

Abstract
Perfluorinated compounds, such as perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA), have been shown to alter various immune functions suggesting they are immunotoxic. This study assessed the effects of PFOS and PFOA on interleukin (IL)-2 production in the human Jurkat T-cell line and PFOS in healthy human primary T cells. Jurkat cells were stimulated with phytohemagglutinin (PHA)/phorbol myristate acetate (PMA), anti CD-3/anti CD-28, or anti CD-3, and dosed with 0, 0.05, 0.1, 0.5, 1, 5, 10, 50, 75, or 100 μg ml−1 PFOS or 0, 0.005, 0.01, 0.05, 0.1, 0.5, 1, 5, or 10 μg ml−1 PFOA. Jurkat cells stimulated with PHA/PMA or anti CD-3 exhibited decreased IL-2 production beginning at 50 μg PFOS ml−1 and 5 μg PFOS ml−1 respectively, but stimulation with anti-CD3/anti-CD28 resulted in no changes compared with the control. Addition of the PPAR-alpha antagonist GW6471 to PFOS-dosed cells stimulated with PHA/PMA resulted in decreases in IL-2 production starting at 50 μg PFOS ml−1, which suggests PFOS affected T-cell IL-2 production via PPAR-alpha-independent mechanisms. Exposure to PFOA, PFOA + GW6471, or PFOS + PFOA in Jurkat cells resulted in no significant differences in IL-2 production. In vitro dosing studies using healthy primary human CD4+ T cells were consistent with the Jurkat results. These data demonstrated that PFOA did not impact IL-2 production, but PFOS suppressed IL-2 production in both a human cell line and human primary cells at dose levels within the high end of the human exposure range. A decrease in IL-2 production is characteristic of autoimmune diseases such as systemic lupus erythematosus and should be further investigated.
Keywords: PFOS, PFOA, IL-2 production, immunology, ppar-alpha, perfluorinated compounds, cytokine, human T cells, in vitro, immunosuppression
Introduction
The potential human health effects of perfluorinated compounds such as perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) are becoming an increasing concern in the United States and worldwide. PFOS and PFOA are part of an emerging class of contaminants known as perfluoroalkyl acids (PFAAs). PFOS is a surfactant and historically has been used in a variety of products including surface treatments, paper production, and in performance chemicals such as firefighting foams, floor polishes, photographic film, denture cleaners, shampoos, carpet spot cleaners and as an insecticide in bait stations (OECD, 2002). PFOA is used in manufacturing of non-stick cookware and stain repellant carpet treatments. Monitoring data indicate that the general population may be exposed to PFOS and PFOA via ingestion of contaminated fish, drinking water, and dermal contact with products containing the chemicals (EPA, 2013).
PFAAs are carbon chains that have had the hydrogens replaced with fluorine and they contain various substitution groups (R-groups) on the terminal end that impart their active properties. This substitution with fluorine creates carbon–fluorine bonds that are extremely strong making these compounds very stable in the environment and in the body (Giesy et al., 2001). PFAAs are resistant to typical environmental degradation processes and are, therefore, widely distributed and found in water, soil, and air at sites around the United States. There was enough concern over these compounds that the USEPA nominated PFOS and related fluorochemicals to CDC for inclusion in the National Health and Nutrition Examination Survey (NHANES) in 2003 (EPA, 2003). PFAA plasma concentrations were increasing in adult and children human blood samples in the United States until 2004 (Olsen et al., 1999, 2001a, 2001b, 2003a, 2003b; Harada et al., 2004). Of all the perfluorinated compounds, PFOS is the one that is found at the highest concentration in blood serum (PFOA is found at the second highest concentration). In 2000, 3 M, the primary American producer of PFOS, announced the phase-out of PFOS and PFOS-related products in response to studies related to the toxicity of PFOS (3 M, 2008). PFOA is still being manufactured, but in accordance with the 2010/2015 PFOA Stewardship Program, the eight major fluoropolymer and telomer manufacturers committed to achieve a 95% reduction in PFOA by 2010 and to work towards the elimination of these PFOA and PFOA breakdown products by 2015 (EPA, 2014). PFOS is still detected in the serum of almost all people in the United States, but levels are starting to decrease since the phase-out (Renner, 2008). However, PFOS levels in blood serum in China continue to increase where PFOS and PFOS-related compounds are still being manufactured (Renner, 2008). In addition, a recent study in Dallas children showed perfluorinated compound levels increasing from birth to 12 years of age in spite of discontinued and reduced manufacturing of PFOS and PFOA, respectively (Schecter et al., 2012). These children are still being exposed to perfluorinated compounds years after changes in manufacturing. In addition to these effects, cause for concern about PFOS and PFOA is as a result of the long half-lives of these compounds. In humans, PFOS has an average half-life 5.4 years and PFOA has an average half-life of 2.3 years (Olsen et al., 2007; Bartell et al., 2010). Between 1999–2008, mean serum levels in humans ≥ 12 years of age in the United States are reported at 20.6 ng ml−1 for PFOS and 4.4 ng ml−1 for PFOA (Kato et al., 2011).
Although little data are available on the toxicity of many perfluorinated compounds, much is known about PFOS and PFOA. These compounds both cause peroxisomal proliferation and liver damage, alter estradiol and thyroid hormone pathways and have health effects related to genotoxcity, reproductive and developmental toxicity, and carcinogenity (Kennedy et al., 2004; Lau et al., 2007). In addition to all of these effects, both PFOS and PFOA have been shown to alter various immune functions in mice suggesting that they are immunotoxic. A 28-day oral exposure to PFOS in adult B6C3F1 female mice resulted in increased ex vivo basal production of interleukin (IL)-6 from B-cell and decreased basal production of IL-2 by T-cells (Fair et al. 2011; Peden-Adams et al., 2011; DeWitt et al., 2012,). PFOA (0.02%) was added to the feed of male C57B1/6 mice for 7–10 days and caused a reduced body weight, reduced numbers of thymus and spleen cells, reduced numbers of CD4+ and CD8+ cells in the thymus, an increase in liver weight, and peroxisome proliferation (Yang et al., 2002a, 2002b). PFOS and PFOA exposure have been shown to decrease T-cell-dependent IgM antibody responses in mice (Dewitt et al., 2008, Peden-Adams et al., 2008). PFOS also decreased T-independent IgM production and decreased host resistance to influenza A (Peden-Adams et al., 2008; Guruge et al., 2009). These immune effects have recently been seen in human studies as well. Grandjean et al. (2012) showed that in children between the ages of 5 and 7 years old, PFOS and PFOA concentrations in serum at commonly seen levels is associated with lower antibody responses to childhood immunizations such as diphtheria and tetanus. This is one of the first studies to demonstrate childhood deficits in immune system functions connected to exposure of PFOS and PFOA.
