

Abstract
Mipomersen (Kynamro®), a second-generation 2′-O-methoxyethyl chimeric antisense oligonucleotide (ASO), inhibits the synthesis of apolipoprotein B (apoB) and is indicated in the US as an adjunct therapy for homozygous familial hypercholesterolemia (HoFH) at a dose of 200 mg subcutaneously (SC) once weekly. The pharmacokinetic (PK) properties of mipomersen are generally consistent across all species studied, including mouse, rat, monkey, and humans. After SC administration, mipomersen is rapidly and extensively absorbed. It has an apparent plasma and tissue terminal elimination half-life of approximately 30 days. Mipomersen achieves steady-state tissue concentrations within approximately 4–6 months of once-weekly dosing. It does not exhibit PK-based drug–drug interactions with other concomitant medications, either involving competition for plasma protein binding or alterations in disposition of any evaluated drugs. Furthermore, mipomersen does not prolong the corrected QT (QTc) interval. There have been no ethnic- or gender-related differences in PK observed. In clinical trials, both as a single agent and in the presence of maximal lipid-lowering therapy, mipomersen has demonstrated significant dose-dependent reductions in all measured apoB-containing atherogenic lipoproteins. Overall, mipomersen has well-characterized PK and pharmacodynamic properties in both animals and humans, and is an efficacious adjunct treatment for patients with HoFH.
Key Points
| Mipomersen is an antisense oligonucleotide inhibitor of apolipoprotein B-100 synthesis, approved in the US for the treatment of patients with homozygous familial hypercholesterolemia. |
| Mipomersen has consistent and well-characterized pharmacokinetic and pharmacodynamic properties in both animals and humans. |
| Mipomersen reduces levels of all apolipoprotein-B containing atherogenic particles in a dose-dependent manner as a single agent and in the presence of maximal lipid-lowering therapy. |
Introduction
Antisense oligonucleotides (ASOs) are single-stranded synthetic oligonucleotides, typically comprised of 16–20 nucleotides, which bind to RNA through sequence-specific Watson–Crick base-pair interactions [1]. Once bound, ASOs can affect the metabolism of the target RNA by a number of mechanisms, including degradation via the recruitment of endogenous RNase H1 [2, 3]. Reduction in target messenger RNA (mRNA) levels by ASOs that support the RNase H1 mechanism has proven to correlate directly with reduction in target protein levels [4, 5].
There is significant growth and maturity in the number and type of RNA-targeted drugs being studied in clinical trials today. Most RNA targeted drugs in the clinic today are single-stranded ASOs that are chemically modified for enhanced pharmacokinetic (PK) and pharmacodynamic (PD) properties, and utilize a chimeric design to support RNase H1 activity [1]. These modifications include the phosphorothioate (PS) backbone, where one of the two non-bridging oxygens of the natural phosphodiester linkage is replaced with sulfur, and partial sugar modifications using 2′-O-methoxyethyl (MOE). The PS modification reduces the rate of degradation by nucleases, enhances distribution to tissues by binding to plasma proteins, and also supports RNase H1 activity when combined with a 2′-deoxy sugar residue. The 2′-MOE sugar modification imparts additional drug stability and also increases the affinity for the target RNA to achieve up to tenfold greater potency [6].
ASOs are distinguished from other classes of pharmaceutical agents by both the mechanism of action and physical–chemical or pharmaceutical properties. These distinguishing features provide an opportunity to pursue a large spectrum of first-in-class or best-in-class therapeutic agents for molecular targets that are validated but unreachable by other classes of agents, e.g. small molecules and antibodies, which predominantly target proteins. Apolipoprotein B (apoB)-100 is a validated molecular target, where both the gene expression profile and protein structure function are well-suited for application of the antisense technology. ApoB is an essential structural component of low-density lipoprotein (LDL) and its metabolic precursor, very-low-density lipoprotein (VLDL) [7]. LDL cholesterol (LDL-C) levels bear a direct relationship to major cardiovascular events, including death [8–10]. Therefore, reduction in LDL-C is the primary goal for most lipid-modifying interventions [11, 12]. Individuals with familial hypercholesterolemia (FH) frequently fail to meet LDL-C targets while taking the maximum tolerated lipid-lowering therapy, including high-dose statins, particularly those who are homozygous [13, 14]. Thus, additional effective treatments are needed [15, 16].
Mipomersen (Kynamro®; Genzyme, A Sanofi Corporation, Cambridge, MA, USA) is a synthetic second-generation ASO containing 20 nucleotides complementary to the coding region of the apoB mRNA, which is approved in the US as an adjunct to lipid-lowering medications and diet to reduce LDL-C, apoB, total cholesterol (TC), and non-high-density lipoprotein cholesterol (non-HDL-C) in patients with homozygous FH [17, 18]. ApoB and apoB-containing lipoproteins, such as LDL, and lipoprotein(a) [Lp(a)], are independent risk factors of cardiovascular disease [19, 20]. Patients with homozygous FH have a marked elevation in each of these risk factors compared with other high-risk populations [21]. Further to reductions in LDL-C levels, mipomersen produced significant reductions in all measured apoB-containing lipoproteins, including Lp(a), in the pivotal phase III trial [18].
Mipomersen utilizes a chimeric gapmer design whereby regions of 2'-O-MOE modified residues (5-mer ‘wings’) on either end of the molecule flank a central DNA region (10-mer ‘gap’). This design provides enhanced metabolic stability and potency, and favorable reductions in non-hybridization-based toxicities, while supporting RNase H-mediated reduction of the target mRNA. The preclinical and clinical PK properties of chimeric second-generation ASOs have been reviewed previously [22–25]. This article is an updated review of the PK–PD properties of mipomersen [26].
Pharmaceutical Properties of Mipomersen
Mipomersen sodium is a synthetic PS oligonucleotide sodium salt, 20 nucleotides in length, with the following sequence: 5′-GCCUC AGTCTGCTTC GCACC-3′, where the underlined residues are 2′-O-MOE nucleosides and all other residues are 2′-deoxynucleosides (Fig. 1) [17]. Cytosine (C) and uracil (U) bases are modified at the 5 position with a methyl group. The molecular formula of mipomersen sodium is C230H305N67O122P19S19Na19, and its molecular weight is 7,594.9 g/mol [17].
Fig. 1.
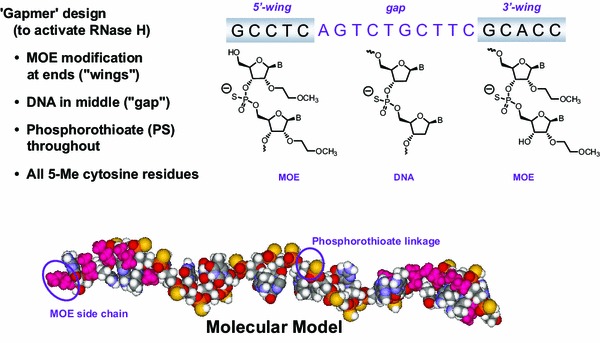
Structural and molecular formulae of mipomersen sodium. The binding site for mipomersen is located in the coding region of the apoB mRNA at position 3249–3268 (Acc. No. NM_000384.1). Reproduced from Crooke et al. [26], with permission. apoB apolipoprotein B, mRNA messenger RNA, MOE methoxyethyl
For clinical use, mipomersen is formulated in a pre-filled syringe containing 200 mg of mipomersen sodium in 1 mL of aqueous solution (pH 7.5–8.5) without any added preservative [17].
