

Abstract
Identifying the genetic input for fetal growth will help to understand common, serious complications of pregnancy such as fetal growth restriction. Genomic imprinting is an epigenetic process that silences one parental allele, resulting in monoallelic expression. Imprinted genes are important in mammalian fetal growth and development. Evidence has emerged showing that genes that are paternally expressed promote fetal growth, whereas maternally expressed genes suppress growth. We have assessed whether the expression levels of key imprinted genes correlate with fetal growth parameters during pregnancy, either early in gestation, using chorionic villus samples (CVS), or in term placenta. We have found that the expression of paternally expressing insulin-like growth factor 2 (IGF2), its receptor IGF2R, and the IGF2/IGF1R ratio in CVS tissues significantly correlate with crown–rump length and birthweight, whereas term placenta expression shows no correlation. For the maternally expressing pleckstrin homology-like domain family A, member 2 (PHLDA2), there is no correlation early in pregnancy in CVS but a highly significant negative relationship in term placenta. Analysis of the control of imprinted expression of PHLDA2 gave rise to a maternally and compounded grand-maternally controlled genetic effect with a birthweight increase of 93/155 g, respectively, when one copy of the PHLDA2 promoter variant is inherited. Expression of the growth factor receptor-bound protein 10 (GRB10) in term placenta is significantly negatively correlated with head circumference. Analysis of the paternally expressing delta-like 1 homologue (DLK1) shows that the paternal transmission of type 1 diabetes protective G allele of rs941576 single nucleotide polymorphism (SNP) results in significantly reduced birth weight (−132 g). In conclusion, we have found that the expression of key imprinted genes show a strong correlation with fetal growth and that for both genetic and genomics data analyses, it is important not to overlook parent-of-origin effects.
Keywords: genomic imprinting, fetal growth restriction, placenta, chorionic villus sampling, birth weight, type 1 diabetes
1. Background and results
(a). Fetal growth
Birthweight and its relationship to mortality show one of the strongest links observed in epidemiology, illustrated by a reverse-J-shaped curve with the highest mortality observed in the lightest and heaviest groups [1]. Growing appropriately in utero is essential for a long and healthy life. Fetal growth restriction (FGR) affects approximately 6% of pregnancies, and is identified in approximately half of stillborn fetuses without malformations [2,3]. While the majority of FGR babies demonstrate catch-up growth, the combination of suboptimal intrauterine growth followed by accelerated childhood growth can increase their susceptibility to adult-onset diseases, including type 2 diabetes, hypertension and coronary artery disease [4]. Each baby's unique growth potential in utero is determined by the successful nutritional and respiratory support from the mother to the fetus via a placenta, and disturbing this balance could lead to FGR [5]. Fetal growth is influenced by both genetic and environmental factors, although the relevant molecular pathways are still poorly defined. Identifying key genes and pathways that regulate fetal growth will allow for better monitoring of intrauterine growth, maximizing healthy outcomes.
(b). Genomic imprinting
Genomic imprinting is a process of epigenetic modification on the genome that causes silencing of one allele according to its parental origin, resulting in monoallelic expression, without changing the DNA sequence [6–8]. Sex-specific imprint marks are heritable to daughter cells, but are erased and re-established in the germline during gametogenesis [9]. Evidence from mouse models and rare human imprinting disorders suggests that genes that are paternally expressed tend to increase fetal growth, whereas maternally expressed genes restrict fetal growth. For example, mice knockouts for paternally expressed genes Igf2, mesoderm-specific transcript (Mest) and paternally expressed gene 3 (Peg3) result in FGR, whereas mice deficient for maternally expressed genes insulin-like growth factor 2 receptor (Igf2r), H19 and Grb10 show an overgrowth phenotype [10–14] (table 1). Rare imprinting disorders such as the growth-restricted phenotype of Silver–Russell syndrome (SRS) may implicate complex roles involving both absence of growth promoters such as IGF2 and potential increase of growth restrictors such as GBR10 (reviewed in [26]).
Table 1.
Imprinted genes highly expressed in the placenta. Origin, parental origin of the expressed allele; M, maternally expressed; P, paternally expressed; ncRNA, non-coding RNA; FGR, fetal growth restriction; Dup, duplication; UPD, uniparental disomy; ICR, imprinting control region; LBW, low birthweight; BW, birthweight; HC, head circumference; CVS, chorionic villus sampling tissues; CRL, crown–rump length; PIP, phosphatidylinositol phosphate lipid; mat del, maternally inherited deletion; pat del, paternally inherited deletion; T1D, type 1 diabetes; TNDM, transient neonatal diabetes mellitus; BWS, Beckwith–Wiedemann syndrome; SRS, Silver–Russell syndrome; CNV, copy number variation; asterisk, findings from this study.
| locus | gene | origin | description | mouse KO phenotypes | human growth phenotypes |
|---|---|---|---|---|---|
| 6q24 | PLAGL1 | P | zinc finger protein | FGR, bone malformation, high neonatal lethality [15] | TNDM (pUPD6, pDup6q24, ICR hypomethylation) [16] |
| 6q25 | IGF2R | M/biallelic | clearance of IGF2 | fetal and placental overgrowth, organ and skeletal abnormalities [11] | CVS expression positively correlated to BW [17] and CRL* |
| 7p12 | GRB10 | M/P | GF receptor-bound protein | fetal and placental overgrowth [10] | implicated in SRS (mDup7p11.2–13) [18]; term placenta expression negatively associates with HC* |
| 7q21.3 | PEG10 | P | retrotransposon derived | embryonic lethal due to placental malformation [19] | hypermethylation at ICR and reduced expression in LBW cord blood [20]; upregulated in FGR placenta [21] |
| 7q32.2 | MEST | P/biallelic | α/β hydrolase fold family | fetal and placental growth restriction, high postnatal lethality, abnormal maternal behaviour [12] | implicated in SRS (mUPD 7q31-qter) [22] |
| 11p15 | H19 | M | long ncRNA | fetal and placental overgrowth [13,23] | ICR1 hypomethylation [24] and CNV [25] in SRS |
| IGF2 | P | growth factor | fetal and placental growth restriction | CVS expression positively correlated to BW [17] and CRL*; implicated in BWS and Wilm's tumour [26] | |
| CDKN1C | M | tumour suppressor | gestational fetal and placental overgrowth [27] | mutated in IMAGe [28], BWS [29] and SRS [30] patients | |
| SLC22A18 | M | organic cation transporter | not reported | term placenta expression associated with HC [31] | |
| PHLDA2 | M | PH domain, PIP binding | placental overgrowth [32] | highly expressed in lower BW and FGR placenta [21,33–35]; promoter variant associated with BW [36] | |
| 14q32 | DLK1 | P | transmembrane glycoprotein | pre- and postnatal growth restriction, high perinatal lethality, obese postnatally [37] | associated with T1D [38], UPD14 syndromes [26], T1D SNP correlated to BW* |
| MEG3 | M | ncRNA | postnatal lethal (mat del), pre- and postnatal growth restriction, high perinatal lethality (pat del) [39] | associated with T1D [38], reduced expression in FGR placenta [35] | |
| 19q13.4 | PEG3 | P | zinc finger protein | placental and fetal growth restriction, abnormal maternal behaviour [14] | tumour suppressor [40] |
The kinship theory or parental ‘conflict theory’ predicts that imprinting may have evolved as a result of competition between the paternal and maternal genome for maternal nutrient provision. The paternal genome encourages fetal growth by extracting nutrients from the mother, whereas the maternal genome counterbalances this by limiting resources to the offspring to ensure not only her survival, but also the equal provision of nutrients among her offspring [41]. Genomic imprinting is observed predominantly in placental mammals, and it is, indeed, the placenta which serves as the key regulatory site for this genomic conflict.