IL-2 is a very important cytokine as it is required for generation and maintenance of regulatory T cells (Tregs), which are needed to provide lifelong protection from autoimmune disease (Malek, 2003). A decrease in T cell IL-2 production was seen after PFOS exposure in two different studies. Dong et al. (2011) showed decreases in numbers of T-cells secreting IL-2 (ELISPOT) at 50 mg kg−1 total administered dose (TAD) over 60 days and Zheng et al. (2011) showed decreased numbers of T-cells producing IL-2 after a 7-day exposure to 20 mg kg−1 day−1 PFOS (both in male C57Bl/6 mice). These same studies also showed that after both short-term and subchronic PFOS exposure, the cytokine balance favored T-helper (Th)-2 responses (Dong et al., 2011, Zheng et al., 2011). PFOA has not be been previously investigated for modulation of IL-2 production.
The current study assessed the effects of PFOS and PFOA on IL-2 production in the human Jurkat T-cell line and primary human cells. Because a decrease in IL-2 production was seen in mice exposed to PFOS in vivo, it was hypothesized that similar effects would be seen in in vitro human studies. In addition, because immunotoxicity of PFAAs has been suggested to be related to proliferator-activated receptor (PPAR)-α activation (Yang et al., 2000, 2002a, 2002b), this study assessed PPARα as a possible mechanism for decreased IL-2 production.
Materials and Methods
Chemicals, Antibodies, and Supplies
Perfluorooctane sulfonic acid potassium salt (stated purity >98%) was obtained from Fluka Chemical (via Sigma, St. Louis, MO, USA; CAS No. 2795-39-3). PFOA was obtained from Sigma-Aldrich/Fluka (Steinheim, Switzerland). The PPARα antagonist, GW6471, was purchased from Tocris Bioscience (Bristol, United Kingdom). Human IL-2 enzyme-linked immunosorbent assay (ELISA) sets, assay diluent, coating buffer (pH 9.5), wash concentrate, stop solution, and substrate reagents A and B were obtained from BD Biosciences (San Jose, CA, USA). Anti-human CD3 and Anti-human CD28 were purchased from BD Pharmingen (San Diego, CA, USA). Phytohemagglutinin (PHA-P) and phorbol 12-myristate 13-acetate for molecular biology, ≥ 99% (TLC)-(PMA) were purchased from Sigma (St. Louis). Phosphate-buffered saline (without Ca2+ and Mg+) and RPMI-1640 medium (with l-glutamine and sodium bicarbonate) were purchased from Cellgro (Mediatech, Herndon, VA, USA). Non-essential amino acids (10 mM 100×), sodium pyruvate (100 mM), and antibiotic/antimycotic (100×) were obtained from Invitrogen (Gibco brand; Carlsbad, CA, USA). Fetal bovine serum was purchased from Gemini Bio-Products (West Sacramento, CA, USA). N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), flat bottom 96-well plates, and other disposables were obtained from Fisher Scientific (Atlanta, GA, USA). The naïve CD4+ T cell isolation kit (human) and LS columns used for magnetic isolation in the whole blood assay were purchased from Miltenyi Biotec (San Diego, CA, USA).
Cells
Jurkat cells were received from the American Type Culture Collection (ATCC, Manassas, Virginia). For all experiments using the Jurkat human T-cell line, the cells were maintained using standard tissue culture protocols. Cells were cultured in 75-cm2 tissue culture flasks in supplemented RPMI-1640 medium (RPMI, 10% FBS, 1% antibiotic/antimycotic) and incubated under a humidified atmosphere of 5% CO2/95% air at 37 °C. Growth medium was changed every 2 days.
Dosing-Jurkat Cell Line
Jurkat cells were plated in triplicate per dose on 96-well plates at 1 × 105 cells per well and stimulated with the combination of 1 μg ml−1 PHA and 1 μg ml−1 PMA, the combination of 1 μg ml−1 anti-CD3 and 1 μg ml−1 anti-CD28, or 1 μg ml−1 anti-CD3 after optimization experiments. Cells were dosed with 0, 0.05, 0.1, 0.5, 1, 5, 10, 50, 75, or 100 μg ml−1 PFOS only or 0, 0.005, 0.01, 0.05, 0.1, 0.5, 1, 5, or 10 μg ml−1 of PFOA only. These doses were chosen based on both exposure levels seen in humans and dose levels used in animal experiments (DeWitt et al., 2009). As PFOS and PFOA do not dissolve readily in medium, dimethyl sulfoxide (DMSO) was used as a vehicle during the experiments (0.05% DMSO) and this constituted the vehicle control for the experiments (0 μg ml−1 PFOS and 0 μg ml−1 PFOA). In addition to single-component dosing, cells were also dosed with a combination of PFOS plus PFOA (0, 0.005, 0.01, 0.05, 0.1, 0.5, 1, 5, or 10 μg ml−1 PFOA + PFOS) to determine possible interactions as these two PFAAs are typically found in the environment together. In order to explore the role of the PPARα mechanism, cells were also dosed with a PPARα antagonist, GW6471 (5 μmol). DMSO was used as a vehicle for GW6471 (0.05% DMSO). Cells were incubated for 18 h at 37 °C (Cattan et al., 2000). The supernatant from each triplicate was pooled by dose for each treatment, aliquoted, and stored at −80 °C until IL-2 analysis was performed using an IL-2 ELISA kit. Each experiment was repeated on three different days.
