

Abstract
The pathogenesis of pyelonephritis caused by uropathogenic Escherichia coli (UPEC) is not well understood. Here, we show that besides UPEC virulence, the severity of the host innate immune response and invasion of renal epithelial cells are important pathogenic factors. Activation of endogenous anti-inflammatory mediator cAMP significantly attenuated acute pyelonephritis in mice induced by UPEC. Administration of forskolin (a potent elevator of intracellular cAMP) reduced kidney infection (ie, bacterial load, tissue destruction); this was associated with attenuated local inflammation, as evidenced by the reduction of renal production of proinflammatory mediators, renal infiltration of inflammatory cells, and renal myeloperoxidase activity. In primary cell culture systems, forskolin not only down-regulated UPEC-stimulated production of proinflammatory mediators by renal tubular epithelial cells and inflammatory cells (eg, monocyte/macrophages) but also reduced bacterial internalization by renal tubular epithelial cells. Our findings clearly indicate that activation of endogenous anti-inflammatory mediator cAMP is beneficial for controlling UPEC-mediated acute pyelonephritis in mice. The beneficial effect can be explained at least in part by limiting excessive inflammatory responses through acting on both renal tubular epithelial cells and inflammatory cells and by inhibiting bacteria invasion of renal tubular epithelial cells.
Urinary tract infections (UTIs) are among the most common infectious diseases, particularly in women, babies, and the elderly, and are the most common hospital-acquired infections in the developed world, which contribute to high financial burden worldwide. Although antibiotics are available to treat the disease, there remain a number of challenges, including frequent recurrence, persistence of infection, and the increasing risk of resistance to antibiotics.1,2 For these reasons, it is imperative to improve our understanding of the pathogenesis of UTIs and to develop novel treatment strategies that could be used to improve current treatment.
UTI usually starts as a bladder infection (cystitis) but can develop to kidney infection (pyelonephritis) that is an organ- and even life-threatening condition, because it may lead to renal scarring, chronic renal failure, sepsis, and severe systemic infection with multiorgan failure.3 Uropathogenic Escherichia coli (UPEC) is the primary cause of UTI. The pathogenesis of UTIs can be influenced both by properties of the infecting pathogens and host responses to pathogens, in addition to other factors such as anatomical abnormality. Most UPEC strains express a variety of fimbriae (eg, P, type 1, Dr) that enable them to attach to uroepithelial cells (a critical step in colonization). On contact with uroepithelial cells, UPEC liberates toxins (eg, hemolysin and cytotoxic necrotizing factor-1) which mediate direct injury to the cells, disrupting the mucosal barrier, and opening access to the underlying tissue.4 UPEC is also able to transiently invade, survive, and multiply within uroepithelial cells, which is thought to enable organisms to evade host defenses and to act as a reservoir for further infection. In addition, most human UPEC strains are resistant to complement-mediated killing5,6 but exploit complement for entering epithelial cells that contribute to kidney infection.7,8 UPEC has evolved mechanisms through carrying intracellular Toll/IL-1 receptor domain homologous sequences to impair immune protection while promoting inflammation and tissue damage.9
Besides the properties of UPEC, the host immune response to pathogens has a big effect on the pathogenesis of UTI. Although innate immune responses play essential roles in the first line of host defense against pathogens, they also cause harm when present in excess or are dysregulated. For example, in the acute condition, uroepithelial cells and inflammatory cells, in response to UPEC stimulation, produce a number of proinflammatory mediators [eg, IL-6, tumor necrosis factor (TNF)-α, and IL-8], which (if present in excess) cause epithelial inflammation/damage, allowing bacteria to enter the underlying tissue.10 In addition, if the activation of neutrophils is not tightly regulated, the reactive oxygen species (ROS) and cytotoxic enzymes and ingested bacteria could be released into the surrounding area, causing tissue destruction and pathogen survival/dissemination.11 Previous studies in acute pyelonephritis have found that paradoxically Tlr4−/−, Il-1β−/−, or C3−/− mice had less severe acute kidney infection and inflammation, suggesting that innate immune responses driven by TLR-4 signaling, proinflammatory cytokine IL-1β, or complement effector molecules are harmful, instead of beneficial for the host.7,12
More than half a century after its discovery cAMP remains the object of intense scientific interest.13 cAMP is an intracellular second messenger induced by extracellular signals such as hormones, inflammatory mediators, and cytokines. It induces intracellular signal transduction conveying the cAMP-dependent pathways to regulate diverse cellular responses.14 Among the diverse functions, cAMP is known as a potent anti-inflammatory mediator, having inhibitory effects on production of proinflammatory cytokines in immune cells,15 leukocyte chemotaxisis,16 and release of bioactive molecules and cytotoxic agents from granulocytes.17 In addition, cAMP is found to regulate exocytic process in a variety of cells18 and to induce exocytosis of E. coli from bladder epithelial cells, thus reducing E. coli colonization of bladders.19
Forskolin, a potent activator of cAMP, is used for investigating the role of cAMP in various cellular processes and diseases. Previous human and animal studies have found that administration of forskolin is beneficial in several clinical disorders, including UTI, bronchoconstriction, ischemia reperfusion injury, high blood pressure, and leukemia.19–23 Although forskolin was studied in experimental UTI and showed that forskolin treatment reduces bladder infection (cystitis),19 it is unknown whether forskolin treatment would be effective in kidney infection (pyelonephritis). Here, we evaluated the effect of forskolin on protection against murine acute pyelonephritis and explored cellular and molecular mechanisms by which forskolin confers the protection. Our results indicate that forskolin treatment significantly reduced kidney infection and suggest that forskolin mediated the suppression of excessive inflammatory responses and inhibition of E. coli invasion of renal tubular epithelial cells (RTECs) that are responsible for the protection.
Materials and Methods
Materials
Forskolin, hydroxyurea, hydrocortisone, tri-iodothyronine, and thioglycollate were from Sigma-Aldrich (Shanghai, China). Collagenase D was from Roche (Welwyn, UK). The antibodies for CD45 (30-F11, allophycocyanin), Gr-1 (RB6-8C5, phosphatidylethanolamine), F4/80 (BM8, phosphatidylethanolamine), Ly6G (1A8, phosphatidylethanolamine), and CD11b (M1/70, fluorescein isothiocyanate) were from BioLegend (San Diego, CA). Horseradish peroxidase-conjugated rabbit anti-rat polyclonal antibody was from Dako (Cambridge, UK). FcR blocking antibody (CD16/32, 2.4G2) and mouse ELISA kits for TNF-α, IL-6 were from BD Biosciences (San Jose, CA). Myeloperoxidase (MPO) assay kit and mouse ELISA kit for keratinocyte chemoattractant (KC) and Parameter cAMP assay kit were from R&D Systems (Shanghai, China). Cell culture medium, fetal calf serum, insulin-transferring-selenium solution, gentamicin, and CountBright absolute counting beads were purchased from Invitrogen China Limited (Beijing, China).