Mipomersen is well absorbed from subcutaneous (SC) injection sites and, once in the systemic or vascular circulation, is loosely bound to plasma proteins and rapidly taken up by tissues through interactions with cell-surface proteins and endocytosis without usage of specialized formulations for delivery [25]. Once taken up by tissues, mipomersen is slowly eliminated by nuclease metabolism and, ultimately, excretion of predominantly nuclease digested metabolites in urine.
Translational Pharmacokinetic (PK) Properties of Mipomersen
Preclinical PK
Mipomersen has demonstrated consistent absorption, distribution, metabolism and elimination (ADME) properties across all animal species studied to date, including mouse, rat, and monkey [5, 27], which ultimately translate well to man.
Absorption and Distribution
Following SC administration, mipomersen is rapidly absorbed from the injection site into the circulation, with peak plasma concentrations consistently reached within 3–4 h [27]. Nearly-complete absolute bioavailability was observed after SC administration in monkeys. Following either intravenous (IV) or SC administration, mipomersen plasma concentrations rapidly decline from peak concentrations in a multi-exponential fashion—characterized by a dominant initial rapid distribution phase (half-life of a few hours or less), followed by a much slower terminal elimination phase (half-life of several weeks). The apparent terminal elimination rate in monkey plasma was consistent with the slow elimination of mipomersen from monkey tissues, indicating equilibrium between post-distribution-phase plasma concentrations and tissue concentrations (Fig. 2) [5]. The partition ratios between liver and post-distribution plasma levels were similar across animal species (approximately 6,000:1), and therefore post-distribution plasma concentrations are also expected to provide a surrogate for tissue exposure in humans [5]. Plasma PK parameter estimates for mipomersen across species (animals and human) after single-dose IV or SC injection are provided in Table 1 [27].
Fig. 2.

Post-distribution-phase plasma and liver concentrations of mipomersen in monkeys. Each tissue data point represents average concentrations in two animals. Note that both plasma and tissue concentrations decay similarly over time following cessation of intravenous administration. Reproduced from Yu et al. [5], with permission
Table 1.
Plasma pharmacokinetic parameter estimates for mipomersen compared across species [27]
| Parameter | Mouse | Rata | Monkey | Human |
|---|---|---|---|---|
| 5 mg/kg SC | 5 mg/kg IV bolus | 4 mg/kg 1-h IV infusion | 200 mg 2-h IV infusion | |
| C max (µg/mL) | 3.8 (0.57) | 73.9 (2.4) | 39.8 (6.7) | 21.5 (4.2) |
| t max (h) | 0.5 | 2 min | 1 (0.1) | 1.98 (0.21) |
| AUC (µg·h/mL) | 7.41 | 27.7 | 82.0 (19.1) | 68.2 (13.5) |
| t ½α (h) | 0.33 | 0.39 | 0.68 (0.19) | 1.26 (0.16) |
| t ½β (day) | NM | 4.7b | 16c | 31.1 (11.4)d |
| CLp (mL/h/kg)e | 674 | 181 | 51.1 (11.1) | 40.9 (5.12) |
| V ss (L/kg)e | NM | 1.0 | 7.7c | 48.3 (14.7)d |
Standard deviation of the estimates is shown in parentheses
Reproduced from Yu et al. [27], with permission
AUC area under the plasma concentration–time curve, C max maximum plasma concentration, CL p plasma clearance, IV intravenous, SC subcutaneous, t max time to reach C max, t ½α distribution half-life, t ½β terminal half-life, V ss apparent volume of distribution at steady state
aMipomersen concentrations were measured using cold a ssay, hybridization ELISA method
bPlasma concentration–time profile seemed triphasic, with a half-life of 2.9 h in the second phase; therefore, this half-life represents the terminal half-life. Additionally, the terminal half-life may be underestimated because of limited time points
c N = 2
dDetermined following SC administration
eCLp/F and Vz/F reported for SC dosing
For all animal species evaluated, mipomersen distributes broadly into most tissues, with the exception of the brain, as is the case with all second-generation ASOs [22, 23]. The major tissues of distribution include liver, kidney, bone marrow, adipose tissue (cell body but not lipid fraction), and lymph nodes [5, 27].
Mipomersen is extensively bound to plasma proteins (≥85 %) across all species [27], with albumin being the protein that appears to bind the greatest amount of the drug in most species, including man. Protein binding to albumin is of relatively low affinity, and thus prevents loss of drug to renal filtration but does not prohibit uptake in tissues (K d approximately 150 µM) [22]. Plasma protein binding is not saturated at clinically relevant doses due to the large capacity of binding to proteins in plasma.
Metabolism
The metabolism of mipomersen to chain-shortened metabolites within tissues occurs initially by endonucleases that cleave the molecule in the gap, resulting in the production of short oligonucleotides that no longer retain pharmacological activity and have less binding affinity and capacity with proteins (Fig. 3). These smaller oligonucleotides can be further digested by exonucleases to produce a cascade of shorter oligonucleotides that is similar across species [27, 28]. Nucleases are ubiquitously expressed in most tissues, and therefore explain why potential hepatic insufficiencies do not incur a measurable effect on the overall clearance of mipomersen.
Fig. 3.

Mipomersen metabolism is similar across species. a Schematic of mipomersen metabolism by nucleases [26]. b Intact mipomersen and its metabolites identified in urine by liquid chromatography–electrospray mass spectrometry [27]. c Representative chromatograms of urine samples collected from monkeys and humans treated with mipomersen. IS internal standard, MOE methoxyethyl, i.v. intravenous [27]
Mipomersen is not metabolized by traditional drug-metabolizing enzymes, such as cytochrome P450 (CYP), and therefore does not interact with small molecules that are predominantly cleared through oxidative metabolic pathways [29].
Elimination
The clearance of mipomersen from tissues is slow in all species studied and involves both metabolism in tissues (via nucleases) and predominantly urinary excretion of both parent drug and its chain-shortened metabolites. The tissue elimination half-life for mipomersen in mice and rats was ≥13 days, and in monkeys ranged from 18 to 35 days, and were not affected by dose [27]. Urinary excretion of mipomersen and its chain-shortened metabolites is the major route of whole-body clearance of the drug. Urinary excretion of total oligonucleotide (mipomersen + chain-shortened metabolites) within just the first 24 h after a single dose, accounted for only a small percentage of the administered dose in a mouse, rat, and monkey (less than 10 %), consistent with extensive distribution of the bulk of mipomersen to tissues after dosing [27].