Human Blood Collection of CD4+ T Cells
Samples were collected from 11 healthy volunteer subjects under an approved Institutional Review Board (IRB) protocol. Criteria for exclusion were medication known to affect the immune system (i.e. steroids and non-steroidal anti-inflammatory drugs, or subjects suffering from an autoimmune disease). All subjects signed a consent form and were informed about methods and aims of the study. Six females and five males participated in the study. Blood samples (10 ml) were taken by venous puncture with heparin as the anticoagulant. Heparin was chosen over EDTA since EDTA can interfere with cell activation (Brunialti et al., 2002).
Dosing-Human CD4+ T Cells
Red blood cells were lysed, white blood cells were collected, and CD4+ T cells were isolated from the white blood cell layer using magnetic separation. Cells were resuspended in supplemented RPMI-1640 media (RPMI, 10% FBS, 1% antibiotic/antimycotic) at 1 × 105 cells per well, stimulated with 1 μg ml−1 PHA and 1 μg ml−1 PMA, dosed with five different doses of PFOS: 0, 0.1, 1, 10, or 100 μg ml−1, and plated in triplicate per dose per individual on a 96-well plate. DMSO was used as a vehicle during the experiments (0.05% DMSO). Cells were incubated for 18 h at 37 °C. The supernatant from each triplicate was then pooled for each treatment by each individual, aliquoted, and stored at −80 °C until IL-2 analysis was performed using an IL-2 ELISA kit.
Cell Viablity
Cells were dosed as described above, incubated, and viability was assessed with Trypan blue dye via a hemocytometer (Strober, 2001). Living cells excluded the dye while dead cells were stained blue owing to the damaged cell membrane. Five squares were counted on the hemocytometer and viability was expressed as the percentage of living cells per total number of cells counted.
Statistical Analysis
All experiments with Jurkat cells were repeated at least three times, with representative results shown. Data are expressed as mean ± standard error of the mean (SEM). Data were tested for normality (Shapiro–Wilk’s W-test) and homogeneity (Bartlett’s test for unequal variances). If need be, transformations were made. Statistical significance was determined using a one-way ANOVA (P ≤ 0.05). Dunnett’s comparison was used to compare treatment groups to controls. Statistical analysis was performed using JMP version 10 (SAS Institute Inc., Cary, NC, USA).
Results
Effects of PFOS and PFOA on the Human Jurkat T Cell Line
Jurkat cells stimulated with PHA + PMA exhibited decreased IL-2 production after exposure to 50, 75, and 100 μg PFOS ml−1 (38%, 50%, and 61% decrease compared with the control, respectively, Fig. 1). PFOA exposure in cells stimulated with PHA/PMA resulted in no significant differences as compared with the control (Fig. 2). PFOS + PFOA exposure in Jurkat cells also resulted in no significant differences in IL-2 production up to 10 μg PFOS + 10 μg PFOA ml−1 (data not shown). Addition of the PPARα antagonist GW6471 to PFOS dosed cells stimulated with PHA + PMA resulted in decreased in IL-2 production after exposure to 50, 75, and 100 μg PFOS ml−1 + 5 μmol GW6471 (66%, 87%, and 94% decrease compared with the control, respectively, Fig. 1). Addition of the PPARα antagonist to PFOA-dosed cells stimulated with PHA + PMA resulted in no significant differences up to 10 μg PFOA ml−1 + 5 μmol GW6471 (Fig. 2). Jurkat cells stimulated with anti-CD3 + anti-CD28 showed no significant changes in IL-2 production after exposure to PFOS, PFOA, PFOS + PFOA, PFOS + GW6471, and PFOA + GW6471 (data not shown). Jurkat cells stimulated with anti-CD3 only showed decreased IL-2 production at 5, 10, 50, and 100 μg PFOS ml−1 (64%, 80%, 63%, and 96% decrease as compared with the control, respectively, Fig. 3). Exposure to PFOA, PFOS + PFOA, and PFOA + GW6471 with stimulation by anti-CD3 only was not examined as no significant differences were seen in PHA + PMA or anti-CD3 + anti-CD28 stimulation in Jurkat cells. Jurkat cell viability was not significantly different from the control at any of the in vitro exposure concentrations for PFOS (highest dose group exhibited 97% viability) or PFOA (highest dose group exhibited 100% viability).
Figure 1.

In vitro effect of (A) perfluorooctane sulfonate (PFOS) and (B) PFOS plus a PPAR-α antagonist (GW6471) on interleukin (IL)-2 production in the Jurkat cell line stimulated with 1 μg ml−1 phytohemagglutinin (PHA) and 1 μg ml−1 phorbol myristate acetate (PMA). Each value represents the mean ± standard error of the mean (SEM). The sample size for all treatments is six. *Significantly different from the control (P < 0.05). This experiment was conducted three times. Data from a single experiment are shown, as results are representative of experiments.
Figure 2.

In vitro effect of (A) perfluorooctanoic acid (PFOA) and (B) PFOA plus a PPAR-α antagonist (GW6471) on interleukin (IL)-2 production in the Jurkat cell line stimulated with 1 μg ml−1 phytohemagglutinin (PHA) and 1 μg ml−1 phorbol myristate acetate (PMA). Each value represents the mean ± standard error of the mean (SEM). The sample size for all treatments is six. *Significantly different from the control (P <0.05). This experiment was conducted three times. Data from a single experiment are shown, as results are representative of experiments.
Figure 3.
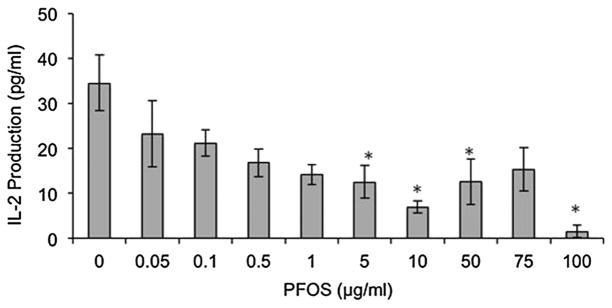
In vitro effect of perfluorooctane sulfonate (PFOS) on interleukin (IL)-2 production in the Jurkat cell line stimulated with 1 μg ml−1 anti-CD3. Each value represents the mean ± SEM. Sample size for all treatments is six. *Significantly different from control (P <0.05). This experiment was conducted three times. Data from a single experiment are shown, as results are representative of experiments.