Induction of Pyelonephritis and Forskolin Administration
Murine UTI, a model of ascending urinary tract infection leading to pyelonephritis, was induced in female C57BL/6 mice (8 to 10 weeks old) by bladder inoculation of human UPEC strain J96 [108 colony-forming unit (CFU) in 50 μL of phosphate-buffered saline (PBS); serotype O4; K6, it is a serum-resistant, hemolysin-secreting E. coli strain that expresses both type 1 and P fimbriae)] per urethra as previously described.7,24 Forskolin (10 mg/kg) or vehicle control (25% of dimethyl sulfoxide in PBS) was given daily by i.p. injection, starting at 1 hour before or 1 hour after the inoculation. The effect of the forskolin treatment was confirmed by measuring tissue cAMP amounts in the kidneys 2 hours after the administration. Results indicate that kidney cAMP amounts were clearly increased in forskolin-treated mice compared with that in control-treated mice [forskolin versus control (mean ± SEM): 1176 ± 50 versus 926 ± 139 pmoL/mg wet wt; n = 3; P < 0.05]. In some experiments, before the forskolin administration and induction of UTI, mice were treated by i.p. injection of 20 mg of hydroxyurea per mouse daily for 7 days and 100 μg of anti-mouse Gr-1 antibody per mouse on the last day of treatment to deplete neutrophils as previously described.25 C57BL/6 mice were purchased from Vital River Laboratories (Beijing, China). The Ethics Review Committee for Animal Experimentation at Xi'an Jiaotong University approved and oversaw all mouse experiments.
Assessment of Bacterial Load in the Kidney
Total bacterial load in kidney tissues was analyzed by the agar plate assay as previously described with modifications.26 In brief, the kidney was weighed and subsequently homogenized in 2 mL of PBS. One hundred microliters of a series dilution of homogenates was plated on cysteine lactose electrolyte deficient (CLED) plates. After incubation for 24 hours, bacterial CFU on agar plates were manually counted and expressed as CFU per gram of kidney tissue.
Assessment of Renal Pathology
To assess renal pathology, the kidney was fixed in a solution of 4% formalin in PBS for 24 hours and embedded in paraffin. Sections (3 μm) were stained with hematoxylin and eosin. The severity of renal pathology (ie, cellular infiltration, tissue destruction, bacterial patchiness, abscess) was graded in a blinded fashion by two investigators (N.W. and B.-S.G.) with the use of a 7-point scale as described previously with modifications,27 in which 0, 1, 2, and 3 indicated normal, mild, moderate, and severe pyelitis, respectively (pathologic changes were mainly located at papilla and medulla); whereas 4, 5, and 6 indicated mild, moderate, and severe pyelonephritis, respectively (pathologic changes were distributed to papilla, medulla, and cortex).
Assessment of Inflammatory Cell Infiltration in the Kidney
Single renal cell suspension was prepared with the method described previously with modifications.28 Kidneys were weighed, minced, and incubated with 0.75 mg/mL collagenase D for 10 minutes at 37°C with gentle agitation. The collagenase was inactivated with an equal volume of Dulbecco’s modified Eagle’s medium (DMEM)-F12 that contained 10% fetal calf serum. The digested tissue mixture was then passed through a 40-μm nylon sieve to remove tissue debris. The cell segments were collected and treated with red cell lysis buffer to remove remaining red blood cells. The cell pellet was washed and re-suspended in PBS that contained 1% bovine serum albumin and was followed by flow cytometric analysis. The cells were pre-incubated with FcR blocking antibody (CD16/32) and then stained with fluorochrome-conjugated monoclonal antibodies or the appropriate isotype control antibodies at 4°C for 20 minutes. To quantify absolute cell counts in kidney tissue, we used CountBright absolute counting beads in our flow cytometric assays according to the instructions of the manufacturer. All flow cytometric analysis was performed with Calibur Flow Cytometer (BD Biosciences) and FlowJo software version 7.6.5 (TreeStar Inc., Ashland, OR).
Immunohistochemical Staining
Frozen sections (4 μm) of OCT-embedded kidneys were air-dried for a minimum of 20 minutes at room temperature and then acetone fixed. Sections were incubated with rat anti-mouse Gr-1 or F4/80 antibody and followed by horseradish peroxidase-conjugated rabbit anti-rat antibody.
Cell Cultures and Treatment
Primary tubular epithelial cell cultures were prepared from kidneys of naive male C57BL/6 mice as described previously.29 In brief, minced cortex was digested with 0.1% collagenase II and passed through a 40-μm nylon sieve. The cells and tubules were collected and cultured in a DMEM-12 medium that contained 2% fetal calf serum, 5 μg/mL insulin, 5 μg/mL transferrin, 5 ng/mL selenium, 40 ng/mL hydrocortisone, and 10−12 mol/L tri-iodothyronine. Nonpassaged 7-day cultured cells were used. Neutrophils and macrophages were prepared from peritoneal lavage of C57BL/6 mice after i.p. injection of 1 mL of 3% thioglycollate. For neutrophils, peritoneal cells were harvested at 24 hours after thioglycollate injection and cultured for 1 hour to discard adherent cells and to collect suspension cells for further analysis. For macrophages, peritoneal cells were harvested at 5 days after thioglycollate injection and cultured for 1 hour to discard suspension cells and to collect adherent cells for further analysis.
To assess the effect of forskolin on mRNA expression of proinflammatory mediators in these cells, confluent layers of RTECs grown on 24-well plates or 2 × 106/well neutrophils or macrophages (prepared as described in the previous paragraph) seeded in 24-well plates were incubated with or without 10 μmol/L forskolin, in the presence of 2 × 106/well of heat-killed J96, for 4 hours at 37°C, in 1 mL of total volume.
Semiquantitative Real-Time RT-PCR
Total RNA extraction from kidney and cell samples, reverse transcription reaction, and the PCR were performed as previous described.30 The relative gene expression was analyzed with the 2−ΔΔCT method31 and expressed as 2−ΔΔ(Ct), where Ct is cycle threshold,
| (1) |
| (2) |
The control samples were either normal kidney tissues or untreated cells. The testing samples were infected kidney tissues or forskolin-treated cells. The information for primer sequences is given in Table 1.
Table 1.
PCR Primer Sequences and Product Sizes
| Primer∗ | Oligonucleotide sequence | Product size (bp) | GeneBank code |
|---|---|---|---|
| 18S-1 | 5′-ATCCCTGAGAAGTTCCAGCA-3′ | 153 | NM_011296.1 |
| 18S-2 | 5′-CCTCTTGGTGAGGTCGATGT-3′ | ||
| TNF-α-1 | 5′-TGAGCACAGAAAGCATGATCC-3′ | 200 | NM_013693.3 |
| TNF-α-2 | 5′-GCCATTTGGGAACTTCTCATC-3′ | ||
| IL1β-1 | 5′-GCCCATCCTCTGTGACTCAT-3′ | 230 | NM_008361 |
| IL1β-2 | 5′-AGGCCACAGGTATTTTGTCG-3′ | ||
| KC-1 | 5′-TGAAGCTCCCTTGGTTCAGA-3′ | 361 | NM_008176.3 |
| KC-2 | 5′-TGCACTTCTTTTCGCACAAC-3′ | ||
| IFNγ-1 | 5′-ACTGGCAAAAGGATGGTGAC-3′ | 237 | NM_008337.3 |
| IFNγ-2 | 5′-TGAGCTCATTGAATGCTTGG-3′ | ||
| IL6-1 | 5′-GTTCTCTGGGAAATCGTGGA-3′ | 339 | NM_031168 |
| IL6-2 | 5′-GAAATTGGGGTAGGAAGGA-3′ | ||
| MCP-1-1 | 5′-GGCTCAGCCAGATGCAGTTA-3′ | 219 | NM_011333.3 |
| MCP-1-2 | 5′-ATTTGGTTCCGATCCAGGTT-3′ | ||
| MIP-1α-1 | 5′-CACTGCCCTTGCTGTTCTTC-3′ | 262 | NM_011337.2 |
| MIP-1α-2 | 5′-GGCATTCAGTTCCAGGTCAG-3′ | ||
| MIP-2α-1 | 5′-CCCAGCTCTGTGCAAACCTA-3′ | 248 | NM_009140 |
| MIP-2α-2 | 5′-TCTGCCTCTTTTGGTCAGGA-3′ | ||
| RANTES-1 | 5′-GTGCCCACGTCAAGGAGTAT-3′ | 186 | NM_013653 |
| RANTES-2 | 5′-GGGAAGCGTATACAGGGTCA-3′ |
KC, keratinocyte chemoattractant; IFN, interferon; MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; RANTES, regulated on activation normal T cell expressed and secreted; TNF, tumor necrosis factor.