Clinical PK Properties
The clinical PK properties of mipomersen (30–400 mg SC or IV dosing) have been studied and reported from several phase I, II, and III studies [5, 18, 27, 30–38], and compare nearly identically with other similar 2′-O-MOE second-generation ASOs [22–25, 39, 40]. The PK characteristics of mipomersen in humans appear generally consistent with those observed in animals, again similar to what has been published for this chemical class of ASOs overall. The FDA-approved dosing regimen for mipomersen is 200 mg SC once weekly. This dosing regimen was extensively studied in four phase III pivotal studies and a respective phase III extension study [18, 36–38, 41].
The PK properties of mipomersen in humans were initially evaluated in a randomized, double-blind, placebo-controlled, dose-escalation phase I study involving volunteers with mild dyslipidemia [30]. Summaries of reported plasma PK parameters from 200 mg IV single dosing and 50–400 mg SC single and repeat (4 weeks of treatment) dosing are provided in Tables 1 and 2. The study consisted of a single and multiple SC dose of mipomersen 50, 100, 200 or 400 mg. The multiple-dose phase of this study was characterized by three alternate-day IV infusions of antisense designed to reach ~70 % of hepatic tissue steady-state levels in the first week, followed by three once-weekly SC doses. Dose-dependent maximum plasma concentrations (C max) ranged from 4.8 to 21.5 μg/mL (50–200 mg) at the end of the 2-h IV infusion. C max were also dose-dependent after SC administration, but approximately fivefold lower than the equivalent IV dose (range 1.0–2.7 µg/mL). The time of peak concentrations (t max) of mipomersen were typically observed 3–4 h after SC dosing. Importantly, IV and SC routes of administration showed similar plasma area under the concentration–time curve (AUC), with reported absolute bioavailability (relative to 2-h IV infusion) that ranged from 54 to 78 %. These plasma exposure estimates of absolute bioavailability somewhat underestimate total bioavailability. The terminal elimination plasma half-life ranged from 23 (±1) days in the 50 mg group to 31 (±11) days in the 200 mg group. While it appears that the terminal elimination half-life (t ½) may increase as dose increases, this is likely an anomaly owing to the detection limit of the bioanalytical assay and the concentrations in plasma dropping below detectable levels at earlier time points at the lowest dose tested.
Table 2.
Mipomersen pharmacokinetic summary from a single and multiple subcutaneous dose-escalation study in healthy human volunteers [26]
| Cohort | 50 mg (A) | 100 mg (B) | 200 mg (C) | 400 mg (D) | ||||
|---|---|---|---|---|---|---|---|---|
| Dose | First | Last | First | Last | First | Last | First | Last |
| n | 3 | 6 | 3 | 3 | 3 | 6 | 4 | 2 |
| AUC(0-48 h) (µg·h/mL) | 7.01 (0.49) | 7.68 (1.8) | 14.5 (2.91) | 19.6 (2.65) | 38.3 (29.8) | 35.4 (8.0) | 106 (17) | 113 |
| C max (µg/mL) | 1.40 (0.29) | 1.0 (0.31) | 1.45 (0.83) | 2.58 (1.41) | 2.73 (1.66) | 2.06 (0.95) | 7.60 (0.81) | 7.15 |
| t max (h) | 2.67 (0.58) | 3.75 (2.23) | 3.00 (1.00) | 3.33 (1.15) | 4.17 (3.40) | 4.64 (3.97) | 4.00 (1.63) | 7.50 |
| Apparent distribution t ½ (h) | 1.84 (0.49) | 3.44 (1.38) | 4.22 (2.71) | 2.96 (1.30) | 3.59 (1.22) | 7.60 (2.13) | 4.33 (0.49) | 5.48 |
| %BAV | 69.2 (9.63) | 77.0 (12.1) | 53.0 (17.2) | 74.5 (4.7) | ||||
Data are presented as mean (standard deviation), except for cohort D at last dose
AUC(0–48 h) area under the plasma concentration–time curve from time zero to 48 h, C max maximum plasma concentration, t max time to reach C max, t ½ terminal elimination half-life, %BAV plasma bioavailability (%) following subcutaneous administration
Adapted from Crooke and Geary [26]
In another short-term, repeat dosing (3 weeks of treatment), phase I, healthy male and female volunteer study, three different mipomersen dosing regimens were compared [31]. All three regimens were designed to deliver approximately 200 mg cumulative dose each week (30 mg daily vs. 70 mg three times weekly vs. 200 mg once weekly). With repeated administration, and little to no accumulation in peak (C max) and total (AUC) plasma exposure measures, no gender differences were observed, and time-invariant kinetics were observed for all treatment regimens. While C max and AUC of individual doses were dose-dependent, the relative bioavailabilities of the 30 mg once daily and 70 mg three times a week treatments were similar to that of the reference 200 mg every week treatment based on the post-distribution-phase plasma exposure measures. This latter observation reflects a comparable total exposure to mipomersen across treatments, and is consistent with the post-distribution plasma concentrations being in equilibrium with tissue concentrations. It also provides evidence of similar tissue concentrations achieved with each treatment after 3 weeks.
The PK properties of mipomersen were also investigated in healthy first-generation Japanese male subjects in a phase I, single SC dose, dose-escalation study, and compared with previously obtained results from healthy Western subjects [32]. Twenty healthy first-generation Japanese male subjects were enrolled into one of three treatment cohorts (mipomersen 50, 100 and 200 mg SC). The median t max varied between 2 and 3 h. Mean C max and AUC values increased in a dose-dependent manner. The mean PK exposure measures of mipomersen observed in Japanese subjects were similar to those previously observed in Western subjects, supporting the ‘no need for dose adjustment in the Japanese population’ in future clinical development.
Additional phase I studies involving drug–drug interaction evaluations with mipomersen are described in Sect. 4.
In multiple randomized, double-blind, placebo-controlled, dose-escalation phase II studies in subjects with varying degrees of a hyperlipidemia phenotype, the PK properties of mipomersen as a single agent (50–400 mg/week) or as an add-on to maximum lipid-lowering therapy were evaluated [33–35]. The PK characteristics in patients were shown to be similar to those observed in healthy volunteers, even when added to maximum lipid-lowering therapy [34]. In patients receiving weekly doses as monotherapy without a loading period (200, 300 and 400 mg/week dose groups), the plasma trough concentrations continued to increase throughout the treatment period consistent with the drug elimination half-life, dose interval, and time needed to reach steady-state, while C max and AUC were again unchanged over time, supporting time-invariant kinetics [33]. In patients receiving maximum lipid-lowering therapy, mipomersen plasma t ½ of approximately 30 days was reported across all dose groups, with no significant change seen with extension of dosing from 5 to 13 weeks [34].