Effects of PFOS on Primary Human CD4+ T Cells
Primary human CD4+ T cells were isolated and exposed to PFOS to determine if similar results were seen as with the immortalized Jurkat cell line. Only PFOS was examined, as no significant differences were noted with PFOA exposure at any dose level with Jurkat cells. IL-2 production was significantly decreased at 100 μg PFOS ml−1, the highest PFOS dose in the primary human CD4+ T cells (86% decrease as compared with the control, Fig. 4). Cell viability was not significantly different from the control at any of the in vitro exposure concentrations (highest dose group exhibited 94% viability).
Figure 4.
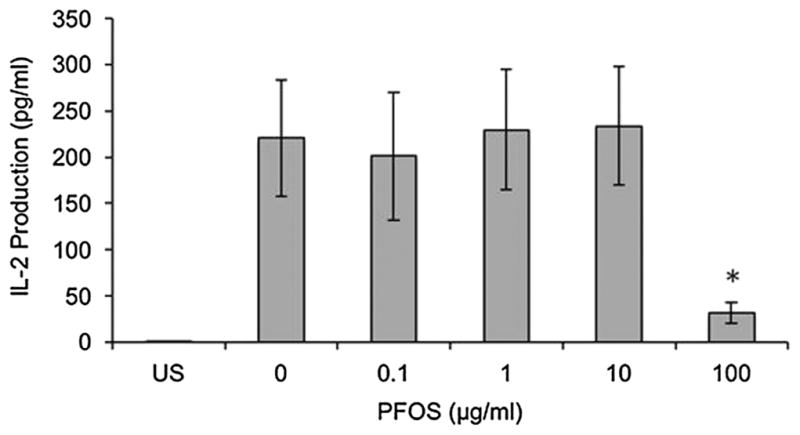
In vitro effect of perfluorooctane sulfonate (PFOS) on interleu-kin (IL)-2 production in healthy human primary CD4+ T cells stimulated with 1 μg ml−1 phytohemagglutinin (PHA) and 1 μg ml−1 phorbol myristate acetate (PMA). Each value represents the mean ± SEM. The sample size for all treatments is 11 (6 females and 5 males). *Significantly different from the control (P <0.05). US, unstimulated.
Discussion
Perfluorinated compounds have been shown to be immunotoxic (DeWitt et al., 2009, 2012). PFOS has been shown to effect cytokine secretion in multiple studies. Corsini et al. (2011) demonstrated suppressed lipolysaccharide (LPS)-induced tumor necrosis factors (TNF)-α and IL-6 secretion in human peripheral blood leukocytes starting at 0.1 μg ml−1 PFOS. LPS-induced release of TNF-α and IL-8 was also significantly reduced starting at 1 μg PFOS ml−1 in the human promyelocytic cell line THP-1. In this same study, PHA-stimulated peripheral blood leukocytes were examined and the addition of PFOS at 0.1, 1, and 10 μg PFOS ml−1 significantly decreased IL-4, IL-10, and IFN-γ production. Dong et al. (2011) showed decreases in numbers of T-cells secreting IL-2 (ELISPOT) at 50 mg kg−1 TAD over 60 days and Zheng et al. (2011) showed decreased numbers of T-cells producing IL-2 after a 7-day exposure to 20 mg kg−1 day−1 PFOS (both in male C57Bl/6 mice). Peden-Adams et al. (2011) showed a significant decrease in IL-2 production in female B6C3F1 mice stimulated with anti-CD3 at 0.1, 0.5, and 5 PFOS (mg kg−1 TAD). In the current study, a significant decrease in IL-2 production was observed at 50, 75, and 100 μg PFOS ml−1 in PHA + PMA-stimulated Jurkat cells and a significant decrease at 5, 10, 50, and 100 μg PFOS ml−1 in anti-CD3-stimulated Jurkat cells. No other PFOS studies assessed this and are available for comparison, however, our studies are consistent with the studies of Zheng et al. (2011), Dong et al. (2011) and Peden-Adams et al. (2011) which all demonstrated that IL-2 production was significantly decreased after PFOS exposure.
In addition to the Jurkat cell line, IL-2 production after PHA/PMA combined stimulation was examined in primary human CD4+ T cells and a significant reduction in IL-2 secretion was seen at the highest dose, 100 μg PFOS ml−1. Therefore, the lowest observed effect level (LOEL) in primary human cells was 100 μg PFOS ml−1 and the no observed effect level (NOEL) was 10 μg PFOS ml−1. This NOEL corresponds to the NOEL from the Jurkat cell line after PHA/PMA combined stimulation. The LOEL in the Jurkat cell line after PHA/PMA combined stimulation was 50 μg PFOS ml−1. This concentration was, however, not assessed in the primary human CD4+ cells. As a result of CD4+ cell isolation per individual in the primary CD4+ cell study the number of PFOS concentrations assessed was reduced to accommodate for less available cells for exposure. It is probable that the true LOEL in the primary cells may be between 10 and 100 μg PFOS ml−1. Future studies should determine this. The overall LOEL of the study was 5 μg PFOS ml−1 in Jurkat cells after being stimulated with Anti-CD3 with a NOEL of 1 μg PFOS ml−1.
Although averaged PFOS serum levels over a 9-year period (1999–2008) are reported at 20.6 ng ml−1 (0.021 ppm), this number may be deceiving (Kato et al., 2011). This study excluded children under 12 years even although a recent study showed perfluorinated compound levels increasing from birth to 12 years of age in spite of discontinued manufacturing of PFOS and decreased manufacturing of PFOA (Schecter et al., 2012). In addition, the Kato et al. (2011) study did not take into account humans exposed to higher concentrations of PFOS either occupationally or through other sources. The highest reported geometric mean for PFOS in the Kato et al. (2011) study was 30.4 ng ml−1 (0.03 ppm) between 1999–2000, but PFOS serum concentrations have been reported to range up to 12 ppm (Fromme et al., 2009). The current study demonstrated decreased IL-2 production starting at 5 μg PFOS ml−1 (5 ppm), which suggests a possible risk of decreased IL-2 production owing to PFOS exposure in human populations at the higher end of the exposure range (up to 12 ppm). A significant decrease in IL-2 production could be detrimental as IL-2 is required for generation and maintenance of Tregs, which are needed to provide life-long protection from autoimmune disease (Malek, 2003).