More than 100 imprinted genes have been identified in mice and approximately half of them are conserved in humans. In addition to this, many more tissue-specific human-imprinted loci are being discovered (http://igc.otago.ac.nz/; http://www.har.mrc.ac.uk/) [42]. In the current project, we have studied 13 imprinted genes that are highly expressed in human term placenta and are known to lead to growth phenotypes when deficient in mice (table 1). In addition, we included three non-imprinted genes that were critical to the action of IGF2, which is a key paternally expressed imprinted growth promoter (table 2). We have investigated the expression of these genes in both early and late gestation using the King's College London (KCL) CVS cohort (11–13 weeks of gestation) and the Moore term placenta cohort, respectively, and correlated these data with important growth parameters such as birthweight, placental weight and head circumference.
Table 2.
Non-imprinted genes highly expressed in the placenta.
| locus | gene | description | mouse KO phenotypes | human growth phenotypes |
|---|---|---|---|---|
| 7p12 | IGFBP3 | carrying protein for IGF1 and IGF2 | retinal vessel loss [43] | implicated in common cancers [44] |
| 12q23.2 | IGF1 | growth promoter | pre- and postnatal growth restriction, infertile [45] | pre- and postnatal growth restriction [46] |
| 15q26.3 | IGF1R | IGF1 and IGF2 receptor | fetal growth restriction and perinatal lethal | pre- and postnatal growth restriction [47] |
Also, in a separate analysis reported here, the potential influence of other variables such as the baby's sex, gestational age, parity, maternal weight/body mass index (BMI) and maternal smoking were tested against gene expression. In some situations, loss of imprinting (LOI) can occur, leading to biallelic expression of the gene. Because this could potentially influence the overall gene dosage, term placenta and CVS samples used in these expression studies were also investigated to see whether they retained a normal imprinting pattern, or showed monoallelic expression. In this hybrid review/research article, we summarize our previous findings together with new data.
(c). Insulin-like growth factor axis and IGF2/H19 locus
The insulin/IGF growth factor ‘axis' constitutes key regulatory endocrine factors of pre- and postnatal growth. These include insulin (INS), IGF1, IGF2 and their corresponding receptors (IR, IGF1R and IGF2R), and six binding proteins (IGFBP1–6) [48]. INS and IGF1 exclusively bind to IR and IGF1R, respectively, whereas IGF2 can bind to IGF1R, IGF2R and IR 11-isoform [49]. IGF2R is located on human chromosome 6q25.3 and shows maternal expression in only 10% of term placentas and CVS [17,50]. One of its major functions is the lysosomal targeting and degradation of IGF2, thus acting as a growth suppressor [51]. IGF2 and H19 map to one of the most intensely studied imprinted gene clusters on human chromosome 11p15. Their reciprocal imprinting is controlled by differential methylation of imprinting control region 1 (ICR1) which is normally only methylated on the paternal allele [52]. The unmethylated maternal ICR1 allows the binding of the CTCF transcription factor, blocking the access of IGF2 promoters to the H19 downstream enhancers, resulting in the activation of H19 expression. Conversely, the CTCF protein is prevented from binding to the paternal methylated ICR1, resulting in monoallelic paternal IGF2 expression owing to IGF2 promoter interaction with the enhancers. Approximately 50% of the growth-restricted SRS cases show loss of methylation at ICR1, which could lead to decreased IGF2 expression [24] and that may well contribute to SRS growth restriction.
In our previous studies, we have shown that IGF2 and IGF2R expression in term placenta has no correlation with baby's birth size parameters. However, their expression levels in CVS tissues showed a strong positive correlation with birthweight [17,33], indicating their role as ‘early growth effectors'. In addition to this study, the expression levels of H19 (n = 104) relative to the ribosomal protein L19 (L19) endogenous control gene in CVS tissues was measured by RT-quantitative polymerase chain reaction (qPCR). The relative expression levels of H19 were correlated to birth weight in a regression model adjusted for baby's sex, parity, gestational age at birth, maternal BMI and smoking habits. The CVS expression data for IGF2, IGF2R, H19, PHLDA2, IGF1 and IGF1R were also correlated to CRL at the gestational age of 12 weeks, using the same regression model, except this time the gestational age at CRL measurement was used instead of gestational age at birth. Correlation between H19 expression and birthweight was not statistically significant (p = 0.07). However, there was significant evidence for positive association between CRL at 12 weeks and IGF2 expression (p = 0.004; figure 1a), IGF2R expression (p = 0.03; figure 1b), IGF2/IGF1R ratio (p = 0.03; figure 1c) and H19 expression (p = 0.04; figure 1d and table 3). These results suggest that the many members of the IGF axis (IGF2, IGF2R and IGF1R), and the closely associated H19, shape the growth trajectory early in pregnancy.
Figure 1.

Correlation between imprinted gene expression in CVS and CRL. Expression levels of each gene relative to the L19 endogenous control gene were correlated to crown–rump length (CRL: mm) using a multiple linear regression model adjusted for maternal BMI, baby's sex, parity, gestational age when CRL was measured and maternal smoking habit. Positive correlations with CRL and (a) IGF2 expression (r = 0.77; p = 0.004), (b) IGF2R expression (r = 0.76; p = 0.03), (c) IGF2/IGF1R ratio (r = 0.74; p = 0.03) and (d) H19 expression (r = 0.74; p = 0.04) were observed. (Online version in colour.)
Table 3.
The association between mRNA levels and fetal growth in term placenta and CVS. Shading indicates previously published results [17,33]. The correlation significance is indicated by p-values. Correlation coefficient (r) is presented underneath the p-values for the associations reaching significance. n, number of samples; BW, birth weight; PW, placental weight; HC, head circumference; CRL, crown–rump length; NT, not tested.
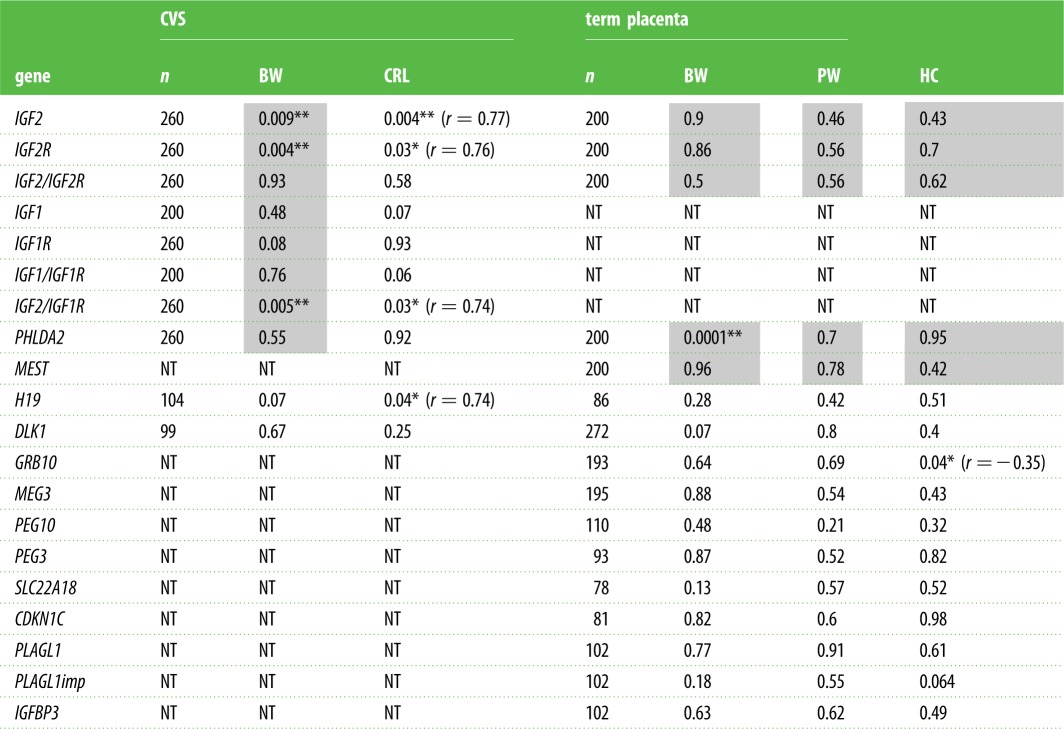 |
There was no correlation between maternal smoking and the expression in CVS of the genes tested (those listed above) in our samples. Nevertheless, we observed an association between IGF2 expression and parity, whereby IGF2 expression is higher in the ‘parity greater than one’ group of babies (p = 0.03; electronic supplementary material, figure S1a); this is consistent with the role of IGF2 as a positive growth regulator. This observation is interesting as the majority of second born babies are bigger [36]. We also found evidence that the maternal BMI was positively correlated with IGF2R expression (p = 0.03; electronic supplementary material, figure S1b) and negatively correlated with IGF1 (p = 0.046; electronic supplementary material, figure S1c). This suggests that it is important to allow for correction for maternal BMI/weight when investigating the gene expression in association with fetal growth. Interestingly, H19 was expressed significantly higher in males (p = 0.006; electronic supplementary material, figure S1e and table S2). The observed sex bias cannot be explained by LOI (i.e. biallelic expression of H19 in males only), because all of the CVS tissues tested retained monoallelic expression (table 4), therefore it is likely to result from upregulation of the active maternal copy. Males are normally born bigger than females [36], and the sexual dimorphism in antenatal biometry has been reported to be evident around 8–12 weeks of gestation [53]. As H19 is a negative growth regulator, the higher expression may help prevent male babies from growing too large.