Furthermore, PK measures were determined in FH patients. In a randomized, double-blind, placebo-controlled, dose-escalation phase II study in 44 patients with FH, mipomersen SC doses ranging from 50 to 300 mg/week (eight doses given over a 6-week treatment period, or 13 doses over 13 weeks at 300 mg) were evaluated and some PK measures determined [35]. Patients received stable conventional lipid-lowering therapy for at least 4 weeks and consumed a low-fat diet for at least 8 weeks before the first dose of the study drug and throughout the study. Once again, the mean plasma t ½ ranged from 25 to 34 days across the evaluated dose groups.
Finally, PK results have recently been reported from a phase III, open-label extension study with mipomersen [41]. In this open-label extension study, 141 patients with FH were treated with mipomersen 200 mg SC weekly for up to 2 years. Most patients were reported to achieve steady-state plasma trough levels during the study, with no apparent gender differences noted. In this study, the reported median t ½ was 35.1 days, which is generally consistent with that reported from phase I and II studies.
Drug–Drug Interaction Evaluations with Mipomersen
Mipomersen was evaluated in vitro for its effect on CYP enzyme activity using cryopreserved human hepatocytes [29]. Activity was inhibited by less than 10 % at the concentrations tested, including up to 800 μg/mL (~100 μmol/L), approximately ten times the expected liver tissue concentrations. Based on the results of this study, mipomersen is not considered an inhibitor of CYP1A2, CYP2C9, CYP2C19, CYP2D6 or CYP3A4. Given that mipomersen does not interact with CYP enzymes, no clinically relevant PK interactions were anticipated between mipomersen and medications such as simvastatin, ezetimibe or warfarin.
This lack of effect in vitro was demonstrated in two phase I clinical studies [29, 42]. As expected, no clinically significant PK interactions were observed in the phase I clinical study which evaluated the effects of co-administration of mipomersen (200 mg 2-h IV infusion) with simvastatin (40 mg oral) or ezetimibe (10 mg oral) [29]. In this study, ten healthy male subjects were enrolled per cohort and received a single oral dose of simvastatin or ezetimibe alone, followed by four 2-h IV doses of mipomersen over an 8-day period, with simvastatin or ezetimibe being administered again with the last dose of mipomersen. The AUC from time zero to 24 h (AUC24), C max and t ½ values for mipomersen were similar when administered alone and in combination with oral simvastatin or oral ezetimibe, providing evidence that neither simvastatin nor ezetimibe exhibits a clinically relevant PK interaction with the disposition of mipomersen. The AUC24 and t ½ values for simvastatin and its metabolite were similar when administered alone or in combination with mipomersen. The C max for simvastatin when coadministered with mipomersen was in close agreement with literature values. No statistically significant differences were observed in free or total ezetimibe in t max, AUC from time zero to 72 h (AUC72) and t ½ when ezetimibe was administered alone or in combination with mipomersen. The observed C max values for free ezetimibe when administered alone or in combination with mipomersen were also similar to previously reported literature values.
In an open-label, single-sequence, two-period, phase I study in 18 healthy subjects, coadministration of warfarin (25 mg orally) with mipomersen (200 mg SC) had no effect on the PK and PD properties of warfarin [42]. Eighteen subjects were enrolled and received a single oral 25 mg warfarin dose on day 1 and, after a 7-day washout period, received 200 mg mipomersen SC every other day on days 8–12. They then received both warfarin and mipomersen concurrently on day 14. There were no clinically significant changes in the PK of mipomersen with concurrent administration of warfarin. As expected, all R- and S- warfarin PK parameters evaluated (AUC from time zero to the last measurable concentration [AUClast], AUC from time zero to infinity [AUCinf], C max, t max and t ½) were similar when warfarin was given alone or with mipomersen. Furthermore, mipomersen did not cause any clinically meaningful differences in international normalized ratio (INR), prothrombin time (PT) or activated partial thromboplastin time (aPTT) effects of warfarin.
Lack of Corrected QT (QTc) Interval Effects and Other Cardiovascular Events with Mipomersen
Mipomersen does not prolong the corrected QT (QTc) interval. This was definitively demonstrated in a randomized, double-blind, 4-way, crossover phase I study conducted in 60 enrolled healthy volunteers [43]. The primary study objective was to assess effects of mipomersen on ECG following administration as single 200 mg therapeutic (SC) or supratherapeutic (2-h IV infusion) doses. Study treatments included mipomersen IV/placebo SC, mipomersen SC/placebo IV, moxifloxacin (positive control; 400 mg 2-h IV infusion)/placebo SC, and placebo IV/placebo SC). The primary endpoint was the time-matched placebo- and baseline-corrected (ΔΔ) QTcF interval. Assay sensitivity in the study was established from a positive moxifloxacin response. The effect on cardiac repolarization [maximum mean (90 % CI) ΔΔQTcF] was just 0.6 ms (−1.5 to 2.7) and 1.0 ms (−1.2 to 3.2) for the mipomersen 200 mg SC and IV doses, respectively. Mipomersen mean peak plasma exposure was 3.8-fold higher after IV versus SC treatment, consistent with the former being used as a supratherapeutic dose.
In a long-term, open-label study with mipomersen, there were also no significant changes noted in QTc measurements in patients being treated for up to 2 years [41].
Pharmacodynamic Properties of Mipomersen
Preclinical PD and PK/PD Relationship
In all animal species studied, mipomersen has demonstrated a dose-, concentration- and time-dependent reduction of apoB-100 mRNA in the liver, which correlates with the reduction of apoB-100 lipoprotein and apoB-100 containing lipid particles in the blood [44, 45]. In a human apoB transgenic mouse model, the relationship between human apoB hepatic mRNA expression and concentrations of mipomersen in the mouse liver and trough plasma was reported with estimated EC50 values of 119 ± 15 μg/g in liver and 18 ± 4 ng/mL in plasma [5]. Furthermore, the apoB mRNA reductions in human transgenic mice treated with mipomersen were specific for human apoB mRNA, as no change in murine apoB mRNA was observed in this model. Additionally, corresponding reductions in liver apoB protein, serum apoB protein, LDL-C, and TC were reported in the animals with reduced hepatic apoB mRNA. In these experimental evaluations, control ASOs had no effect on apoB mRNA in the liver, indicating that inhibition of apoB expression was both target- and sequence-dependent.
Clinical PD and PK/PD Relationship
In the first-in-man phase I study conducted in volunteers with mild dyslipidemia, the 200 mg dose produced an approximate 50 % reduction in circulating apoB following 4 weeks of treatment. The trough plasma mipomersen drug concentrations increased during weekly administration and slowly cleared from plasma with an approximate 31-day half-life following cessation of dosing. Consistent with the drug levels of mipomersen, apoB levels decreased during mipomersen treatment, reaching its nadir shortly after the last administered dose (Fig. 4) [5]. Upon cessation of dosing, apoB levels remained low for several weeks and slowly returned to baseline over several months, consistent with the slow clearance of the drug. These early data confirmed animal PK/PD relationships and the understanding that drug levels correlate directly with drug activity.
Fig. 4.
