E, Bellido D, Giralt E, Vila J. Proteomic analysis of a fraction enriched in cell envelope proteins of Acinetobacter baumannii. Proteomics. 2006;6(Suppl 1):S82–87. doi: 10.1002/pmic.200500323. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bueno LC, Hansen EJ, DiRienzo JM. Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect Immun. 1999;67:1227–1237. doi: 10.1128/iai.67.3.1227-1237.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz KP. Identification of an extracellular matrix protein adhesin, EmaA, which mediates the adhesion of Actinobacillus actinomycetemcomitans to collagen. Microbiology. 2004;150:2677–2688. doi: 10.1099/mic.0.27110-0. [DOI] [PubMed] [Google Scholar]
- Mintz KP, Fives-Taylor PM. Adhesion of Actinobacillus actinomycetemcomitans to a human oral cell line. Infect Immun. 1994;62:3672–3678. doi: 10.1128/iai.62.9.3672-3678.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen I, Shah HN, Gharbia SE. Taxonomy and biochemical characteristics of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Periodontol 2000. 1999;20:14–52. doi: 10.1111/j.1600-0757.1999.tb00156.x. [DOI] [PubMed] [Google Scholar]
- Pao SS, Paulsen IT, Saier MH., Jr Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paturel L, Casalta JP, Habib G, Nezri M, Raoult D. Actinobacillus actinomycetemcomitans endocarditis. Clin Microbiol Infect. 2004;10:98–118. doi: 10.1111/j.1469-0691.2004.00794.x. [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Postma PW, Lengeler JW, Jacobson GR. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Rev. 1993;57:543–594. doi: 10.1128/mr.57.3.543-594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabilloud T. Membrane proteins and proteomics: love is possible, but so difficult. Electrophoresis. 2009;30(Suppl 1):S174–180. doi: 10.1002/elps.200900050. [DOI] [PubMed] [Google Scholar]
- Rahamat-Langendoen JC, van Vonderen MG, Engstrom LJ, Manson WL, van Winkelhoff AJ, Mooi-Kokenberg EA. Brain abscess associated with Aggregatibacter actinomycetemcomitans: case report and review of literature. J Clin Periodontol. 2011;38:702–706. doi: 10.1111/j.1600-051X.2011.01737.x. [DOI] [PubMed] [Google Scholar]
- Rigel NW, Silhavy TJ. Making a beta-barrel: assembly of outer membrane proteins in Gram-negative bacteria. Curr Opin Microbiol. 2012;15:189–193. doi: 10.1016/j.mib.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JE, Meyer DH, Fives-Taylor PM. Aae, an autotransporter involved in adhesion of Actinobacillus actinomycetemcomitans to epithelial cells. Infect Immun. 2003;71:2384–2393. doi: 10.1128/IAI.71.5.2384-2393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rylev M, Abduljabar AB, Reinholdt J, Ennibi OK, Haubek D, Birkelund S, Kilian M. Proteomic and immunoproteomic analysis of Aggregatibacter actinomycetemcomitans JP2 clone strain HK1651. J Proteomics. 2011;74:2972–2985. doi: 10.1016/j.jprot.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Ryter A. Association of the nucleus and the membrane of bacteria: a morphological study. Bacteriol Rev. 1968;32:39–54. doi: 10.1128/br.32.1.39-54.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH., Jr Families of transmembrane transporters selective for amino acids and their derivatives. Microbiology. 2000;146(Pt 8):1775–1795. doi: 10.1099/00221287-146-8-1775. [DOI] [PubMed] [Google Scholar]
- Saier MH, Jr, Yen MR, Noto K, Tamang DG, Elkan C. The Transporter Classification Database: recent advances. Nucleic Acids Res. 2009;37:D274–278. doi: 10.1093/nar/gkn862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannapieco FA. Role of oral bacteria in respiratory infection. J Periodontol. 1999;70:793–802. doi: 10.1902/jop.1999.70.7.793. [DOI] [PubMed] [Google Scholar]
- Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol. 2010;2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis N, Cordwell SJ. Current methodologies for proteomics of bacterial surface-exposed and cell envelope proteins. Proteomics. 2011;11:3169–3189. doi: 10.1002/pmic.201000808. [DOI] [PubMed] [Google Scholar]
- Sreenivasan PK, Meyer DH, Fives-Taylor PM. Factors influencing the growth and viability of Actinobacillus actinomycetemcomitans. Oral Microbiol Immunol. 1993;8:361–369. doi: 10.1111/j.1399-302x.1993.tb00612.x. [DOI] [PubMed] [Google Scholar]
- Syrjanen SM, Alakuijala L, Alakuijala P, Markkanen SO, Markkanen H. Free amino acid levels in oral fluids of normal subjects and patients with periodontal disease. Arch Oral Biol. 1990;35:189–193. doi: 10.1016/0003-9969(90)90054-e. [DOI] [PubMed] [Google Scholar]
- Tang G, Kawai T, Komatsuzawa H, Mintz KP. Lipopolysaccharides mediate leukotoxin secretion in Aggregatibacter actinomycetemcomitans. Mol Oral Microbiol. 2012;27:70–82. doi: 10.1111/j.2041-1014.2011.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Kitten T, Munro CL, Wellman GC, Mintz KP. EmaA, a potential virulence determinant of Aggregatibacter actinomycetemcomitans in infective endocarditis. Infect Immun. 2008;76:2316–2324. doi: 10.1128/IAI.00021-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Tokuda H, Matsuyama S. Sorting of lipoproteins to the outer membrane in E. coli. Biochim Biophys Acta. 2004;1693:5–13. doi: 10.1016/j.bbamcr.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Tomich M, Planet PJ, Figurski DH. The tad locus: postcards from the widespread colonization island. Nat Rev Microbiol. 2007;5:363–375. doi: 10.1038/nrmicro1636. [DOI] [PubMed] [Google Scholar]
- Tseng TT, Tyler BM, Setubal JC. Protein secretion systems in bacterial-host associations, and their description in the Gene Ontology. BMC Microbiol. 2009;9(Suppl 1):S2. doi: 10.1186/1471-2180-9-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu A, Chen C. Genetic basis for conversion of rough-to-smooth colony morphology in Actinobacillus actinomycetemcomitans. Infect Immun. 2005;73:3749–3753. doi: 10.1128/IAI.73.6.3749-3753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MP. Utilisation of proteomics datasets generated via multidimensional protein identification technology (MudPIT) Brief Funct Genomic Proteomic. 2004;3:280–286. doi: 10.1093/bfgp/3.3.280. [DOI] [PubMed] [Google Scholar]
- Wu S, Zhu Z, Fu L, Niu B, Li W. WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics. 2011;12:444. doi: 10.1186/1471-2164-12-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CS, Lin CJ, Hwang JK. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004;13:1402–1406. doi: 10.1110/ps.03479604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26:1608–1615. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambon JJ, Slots J, Genco RJ. Serology of oral Actinobacillus actinomycetemcomitans and serotype distribution in human periodontal disease. Infect Immun. 1983;41:19–27. doi: 10.1128/iai.41.1.19-27.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Rudd KE. EcoGene 3.0. Nucleic Acids Res. 2013;41:D613–624. doi: 10.1093/nar/gks1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zijnge V, Kieselbach T, Oscarsson J. Proteomics of protein secretion by Aggregatibacter actinomycetemcomitans. PLoS One. 2012;7:e41662. doi: 10.1371/journal.pone.0041662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.