Table 4.
Summary of imprinting analysis in CVS tissues and term placenta. M, maternal expression; P, paternal expression. %, percentage of samples with monoallelic expression within informative samples; n.a., not available.
| gene | parental origin | imprinting in term placenta | imprinting in CVS | polymorphic site |
|---|---|---|---|---|
| IGF2 | P | 67/67 (100%) | 40/40 (100%) | rs680 |
| IGF2R | M/biallelic | n.a. | 3/24 (12%) | rs1805075 |
| PHLDA2 | M | 11/11 (100%) | 21/21 (100%) | rs13390, rs1056819 |
| MEST | P/biallelic | 34/42 (81%) | n.a. | rs10863 |
| H19 | M | 19/19 (100%) | 33/33 (100%) | rs2067051 |
| DLK1 | P | 30/30 (100%) | n.a. | rs1802710 |
| MEG3 | M | 9/9 (100%) | n.a. | rs45617834, rs941575 |
| PEG3 | P | 14/16 (88%) | n.a. | rs1055359 |
| PEG10 | P | 42/42 (100%) | n.a. | rs13073, rs13226637 |
| GRB10 | M (placenta), P (brain) | n.a. | n.a. | n.a. |
| SLC22A18 | M | 23/23 (100%) | n.a. | rs1048046, rs1048047 |
| PLAGL1 | P | 11/11 (100%) | n.a. | rs2076684 |
| CDKN1C | M | 24/24 (100%) | n.a. | PAPn repeat |
(d). GBR10
GRB10 is located in the human chromosome 7q12 imprinted region. Chromosome 7 is implicated in causality for SRS, because 10% of patients show maternal uniparental disomy of chromosome 7. FGR is a key feature of SRS, which has been suggested to result either from the overexpression of a maternally expressed gene or loss of a paternally expressed growth-promoting gene. GRB10 encodes a growth factor receptor binding protein that can interact with receptor tyrosine kinases and intracellular proteins [54]. GRB10 is imprinted in an isoform- and a tissue-specific manner [55]. In humans, GRB10 shows biallelic expression in most tissues, while exhibiting isoform-specific paternal expression in the brain but with maternal expression confined to the placental villous trophoblast [56]. In mice, Grb10 is paternally expressed in the brain, but shows ubiquitous maternal expression in other tissues [55]. This pattern is roughly the opposite of what is seen for Igf2, where it is preferentially maternally expressed in the adult mouse brain but paternally expressed in other tissues [57]. Inactivation of the maternal copy of Grb10 results in fetal and placental overgrowth, indicative of its role as a potent growth suppressor [10]. In contrast, mice with a disrupted paternal copy showed normal growth but increased social dominance behaviour, illustrated by increased facial barbering (whisker removal) on cage-mates [58].
In this study, we have observed a significant negative association between GRB10 expression (all isoforms) and head circumference (figure 2, p = 0.04), but no significant correlation with birthweight (p = 0.64) or placental weight (p = 0.69; table 3). The direction of association is consistent with the role of GRB10 as a negative growth regulator. It is interesting that the observed association is specific to head circumference, because it is oppositely imprinted in the brain. There was no correlation between maternal smoking and expression of the genes tested in term placenta samples (electronic supplementary material, table S3). Interestingly, GRB10 expression showed a positive association with increasing gestational age (p = 0.03; electronic supplementary material, figure S2a). This suggests that GRB10 is acting to suppress the head circumference of the baby close to birth, because a head size too large for the birth canal would be detrimental for the mother.
Figure 2.
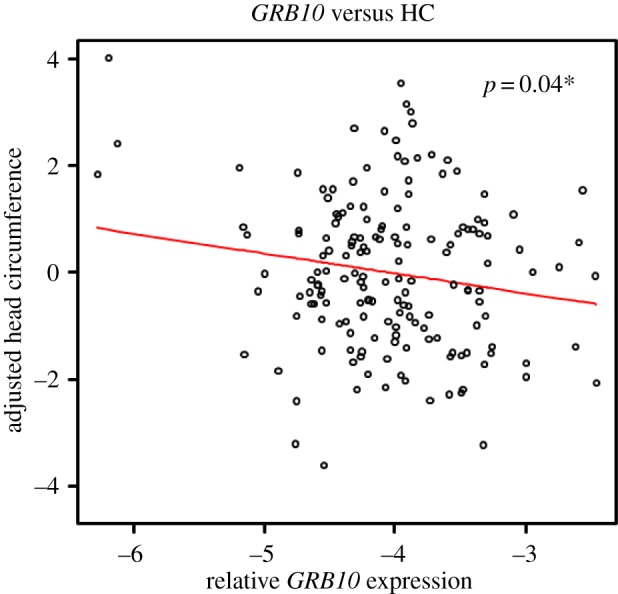
Negative correlation between GRB10 term placental expression and head circumference. The expression level of GRB10 relative to the L19 housekeeping gene was correlated to head circumference (cm) using a multiple linear regression model adjusted for baby's sex, parity, gestational age at birth, maternal weight and smoking habits. GRB10 expression values in logarithmic scale was used. Significant negative association was observed for GRB10 term placenta expression and head circumference (r =−0.35; p = 0.04). (Online version in colour.)
(e). PHLDA2
PHLDA2 is a maternally expressed gene located on the centromeric domain of the Chr11p15 imprinting cluster, along with other maternally expressed genes CDKN1C and SLC22A18. PHLDA2 encodes a small (144 amino acid) protein with a Pleckstrin-homology (PH) domain which has the capacity to bind membrane phosphatidylinositol phosphate lipids (PIPs) [59], suggesting a role for it as a cell signalling protein. In line with the kinship theory, Phlda2-deficient mice have an enlarged placenta, whereas overexpression of Phlda2 in transgenic mice results in placental stunting with a modest reduction in fetal weight [60,61]. We have previously shown that birth weight is not correlated with PHLDA2 expression levels in CVS tissues, but has a significant negative correlation in term placenta [17,33], indicative of a function as a ‘late growth effector’. Other studies have observed upregulation of PHLDA2 in FGR placentas [21,34,35], and in first and second trimester miscarriage placentas [62]; these data all support the hypothesis that PHLDA2 is an important negative regulator of growth.
More recently, upregulation of placental PHLDA2 expression among mothers who smoke during pregnancy has been reported [63]. In our study, however, we did not observe any correlation between maternal smoking and CVS or term placental expression of PHLDA2 (electronic supplementary material, table S3). PHLDA2 expression in CVS and term placenta did not show correlation with any of the confounding variables used in the model, except for gestational age. We identified that reduced PHLDA2 expression in CVS tissues was associated with advancing gestational age at birth (p = 0.0092; electronic supplementary material, figure S1e). Because a shorter gestation results in smaller babies, its high expression in CVS fits its role as a growth suppressor.