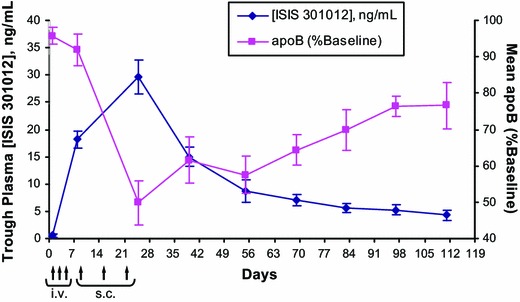
Exposure–response relationships between ISIS 301012 trough plasma concentrations and serum apoB levels (% baseline) in healthy human volunteers during and following six doses of mipomersen at 200 mg per injection. Trough plasma concentrations of ISIS 301012 were measured at ≥72 h from the last dose. Data are presented as mean ± standard error. Arrows represent dosing days. Reproduced from Yu et al. [5], with permission. apoB apolipoprotein B, i.v. intravenous infusion, s.c. subcutaneous injection
Mipomersen has a long t ½, with post-distribution-phase plasma concentrations best predicting tissue (e.g. liver) concentrations and therefore pharmacological activity. The post-distribution (trough) plasma concentration-response relationships for mipomersen in healthy volunteers, hypercholesterolemic patients, and hypercholesterolemic patients taking statins are shown in Fig. 5, and demonstrate a consistent EC50 of 10–17 ng/mL [26].
Fig. 5.
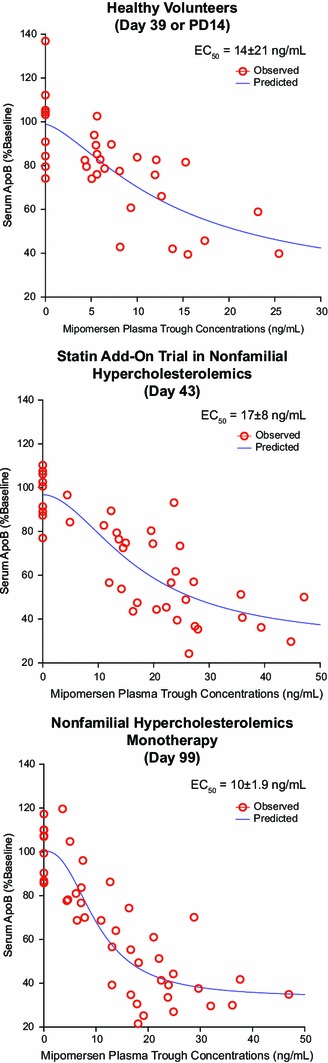
Plasma concentration-response relationships for mipomersen. The individual points represent measurements of apoB or mipomersen in serum of (a) healthy volunteers or (b) phase II studies in hypercholesterolemic patients in combination with statins or (c) as a single agent. Reproduced from Crooke et al. [26], with permission. apoB apolipoprotein B, PD14 14 days after the last dose
In the four previously described phase I and II studies, the PD properties of mipomersen (30–400 mg/week) were evaluated [30–35]. These studies included patients (N = 33) with mild dyslipidemia (who were not on lipid-lowering medication) [30], patients (N = 50) with mild-to-moderate hyperlipidemia (who were not on lipid-lowering medication) [33], patients (N = 74) with hypercholesteremia receiving stable statin therapy [34], and patients (N = 44) with FH (who were on stable lipid-lowering medication for at least 4 weeks and a low-fat diet for at least 8 weeks before the first study dose) [35]. Across all of these studies, mipomersen produced dose-dependent reductions in all apoB-containing lipoproteins. Data shown in Fig. 6 are representative of the dose-dependent and prolonged effects of mipomersen treatment on apoB and LDL-C levels over time [33].
Fig. 6.
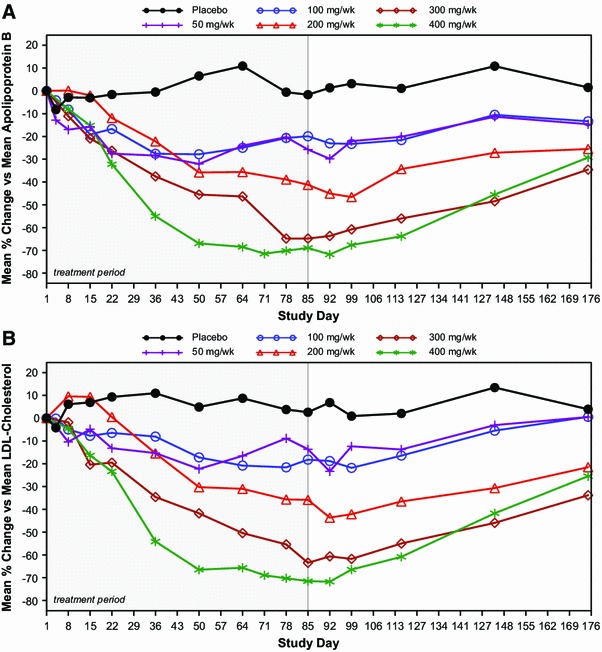
Dose-dependent effects of mipomersen as a single agent on (a) apoB and (b) LDL cholesterol in patients with mild to moderate hyperlipidemia. Patients (N = 50) were treated for 13 weeks with mipomersen at a dose of 50–400 mg, or placebo [34]. Data presented are the median percentage change from baseline from day 1 to day 175. apoB apolipoprotein B, LDL low-density lipoprotein
Mipomersen was also evaluated in 34 high-risk patients with hypercholesterolemia who were statin intolerant [46]. In this randomized, double-blind, placebo-controlled phase II study, patients received mipomersen at 200 mg/week SC for 26 weeks. As observed across all other study populations, treatment with mipomersen resulted in significant reductions in LDL-C (−47 %, p < 0.001 vs. placebo) and apoB (−46 %, p < 0.001 vs. placebo). Patients who were treated with mipomersen also had significantly lowered TC, triglycerides, and lipoprotein (a), with no effect on HDL-C and apo-A1.
Based on the extensive evaluations of dose and drug plasma concentration-response relationships in phase I and II studies, a weekly SC dose of mipomersen 200 mg was selected for phase III evaluations. Four randomized, placebo-controlled, phase III trials have been conducted in patients who were taking maximally tolerated doses of lipid-lowering drugs. In these trials, patients were dosed weekly with either placebo or mipomersen 200 mg SC for 26 weeks (Table 3) [18, 36–38]. Consistent reductions in apoB and apoB-containing lipoproteins were observed across these four phase III trials with this dose regimen.
Table 3.