Primer-1 is identical to the coding strand; primer-2 is complementary to the coding strand.
ELISA and MPO Assays
TNF-α, IL-6, and KC amounts in urine and kidney homogenate were measured by a 96-well plate method with murine IL-6 and KC ELISA kits. Renal MPO activity was measured in kidney homogenate by a specific ELISA method with MPO assay kit according to the instructions of the manufacturer. Mouse kidney was weighed and homogenated in PBS that contained 1% Triton X-100, 1 mmol/L EDTA, and 1% protease inhibitor cocktail (Sigma-Aldrich) and spin cleared at 10,000 × g for 10 minutes as previously described.32 The resulting supernatants were subjected to ELISA and MPO assays. Urine samples were collected at the end of the experiment when mice were sacrificed.
Bacterial Binding and Internalization Assays
The assays were performed as we have described previously.8 Briefly, mouse RTECs were primarily cultured on 24-well plates and grown to confluence. RTEC monolayers were preincubated with or without forskolin for 2 hours; bacteria (J96, 1 × 107 CFU/well) were then added and incubated for an additional 1 hour at 37°C. For binding assay, cells were vigorously washed to remove unattached bacteria and lyzed with sterile H2O, and the lysate was plated onto CLED plates. The colonies were counted, and results were expressed as CFU per well. For internalization, after washing, the cells were incubated for 1 hour with 100 μg/mL gentamicin to kill extracellular bacteria and lyzed. The lysate was plated onto CLED plates, and the colonies were counted.
Bacterial Exocytosis Assay
Bacterial exocytosis assay was performed as described previously.19 Briefly, RTEC monolayers were incubated with bacteria for 1 hour at 37°C. After washing, the cells were incubated for 1 hour with 100 μg/mL gentamicin to kill extracellular bacteria. After additional washes, the cells were incubated with medium with or without forskolin for an additional 2 hours at 37°C. The supernatant was plated onto CLED plates. The percentage of bacterial exocytosis was calculated as the number of bacteria in the supernatants divided by the total number of bacteria surviving incubation with gentamicin (including intracellular and exited bacteria). In some experiments, 5 μmol/L H89 was added to the medium as an inhibitor of cAMP signal pathway.
Statistical Analysis
Data are expressed as means ± SEM or the readout of individual mice. Mann-Whitney test, Student's t-test, or analysis of variance was used when appropriate to determine significant differences between samples. All of the analyses were performed with GraphPad Prism software version 5 (GraphPad Inc., San Diego, CA).
Results
Forskolin Reduces Bacterial Load in Kidneys and Attenuates Renal Pathology
Previous studies in murine acute nephritis have found that renal infection (detecting viable bacteria in kidneys and renal tissue damage) appears as early as 4 hours after bladder inoculation of UPEC and peaks within 48 to 72 hours.26 We therefore assessed the effects of forskolin on renal infection at three time points (6, 24, and 48 hours after infection). We initially pretested two protocols of forskolin administration, which involved the start of administration at 1 hour before or 1 hour after the onset of infection, aiming to assess the possible roles for forskolin in prevention and treatment of infection, respectively. We found that both protocols of forskolin administration provided similar protection against kidney infection at 6 and 24 hours after infection. Because the protocol for administering forskolin after the onset of infection is more relevant to clinical practice, it was used in the rest of the experiments. Bacterial loads in the kidneys were significantly reduced in the forskolin-treated group by 10- to 100-fold compared with the control group at all time points (6, 24, and 48 hours) (Figure 1). In addition to bacterial loads, we also assessed the effect of forskolin treatment on renal histopathology. Renal histopathologic changes, including cellular infiltration, tissue inflammation, and mild tissue destruction, were detected at 6 hours after infection, which was mainly located at papilla and medulla. At 24 hours after infection, renal histopathologic changes became manifest. Cellular infiltration, bacterial patchiness, abscess, and tubule destruction were observed at papilla/medulla and cortex. At 48 hours after infection, cellular infiltration and bacterial patchiness were reduced or cleared, which left some cavities at papilla/medulla and cortex. On the basis of these histopathologic changes, we performed histologic scoring in individual mice of the two groups. Our results showed that, compared with the control group, the forskolin-treated group exhibited attenuated renal histopathology at all time points (6, 24, and 48 hours) (Figure 2). We also assessed renal function in the two groups of mice (control and forskolin treated) at all studied time points (6, 24, and 48 hours) after infection by measuring blood urea nitrogen concentrations. Results show that blood urea nitrogen concentrations were only slightly elevated in some infected mice compared with naive mice but did not differ significantly between the control and forskolin-treated groups (Supplemental Figure S1). These results suggest that renal functional impairment may not be an important index for renal pathology in this model; in consequence the beneficial effect of forskolin cannot be identified in this context.
Figure 1.
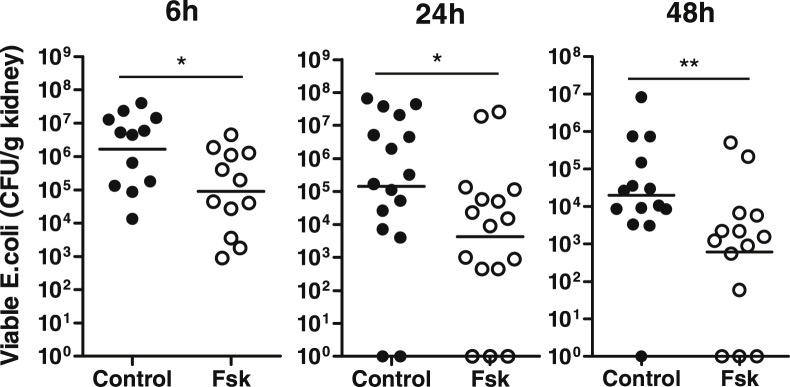
Forskolin administration reduces bacterial load in the kidneys. Bacterial loads in kidneys were examined at 6, 24, and 48 hours after inoculation of J96. For the 6-and 24-hour groups, a single administration of forskolin or vehicle control was given at 1 hour after the inoculation. For the 48-hour group, an additional administration of forskolin was given at 24 hours after the inoculation. Each dot represents a single animal and is shown as the average number of two replicate agar plates; horizontal lines represent the means of CFU counts. ∗P < 0.05, ∗∗P < 0.01, Mann-Whitney test. CFU, colony-forming unit; Fsk, forskolin.
Figure 2.

Forskolin administration attenuates renal pathology after the infection. A: Representative images of hematoxylin and eosin–stained kidneys from the control and forskolin-treated mice show renal histological changes including cellular infiltration, bacterial patchiness, abscess, and tubule destruction (arrows). B: Histologic scores in the control (black dots) and forskolin-treated (white dots) mice. Each dot represents a single animal; horizontal lines represent the means of renal pathological scores. ∗P < 0.05, ∗∗P < 0.01, Mann-Whitney test. Original magnification, ×100. Fsk, forskolin.