All the samples used in the analysis showed monoallelic expression of PHLDA2, demonstrating that LOI cannot account for the increased expression seen in the smaller birth weight babies [33]. To further investigate this correlation, we successively interrogated the nearby region for potential genetic variations that correlate with fetal growth. We identified a rare 15 bp repeat sequence variant (RS1) in the PHLDA2 promoter region, which has been shown to reduce the PHLDA2 promoter efficiency [36]. Maternal inheritance of RS1 resulted in a 93 g increase in birthweight, and when the mother is homozygous for RS1, the effect on birthweight is 155 g, suggesting a grand-maternal influence. Paternal inheritance of RS1 does not influence fetal growth as the variant lies on the epigenetically silenced paternal allele, emphasizing the importance of taking into account parent-of-origin effects when analysing genetic variants. Taken together, these data show that PHLDA2 is a strong negative growth suppressor and provide a potential pre-pregnancy test, using the RS1 variant, to predict birthweight.
(f). DLK1
DLK1 (PREF1 and FA1) is a paternally expressed gene located in the human chromosome 14q32 imprinting cluster, approximately 90 kb away from the maternally expressed non-coding RNA gene MEG3 (also called GTL2). DLK1 encodes a transmembrane glycoprotein with six epidermal growth factor-like repeat motifs [64], known to be involved in adipogenesis [65]. Dlk1-null mice show high perinatal lethality, pre- and postnatal growth restriction followed by an obese phenotype [37], suggesting that it acts as a growth promoter.
In this study, the expression levels of DLK1 (all isoforms) in CVS (n = 99) and term placenta (n = 272) were correlated to fetal growth parameters. For the CVS analysis, only the tissues from extreme birthweight babies (less than 10th centile and more than 90th centile) were used. Using the regression model as described for H19, we did not observe any association between DLK1 expression and birthweight (p = 0.23) or with CRL (p = 0.16). However, term placental DLK1 expression did show a weak positive association with birthweight (p = 0.07; table 3). Although this trend did not reach statistical significance, the direction of influence is consistent with its role as a growth promoter. Interestingly, DLK1 expression showed a positive correlation with increasing parity (p = 0.05; electronic supplementary material, figure S2b and table S3), possibly increasing the size of the later parity babies.
The rs941576 (G/A) SNP of the DLK1-MEG3 gene region on human chromosome 14 has previously been identified as a type 1 diabetes (T1D) susceptibility locus [38]. A reduced paternal, but not maternal, transmission of the protective G allele was observed in the T1D-affected individuals, showing a clear parent-of-origin effect. It was suggested that the rs941576 variant may affect nearby paternally expressed genes, including DLK1. Notably, higher birthweight has been linked to increased T1D risk [66–68]. This prompted us to test whether paternal transmission of the protective G allele is associated with (i) lower DLK1 expression and/or with (ii) reduced birthweight using the DNA samples from the Moore cohort. Because this is located within intron 6 of MEG3 and 105 kb downstream of DLK1, its potential influence on MEG3 expression was also tested.
In this study, 295 trio DNA samples from the Moore cohort were used for genotyping the rs941576 SNP. The resulting frequencies of the three genotypes were GG: 24%, AG: 45% and AA: 31%. 112 and 141 babies inherited paternal G and A bases, respectively, and 119 and 132 babies inherited maternal G and A, respectively. Using multiple linear regression analysis, we found that paternal or maternal transmission of the G allele is not correlated with DLK1 expression (p = 0.47 and p = 0.63, respectively) or with MEG3 expression (p = 0.7 and p = 0.085, respectively).
Next, the association between the inheritance of a paternal G allele with fetal growth was investigated, using a multiple linear regression model, adjusted for sex of the baby, parity, gestational age and maternal weight and smoking habit. Paternal transmission of the G allele was significantly associated with an average decrease of birthweight by 132 g (p = 0.01, 95% CI− 232 to −32; figure 3a), and a 0.5 cm reduction in head circumference of the baby (p = 0.01, 95% CI −0.85 to −0.11; figure 3b), but not with placental weight (−0.45 g; p = 0.98, 95% CI −35 to 35; figure 3c). Importantly, the scale of birthweight reduction (−132 g) associated with paternal G transmission is similar to that of the maternal smoking (−152 g). Maternal inheritance of the G or A allele was not associated with birthweight (p = 0.8), head circumference (p = 0.62) or placental weight (p = 0.86), consistent with the observed paternal effect of the protective G allele in T1D susceptibility.
Figure 3.

The association between paternal A/G SNP rs941576 at the DLK1 locus and fetal growth. Partial residual plots illustrating the correlation between paternal inheritance of the A or G allele and (a) birthweight (g), (b) head circumference (cm) and (c) placental weight (g), corrected for baby's sex, parity, gestational age, maternal weight and smoking habit in the multiple regression model. In comparison to the A allele, paternal G allele inheritance is associated with significantly reduced birthweight (p = 0.01, 95% CI−232 to −32) and head circumference (p = 0.01, 95% CI −0.85 to −0.11) but not with placental weight (p = 0.98, 95% CI −35 to 35). Paternal A/G, paternal transmission of A/G SNP rs941576; A, paternal transmission of the A allele; G, paternal transmission of the G allele; BW, birthweight; HC, head circumference; PW, placental weight. (Online version in colour.)
(g). Other imprinted genes studied
No evidence of correlation between H19, MEG3, PEG10, PEG3, SLC22A18, CDKN1C, PLAGL1_imp (imprinted transcript), PLAGL1_all (all transcripts) or IGFBP3 expression, in term placenta, with fetal growth was observed (summarized in table 3). In addition, we were unable to corroborate a previously reported association between SLC22A18 expression and head circumference [31]. We did not observe any LOI in our samples, except for PEG3, where 2/16 (12%) samples showed biallelic expression in term placenta. Table 4 details the polymorphic variants used for each gene and imprinting analysis results.
It was not possible to test all the candidate genes in both term placenta and for CVS tissues, owing to the limited availability of material for the latter, whereas some candidates also showed a level of expression undetected by quantitative PCR. Therefore, the candidate genes have been prioritized according to their functional relevance. Although it would have been interesting to test GRB10 expression in CVS, head circumference measurements were not available for the KCL CVS cohort.
2. Discussion
Suboptimal or excessive intrauterine growth leads to perinatal morbidity and mortality, as well as an increased risk for adulthood diseases [4]. Finding genetic factors that regulate normal fetal growth will potentially provide more precise monitoring of intrauterine growth. Genomic imprinting epigenetically silences one parental allele resulting in monoallelic expression. It is now accepted that paternally expressed genes tend to encourage fetal growth, whereas maternally expressed genes restrict this. In this paper, the role of imprinted genes on fetal growth was explored by summarizing and connecting our previous and current findings. Although DNA methylation plays a key regulatory role in imprinted gene expression, their methylation statuses were not assessed in our samples as CVS is a limited resource. RNA expression variation is downstream of DNA methylation or other possible DNA regulatory factors, and therefore potentially more functionally relevant. An additional DNA methylation status assessment would be an interesting aspect for the future study.
(a). The early and late effectors of fetal growth
Combining past and present studies, we have investigated the correlation between fetal growth measurements and expression levels of 13 imprinted and three non-imprinted genes highly expressed in CVS tissues and term placenta (tables 1 and 2). Our candidate gene approach has identified some early and late effectors of fetal growth. We have shown that the CVS expression of IGF2 and IGF2R is positively correlated to birthweight, whereas this correlation disappears in term placenta (table 3). Conversely, PHLDA2 expression in CVS is not correlated to birthweight, whereas PHLDA2 expression at term is strongly negatively correlated to birthweight. Although GRB10 expression in CVS was not tested, its expression in term placenta showed a strong negative association with head circumference. These observations suggest that IGF2 and IGF2R can act to set the growth potential of the baby early in the pregnancy, and two maternally expressed growth suppressing genes, PHLDA2 and GRB10, act to fine tune growth in late pregnancy, potentially to avoid the risk of giving birth to a macrosomic baby. Importantly, mouse studies indicate that both Phlda2 and Grb10 control placental growth by mechanisms independent of Igf2 [10,32], implying the evolution of separate pathways to control overall fetal size, possibly reflected by the difference in timing of their functional action.