Moreover, PFOS binds strongly to bovine serum albumin and studies have shown that the concentrations of PFOS required to saturate albumin would be in excess of 50–100 mg/l (ppm) possibly because PFOS is not available to other sites of action until the pool of available binding sites on albumin are occupied (Jones et al., 2003). This is consistent with our study where most of the significant differences in IL-2 production were seen at PFOS concentrations above 50 ppm. One issue with in vitro studies in this case is that adverse effects of PFOS may not be seen until these binding sites are saturated. PFOS has been shown to have a high binding capacity for serum albumin in in vitro studies (Zhang et al., 2009) and a study by Wambaugh et al. (2013) indicates that in in vitro systems studying PFAAs binding to proteins and lipids in medium along with portioning to the well of the wall may result in differences in chemical concentration between the administered concentration and the concentration at the site of action the cell. In fact, Levitt and Liss (1986) concluded that in vitro toxic effects of PFOA and nonadecafluoro-n-decanoic acid (NDFDA) are reduced when serum is added to cell cultures. Therefore, if effects are seen in in vitro systems containing serum, it is likely they are truly caused by lower concentrations of PFAAs than the final well concentration indicates potentially making this study even more environmentally relevant.
While PFOS and PFOA are almost always found together in human serum samples, much less research is available on the effects of PFOA on cytokine production. Corsini et al. (2011) found that PFOA significantly reduced LPS-induced release of TNF-α in peripheral blood leukocytes at 1 and 10 μg PFOA ml−1 and at 10 and 100 μg PFOA ml−1 in THP-1 cells. In THP-1 cells, IL-8 was also significantly reduced at 100 μg PFOA ml−1. In the same study, PHA-induced IL-4 and IL-10 was significantly decreased at 10 μg PFOA ml−1 in peripheral blood leukocytes (Corsini et al., 2011). To our knowledge, no studies have assessed the effect of PFOA on IL-2 production. In the current study, PFOA did not significantly reduce IL-2 production up to 10 μg PFOA ml−1. Human exposure to PFOA is typically 10-fold lower than PFOS in the general public (Olsen et al., 2007). Corsini et al. (2011) exposed a variety of human cell types to PFOS and PFOA and found that in all cases PFOS was able to inhibit cytokine production more than PFOA. In addition, a 2012 study by Corsini et al. (2012) found that of the six different perfluorinated compounds they examined, PFOA was the least active in terms of effects on cytokines. At current human exposure levels of PFOA, the present data would suggest that alterations in cytokines, specifically in IL-2 production, might not be seen. However, this could be confounded with known PFOS levels or by probable differences in final well concentrations and concentrations in the cells (as noted above). Our current study indicates that there is not any chemical interaction evident between the two in relation to in vitro T-cell IL-2 production in the Jurkat cell line at the concentrations utilized.
One possible mechanism of the decreased IL-2 production observed may be through the activation of the nuclear receptor peroxisome proliferator-activated receptor (PPAR)-α. This ligand activated nuclear receptor is a regulator of immune function, particularly inflammation (Daynes and Jones, 2002). Previous studies have shown that PFOS and PFOA can activate PPAR-α in humans and mice (Sohlenius et al., 1992; Shipley et al., 2004; Vanden Heuvel et al., 2006). However, this mechanism may apply more to PFOA than to PFOS. PFOS has been shown to be less effective than PFOA at activating PPAR-α and neither PFOS nor PFOA were shown to have a significant activating effect on PPAR-γ (Takacs and Abbott, 2007). Peden-Adams et al. (2012) also showed that at environmentally relevant concentrations, PFOS does not upregulate expression of PPAR-α, γ, or δ genes. Corsini et al. (2011) showed that the effects of PFOA were dependent upon PPAR-α activation where effects of PFOS were independent of PPAR-α activation. Our results for PFOS were consistent with both of these studies where PFOS-induced suppression of IL-2 production was PPAR-α-independent and, therefore, a significant decrease in IL-2 production remained. Additional immune effects of PFOS may exist that are independent of PPAR-α. This is consistent with a previous study, which suggested that a PPAR-α-independent mechanism might contribute to the PFOS-induced suppression of IgM responses (DeWitt et al., 2012) and production of TNF-α and IL-8 (Corsini et al., 2011). Corsini et al. (2012) has recently shown that the inhibitory effect of PFAAs on in vitro cytokine production (IL-6, TNF-α, IL-10 and IFN-γ) by human leukocytes can occur independently of PPARα, and involves inhibition of NF-κB activation.