Forskolin Reduces Renal Production of Proinflammatory Mediators after the Infection
Kidney infection induced by UPEC triggers the production of proinflammatory mediators by epithelial cells and inflammatory cells rapidly. We therefore evaluated the effects of forskolin on renal production of proinflammatory cytokines and chemokines in response to bacterial stimulation. Intrarenal mRNA expression of inflammatory cytokines [ie, IL-6, IL-1β, interferon (IFN)-γ, TNF-α] and chemokines [ie, KC, regulated on activation normal T cell expressed and secreted (RANTES), monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP-)1α, MIP-2α] was significantly reduced in the forskolin-treated group compared with the control group at 6 hours after infection (Figure 3A). Intrarenal protein expression of TNF-α, IL-6, and KC was also reduced in the forskolin-treated group compared with the control group after the infection, particularly at 6 and 24 hours (Figure 3B). In addition to renal tissue, we also analyzed the concentrations of these proteins in the urine. TNF-α, IL-6, and KC were not detectable in normal mouse urine but were clearly detected in the infected mouse urine at 6 and 24 hours after infection. Compared with the control group, the forskolin-treated group had lower concentrations of TNF-α, IL-6, and KC (Figure 3C). Together, these results indicate that forskolin treatment down-regulated renal production of an array of proinflammatory mediators in response to bacteria stimulation.
Figure 3.

Forskolin administration reduces renal production of proinflammatory mediators after the infection. A: Semiquantitative analysis of proinflammatory mediator mRNA expression in the kidney tissue from the control and forskolin-treated mice at 6 hours after infection. The relative changes in gene expression were analyzed with the 2−ΔΔCT method. The dashed lines indicate basal levels of mRNA expression in normal tissue. B: ELISA shows TNF-α, IL-6, and KC protein expression in the kidney tissue from the control (black dots) and forskolin-treated (white dots) mice at different time points after infection. C: ELISA shows urine concentrations of TNF-α, Il-6, and KC in the control and forskolin-treated mice at 6 and 24 hours after infection. Each dot represents a single animal and is shown as the mean of two replicate PCR results; horizontal lines represent the means of arbitrary units of mRNA expression (A). Data are expressed as means ± SEM (B and C). n = 6 mice per group (A); n = 4 to 8 mice per group at each time point (B and C). ∗P < 0.05, ∗∗P < 0.01, Student's t-test. Ctrl, control; Fsk, forskolin; KC, keratinocyte chemoattractant; MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; RANTES, regulated on activation normal T cell expressed and secreted; TNF-α, tumor necrosis factor-α.
Forskolin Reduces Renal Inflammatory Cell Infiltration after the Infection
In addition to renal tissue destruction, renal inflammatory cell infiltration, particularly neutrophils, is a characteristic pathologic change in acute pyelonephritis. To assess the effect of forskolin on cellular infiltration, we performed immunohistochemistry for Gr-1 and F4/80 on kidney tissues at 24 hours after infection. Although Gr-1+ and F4/80+ cells were found in the kidneys of both the control and forskolin-treated mice, the number of these cells, particularly Gr-1+ cells, was less prominent in the kidneys of the forskolin-treated mice than that of the control mice (Figure 4A). We also performed renal tissue MPO assay in both the control and forskolin-treated mice, at various time points (0, 6, 24, and 48 hours) after the infection. MPO activity was significantly lower in the forskolin-treated group than in the control group (Figure 4B), which was consistent with the immunohistochemistry data. To quantify cellular infiltration in the kidney tissues, we performed flow cytometric analysis for CD45, Gr-1, and F4/80 in single-cell suspension prepared from individual kidneys of the control and forskolin-treated mice. At 6 hours after infection, forskolin had no apparent effects on the numbers of infiltrating cells. However, at 24 and 48 hours after infection, the numbers of CD45+, CD45+Gr-1+, and CD45+F4/80+ were significantly reduced in the forskolin-treated group compared with the control group (Figure 4C). Because Gr-1+ and F4/80+ may not be absolutely specific for neutrophils and monocytes/macrophages, respectively, we performed more specific analysis of renal cellular infiltration by using several additional antibodies and relevant gating strategies to assess the influence of forskolin on renal neutrophil and monocytes/macrophage infiltration. Results show that the number of CD45+Ly6G+ cells (highly specific for neutrophils) and CD45+Ly6G-CD11b+ cells (highly specific for monocytes/macrophages) was significantly reduced in the kidneys of the forskolin-treated mice than that of the control mice at 24 hours after infection (Figure 4D). These results are in agreement with the observations that the number of Gr1+ and F4/80+ cells was reduced in the kidneys of the forskolin-treated mice (Figure 4C), indicating that forskolin treatment reduced renal neutrophil and macrophage infiltration after infection.
Figure 4.

Forskolin administration reduces renal leukocyte infiltration after the infection. A: Representative images of immunohistochemical staining for Gr1 and F4/80 in the kidneys from control and forskolin-treated mice at 24 hours after infection. B: MPO activity in the kidney tissue from control and forskolin-treated mice at different time points after infection. C: Flow cytometric analysis of cellular infiltration in the kidneys from control and forskolin-treated mice at different time points after infection by gating CD45+, CD45+Gr-1+, and CD45+F4/80+ cells. The kidney tissue from uninfected mice was used to indicate a basal amount of renal inflammatory cells. Representative flow cytometric dot plot of CD45, Gr-1, and F4/80 expression in renal cell suspension prepared from the control-treated mice. Quantified numbers of CD45+, Gr-1+, and F4/80+ cells in the kidneys from control and forskolin-treated mice. D: Flow cytometric analysis of cellular infiltration in the kidneys from control and forskolin-treated mice at 24 hours after infection by gating CD45+Ly6G+ and CD45+Ly6G−CD11b+ cells. A diagram of gating strategy, quantified numbers of CD45+Ly6G+ and CD45+Ly6G−CD11b+ cells in the kidneys from control and forskolin-treated mice. Data are expressed as means ± SEM (B); horizontal lines represent the means of cell numbers (C and D). n = 5 to 7 mice per group at each time point (B); each dot represents a single animal (C and D). ∗P < 0.05 compared with the control treatment, Student's t-test (B); ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 (C and D). Original magnification, ×100. Ctrl, control; Fsk, forskolin; MPO, myeloperoxidase; Normal, uninfected; SSC, side scatter.
Forskolin Down-Regulates Proinflammatory Mediator Production by RTECs and Inflammatory Cells
Our data presented so far indicate that forskolin administration protected mice from acute pyelonephritis, as evidenced by the reduction of bacterial load, tissue damage, and inflammatory responses within the kidneys. To explore cellular mechanism by which forskolin restricts production of proinflammatory mediators within kidneys, we used primary cell culture systems for RTECs and inflammatory cells to assess the effects of forskolin on proinflammatory mediator production by these cells in response to bacteria stimulation. Incubation with heat-killed E. coli markedly up-regulated mRNA expression of all examined cytokines (ie, IL-1β, TNF-α, IFN-γ, and IL-6) and chemokines (ie, KC, MCP-1, RANTES, MIP-1α, and MIP-2α) by RTECs and macrophages and neutrophils. In the presence of forskolin, mRNA expression amounts for several key proinflammatory mediators were significantly reduced in RTECs (ie, KC, IL-1β, TNF-α, MCP-1, MIP-1α, and RANTES) and macrophages (ie, KC, IL-1β, TFN-α, MCP-1, MIP-1α, and IFN-γ) compared with that in the absence of forskolin. In addition, forskolin treatment also reduced IL-1β and TNF-α mRNA expression in neutrophils (Figure 5). Thus, our data indicate that forskolin has an inhibitory effect on the production of proinflammatory mediators by RTECs and inflammatory cells in response to E. coli; the effects were more prominent in RTECs and macrophages, in terms of the range of proinflammatory mediators that is affected.
Figure 5.