The first half of placental development is characterized by a series of important trophoblast proliferation and differentiation processes, forming mature villous and extravillous structures. The second half of gestation results in an extensive vascularization and placental mass expansion [69]. Early gestational insults such as maternal diabetes have been associated with long-term effects on the fetus, owing to their influence on the initial structural formation of the placenta. It is possible that IGF2 and IGF2R are key regulators of early formation of the placenta, which then sets the growth capacity of the fetus and placenta for the rest of gestation. Interestingly, overexpression of mouse Phlda2 results in placental size reduction, with decreased glycogen storage and failed mobilization, accompanied by progressive fetal weight loss in late gestation [61]. It has been suggested that halving Phlda2 expression by silencing the paternal allele later in gestation may promote energy provision for the fetus at this time, by increasing the glycogen stores that will be used in late gestation when there is a particularly high nutrient demand from the fetus [61].
(b). Environment and other physiological effectors on gene expression
Although placenta is fetal in origin, it is under the influence of both maternal and fetal circulation. The placental villi consist of syncytiotrophoblasts facing the maternal blood, with cytotrophoblasts in the middle and endothelial cells facing the fetal circulation [69]. Therefore, the mRNA measured in the placenta could be a result of response to the hormones and growth factors present in both maternal and fetal circulation. In this study, potential influences of environmental variations (maternal weight/BMI and maternal smoking) and physiological variation (baby's gender, gestational age and parity) on gene expression were tested.
We did not observe a correlation between maternal smoking and gene expression levels with all genes tested in both CVS and term placenta (electronic supplementary material, table S2). This result contradicts the previous report where the upregulation of placental PHLDA2 in smokers (n = 12) compared with non-smokers (n = 64) was observed in a microarray experiment [63]. This could be due to different sensitivities between the two techniques. However, our cohorts contained more smokers (n = 27, Moore cohort and n = 33, CVS cohort; electronic supplementary material, table S1), which allows for more accurate measure of expression. IGF2R expression in CVS showed a positive association with maternal BMI (electronic supplementary material, figure S1b and table S2). This is interesting, because IGF2R has been found in the syncytiotrophoblast, which is in direct contact with the maternal blood circulation, and therefore possibly regulating the effect of fetal IGF2 levels on the mother [70].
Notably, we have found a sex-biased expression of H19 in CVS tissues, where it is expressed more highly in males (electronic supplementary material, figure S1d). H19 has previously been reported to show female-biased expression in mouse eyes [71]. Therefore, H19 expression could be dually regulated according to the sexes of the parent (imprinting) and also the baby (sexual dimorphism), in a tissue- and time-specific manner. Moreover, downregulation of PLAGL1 in FGR placenta of females, but not males, has been reported [72]. This was not evident in our normal term placenta samples, implying FGR-specific effects. Insight into the effect of sexual dimorphism is important for understanding both normal molecular mechanisms and sex-biased disease conditions.
(c). DLK1, type 1 diabetes and parent-of-origin effect on fetal growth
Type 1 diabetes (T1D) is caused by autoimmune destruction of pancreatic beta cells, resulting in insulin deficiency, although its aetiology is not fully understood [68]. The DLK1-MEG3 imprinting locus has recently been identified as a T1D susceptibility region, marked by the rs941576 SNP in which paternal inheritance of a G allele was associated with reduced risk [38]. DLK1 is highly expressed in pancreatic islet cells and is involved in differentiation of pancreatic beta cells, suggesting its strong functional candidacy [73].
In this study, we found that paternal transmission of the protective G allele results in a significant decrease of birthweight, by 132 g (figure 3a), and head circumference, by 0.5 cm (figure 3b). Of note, higher birthweight has been linked to increased T1D risk [66–68]. Therefore, paternal inheritance of the G allele may give protective effect from T1D via its association with reduced birthweight. This could also be associated with a decrease in DLK1 expression although this association did not reach statistical significance. Importantly, the magnitude of birthweight reduction (−132 g) and head circumference (−0.5 cm) related to the paternal G allele inheritance was similar to that observed for the increase in birthweight (+155 g) and in head circumference (+0.23 cm) caused by inheriting a PHLDA2 promoter RS1 allele from a RS1 homozygous mother [36]. Our current working hypothesis regarding the relationship between the role of DLK1 in fetal growth and T1D is described in figure 4 [37,64,65].
Figure 4.
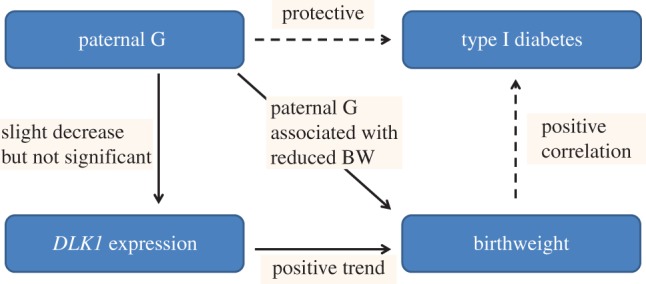
Current hypothesis on the association between paternal G SNP rs941576 and fetal growth. Solid lines indicate results from this study and dotted lines indicate published data [41–43]. Paternal inheritance of the G allele is associated with an average reduction in birthweight by 132 g (p = 0.01). The paternal G allele is also correlated with reduction in DLK1 expression although not significantly (p = 0.47). There was a trend of positive association between DLK1 expression in term placenta and birthweight (p = 0.07). Our hypothesis suggests that the paternal G allele reduces DLK1 expression which causes reduction in birthweight and risk of type 1 diabetes. (Online version in colour.)
3. Conclusion
We have identified that expression of IGF2 and IGF2R in early placenta (CVS) are positively correlated to CRL and birthweight, but not in term placenta when the oppositely maternally expressed genes PHLDA2 and GRB10 act to negatively regulate growth. We have also identified that the paternal transmission of the T1D protective G allele of rs941576 SNP results in a significant reduction in birthweight (p = 0.01, 95% CI−232 to −32), emphasizing the importance of accounting for parent-of-origin effects when analysing genomic data. Characterization of genes important in intrauterine growth will allow a more accurate surveillance of fetal growth and help identify targets for clinical intervention in suboptimal pregnancies. During pregnancy, a combination of different levels of imprinted genes or genetic predispositions will affect the baby's birthweight. An additional environmental layer is added by maternal smoking. Further investigation of all these candidates is warranted in larger cohorts to identify further genetic variants that exhibit parent-of-origin associated growth regulation and to find gene expression variations. Together with previously known genetic variants associated with fetal growth (reviewed in [26]), and expression studies, these may be used as an effective, combined diagnostic tool to identify and predict growth-restricted and macrosomic babies, which would provide huge benefits for the short- and long-term health of both mother and baby.
4. Materials
(a). King's College London chorionic villus sample cohort
CVS was carried out between 11 and 13 weeks of gestation in 355 singleton pregnancies that were followed by normal live birth at term. Participants were undergoing CVS for prenatal diagnosis for chromosomal abnormality at King's College Hospital London. The samples used in this study were obtained from excess CVS tissues from fully ethically consented women, and the research was approved by the King's College Hospital Ethics Committee. The medical records of this cohort are summarized in the electronic supplementary material, table S1 [17].
(b). Moore cohort
The Moore cohort consists of 302 consented white European trios recruited at Queen Charlotte's and Chelsea Hospital between 2003 and 2004 [33]. The placental samples were collected from ultrasound dated, live birth singleton pregnancies. Each placental sample was dissected into four pieces near the umbilical cord insertion point, washed in phosphate-buffered saline, snap-frozen in liquid nitrogen and stored at −80°C. Parental blood samples (10 ml) were collected in EDTA tubes. The medical records and characteristics of the Moore cohort are summarized in the electronic supplementary, table S1.