Key results from randomized placebo-controlled phase III clinical trials
| % Change from Baseline | Drug | HoFH [18] Mipomersen (n = 34) Placebo (n = 17) [%] |
Severe HC [36] Mipomersen (n = 39) Placebo (n = 18) [%] |
HeFH with CAD [37] Mipomersen (n = 82) Placebo (n = 41) [%] |
HC at high risk [38] Mipomersen (n = 101) Placebo (n = 50) [%] |
|---|---|---|---|---|---|
| LDL-C* | Mipomersen | −25 | −36 | −28 | −37 |
| Placebo | −3 | +13 | +5 | −5 | |
| apoB* | Mipomersen | −27 | −36 | −26 | −38 |
| Placebo | −3 | +11 | +7 | −4 | |
| Lp(a)* | Mipomersen | −31 | −33 | −21 | −26 |
| Placebo | −8 | −2 | 0 | −0 | |
| TG** | Mipomersen | −17 | −9 | −14 | −25 |
| Placebo | +0.4 | +27 | +1 | +11 | |
| HDL-C*** | Mipomersen | +15.1 | +6 | +3 | +2 |
| Placebo | +3.9 | +3 | +6 | +2 |
Data shown is based on the analysis of the intent-to-treat population (n), defined as those who received at least one dose of study drug and had at least one post-baseline LDL-C measurement
apoB apolipoprotein B, CAD coronary artery disease, HC hypercholesterolemia, HDL-C high-density lipoprotein cholesterol, HeFH heterozygous familial hypercholesterolem ia, HoFH homozygous familial hypercholesterolemia, Lp(a) lipoprotein(a), LDL-C low-density lipoprotein cholesterol, TG triglyceride
p-Values (vs. placebo): * p < 0.001 all populations, ** p < 0.05 all populations, *** p < 0.05 in HoFH population
The four phase III studies enrolled patients with homozygous FH (N = 51) [18], severe hypercholesterolemia (N = 58) [36], heterozygous FH (N = 124) [37] and severe hypercholesterolemia in patients at high cardiovascular risk (N = 158) [38]. All patients in these four trials were on maximally tolerated lipid-lowering therapy and had an average of more than two prior cardiovascular events. The percentage change from baseline in LDL-C in patients treated with mipomersen 200 mg SC weekly ranged from −25 to −37 %, while those treated with placebo ranged from −5 to +13 % (Table 3). The effects on LDL-C, apoB and Lp(a) levels in homozygous FH patients after 26 weeks of treatment with mipomersen compared with placebo are shown in Fig. 7.
Fig. 7.
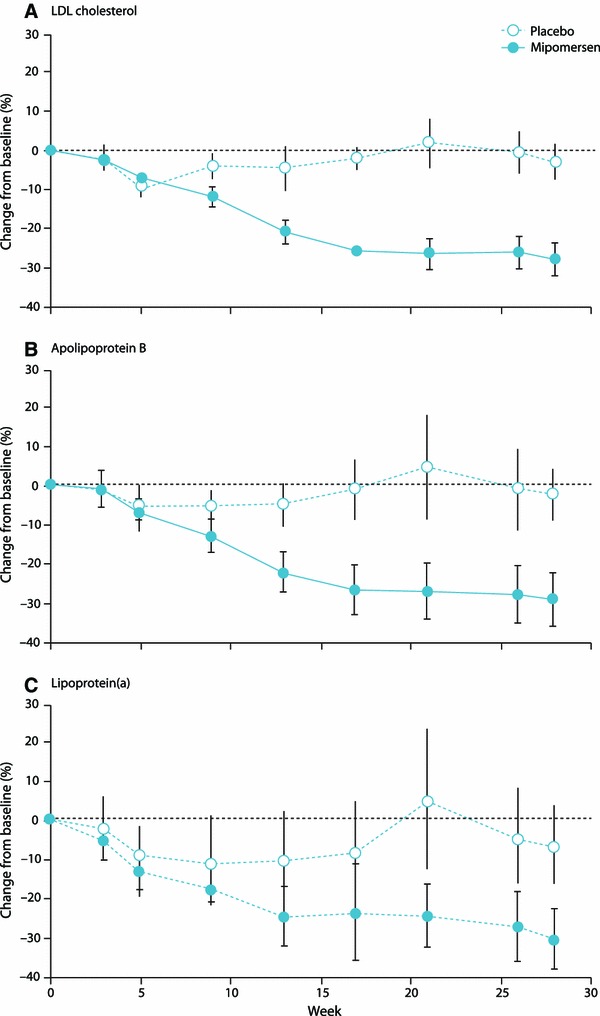
Mean percentage change from baseline (week 0) to primary efficacy timepoint for (a) LDL cholesterol, (b) apoB, and (c) Lipoprotein(a). Error bars indicate 95 % CI. Reproduced from Raal et al. [18], with permission. apoB apolipoprotein B, LDL low-density lipoprotein
Sustained reductions in LDL-C, apoB, TC, non-HDL-C and Lp(a) were observed in the corresponding phase III open-label extension study, which involved 141 patients with FH [41]. Figure 8 shows the results for apoB and LDL-C. Five of the patients in this study were treated with mipomersen 200 mg SC weekly for a mean duration of 48 months (range 41–52 months) [47]. LDL-C decreased from a mean baseline of 174 mg/dL to a mean of 120 mg/dL (−31 % vs. baseline, p = 0.003) at last assessment. Similar reductions also occurred in apoB.
Fig. 8.
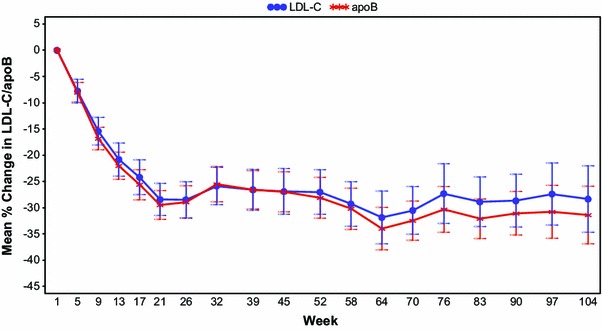
Sustained reductions in LDL-C and apoB during long-term mipomersen treatment in a phase III, open-label extension study. Results shown are from 2-year interim analysis [44]. apoB apolipoprotein B, LDL-C low-density lipoprotein cholesterol
Pediatric patients (age range from 12 to 18 years) with homozygous FH were also examined separately from the phase III study population [48]. In this subpopulation, improvements in LDL-C ranged from −30.8 to −62.0 % in the mipomersen-treated patients (n = 3), and from −7.9 to +43.1 % in patients receiving placebo (n = 4). After completion of the index study, all of the placebo-treated pediatric patients entered an open-label extension study and received mipomersen. The efficacy of mipomersen in these pediatric patients was consistent with that observed in adult patients.
Finally, mipomersen treatment can result in elevations in hepatic fat in man with or without increases in transaminases [41]. The effect on liver fat increases does not appear to be associated with drug concentration but rather a reciprocal association with the rate and extent of apoB and LDL-C lowering [30, 37]. This suggests that changes in lipid excretion from the liver may alter storage and possibly extend to increases in transaminases. Importantly, in long-term, open-label clinical trials, investigators have reported that the increases in liver fat observed in some patients decrease with continued dosing, suggesting that the liver may adapt to the change in lipid metabolism [41].