Forskolin down-regulates proinflammatory mediator production by RTECs and inflammatory cells in response to bacteria stimulation. Semiquantitative analysis of proinflammatory mediator mRNA expression in the control and forskolin-treated primary cultured RTECs (A) and freshly prepared peritoneal macrophages (B) and neutrophils (C). The relative changes in gene expression were analyzed with the 2−ΔΔCT method. The dashed line indicates a basal amount of mRNA expression in the cells not being exposed to J96 and forskolin. Data are expressed as means ± SEM. n = 4. ∗P < 0.05, ∗∗P < 0.01, Student's t-test. Ctrl, control; Fsk, forskolin; KC, keratinocyte chemoattractant; MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; RANTES, regulated on activation normal T cell expressed and secreted; RTEC, renal tubular epithelial cell; Tnf-α, tumor necrosis factor-α.
Critical Role for Forskolin in Limiting E. coli Invasion of RTECs
In addition to the mechanism of restricting inflammatory responses within the kidney, we also hypothesized that forskolin may play important roles in limiting E. coli invasion of RTECs which would confer the protective effects. To investigate this, using primary cultured RTECs, we assessed the effects of forskolin on bacterial binding to and internalization by RTECs. RTECs were preincubated with forskolin and then cocultured with bacteria (J96) and followed by binding and internalization assays. Forskolin treatment had no apparent effects on bacteria binding, but it significantly reduced the number of viable intracellular bacteria and enhanced the number of viable bacteria that had exited the cells (exocytosis) (Figure 6, A–C). Because the above experiments were performed under a condition that forskolin was present in the coculture of bacteria and RTECs, although the RTECs were pretreated with forskolin, the effects of forskolin may not be entirely attributed to the action on RTECs. To assess the possibility of forskolin having direct effects on bacteria to inhibit their ability to invade epithelial cells, bacteria (J96) were preincubated with forskolin, and the washed bacteria were then used for analyzing their growth and ability to invade RTECs. Results show that forskolin pretreatment did not affect such prosperities of bacteria (Supplemental Figure S2). We also assessed the possibility of forskolin having an effect on viability of the host cells, which may influence the cell functions. RTECs were preincubated with forskolin, and the washed cells were then used for analyzing their viability and uptake capacity. Results show that the viability of forskolin-treated RTECs was comparable with that of control-treated RTECs, whereas the uptake of bacteria was significantly reduced in forskolin-treated RTECs compared with control-treated RTECs (Supplemental Figure S3). Collectively, these data suggest that forskolin-mediated reduction of E. coli invasion of RTECs is mainly through their specific actions on RTECs rather than acting on bacteria or causing RTEC death.
Figure 6.

Critical role for forskolin in limiting UPEC invasion of RTECs. A: Effect of forskolin on bacterial binding to RTECs. B: Effect of forskolin on intracellular population of E. coli in RTECs. C: Effect of forskolin on bacterial exocytosis by RTECs. D: Effect of forskolin on intracellular cAMP in RTECs. E: Effect of H89 on intracellular population of E. coli in RTECs. F: Effect of H89 on bacterial exocytosis by RTECs. Data are expressed as means ± SEM. n = 4 (A–D). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, Student's t-test. CFU, colony-forming unit; Fsk, forskolin; RTEC, renal tubular epithelial cell; UPEC, uropathogenic Escherichia coli.
To further investigate whether the effects of forskolin on bacteria internalization by RTECs are through activating cAMP, we measured intracellular cAMP amounts of RTECs after forskolin stimulation and assessed whether intracellular bacteria and exocytosis of bacteria by RTECs depend on cAMP signals. As we expected, the cAMP amounts were increased by forskolin treatment in a dose-dependent manner (Figure 6D) and inhibition of cAMP signal pathway by H89 [a potent protein kinase A (PKA; cAMP-dependent) inhibitor] abolished the effects of forskolin on intracellular population of E. coli and exocytosis (Figure 6, E and F).
Forskolin Mediates Protective Effect on Kidney Infection through Acting on Renal Parenchymal Cells
Data from cell culture experiments suggested important roles for forskolin in limiting E. coli invasion of RTECs and in down-regulating production of proinflammatory mediators by RTECs. To assess the possibility that forskolin mediates its protective effect on kidney infection through acting on renal parenchymal cells, we evaluated the effects of forskolin on kidney infection in neutrophil-depleted mice. After neutrophil depletion, peripheral blood neutrophil count was significantly reduced (by 85% to 90% reduction), and no neutrophils were detected in kidney tissues (Figure 7A). Depletion of neutrophils not only reduced bacterial load in the kidneys (Figure 7B) but also attenuated tissue destruction (Figure 7, C and D). Administration of forskolin in neutrophil-depleted mice further reduced bacterial load in the kidneys and tissue destruction, compared with the control treatment (Figure 7, B–D).
Figure 7.

Forskolin mediates protective effect on kidney infection through acting on renal parenchymal cells. A: Representative flow cytometry dot plot of Gr-1+ cells in peripheral blood and renal cell suspension from neutrophil-depleted and nondepleted mice. B: Bacterial loads in the kidneys of non–neutrophil-depleted mice and neutrophil-depleted mice that received forskolin or control treatment at 24 hours after infection. C: Representative images of kidneys of the mice mentioned in B stained with hematoxylin and eosin (papilla/medullar) or periodic acid-Schiff (cortex), showing lesions (arrows). D: Histologic scores of the kidneys mentioned in B. Each dot represents a single animal and is shown as the average number of two replicate agar plates; horizontal lines indicate the means of CFU counts. ∗P < 0.05, ∗∗P < 0.01, Mann-Whitney test. Scale bar = 100 μm (D). Original magnification, ×200. Fsk, forskolin; N depl, neutrophil-depleted; non-depl, non–neutrophil-depleted.
Discussion
Innate immunity is the first line of immunity against infection; however, excessive innate immune responses are implicated in the pathogenesis of a number of clinical disorders, including inflammatory and infectious diseases.12,30,33,34 Thus, therapies to prevent severe acute and chronic inflammatory states may have beneficial effects in such clinical disorders. In UTI, previous studies found that treatment of UPEC-infected mice with forskolin protected mice from bladder infection.19 The present study not only extends the previous findings by showing that forskolin administration protected mice from acute pyelonephritis (a more severe type of UTI) but also reveals potential cellular and molecular pathways by which forskolin confers the protection.
To understand how forskolin confers the protection against murine acute pyelonephritis, we performed a series of in vivo/ex vivo and in vitro cell culture experiments to dissect the effects of forskolin on inflammatory cells and RTECs. For inflammatory cells, consistent with the known anti-inflammatory property of forskolin/cAMP in leukocytes, our in vivo/ex vivo data indicate that forskolin treatment significantly reduced the number of leukocytes, particularly neutrophils within the kidneys after the infection, and our in vitro cell culture data indicate that forskolin had inhibitory effects on the production of proinflammatory mediators by inflammatory cells, particularly macrophages in response to bacteria stimulation. Furthermore, the data from the neutrophil depletion experiment indicate that depletion of neutrophils not only attenuated tissue destruction but also to a less extent reduced bacterial load in the kidneys. Together, our data suggest that neutrophils are an important pathogenic factor in this model, and forskolin-mediated inhibitory effects on leukocytes chemotaxis and production of proinflammatory mediators will have favorable effects in tissue damage and bacteria access to the underlying tissue during acute pyelonephritis.