5. Methods
(a). DNA and RNA extraction
Total RNA from term placental tissue was extracted using Trizol reagent (Life Technologies), and treated with TURBO DNase (Ambion) according to the manufacturers guidelines. Fetal DNA from 1 g of term placental tissue and parental DNA from 2 ml of whole blood were isolated using a standard phenol–chloroform protocol. RNA and DNA from CVS tissues were extracted by the iPrep PureLink total RNA and TrizolPlus RNA kit, including the DNase treatment and iPrep ChargeSwitch gDNA tissue kit using the iPrep purification instrument (Life Technologies) following the manufacturer's instructions. The quantity and purity of nucleic acid was measured by NanoDrop ND-1000 spectrophotometer (Thermo Scientific). Only RNA samples with the 260/260 ratio in the range of 2 ± 0.2 were used for further study.
(b). Reverse transcription
A first strand of complementary DNA (cDNA) was synthesized from 1 μg (term placenta) and 100 ng (CVS) of RNA with Moloney murine leukemia virus reverse transcriptase (M-MLV RT) according to the manufacturer's instructions (Promega). Duplicate sets of samples without reverse transcriptase were made as negative controls to detect any genomic contamination in RNA samples. The conversion of RNA to cDNA was confirmed by polymerase chain reaction (PCR) with Taq DNA polymerase (Bioline) beta-actin (ACTB) primers (electronic supplementary material table S5).
(c). Quantitative polymerase chain reaction
qPCR was performed using the Power SYBRGreen PCR master mix (Life Technologies). Each sample was tested in triplicate, and each plate contained a no-template-control and a cDNA pool as a reference sample to control for interplate variations. The reaction plate was placed on the StepOne plus real-time PCR systems, analysed in the comparative Ct mode. Ribosomal protein L19 (L19) housekeeping gene was used as an endogenous control throughout the experiments. Thermal cycle conditions consist of initial incubation at 50°C for 2 min for one cycle, polymerase activation at 95°C for 10 min for one cycle and 40 cycles of denaturation at 95°C for 15 s, and annealing and extension at 60°C for 1 min. The efficiency of the primers was determined by running a standard curve and calculated by 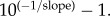 The qPCR primer sequences are provided in the electronic supplementary material, table S4. The resulting data were analysed with the StepOne v. 2.1 software to obtain relative quantification (RQ) values, using the formula RQ = 2−ΔΔCt.
The qPCR primer sequences are provided in the electronic supplementary material, table S4. The resulting data were analysed with the StepOne v. 2.1 software to obtain relative quantification (RQ) values, using the formula RQ = 2−ΔΔCt.
(d). Imprinting analysis
Monoallelic expression of genes was investigated by sequencing gene-specific amplicons from cDNA samples that corresponded to genomic DNA heterozygous for selected SNPs. Parental DNA was available for term placental samples, and was used for sequencing to check the parental origin of the expressed allele. SNPs with relatively high average heterozygosity were chosen for each gene within the exon covering all isoforms. PCR primer sequences are summarized in the electronic supplementary material, table S5, and the list of selected SNPs is found in table 4. Sequencing was carried out using the BigDye terminator v. 1.1 cycle sequencing kit (Life Technologies), and the read-out was analysed with Sequencher v. 4.8 (Gene Codes Corporation).
(e). Statistical analysis
All statistical analyses were performed using the R software (R Foundation for Statistical Computing). The relative expression of the candidate genes in term placenta was correlated to the baby's birth weight, placental weight and head circumference using a multiple linear regression model adjusted for baby's sex, gestational age, parity, maternal weight/BMI and smoking habits. These variables used in the model have previously been established as confounding factors in our previous studies in the same cohort [33,36]. A logarithmic scale was used for the expression values when appropriate, and BIC test was performed to check the fit of the models. A significance threshold of 5% was used in the analysis.
Supplementary Material
Acknowledgements
We thank Drs Helen Stevens and Chris Wallace for genotyping data and performing initial statistical analysis on the Moore cohort DNA. We are extremely grateful to all the families who took part in this study, the midwives for their help in recruiting the families. G.E.M. co-wrote the manuscript, contributed the concept and design of the experiments, and provided intellectual contributions. M.I. co-wrote the manuscript, performed RT-qPCR and sequencing experiments, collated the experimental data and performed statistical analysis. C.D., L.A., L.J.L, J.L.S., Y.C., R.B., M.B., I.I., O.O., R.A. and D.M. prepared RNA samples, performed RT-qPCR and sequencing experiments and analysed the data. A.J.D. prepared RNA samples and performed RT-qPCR experiments and analysed the data. S.A. and A.C.T. prepared RNA and DNA samples, performed RT-qPCR and sequencing experiments and analysed the data. S.A.A. prepared RNA and DNA samples and provided intellectual contributions. J.M.F. prepared RNA and DNA samples and designed RT-PCR primers. L.R. provided obstetric advice and discussion. A.S. helped collation of the CVS data. K.H.N. provided the CVS samples. J.C.W. performed statistical analysis. P.S. provided intellectual contributions and revised the manuscript.
Ethics statement
The study was approved by the Hammersmith and Queen Charlotte's and Chelsea Hospitals' Trust Research Ethics Committee (registration no. 2001/6029).
Funding statement
G.E.M. Fetal growth and development research team is funded by the MRC, Wellbeing of Women, March of Dimes, SPARKS, Wellcome Trust and the Great Ormond Street Hospital Children's Charity (GOSHCC). M.I. was funded for her PhD by the Child Health Research Appeal Trust (the Institute of Child Health and the Great Ormond Street Hospital for Children), Overseas Research Studentship, the Medical Research Council (MRC). C.D. is funded by Save the Baby Unit, P.S. is supported by GOSHCC.
Competing interests
We have no competing interests.
References
- 1.Basso O, Wilcox AJ, Weinberg CR. 2006. Birth weight and mortality: causality or confounding? Am. J. Epidemiol. 164, 303–311. ( 10.1093/aje/kwj237) [DOI] [PubMed] [Google Scholar]
- 2.Brodsky D, Christou H. 2004. Current concepts in intrauterine growth restriction. J. Intensive Care Med. 19, 307–319. ( 10.1177/0885066604269663) [DOI] [PubMed] [Google Scholar]
- 3.Shankar M, Navti O, Amu O, Konje JC. 2002. Assessment of stillbirth risk and associated risk factors in a tertiary hospital. J. Obstet. Gynaecol. 22, 34–38. ( 10.1080/01443610120101682) [DOI] [PubMed] [Google Scholar]
- 4.Simmons RA. 2009. Developmental origins of adult disease. Pediat. Clin. N. Am. 56, 449–466. ( 10.1016/j.pcl.2009.03.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abu-Amero S, Monk D, Apostolidou S, Stanier P, Moore G. 2006. Imprinted genes and their role in human fetal growth. Cytogenet. Genome Res. 113, 262–270. ( 10.1159/000090841) [DOI] [PubMed] [Google Scholar]
- 6.Barton SC, Surani MA, Norris ML. 1984. Role of paternal and maternal genomes in mouse development. Nature 311, 374–376. ( 10.1038/311374a0) [DOI] [PubMed] [Google Scholar]
- 7.McGrath J, Solter D. 1984. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 37, 179–183. ( 10.1016/0092-8674(84)90313-1) [DOI] [PubMed] [Google Scholar]