T cells may be activated by a number of different agents. This study examined IL-2 production through stimulation of Jurkat cells using soluble PHA + PMA, soluble anti-CD3 + anti-CD28, or soluble anti-CD3 only. Significant decreases in IL-2 production were seen at increasing PFOS concentrations after stimulation with either PHA + PMA or anti-CD3. These significant decreases were also seen with PHA + PMA stimulation even when the PPARα antagonist was added. Interestingly, no significant differences were seen after exposure to PFOS, PFOA, PFOS + PFOA, PFOS + GW6471, and PFOA + GW6471 using stimulation with anti-CD3 + anti-CD28. Activation of the T cell receptor (TCR)-CD3 complex results in signal 1 for T-cells which includes modulation of Lck, Fyn, Zap70, PLCγ, and activation of ERK, JNK, NF-κB, and NFAT pathways. PHA stimulation results in crosslinking of the TCR. PMA stimulates PKCΘ which targets NF-κB and AP-1 activation, but requires a combination of TCR and CD28 stimulation for effective activation of NF-κB and AP-1. PKCΘ also interacts with calcineurin leading to activation of JNK and NFAT. CD28 provides a co-stimulatory signal to TCR activation providing for augmentation of IL-2 production through additional NF-κB activation, but CD28 also activates Lck. Stimulation with anti-CD3 + anti-CD28 then activates both signal 1 (CD3 and CD28 with modulation of Lck) and 2 (CD28). Thus, each of the stimulants acts in a varied manner on signal 1 and signal 2 in the T-cell with PHA + PMA and anti-CD3 + anti-CD28 providing action in multiple locations along the pathways as compared with anti-CD3 alone. These results then suggest that adding direct co-stimulation of CD28 or PKCΘ attenuates the decrease in IL-2 production from TCR stimulation alone. Corsini et al. (2011) found that PFOS caused inhibition of LPS-induced I-κB degradation and decreased NF-κB binding to DNA, p65 phosphorylation, and p65/p50 nuclear translocation in the THP-1 cell line. However, the current data suggests activation of NF-κB in the Jurkat T-cell line by PHA + PMA or anti-CD3 + anti-CD28 stimulation attenuates the deficit in IL-2 production observed with anti-CD3 stimulation to varying degrees. Whether this is due to overcoming an NF-κB signaling pathway deficit with additional stimulation or is due to other alterations in T-cell signaling is not clear and requires further study. However, this trend has been noted in patients with head and neck squamous cell carcinoma. Patients who were not responsive to anti-CD3 therapy were then given anti-CD3 + anti-CD28 stimulation, which enhanced IL-2 production (Shibuya et al., 2000). Stimulation with anti-CD3 + anti-CD28 reversed immune unresponsiveness and induced a type 1 cytokine response. Therefore, anti-CD3 + anti-CD28 stimulation could be moderating the immunotoxicity of PFOS.
In conclusion, this is, to our knowledge, the first study to assess the effects of PFOS and PFOA on IL-2 production in both the Jurkat human cell line and primary human CD4+ T cells. PFOA did not appear to have any effect on IL-2 production, even at the highest concentration used in the Jurkat cells. Anti-CD3 was most effective at stimulating the Jurkat T cell line and provided the most sensitive data. A significant decrease in IL-2 production was seen with PFOS in both a human cell line and primary human CD4+ T cells. This suppression was seen at dose levels within the higher end of reported human exposure ranges (Fromme et al., 2009). A decrease in T-cell IL-2 production is characteristic of autoimmune diseases such as systemic lupus erythematosus (SLE). Because PFOS serum concentrations in humans have been reported to range up to 12 ppm (Fromme et al., 2009) and a recent study demonstrated childhood deficits in immune system functions connected to exposure of PFOS and PFOA (Grandjean et al. 2012), further studies utilizing cells from autoimmune patients who have varying blood levels of PFOS and PFOA are underway to investigate the role of PFOS and PFOA as environmental triggers of autoimmune disease.
Acknowledgments
Funding
Research reported in this publication was supported by the National Institute of Environmental Health Sciences and the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Numbers R21 ES017934 (D.L.K.) and P60 AR062755 (G.S.G. and D.L.K.) and supported by the South Carolina Clinical & Translational Research (SCTR) Institute at the Medical University of South Carolina, NIH/NCRR Grant number UL1 RR029881 and TL1 TR000061. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors would like to thank Dr Tammy Nowling from the Medical University of South Carolina (MUSC) for providing the Jurkat cell line used for this research and laboratory space. We would also like to thank the Center for Coastal Environmental Health and Biomolecular Research (CCEHBR) in Charleston, South Carolina for providing laboratory space for this research in collaboration with MUSC.
Footnotes
Conflict of Interest
The Authors did not report any conflict of interest.
References
- Bartell SM, Calafat AM, Lyu C, Kato K, Ryan PB, Steenland K. Rate of decline in serum PFOA concentrations after granular activated carbon filtration at two public water systems in Ohio and West Virginia. Environ Health Perspect. 2010;118:222–228. doi: 10.1289/ehp.0901252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunialti MK, Kallas EG, Freudenberg M, Galanos C, Salomao R. Influence of EDTA and heparin on lipopolysaccharide binding and cell activation, evaluated at single-cell level in whole blood. Cytometry. 2002;50:14–18. [PubMed] [Google Scholar]
- Cattan N, Mary D, Peleraux A, Mari B, Aussel C, Rossi B. Prostaglandin B(2) delivers a co-stimulatory signal leading to T cell activation. Eur Cytokine Netw. 2000;11:293–299. [PubMed] [Google Scholar]
- Corsini E, Avogadro A, Galbiati V, dell’ Agli M, Marinovich M, Galli CL, Germolec DR. In vitro evaluation of the immunotoxic potential of perfluorinated compounds (PFCs) Toxicol Appl Pharmacol. 2011;250:108–116. doi: 10.1016/j.taap.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Corsini E, Sangiovanni E, Avogadro A, Galbiati V, Viviani B, Marinovich M, Galli CL, Dell’Agli M, Germolec DR. In vitro characterization of the immunotoxic potential of several perfluorinated compounds (PFCs) Toxicol Appl Pharmacol. 2012;258:248–255. doi: 10.1016/j.taap.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748–759. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- Dewitt JC, Copeland CB, Strynar MJ, Luebke RW. Perfluorooctanoic acid-induced immunomodulation in adult C57BL/6 J or C57BL/6 N female mice. Environ Health Perspect. 2008;116:644–650. doi: 10.1289/ehp.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt JC, Peden-Adams MM, Keller JM, Germolec DR. Immunotoxicity of perfluorinated compounds: recent developments. Toxicol Pathol. 2012;40:300–311. doi: 10.1177/0192623311428473. [DOI] [PubMed] [Google Scholar]
- DeWitt JC, Shnyra A, Badr MZ, Loveless SE, Hoban D, Frame SR, Cunard R, Anderson SE, Meade BJ, Peden-Adams MM, Luebke RW, Luster MI. Immunotoxicity of perfluorooctanoic acid and perfluorooctane sulfonate and the role of peroxisome proliferator-activated receptor alpha. Crit Rev Toxicol. 2009;39:76–94. doi: 10.1080/10408440802209804. [DOI] [PubMed] [Google Scholar]
- Dong GH, Liu MM, Wang D, Zheng L, Liang ZF, Jin YH. Subchronic effect of perfluorooctanesulfonate (PFOS) on the balance of type 1 and type 2 cytokine in adult C57BL6 mice. Arch Toxicol. 2011;85:1235–1244. doi: 10.1007/s00204-011-0661-x. [DOI] [PubMed] [Google Scholar]
- EPA. Perfluorooctanoic Acid (PFOA), Fluorinated Telomers; Request for Comment, Solicitation of Interested Parties for Enforceable Consent Agreement Development, and Notice of Public Meeting.2003. [Google Scholar]
- EPA. Contaminants of Emerging Concern (CECs) in Fish: Perfluorinated Compounds. 2013 EPA-820-F-13-005. http://water.epa.gov/scitech/cec/upload/cec_pfc.pdf.