In addition to inflammatory cells, we generated a series of data which strongly support the RTEC-dependent mechanism by which forskolin confers the protection of kidney infection. First, our in vitro cell culture data clearly indicate that RTECs produce a wide range and significant amounts of proinflammatory mediators in response to E. coli stimulation; forskolin treatment had inhibitory effects on the production of most key proinflammatory mediators by RTECs, suggesting that inhibition of renal cell production of proinflammatory mediators represents an important mechanism of forskolin effect. Second, our data from the in vitro model of internalization indicate a critical role for forskolin in limiting E. coli invasion of RTECs. Although previous studies have found that forskolin-induced cAMP can down-regulate internalization of UPEC into bladder epithelial cells, thus limiting bacteria entering the bladder; such a role was not previously described in renal epithelial cells. Our results show that forskolin treatment clearly reduced viable intracellular bacteria in RTECs and increased viable bacteria that had exited the cells, suggesting that forskolin has inhibitory effect on E. coli invasion of RTECs, which may offer an explanation of forskolin-mediated reduction of bacterial load in the kidney. Previous studies have suggested that elevated cAMP can reduce bacterial uptake by epithelial cells through inhibiting activation of Rho GTPase Rac-1 in a PKA-dependent manner.35 The Rho GTPase Rac-1 is an important mediator of bacterial invasion, whose activation leads to actin filament accumulation and cytoskeleton rearrangement at sites of bacterial entry and, consequently, bacterial internalization. In addition, the cAMP signaling system is well known to regulate and modulate exocytosis in a variety of secretory cells,18 and the elevated cAMP can trigger bacterial exocytosis through regulating vesicular trafficking in a PKA-Slac2c-GTPase Rab27-dependent manner.19,36 Therefore, forskolin-mediated inhibition of invasion of RTECs could be regarded as resulting from cAMP negatively regulated bacterial uptake and positively regulated exocytosis. Third, the data from the neutrophil depletion experiment indicate that administration of forskolin in neutrophil-depleted mice still reduced bacterial load in kidneys and attenuated tissue destruction, suggesting that forskolin can mediate its protective effect on kidney infection through acting on renal parenchymal cells.
In general, forskolin-inducible cAMP signaling in leukocytes is thought to be associated with inhibition of cellular responses; therefore, we explored the possibility that forskolin could affect antibacterial functions (eg, phagocytosis and ROS activity) of inflammatory cells, which may have unfavorable effects in acute pyelonephritis. Previous studies have found that elevation of intracellular cAMP is able to mediate the inhibition of receptor (FcR, complement receptor)-mediated phagocytosis and ROS activity in human neutrophils.17,37 However, our data in the phagocytosis experiments (in the absence of FcR and complement receptor ligands) indicate that forskolin treatment had no effect on dextran-bead uptake rates of neutrophils and macrophages (data not shown), which do not support the inhibitory role of forskolin in phagocytosis, although we cannot exclude the possibility that in vivo, in the presence of FcR or complement receptor ligands, forskolin may have an inhibitory effect on phagocytosis. Our data in the ROS experiments indicate that forskolin treatment resulted in a small reduction in ROS activity of neutrophils, which is consistent with the previous observation, but forskolin treatment had no apparent effect on ROS activity of macrophages. Although ROS activity of neutrophils is essential to host defense, their improper activation often causes unwanted tissue damage. Therefore, forskolin-mediated mild inhibition of superoxide generation may have a favorable effect in acute pyelonephritis. Nevertheless, on the basis of our current data in vitro and in vivo, it is tempting to speculate that forskolin may not mediate significantly unfavorable effects through acting on inflammatory cells in acute pyelonephritis.
On the basis of our present findings together with the literature on the properties of cAMP/forskolin,15–17,19,38 we propose a mechanism by which forskolin confers the protection against murine acute pyelonephritis. During UTI, elevation of intracellular cAMP by forskolin has a direct influence on the cellular migratory apparatus of inflammatory cells (eg, neutrophils, monocytes/macrophages) and results in the inhibition of their migration (Figure 8). Elevation of intracellular cAMP also has inhibitory effects on the production of most proinflammatory cytokines (eg, KC, IL-1β, TNF-α, and MCP-1) in both RTECs and inflammatory cells. Furthermore, elevation of intracellular cAMP has a suppressive effect on E. coli invasion of RTECs. Consequently, the lowered migratory capacity of inflammatory cells and reduced local production of proinflammatory mediators lead to the attenuation of cellular infiltration and tissue damage; the attenuated tissue damage and bacteria invasion of RTECs contribute to the reduced bacteria load in the kidneys.
Figure 8.

A proposed mechanism by which forskolin confers the protection against murine acute pyelonephritis. Administration of forskolin leads to elevation of intracellular cAMP, which mediates the inhibition of inflammatory cell migration, down-regulation of the production of proinflammatory mediators by RTECs and inflammatory cells, and limitation of bacteria invasion of RTECs. All of these effects contribute to the attenuation of cellular infiltration, bacteria load, and tissue damage in the kidneys of forskolin-treated mice. Mo, monocyte; Mϕ, macrophage; RTEC, renal tubular epithelial cell.
Conclusion
This study finds that activation of the endogenous anti-inflammatory mediator cAMP by forskolin confers protection in murine acute pyelonephritis and highlights the importance of excessive innate immune responses and invasion of renal epithelial cells in the pathogenesis of acute pyelonephritis. Our findings together with previous observations in bladder infection strongly suggest that pharmacotherapies that increase intracellular cAMP could be alternative or additional therapies for improving current treatment of acute pyelonephritis.
Acknowledgments
K.L. and W.Z. designed the study/experiments; Y.W., K.L., G.-D.C., and Y.L. performed in vivo animal experiments; N.W., B.-S.G., E.-Q.L., and Z.-F.L. performed renal histology and immunochemical staining and assessed renal scores; Y.W. and T.Z. performed in vitro experiments and analyzed data; K.L. and W.Z. prepared the manuscript; the other authors read and commented on the manuscript.
Footnotes
Funded by National Natural Science Foundation of China grant NSFC 81170644 (K.L.), Henry Fok Education Foundation grant 131040 (K.L.), and Medical Research Council of the UK grant G1001141 (W.Z.).
Y.W. and K.L. contributed equally to this work.
K.L. and W.Z. both contributed equally to this work as senior authors.
Disclosures: None declared.
Contributor Information
Ke Li, Email: ke.li@mail.xjtu.edu.cn.
Wuding Zhou, Email: wuding.zhou@kcl.ac.uk.
Supplemental Data
BUN concentrations do not differ significantly between the control and forskolin-treated groups. Serum samples were collected from control and forskolin-treated mice at different time points after infection. BUN was measured with a standard urease kit, based on the measurement of coupled enzyme reactions that involved urease and glutamate dehydrogenase. The dashed line indicates a basal concentration of BUN in naive mice. Data are expressed as means ± SEM. n = 5 to 6 mice per group at each time point. BUN, blood urea nitrogen; Ctrl, control; Fsk, forskolin.
Forskolin treatment has no effect on bacteria (J96) growth and their ability to invade RTECs. Bacteria (J96, 1 × 107 CFU) were preincubated with or without 10 mmol/L forskolin for 1 hour and then thoroughly washed three times with PBS. The washed bacteria were used for assessing their growth and ability to invade RTECs. A: Bacteria growth was determined by counting CFUs on agar plates. B: Bacterial ability to invade RTECs was determined by internalization assay. For this assay, control or forskolin-treated bacteria were incubated with RTECs for 1 hour at 37°C; after washing, RTECs were incubated for 1 hour with 100 μg/mL gentamicin to kill extracellular bacteria and lyzed. The lysate was plated onto CLED plates, and the colonies were counted. Data are expressed as means ± SEM. n = 5. CFU, colony-forming unit; CLED, cysteine lactose electrolyte deficient; Ctrl, control; Fsk, forskolin; PBS, phosphate-buffered saline; RTEC, renal tubular epithelial cell.