Summary
Mipomersen, as seen with many 2′-O-MOE modified second-generation ASOs, has highly predictable PK properties that are generally consistent across all species studied. Single- and multiple-dose PK of mipomersen in healthy volunteers and in patients with FH and non-FH has shown that mipomersen plasma exposure increases with increasing dose in the range of 30–400 mg, with similar exposures at equivalent administered SC doses independent of patient gender, ethnicity, lipidemic state, or concomitant lipid-lowering drug therapy. Given that mipomersen is not metabolized by traditional drug-metabolizing enzymes, such as CYP, it is not surprising that no clinically relevant PK interactions have been reported. Studies designed to evaluate PK interactions between mipomersen and warfarin, or between mipomersen and simvastatin or ezetimibe, have provided additional evidence. Furthermore, no PD interaction was detected in coadministration of mipomersen with warfarin, as determined by INR, aPTT and PT. Importantly, mipomersen does not prolong the QTc interval to any clinically relevant extent.
Conclusion
Mipomersen is dosed once weekly and follows first-order PK accumulation based on plasma trough concentrations associated with overall tissue accumulation and clearance. The half-life of mipomersen is approximately 31 days in monkeys and man. Consistent with the PK, a full PD response as measured by apoB and LDL-C reductions in homozygous FH patients is achieved between 4 and 6 months, at which time steady-state trough plasma concentrations are achieved. Furthermore, chronic studies in monkeys confirm that no further accumulation in liver occurs after achievement of steady-state in the 4- to 6-month timeframe [49]. Maximum reduction in apoB and LDL-C is sustained for up to 4 years with continued dosing. Upon cessation of dosing, the duration of continued reduction in LDL-C levels follows the slow clearance of the drug, returning slowly to baseline levels in approximately 4–6 months.
Mipomersen, a second-generation ASO, has been well-characterized in terms of PK and PD across preclinical and clinical studies. Inhibition of apoB synthesis by mipomersen represents a novel, effective therapy to reduce LDL-C concentrations in patients with hypercholesterolemia who cannot achieve appropriate lipid concentrations with current lipid-lowering agents.
Acknowledgments
The authors thank Starr Grundy of SD Scientific, Inc. for her assistance in drafting this review, and Tracy Reigle of Isis Pharmaceuticals, Inc., for graphics support.
All authors are employees of Isis Pharmaceuticals, Inc., and own stock in Isis Pharmaceuticals, Inc. Stanley T. Crooke serves as CEO and Chairman of the Board at Isis Pharmaceuticals, Inc.
References
- 1.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 2.Crooke ST, Vickers T, Lima W, et al. Mechanisms of antisense drug action, an introduction. In: Crooke ST, et al., editors. Antisense drug technology: principles, strategies, and applications. 2. Boca Raton: CRC Press; 2007. pp. 3–46. [Google Scholar]
- 3.Wu H, Lima WF, Zhang H, et al. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J Biol Chem. 2004;279:17181–17189. doi: 10.1074/jbc.M311683200. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Cook J, Nickel J, et al. Reduction of liver Fas expression by an antisense oligonucleotide protects mice from fulminant hepatitis. Nat Biotechnol. 2000;18:862–867. doi: 10.1038/78475. [DOI] [PubMed] [Google Scholar]
- 5.Yu RZ, Lemonidis KM, Graham MJ, et al. Cross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100. Biochem Pharmacol. 2009;77:910–919. doi: 10.1016/j.bcp.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Moser Altmann KH, Dean NM, Fabbro D, et al. Second generation of antisense oligonucleotides: from nuclease resistance to biological efficacy in animals. Chimia. 1996;50:168–176. [Google Scholar]
- 7.Davis RA. Cell and molecular biology of the assembly and secretion of apolipoprotein B-containing lipoproteins by the liver. Biochim Biophys Acta. 1999;1440:1–31. doi: 10.1016/S1388-1981(99)00083-9. [DOI] [PubMed] [Google Scholar]
- 8.Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60:2631–2639. doi: 10.1016/j.jacc.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 9.Boekholdt SM, Arsenault BJ, Mora S, et al. Association of LDL cholesterol, non-HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta-analysis. JAMA. 2012;307:1302–1309. doi: 10.1001/jama.2012.366. [DOI] [PubMed] [Google Scholar]
- 10.Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- 12.Goldberg AC, Hopkins PN, Toth PP, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:133–140. doi: 10.1016/j.jacl.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262–268. doi: 10.1016/j.atherosclerosis.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 14.Raal FJ, Pilcher GJ, Panz VR, et al. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation. 2011;124:2202–2207. doi: 10.1161/CIRCULATIONAHA.111.042523. [DOI] [PubMed] [Google Scholar]
- 15.Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129:1022–1032. doi: 10.1161/CIRCULATIONAHA.113.001292. [DOI] [PubMed] [Google Scholar]
- 16.Brown WV, Rader DJ, Goldberg AC. JCL roundtable: drug treatment of severe forms of familial hypercholesterolemia. J Clin Lipidol. 2014;8:10–17. doi: 10.1016/j.jacl.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Genyzme Corporation. KYNAMRO® (mipomersen sodium) injection prescribing information. 2013. Available from: http://www.kynamro.com/~/media/Kynamro/Files/KYNAMRO-PI.pdf. Accessed 6 Jun 2014.