- EPA. [26 June 2014];200/2015 PFOA Stewardship Program. 2014 http://www.epa.gov/oppt/pfoa/pubs/stewardship/
- Fair PA, Driscoll E, Mollenhauer MA, Bradshaw SG, Yun SH, Kannan K, Bossart GD, Keil DE, Peden-Adams MM. Effects of environmentally-relevant levels of perfluorooctane sulfonate on clinical parameters and immunological functions in B6C3F1 mice. J Immunotoxicol. 2011;8:17–29. doi: 10.3109/1547691X.2010.527868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromme H, Tittlemier SA, Volkel W, Wilhelm M, Twardella D. Perfluorinated compounds--exposure assessment for the general population in Western countries. Int J Hyg Environ Health. 2009;212:239–270. doi: 10.1016/j.ijheh.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Giesy JP, Kannan K, Jones PD. Global biomonitoring of perfluorinated organics. Scientific World Journal. 2001;1:627–629. doi: 10.1100/tsw.2001.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean P, Andersen EW, Budtz-Jorgensen E, Nielsen F, Molbak K, Weihe P, Heilmann C. Serum vaccine antibody concentrations in children exposed to perfluorinated compounds. JAMA. 2012;307:391–397. doi: 10.1001/jama.2011.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruge KS, Hikono H, Shimada N, Murakami K, Hasegawa J, Yeung LW, Yamanaka N, Yamashita N. Effect of perfluorooctane sulfonate (PFOS) on influenza A virus-induced mortality in female B6C3F1 mice. J Toxicol Sci. 2009;34:687–691. doi: 10.2131/jts.34.687. [DOI] [PubMed] [Google Scholar]
- Harada K, Saito N, Inoue K, Yoshinaga T, Watanabe T, Sasaki S, Kamiyama S, Koizumi A. The influence of time, sex and geographic factors on levels of perfluorooctane sulfonate and perfluorooctanoate in human serum over the last 25 years. J Occup Health. 2004;46:141–147. doi: 10.1539/joh.46.141. [DOI] [PubMed] [Google Scholar]
- Jones PD, Hu W, De Coen W, Newsted JL, Giesy JP. Binding of perfluorinated fatty acids to serum proteins. Environ Toxicol Chem/SETAC. 2003;22:2639–2649. doi: 10.1897/02-553. [DOI] [PubMed] [Google Scholar]
- Kato K, Wong LY, Jia LT, Kuklenyik Z, Calafat AM. Trends in exposure to polyfluoroalkyl chemicals in the U.S. Population: 1999–2008. Environ Sci Technol. 2011;45:8037–8045. doi: 10.1021/es1043613. [DOI] [PubMed] [Google Scholar]
- Kennedy GL, Jr, Butenhoff JL, Olsen GW, O’Connor JC, Seacat AM, Perkins RG, Biegel LB, Murphy SR, Farrar DG. The toxicology of perfluorooctanoate. Crit Rev Toxicol. 2004;34:351–384. doi: 10.1080/10408440490464705. [DOI] [PubMed] [Google Scholar]
- Lau C, Anitole K, Hodes C, Lai D, Pfahles-Hutchens A, Seed J. Perfluoroalkyl acids: a review of monitoring and toxicological findings. Toxicol Sci. 2007;99:366–394. doi: 10.1093/toxsci/kfm128. [DOI] [PubMed] [Google Scholar]
- Levitt D, Liss A. Toxicity of perfluorinated fatty acids for human and murine B cell lines. Toxicol Appl Pharmacol. 1986;86:1–11. doi: 10.1016/0041-008x(86)90394-7. [DOI] [PubMed] [Google Scholar]
- Malek TR. The main function of IL-2 is to promote the development of T regulatory cells. J Leukoc Biol. 2003;74:961–965. doi: 10.1189/jlb.0603272. [DOI] [PubMed] [Google Scholar]
- OECD. (OECD) OfEC-oaD (ed) 2002. Hazard assessment of perfluorooctane (PFOS) and its salts. Joint meeting of the chemicals committee and the working party on chemicals, pesticides, and biotechnology. [Google Scholar]
- Olsen GW, Burris JM, Burlew MM, Mandel JH. Epidemiologic assessment of worker serum perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) concentrations and medical surveillance examinations. J Occup Environ Med/Am College Occup Environ Med. 2003a;45:260–270. doi: 10.1097/01.jom.0000052958.59271.10. [DOI] [PubMed] [Google Scholar]
- Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL, Zobel LR. Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ Health Perspect. 2007;115:1298–1305. doi: 10.1289/ehp.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen GW, Burris JM, Mandel JH, Zobel LR. Serum perfluorooctane sulfonate and hepatic and lipid clinical chemistry tests in fluorochemical production employees. J Occup Environ Med/Am College Occup Environ Med. 1999;41:799–806. doi: 10.1097/00043764-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Olsen GW, Hansen KJ, Burris JM, Lundberg JK, Mandel JH, Zobel LR. USEPA: USEPA Public Docket AR-226. 2001a. Interim Report: Identification of Fluorochemicals in human sera in American Red Cross Blood Donors. [Google Scholar]
- Olsen GW, Hansen KJ, Burris JM, Lundberg JK, Mandel JH, Zobel LR. USEPA: USEPA Public Docket AR-226. 2001b. Interim Report: Identification of Fluorochemicals in Sera of Children in the United States. [Google Scholar]
- Olsen GW, Hansen KJ, Stevenson LA, Burris JM, Mandel JH. Human donor liver and serum concentrations of perfluorooctanesulfonate and other perfluorochemicals. Environ Sci Technol. 2003b;37:888–891. doi: 10.1021/es020955c. [DOI] [PubMed] [Google Scholar]