Forskolin has no effect on RTEC viability but has inhibitory effect on bacterial (J96) uptake by RTECs. Primary RTECs were preincubated with forskolin at different concentrations for 4 hours and washed thoroughly three times with PBS. The washed cells were used for assessing their viability (A) and uptake capacity (B). A: RTEC viability was determined by using MTT. Following the manufacturer's instructions, 100 μL of 5 mg/mL MTT solution was added to each well (containing 400 μL of culture medium) and incubated for 4 hours at 37°C. Thereafter, MTT solution was removed. After adding 600 μL of DMSO the plates were incubated for 15 minutes at 37°C to dissolve the formazan crystals. Absorbance readings of DMSO extracts were performed at 490 nm. B: RTEC uptake capacity was determined by performing internalization assay. For this assay, forskolin-treated RTECs were incubated with bacteria for 1 hour at 37°C; after washing, RTECs were incubated for 1 hour with 100 μg/mL gentamicin to kill extracellular bacteria and lyzed. The lysate was plated onto CLED plates, and the colonies were counted. Data are expressed as means ± SEM. n = 6. ∗∗P < 0.01, Student t-test. CLED, cysteine lactose electrolyte deficient; Ctrl, control; DMSO, dimethyl sulfoxide; Fsk, forskolin; MTT, 3-(4,5-dimethyl-1,3-thiazol-2-yl)-2, 5-diphenyl-2H-tetrazol-3-ium bromide; PBS, phosphate-buffered saline; RTEC, renal tubular epithelial cell.
References
- 1.Shepherd A.K., Pottinger P.S. Management of urinary tract infections in the era of increasing antimicrobial resistance. Med Clin North Am. 2013;97:737–757. doi: 10.1016/j.mcna.2013.03.006. xii. [DOI] [PubMed] [Google Scholar]
- 2.Stamm W.E., Norrby S.R. Urinary tract infections: disease panorama and challenges. J Infect Dis. 2001;183(Suppl 1):S1–S4. doi: 10.1086/318850. [DOI] [PubMed] [Google Scholar]
- 3.Ramakrishnan K., Scheid D.C. Diagnosis and management of acute pyelonephritis in adults. Am Fam Physician. 2005;71:933–942. [PubMed] [Google Scholar]
- 4.Ragnarsdottir B., Lutay N., Gronberg-Hernandez J., Koves B., Svanborg C. Genetics of innate immunity and UTI susceptibility. Nat Rev Urol. 2011;8:449–468. doi: 10.1038/nrurol.2011.100. [DOI] [PubMed] [Google Scholar]
- 5.Taylor P.W. Bactericidal and bacteriolytic activity of serum against gram-negative bacteria. Microbiol Rev. 1983;47:46–83. doi: 10.1128/mr.47.1.46-83.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li K., Zhou W., Hong Y., Sacks S.H., Sheerin N.S. Synergy between type 1 fimbriae expression and C3 opsonisation increases internalisation of E. coli by human tubular epithelial cells. BMC Microbiol. 2009;9:64. doi: 10.1186/1471-2180-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Springall T., Sheerin N.S., Abe K., Holers V.M., Wan H., Sacks S.H. Epithelial secretion of C3 promotes colonization of the upper urinary tract by Escherichia coli. Nat Med. 2001;7:801–806. doi: 10.1038/89923. [DOI] [PubMed] [Google Scholar]
- 8.Li K., Feito M.J., Sacks S.H., Sheerin N.S. CD46 (membrane cofactor protein) acts as a human epithelial cell receptor for internalization of opsonized uropathogenic Escherichia coli. J Immunol. 2006;177:2543–2551. doi: 10.4049/jimmunol.177.4.2543. [DOI] [PubMed] [Google Scholar]
- 9.Cirl C., Wieser A., Yadav M., Duerr S., Schubert S., Fischer H., Stappert D., Wantia N., Rodriguez N., Wagner H., Svanborg C., Miethke T. Subversion of Toll-like receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor domain-containing proteins. Nat Med. 2008;14:399–406. doi: 10.1038/nm1734. [DOI] [PubMed] [Google Scholar]
- 10.Wullt B., Bergsten G., Fischer H., Godaly G., Karpman D., Leijonhufvud I., Lundstedt A.C., Samuelsson P., Samuelsson M., Svensson M.L., Svanborg C. The host response to urinary tract infection. Infect Dis Clin North Am. 2003;17:279–301. doi: 10.1016/s0891-5520(03)00028-x. [DOI] [PubMed] [Google Scholar]
- 11.Kennedy A.D., DeLeo F.R. Neutrophil apoptosis and the resolution of infection. Immunol Res. 2009;43:25–61. doi: 10.1007/s12026-008-8049-6. [DOI] [PubMed] [Google Scholar]
- 12.Yadav M., Zhang J., Fischer H., Huang W., Lutay N., Cirl C., Lum J., Miethke T., Svanborg C. Inhibition of TIR domain signaling by TcpC: MyD88-dependent and independent effects on Escherichia coli virulence. PLoS Pathog. 2010;6:e1001120. doi: 10.1371/journal.ppat.1001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beavo J.A., Brunton L.L. Cyclic nucleotide research – still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3:710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- 14.Lefkimmiatis K., Zaccolo M. cAMP signaling in subcellular compartments. Pharmacol Ther. 2014;143:295–304. doi: 10.1016/j.pharmthera.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zidek Z. Adenosine - cyclic AMP pathways and cytokine expression. Eur Cytokine Netw. 1999;10:319–328. [PubMed] [Google Scholar]
- 16.Burdyga A., Conant A., Haynes L., Zhang J., Jalink K., Sutton R., Neoptolemos J., Costello E., Tepikin A. cAMP inhibits migration, ruffling and paxillin accumulation in focal adhesions of pancreatic ductal adenocarcinoma cells: effects of PKA and EPAC. Biochim Biophys Acta. 2013;1833:2664–2672. doi: 10.1016/j.bbamcr.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin P., Welch E.J., Gao X.P., Malik A.B., Ye R.D. Lysophosphatidylcholine modulates neutrophil oxidant production through elevation of cyclic AMP. J Immunol. 2005;174:2981–2989. doi: 10.4049/jimmunol.174.5.2981. [DOI] [PubMed] [Google Scholar]
- 18.Seino S., Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- 19.Bishop B.L., Duncan M.J., Song J., Li G., Zaas D., Abraham S.N. Cyclic AMP-regulated exocytosis of Escherichia coli from infected bladder epithelial cells. Nat Med. 2007;13:625–630. doi: 10.1038/nm1572. [DOI] [PubMed] [Google Scholar]
- 20.Wajima Z., Yoshikawa T., Ogura A., Imanaga K., Shiga T., Inoue T., Ogawa R. Intravenous colforsin daropate, a water-soluble forskolin derivative, prevents thiamylal-fentanyl-induced bronchoconstriction in humans. Crit Care Med. 2002;30:820–826. doi: 10.1097/00003246-200204000-00017. [DOI] [PubMed] [Google Scholar]
- 21.Murata S., Miniati D.N., Kown M.H., Koransky M.L., Balsam L.B., Lijkwan M.A., Martens J.M., Robbins R.C. Elevated cyclic adenosine monophosphate ameliorates ischemia-reperfusion injury in rat cardiac allografts. J Heart Lung Transplant. 2003;22:802–809. doi: 10.1016/s1053-2498(02)00651-4. [DOI] [PubMed] [Google Scholar]
- 22.Meyer B.H., Stulting A.A., Muller F.O., Luus H.G., Badian M. The effects of forskolin eye drops on intra-ocular pressure. S Afr Med J. 1987;71:570–571. [PubMed] [Google Scholar]
- 23.Styczynski J., Wysocki M. Ex vivo modulation of response to prednisolone in childhood acute lymphoblastic leukaemia. Br J Haematol. 2006;133:397–399. doi: 10.1111/j.1365-2141.2006.06032.x. [DOI] [PubMed] [Google Scholar]