- 18.Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006. doi: 10.1016/S0140-6736(10)60284-X. [DOI] [PubMed] [Google Scholar]
- 19.Sniderman AD, Williams K, Contois JH, et al. A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ Cardiovasc Qual Outcomes. 2011;4(3):337–345. doi: 10.1161/CIRCOUTCOMES.110.959247. [DOI] [PubMed] [Google Scholar]
- 20.Tsimikas S, Hall JL. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease. J Am Coll Cardiol. 2012;60:716–721. doi: 10.1016/j.jacc.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 21.Kraft HG, Lingenhel A, Raal FJ, Hohenegger M, Utermann G. Lipoprotein(a) in homozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2000;20:522–528. doi: 10.1161/01.ATV.20.2.522. [DOI] [PubMed] [Google Scholar]
- 22.Levin AA, Yu RZ, Geary RS. Basic principles of the pharmacokinetics of antisense oligonucleotide drugs. In: Crooke ST, editor. Antisense drug technology: principles, strategies, and applications. 2. Boca Raton: CRC Press; 2007. pp. 183–216. [Google Scholar]
- 23.Geary RS, Yu RZ, Siwkowski A, et al. Pharmacokinetic/pharmacodynamic properties of phosphorothioate 2′-O-(2-methoxyethyl)-modified antisense oligonucleotides in animal and man. In: Crooke ST, et al., editors. Antisense drug technology: principles, strategies, and applications. 2. Boca Raton: CRC Press; 2007. pp. 305–326. [Google Scholar]
- 24.Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2009;5:381–391. doi: 10.1517/17425250902877680. [DOI] [PubMed] [Google Scholar]
- 25.Yu RZ, Grundy JS, Geary RS. Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin Drug Metab Toxicol. 2013;9:169–182. doi: 10.1517/17425255.2013.737320. [DOI] [PubMed] [Google Scholar]
- 26.Crooke ST, Geary RS. Clinical pharmacological properties of mipomersen (Kynamro), a second generation antisense inhibitor of apolipoprotein B. Br J Clin Pharmacol. 2013;76:269–276. doi: 10.1111/j.1365-2125.2012.04469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu RZ, Kim TW, Hong A, et al. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab Dispos. 2007;35(3):460–468. doi: 10.1124/dmd.106.012401. [DOI] [PubMed] [Google Scholar]
- 28.Baek M, Yu RZ, Gaus H, Grundy JS, Geary RS. In vitro metabolic stabilities and metabolism of 2′-O-(methoxyethyl) partially modified phosphorothioate antisense oligonucleotides in reincubated rat or human whole liver homogenates. Oligonucleotides. 2010;20:309–316. doi: 10.1089/oli.2010.0252. [DOI] [PubMed] [Google Scholar]
- 29.Yu RZ, Geary RS, Flaim JD, et al. Lack of pharmacokinetic interaction of mipomersen sodium (ISIS 301012), a 2′-O-methoxyethyl modified antisense oligonucleotide targeting apolipoprotein B-100 messenger RNA, with simvastatin and ezetimibe. Clin Pharmacokinet. 2009;48:39–50. doi: 10.2165/0003088-200948010-00003. [DOI] [PubMed] [Google Scholar]
- 30.Kastelein JJ, Wedel MK, Baker BF, et al. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 2006;114:1729–1735. doi: 10.1161/CIRCULATIONAHA.105.606442. [DOI] [PubMed] [Google Scholar]
- 31.Flaim JD, Grundy JS, Baker BF, et al. Changes in mipomersen dosing regimen provide similar exposure with improved tolerability in randomized placebo-controlled study of healthy volunteers. J Am Heart Assoc. 2014;3:e000560. doi: 10.1161/JAHA.113.000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Hard ML, Andersen G, et al. Pharmacokinetics, safety and tolerability of mipomersen in healthy Japanese volunteers and comparison with Western subjects. Int J Clin Pharmacol Ther. 2014;52:314–320. doi: 10.5414/CP201975. [DOI] [PubMed] [Google Scholar]
- 33.Akdim F, Tribble DL, Flaim JD, et al. Efficacy of apolipoprotein B synthesis inhibition in subjects with mild-to-moderate hyperlipidaemia. Eur Heart J. 2011;32:2650–2659. doi: 10.1093/eurheartj/ehr148. [DOI] [PubMed] [Google Scholar]
- 34.Akdim F, Stroes ES, Sijbrands EJ, et al. Efficacy and safety of mipomersen, an antisense inhibitor of apolipoprotein B, in hypercholesterolemic subjects receiving stable statin therapy. J Am Coll Cardiol. 2010;55:1611–1618. doi: 10.1016/j.jacc.2009.11.069. [DOI] [PubMed] [Google Scholar]
- 35.Akdim F, Visser ME, Tribble DL, et al. Effect of mipomersen, an apolipoprotein B synthesis inhibitor, on low-density lipoprotein cholesterol in patients with familial hypercholesterolemia. Am J Cardiol. 2010;105:1413–1419. doi: 10.1016/j.amjcard.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 36.McGowan MP, Tardif JC, Ceska R, et al. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS One. 2012;7:e49006. doi: 10.1371/journal.pone.0049006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stein EA, Dufour R, Gagne C, et al. Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation. 2012;126:2283–2292. doi: 10.1161/CIRCULATIONAHA.112.104125. [DOI] [PubMed] [Google Scholar]
- 38.Thomas GS, Cromwell WC, Ali S, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2013;62:2178–2184. doi: 10.1016/j.jacc.2013.07.081. [DOI] [PubMed] [Google Scholar]
- 39.Graham MJ, Lee RG, Bell TA, 3rd, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112:1479–1490. doi: 10.1161/CIRCRESAHA.111.300367. [DOI] [PubMed] [Google Scholar]
- 40.Jones NR, Pegues MA, McCrory MA, Singleton W, Bethune C, Baker BF, Norris DA, Crooke RM, Graham MJ, Szalai AJ. A selective inhibitor of human C-reactive protein translation is efficacious in vitro and in C-reactive protein transgenic mice and humans. Mol Ther Nucleic Acids. 2012;1:e52. doi: 10.1038/mtna.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santos RD, Duell PB, East C, et al. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension. Eur Heart J. Epub 23 Dec 2013. [DOI] [PMC free article] [PubMed]
- 42.Li Z, Hard ML, Grundy JS, et al. Lack of clinical pharmacodynamic and pharmacokinetic drug-drug interactions between warfarin and the antisense oligonucleotide mipomersen. J Cardiovasc Pharmacol. 2014;64:164–171. doi: 10.1097/FJC.0000000000000101. [DOI] [PubMed] [Google Scholar]
- 43.Li Z, Yu R, Hard M, et al. The second generation antisense oligonucleotide (ASO) mipomersen does not prolong QT interval in a thorough QT/QTC study in healthy subjects. Clin Pharm Ther. 2014;95:S23. doi: 10.1038/clpt.2014.16. [DOI] [Google Scholar]
- 44.Crooke RM, Graham MJ, Lemonidis KM, et al. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J Lipid Res. 2005;46:872–884. doi: 10.1194/jlr.M400492-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Crooke RM, Baker BF, Wedel MK. Cardiovascular therapeutic applications. In: Crooke ST, editor. Antisense drug technology principles, strategies, and applications. 2. Boca Raton: CRC Press; 2007. pp. 601–639. [Google Scholar]
- 46.Visser ME, Wagener G, Baker BF, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, lowers low-density lipoprotein cholesterol in high-risk statin-intolerant patients: a randomized, double-blind, placebo-controlled trial. Eur Heart J. 2012;33:1142–1149. doi: 10.1093/eurheartj/ehs023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duell PB, Rose JE, Selvey S, et al. Long term efficacy and safety of mipomersen during 4 years of treatment in a cohort of patients with heterozygous familial hypercholesterolemia and coronary artery disease (CAD) J Clin Lipidology. 2013;7:276. doi: 10.1016/j.jacl.2013.03.080. [DOI] [Google Scholar]
- 48.Selvey S, Raal FJ. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces LDL-C in HoFH pediatric patients. J Clin Lipidol. 2014;8:330. doi: 10.1016/j.jacl.2014.02.056. [DOI] [Google Scholar]
- 49.Yu R, Geary R, Kim T, Levin AA. Mouse and monkey toxicokinetics of a second generation antisense oligonucleotide (ASO) targeting human apoB-100, following chronic treatment for up to 1 year [abstract no. T2492]. In: Annual Meeting of American Association of Pharmaceutical Scientists; 11–15 November 2007; San Diego (CA).