- Peden-Adams MM, Alaya N, Young S, Hinckley K, Rueckert J, Taylor L, Chow M, David W, Henry N, Keil DE. Evaluation of possible modes of action for PFOS-induced humoral immunosuppression. The Toxicologist (Supplement to Toxicological Sciences) 2012;126 (S-1):307–311. [Google Scholar]
- Peden-Adams MM, Berger-Ritchie J, Young S, Ayala N, Keil DE. The Role of T-cell IL-2 and Macrophage IL-10 Production in PFOS-induced Humoral Immunosuppression. The Toxicologist (Supplement to Toxicological Sciences) 2011;125 (S-1):225–230. [Google Scholar]
- Peden-Adams MM, Keller JM, Eudaly JG, Berger J, Gilkeson GS, Keil DE. Suppression of humoral immunity in mice following exposure to perfluorooctane sulfonate. Toxicol Sci. 2008;104:144–154. doi: 10.1093/toxsci/kfn059. [DOI] [PubMed] [Google Scholar]
- Renner R. PFOS phaseout pays off. Environ Sci Technol. 2008;42:4618. doi: 10.1021/es0871614. [DOI] [PubMed] [Google Scholar]
- Schecter A, Malik-Bass N, Calafat AM, Kato K, Colacino JA, Gent TL, Hynan LS, Harris TR, Malla S, Birnbaum L. Polyfluoroalkyl compounds in Texas children from birth through 12 years of age. Environ Health Perspect. 2012;120:590–594. doi: 10.1289/ehp.1104325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya TY, Wei WZ, Zormeier M, Ensley J, Sakr W, Mathog RH, Meleca RJ, Yoo GH, June CH, Levine BL, Lum LG. Anti-CD3/anti-CD28 bead stimulation overcomes CD3 unresponsiveness in patients with head and neck squamous cell carcinoma. Arch otolaryngology--head neck surg. 2000;126:473–479. doi: 10.1001/archotol.126.4.473. [DOI] [PubMed] [Google Scholar]
- Shipley JM, Hurst CH, Tanaka SS, DeRoos FL, Butenhoff JL, Seacat AM, Waxman DJ. trans-activation of PPARalpha and induction of PPARalpha target genes by perfluorooctane-based chemicals. Toxicol Sci. 2004;80:151–160. doi: 10.1093/toxsci/kfh130. [DOI] [PubMed] [Google Scholar]
- Sohlenius AK, Andersson K, DePierre JW. The effects of perfluorooctanoic acid on hepatic peroxisome proliferation and related parameters show no sex-related differences in mice. Biochem J. 1992;285(Pt 3):779–783. doi: 10.1042/bj2850779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober W. Trypan blue exclusion test of cell viability. In: Coligan John E, et al., editors. Current protocols in immunology. 2001. Appendix 3: Appendix 3B. [DOI] [PubMed] [Google Scholar]
- Takacs ML, Abbott BD. Activation of mouse and human peroxisome proliferator-activated receptors (alpha, beta/delta, gamma) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicol Sci. 2007;95:108–117. doi: 10.1093/toxsci/kfl135. [DOI] [PubMed] [Google Scholar]
- Vanden Heuvel JP, Thompson JT, Frame SR, Gillies PJ. Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: a comparison of human, mouse, and rat peroxisome proliferator-activated receptor-alpha, –beta, and -gamma, liver X receptor-beta, and retinoid X receptor-alpha. Toxicol Sci. 2006;92:476–489. doi: 10.1093/toxsci/kfl014. [DOI] [PubMed] [Google Scholar]
- Wambaugh JF, Setzer RW, Pitruzzello AM, Liu J, Reif DM, Kleinstreuer NC, Wang NC, Sipes N, Martin M, Das K, DeWitt JC, Strynar M, Judson R, Houck KA, Lau C. Dosimetric anchoring of in vivo and in vitro studies for perfluorooctanoate and perfluorooctanesulfonate. Toxicol Sci. 2013;136:308–327. doi: 10.1093/toxsci/kft204. [DOI] [PubMed] [Google Scholar]
- Yang Q, Abedi-Valugerdi M, Xie Y, Zhao XY, Moller G, Nelson BD, DePierre JW. Potent suppression of the adaptive immune response in mice upon dietary exposure to the potent peroxisome proliferator, perfluorooctanoic acid. Int Immunopharmacol. 2002a;2:389–397. doi: 10.1016/s1567-5769(01)00164-3. [DOI] [PubMed] [Google Scholar]
- Yang Q, Xie Y, Alexson SE, Nelson BD, DePierre JW. Involvement of the peroxisome proliferator-activated receptor alpha in the immunomodulation caused by peroxisome proliferators in mice. Biochem Pharmacol. 2002b;63:1893–1900. doi: 10.1016/s0006-2952(02)00923-1. [DOI] [PubMed] [Google Scholar]
- Yang Q, Xie Y, Depierre JW. Effects of peroxisome proliferators on the thymus and spleen of mice. Clin Exp Immunol. 2000;122:219–226. doi: 10.1046/j.1365-2249.2000.01367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen L, Fei XC, Ma YS, Gao HW. Binding of PFOS to serum albumin and DNA: insight into the molecular toxicity of perfluorochemicals. BMC Mol Biol. 2009;10:16. doi: 10.1186/1471-2199-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Dong GH, Zhang YH, Liang ZF, Jin YH, He QC. Type 1 and Type 2 cytokines imbalance in adult male C57BL/6 mice following a 7-day oral exposure to perfluorooctanesulfonate (PFOS) J Immunotoxicol. 2011;8:30–38. doi: 10.3109/1547691X.2010.537287. [DOI] [PubMed] [Google Scholar]