- 24.O'Hanley P., Lark D., Falkow S., Schoolnik G. Molecular basis of Escherichia coli colonization of the upper urinary tract in BALB/c mice. Gal-Gal pili immunization prevents Escherichia coli pyelonephritis in the BALB/c mouse model of human pyelonephritis. J Clin Invest. 1985;75:347–360. doi: 10.1172/JCI111707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strait R.T., Hicks W., Barasa N., Mahler A., Khodoun M., Kohl J., Stringer K., Witte D., Van Rooijen N., Susskind B.M., Finkelman F.D. MHC class I-specific antibody binding to nonhematopoietic cells drives complement activation to induce transfusion-related acute lung injury in mice. J Exp Med. 2011;208:2525–2544. doi: 10.1084/jem.20110159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagberg L., Engberg I., Freter R., Lam J., Olling S., Svanborg Edén C. Ascending, unobstructed urinary tract infection in mice caused by pyelonephritogenic Escherichia coli of human origin. Infect Immun. 1983;40:273–283. doi: 10.1128/iai.40.1.273-283.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tseng C.C., Huang J.J., Wang M.C., Wu A.B., Ko W.C., Chen W.C., Wu J.J. PapG II adhesin in the establishment and persistence of Escherichia coli infection in mouse kidneys. Kidney Int. 2007;71:764–770. doi: 10.1038/sj.ki.5002111. [DOI] [PubMed] [Google Scholar]
- 28.Dong X., Swaminathan S., Bachman L.A., Croatt A.J., Nath K.A., Griffin M.D. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 2007;71:619–628. doi: 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- 29.Li K., Patel H., Farrar C.A., Hargreaves R.E., Sacks S.H., Zhou W. Complement activation regulates the capacity of proximal tubular epithelial cell to stimulate alloreactive T cell response. J Am Soc Nephrol. 2004;15:2414–2422. doi: 10.1097/01.ASN.0000135974.06478.7B. [DOI] [PubMed] [Google Scholar]
- 30.Peng Q., Li K., Smyth L.A., Xing G., Wang N., Meader L., Lu B., Sacks S.H., Zhou W. C3a and C5a promote renal ischemia-reperfusion injury. J Am Soc Nephrol. 2012;23:1474–1485. doi: 10.1681/ASN.2011111072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Rouschop K.M., Sewnath M.E., Claessen N., Roelofs J.J., Hoedemaeker I., van der Neut R., Aten J., Pals S.T., Weening J.J., Florquin S. CD44 deficiency increases tubular damage but reduces renal fibrosis in obstructive nephropathy. J Am Soc Nephrol. 2004;15:674–686. doi: 10.1097/01.asn.0000115703.30835.96. [DOI] [PubMed] [Google Scholar]
- 33.Ward P.A. New approaches to the study of sepsis. EMBO Mol Med. 2012;4:1234–1243. doi: 10.1002/emmm.201201375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel S.N., Berghout J., Lovegrove F.E., Ayi K., Conroy A., Serghides L., Min-oo G., Gowda D.C., Sarma J.V., Rittirsch D., Ward P.A., Liles W.C., Gros P., Kain K.C. C5 deficiency and C5a or C5aR blockade protects against cerebral malaria. J Exp Med. 2008;205:1133–1143. doi: 10.1084/jem.20072248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song J., Bishop B.L., Li G., Duncan M.J., Abraham S.N. TLR4-initiated and cAMP-mediated abrogation of bacterial invasion of the bladder. Cell Host Microbe. 2007;1:287–298. doi: 10.1016/j.chom.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song J., Bishop B.L., Li G., Grady R., Stapleton A., Abraham S.N. TLR4-mediated expulsion of bacteria from infected bladder epithelial cells. Proc Natl Acad Sci U S A. 2009;106:14966–14971. doi: 10.1073/pnas.0900527106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ydrenius L., Majeed M., Rasmusson B.J., Stendahl O., Sarndahl E. Activation of cAMP-dependent protein kinase is necessary for actin rearrangements in human neutrophils during phagocytosis. J Leukoc Biol. 2000;67:520–528. doi: 10.1002/jlb.67.4.520. [DOI] [PubMed] [Google Scholar]
- 38.Szaszak M., Christian F., Rosenthal W., Klussmann E. Compartmentalized cAMP signalling in regulated exocytic processes in non-neuronal cells. Cell Signal. 2008;20:590–601. doi: 10.1016/j.cellsig.2007.10.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
BUN concentrations do not differ significantly between the control and forskolin-treated groups. Serum samples were collected from control and forskolin-treated mice at different time points after infection. BUN was measured with a standard urease kit, based on the measurement of coupled enzyme reactions that involved urease and glutamate dehydrogenase. The dashed line indicates a basal concentration of BUN in naive mice. Data are expressed as means ± SEM. n = 5 to 6 mice per group at each time point. BUN, blood urea nitrogen; Ctrl, control; Fsk, forskolin.
Forskolin treatment has no effect on bacteria (J96) growth and their ability to invade RTECs. Bacteria (J96, 1 × 107 CFU) were preincubated with or without 10 mmol/L forskolin for 1 hour and then thoroughly washed three times with PBS. The washed bacteria were used for assessing their growth and ability to invade RTECs. A: Bacteria growth was determined by counting CFUs on agar plates. B: Bacterial ability to invade RTECs was determined by internalization assay. For this assay, control or forskolin-treated bacteria were incubated with RTECs for 1 hour at 37°C; after washing, RTECs were incubated for 1 hour with 100 μg/mL gentamicin to kill extracellular bacteria and lyzed. The lysate was plated onto CLED plates, and the colonies were counted. Data are expressed as means ± SEM. n = 5. CFU, colony-forming unit; CLED, cysteine lactose electrolyte deficient; Ctrl, control; Fsk, forskolin; PBS, phosphate-buffered saline; RTEC, renal tubular epithelial cell.
Forskolin has no effect on RTEC viability but has inhibitory effect on bacterial (J96) uptake by RTECs. Primary RTECs were preincubated with forskolin at different concentrations for 4 hours and washed thoroughly three times with PBS. The washed cells were used for assessing their viability (A) and uptake capacity (B). A: RTEC viability was determined by using MTT. Following the manufacturer's instructions, 100 μL of 5 mg/mL MTT solution was added to each well (containing 400 μL of culture medium) and incubated for 4 hours at 37°C. Thereafter, MTT solution was removed. After adding 600 μL of DMSO the plates were incubated for 15 minutes at 37°C to dissolve the formazan crystals. Absorbance readings of DMSO extracts were performed at 490 nm. B: RTEC uptake capacity was determined by performing internalization assay. For this assay, forskolin-treated RTECs were incubated with bacteria for 1 hour at 37°C; after washing, RTECs were incubated for 1 hour with 100 μg/mL gentamicin to kill extracellular bacteria and lyzed. The lysate was plated onto CLED plates, and the colonies were counted. Data are expressed as means ± SEM. n = 6. ∗∗P < 0.01, Student t-test. CLED, cysteine lactose electrolyte deficient; Ctrl, control; DMSO, dimethyl sulfoxide; Fsk, forskolin; MTT, 3-(4,5-dimethyl-1,3-thiazol-2-yl)-2, 5-diphenyl-2H-tetrazol-3-ium bromide; PBS, phosphate-buffered saline; RTEC, renal tubular epithelial cell.