- 8.Surani MA, Barton SC, Norris ML. 1984. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308, 548–550. ( 10.1038/308548a0) [DOI] [PubMed] [Google Scholar]
- 9.Reik W, Dean W, Walter J. 2001. Epigenetic reprogramming in mammalian development. Science 293, 1089–1093. ( 10.1126/science.1063443) [DOI] [PubMed] [Google Scholar]
- 10.Charalambous M, Smith FM, Bennett WR, Crew TE, Mackenzie F, Ward A. 2003. Disruption of the imprinted Grb10 gene leads to disproportionate overgrowth by an Igf2-independent mechanism. Proc. Natl Acad. Sci. USA 100, 8292–8297. ( 10.1073/pnas.1532175100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lau MM, Stewart CE, Liu Z, Bhatt H, Rotwein P, Stewart CL. 1994. Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev. 8, 2953–2963. ( 10.1101/gad.8.24.2953) [DOI] [PubMed] [Google Scholar]
- 12.Lefebvre L, Viville S, Barton SC, Ishino F, Keverne EB, Surani MA. 1998. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat. Genet. 20, 163–169. ( 10.1038/2464) [DOI] [PubMed] [Google Scholar]
- 13.Leighton PA, Ingram RS, Eggenschwiler J, Efstratiadis A, Tilghman SM. 1995. Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature 375, 34–39. ( 10.1038/375034a0) [DOI] [PubMed] [Google Scholar]
- 14.Li L, Keverne EB, Aparicio SA, Ishino F, Barton SC, Surani MA. 1999. Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science 284, 330–333. ( 10.1126/science.284.5412.330) [DOI] [PubMed] [Google Scholar]
- 15.Varrault A, et al. 2006. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell. 11, 711–722. ( 10.1016/j.devcel.2006.09.003) [DOI] [PubMed] [Google Scholar]
- 16.Mackay DJ, Temple IK. 2010. Transient neonatal diabetes mellitus type 1. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 335–342. ( 10.1002/ajmg.c.30272) [DOI] [PubMed] [Google Scholar]
- 17.Demetriou C, et al. 2014. Paternally expressed, imprinted insulin-like growth factor-2 in chorionic villi correlates significantly with birth weight. PLoS ONE 9, e85454 ( 10.1371/journal.pone.0085454) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abu-Amero S, Monk D, Frost J, Preece M, Stanier P, Moore GE. 2008. The genetic aetiology of Silver–Russell syndrome. J. Med. Genet. 45, 193–199. ( 10.1136/jmg.2007.053017) [DOI] [PubMed] [Google Scholar]
- 19.Ono R, et al. 2006. Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat. Genet. 38, 101–106. ( 10.1038/ng1699) [DOI] [PubMed] [Google Scholar]
- 20.Lim AL, et al. 2012. Epigenetic state and expression of imprinted genes in umbilical cord correlates with growth parameters in human pregnancy. J. Med. Genet. 49, 689–697. ( 10.1136/jmedgenet-2012-100858) [DOI] [PubMed] [Google Scholar]
- 21.Diplas AI, Lambertini L, Lee MJ, Sperling R, Lee YL, Wetmur J, Chen J. 2009. Differential expression of imprinted genes in normal and IUGR human placentas. Epigenetics 4, 235–240. ( 10.4161/epi.9019) [DOI] [PubMed] [Google Scholar]
- 22.Hannula K, Lipsanen-Nyman M, Kontiokari T, Kere J. 2001. A narrow segment of maternal uniparental disomy of chromosome 7q31-qter in Silver–Russell syndrome delimits a candidate gene region. Am. J. Hum. Genet. 68, 247–253. ( 10.1086/316937) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esquiliano DR, Guo W, Liang L, Dikkes P, Lopez MF. 2009. Placental glycogen stores are increased in mice with H19 null mutations but not in those with insulin or IGF type 1 receptor mutations. Placenta 30, 693–699. ( 10.1016/j.placenta.2009.05.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gicquel C, et al. 2005. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver–Russell syndrome. Nat. Genet. 37, 1003–1007. ( 10.1038/ng1629) [DOI] [PubMed] [Google Scholar]
- 25.Begemann M, Spengler S, Gogiel M, Grasshoff U, Bonin M, Betz RC, Dufke A, Spier I, Eggermann T. 2012. Clinical significance of copy number variations in the 11p15.5 imprinting control regions: new cases and review of the literature. J. Med. Genet. 49, 547–553. ( 10.1136/jmedgenet-2012-100967) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishida M, Moore GE. 2013. The role of imprinted genes in humans. Mol. Aspects Med. 34, 826–840. ( 10.1016/j.mam.2012.06.009) [DOI] [PubMed] [Google Scholar]
- 27.Tunster SJ, Van de Pette M, John RM. 2011. Fetal overgrowth in the Cdkn1c mouse model of Beckwith–Wiedemann syndrome. Dis. Models Mech. 4, 814–821. ( 10.1242/dmm.007328) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arboleda VA, et al. 2012. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat. Genet. 44, 788–792. ( 10.1038/ng.2275) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choufani S, Shuman C, Weksberg R. 2010. Beckwith–Wiedemann syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 343–354. ( 10.1002/ajmg.c.30267) [DOI] [PubMed] [Google Scholar]
- 30.Brioude F, et al. 2013. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J. Med. Genet. 50, 823–830. ( 10.1136/jmedgenet-2013-101691) [DOI] [PubMed] [Google Scholar]
- 31.Lambertini L, Marsit CJ, Sharma P, Maccani M, Ma Y, Hu J, Chen J. 2012. Imprinted gene expression in fetal growth and development. Placenta 33, 480–486. ( 10.1016/j.placenta.2012.03.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frank D, et al. 2002. Placental overgrowth in mice lacking the imprinted gene Ipl. Proc. Natl Acad. Sci. USA 99, 7490–7495. ( 10.1073/pnas.122039999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Apostolidou S, et al. 2007. Elevated placental expression of the imprinted PHLDA2 gene is associated with low birth weight. J. Mol. Med. (Berl.) 85, 379–387. ( 10.1007/s00109-006-0131-8) [DOI] [PubMed] [Google Scholar]
- 34.Kumar N, Leverence J, Bick D, Sampath V. 2012. Ontogeny of growth-regulating genes in the placenta. Placenta 33, 94–99. ( 10.1016/j.placenta.2011.11.018) [DOI] [PubMed] [Google Scholar]
- 35.McMinn J, Wei M, Schupf N, Cusmai J, Johnson EB, Smith AC, Weksberg R, Thaker HM, Tycko B. 2006. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta 27, 540–549. ( 10.1016/j.placenta.2005.07.004) [DOI] [PubMed] [Google Scholar]
- 36.Ishida M, et al. 2012. Maternal inheritance of a promoter variant in the imprinted PHLDA2 gene significantly increases birth weight. Am. J. Hum. Genet. 90, 715–719. ( 10.1016/j.ajhg.2012.02.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moon YS, Smas CM, Lee K, Villena JA, Kim KH, Yun EJ, Sul HS. 2002. Mice lacking paternally expressed Pref-1/Dlk1 display growth retardation and accelerated adiposity. Mol. Cell. Biol. 22, 5585–5592. ( 10.1128/MCB.22.15.5585-5592.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG. 2010. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat. Genet. 42, 68–71. ( 10.1038/ng.493) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi N, Okamoto A, Kobayashi R, Shirai M, Obata Y, Ogawa H, Sotomaru Y, Kono T. 2009. Deletion of Gtl2, imprinted non-coding RNA, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum. Mol. Genet. 18, 1879–1888. ( 10.1093/hmg/ddp108) [DOI] [PubMed] [Google Scholar]
- 40.Feng W, Marquez RT, Lu Z, Liu J, Lu KH, Issa JP, Fishman DM, Yu Y, Bast RC. 2008. Imprinted tumor suppressor genes ARHI and PEG3 are the most frequently down-regulated in human ovarian cancers by loss of heterozygosity and promoter methylation. Cancer 112, 1489–1502. ( 10.1002/cncr.23323) [DOI] [PubMed] [Google Scholar]
- 41.Moore T, Haig D. 1991. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 7, 45–49. ( 10.1016/0168-9525(91)90230-N) [DOI] [PubMed] [Google Scholar]
- 42.Court F, et al. 2014. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 24, 554–569. ( 10.1101/gr.164913.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lofqvist C, et al. 2007. IGFBP3 suppresses retinopathy through suppression of oxygen-induced vessel loss and promotion of vascular regrowth. Proc. Natl Acad. Sci. USA 104, 10 589–10 594. ( 10.1073/pnas.0702031104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deal C, et al. 2001. Novel promoter polymorphism in insulin-like growth factor-binding protein-3: correlation with serum levels and interaction with known regulators. J. Clin. Endocrinol. Metab. 86, 1274–1280. [DOI] [PubMed] [Google Scholar]
- 45.Baker J, Liu JP, Robertson EJ, Efstratiadis A. 1993. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75, 73–82. ( 10.1016/0092-8674(93)90680-O) [DOI] [PubMed] [Google Scholar]
- 46.Netchine I, Azzi S, Le Bouc Y, Savage MO. 2011. IGF1 molecular anomalies demonstrate its critical role in fetal, postnatal growth and brain development. Best Pract. Res. Clin. Endocrinol. Metab. 25, 181–190. ( 10.1016/j.beem.2010.08.005) [DOI] [PubMed] [Google Scholar]
- 47.Gannagé-Yared M-H, Klammt J, Chouery E, Corbani S, Mégarbané H, Ghoch JA, Choucair N, Pfäffle R, Mégarbané A. 2013. Homozygous mutation of the IGF1 receptor gene in a patient with severe pre- and postnatal growth failure and congenital malformations. Eur. J. Endocrinol. 168, K1–K7. ( 10.1530/EJE-12-0701) [DOI] [PubMed] [Google Scholar]
- 48.Hiden U, Glitzner E, Hartmann M, Desoye G. 2009. Insulin and the IGF system in the human placenta of normal and diabetic pregnancies. J. Anat. 215, 60–68. ( 10.1111/j.1469-7580.2008.01035.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frasca F, et al. 1999. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 19, 3278–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monk D, Arnaud P, Apostolidou S, Hills FA, Kelsey G, Stanier P, Feil R, Moore GE. 2006. Limited evolutionary conservation of imprinting in the human placenta. Proc. Natl Acad. Sci. USA 103, 6623–6628. ( 10.1073/pnas.0511031103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Willison K. 1991. Opposite imprinting of the mouse Igf2 and Igf2r genes. Trends Genet. 7, 107–109. ( 10.1016/0168-9525(91)90441-R) [DOI] [PubMed] [Google Scholar]
- 52.Vu TH, Li T, Nguyen D, Nguyen BT, Yao XM, Hu JF, Hoffman AR. 2000. Symmetric and asymmetric DNA methylation in the human IGF2-H19 imprinted region. Genomics 64, 132–143. ( 10.1006/geno.1999.6094) [DOI] [PubMed] [Google Scholar]
- 53.Bukowski R, et al. 2007. Human sexual size dimorphism in early pregnancy. Am. J. Epidemiol. 165, 1216–1218. ( 10.1093/aje/kwm024) [DOI] [PubMed] [Google Scholar]
- 54.Holt LJ, Siddle K. 2005. Grb10 and Grb14: enigmatic regulators of insulin action--and more? Biochem. J. 388, 393–406. ( 10.1042/BJ20050216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blagitko N, Mergenthaler S, Schulz U, Wollmann HA, Craigen W, Eggermann T, Ropers H-H, Kalscheuer VM. 2000. Human GRB10 is imprinted and expressed from the paternal and maternal allele in a highly tissue- and isoform-specific fashion. Hum. Mol. Genet. 9, 1587–1595. ( 10.1093/hmg/9.11.1587) [DOI] [PubMed] [Google Scholar]
- 56.Monk D, Arnaud P, Frost J, Hills FA, Stanier P, Feil R, Moore GE. 2009. Reciprocal imprinting of human GRB10 in placental trophoblast and brain: evolutionary conservation of reversed allelic expression. Hum. Mol. Genet. 18, 3066–3074. ( 10.1093/hmg/ddp248) [DOI] [PubMed] [Google Scholar]
- 57.Gregg C, Zhang J, Weissbourd B, Luo S, Schroth GP, Haig D, Dulac C. 2010. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 329, 643–648. ( 10.1126/science.1190830) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garfield AS, et al. 2011. Distinct physiological and behavioural functions for parental alleles of imprinted Grb10. Nature 469, 534–538. ( 10.1038/nature09651) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saxena A, Morozov P, Frank D, Musalo R, Lemmon MA, Skolnik EY, Tycko B. 2002. Phosphoinositide binding by the pleckstrin homology domains of Ipl and Tih1. J. Biol. Chem. 277, 49935–49944. ( 10.1074/jbc.M206497200) [DOI] [PubMed] [Google Scholar]
- 60.Salas M, John R, Saxena A, Barton S, Frank D, Fitzpatrick G, Higgins MJ, Tycko B. 2004. Placental growth retardation due to loss of imprinting of Phlda2. Mech. Dev. 121, 1199–1210. ( 10.1016/j.mod.2004.05.017) [DOI] [PubMed] [Google Scholar]
- 61.Tunster SJ, Tycko B, John RM. 2010. The imprinted Phlda2 gene regulates extraembryonic energy stores. Mol. Cell. Biol. 30, 295–306. ( 10.1128/MCB.00662-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Doria S, Sousa M, Fernandes S, Ramalho C, Brandao O, Matias A, Barros A, Carvalho F. 2010. Gene expression pattern of IGF2, PHLDA2, PEG10 and CDKN1C imprinted genes in spontaneous miscarriages or fetal deaths. Epigenetics 5, 444–450. ( 10.4161/epi.5.5.12118) [DOI] [PubMed] [Google Scholar]
- 63.Bruchova H, Vasikova A, Merkerova M, Milcova A, Topinka J, Balascak I, Pastorkova A, Sram RJ, Brdicka R. 2010. Effect of maternal tobacco smoke exposure on the placental transcriptome. Placenta 31, 186–191. ( 10.1016/j.placenta.2009.12.016) [DOI] [PubMed] [Google Scholar]
- 64.Laborda J, Sausville EA, Hoffman T, Notario V. 1993. Dlk, a putative mammalian homeotic gene differentially expressed in small cell lung carcinoma and neuroendocrine tumor cell line. J. Biol. Chem. 268, 3817–3820. [PubMed] [Google Scholar]
- 65.Sul HS. 2009. Minireview: Pref-1: role in adipogenesis and mesenchymal cell fate. Mol. Endocrinol. 23, 1717–1725. ( 10.1210/me.2009-0160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cardwell CR, et al. 2010. Birthweight and the risk of childhood-onset type 1 diabetes: a meta-analysis of observational studies using individual patient data. Diabetologia 53, 641–651. ( 10.1007/s00125-009-1648-5) [DOI] [PubMed] [Google Scholar]
- 67.Harder T, Roepke K, Diller N, Stechling Y, Dudenhausen JW, Plagemann A. 2009. Birth weight, early weight gain, and subsequent risk of type 1 diabetes: systematic review and meta-analysis. Am. J. Epidemiol. 169, 1428–1436. ( 10.1093/aje/kwp065) [DOI] [PubMed] [Google Scholar]
- 68.Stene LC, Magnus P, Lie RT, Sovik O, Joner G. 2001. Birth weight and childhood onset type 1 diabetes: population based cohort study. BMJ 322, 889–892. ( 10.1136/bmj.322.7291.889) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Desoye G, Hauguel-de Mouzon S. 2007. The human placenta in gestational diabetes mellitus. The insulin and cytokine network . Diabetes Care. 30(Suppl. 2), S120–S126. ( 10.2337/dc07-s203) [DOI] [PubMed] [Google Scholar]
- 70.Harris LK, Crocker IP, Baker PN, Aplin JD, Westwood M. 2011. IGF2 actions on trophoblast in human placenta are regulated by the insulin-like growth factor 2 receptor, which can function as both a signaling and clearance receptor. Biol. Reprod. 84, 440–446. ( 10.1095/biolreprod.110.088195) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reinius B, Kanduri C. 2013. Elevated expression of H19 and Igf2 in the female mouse eye. PLoS ONE 8, e56611 ( 10.1371/journal.pone.0056611) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iglesias-Platas I, Martin-Trujillo A, Petazzi P, Guillaumet-Adkins A, Esteller M, Monk D. 2014. Altered expression of the imprinted transcription factor PLAGL1 deregulates a network of genes in the human IUGR placenta. Hum. Mol. Genet. 23, 6275–6285. ( 10.1093/hmg/ddu347) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Laborda J. 2000. The role of the epidermal growth factor-like protein dlk in cell differentiation. Histol. Histopathol. 15, 119–129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.