

Abstract
The human genome reference assembly is crucial for aligning and analyzing sequence data, and for genome annotation, among other roles. However, the models and analysis assumptions that underlie the current assembly need revising to fully represent human sequence diversity. Improved analysis tools and updated data reporting formats are also required.
Background
One of the flagship products of the Human Genome Project (HGP) was a high-quality human reference assembly [1]. This assembly, coupled with advances in low-cost, high-throughput sequencing, has allowed us to address previously inaccessible questions about population diversity, genome structure, gene expression and regulation [2-5]. It has become clear, however, that the original models used to represent the reference assembly inadequately represent our current understanding of genome architecture.
The first assembly models were designed for simple ‘linear’ genome sequences, with little sequence variation and even less structural diversity. The design fit the understanding of human variation at the time the HGP began [6]. The HGP constructed the reference assembly by collapsing sequences from over 50 individuals into a single consensus haplotype representation of each chromosome. Employing a clone-based approach, the sequence of each clone represented a single haplotype from a given donor. At clone boundaries, however, haplotypes could switch abruptly, creating a mosaic structure. This design introduced errors within regions of complex structural variation, when sequences unique to one haplotype prevented construction of clone overlaps. The assembly therefore inadvertently included multiple haplotypes in series in some regions [7-9].
The Genome Reference Consortium (GRC) began stewardship of the reference assembly in 2007. The GRC proposed a new assembly model that formalized the inclusion of ‘alternative sequence paths’ in regions with complex structural variation, and then released GRCh37 using this new model [10]. The release of GRCh37 also marked the deposition of the human reference assembly to an International Nucleotide Sequence Database Collaboration (INSDC) database, providing stable, trackable sequence identifiers, in the form of accession and version numbers, for all sequences in the assembly. The GRC developed an assembly model that was incorporated into the National Centre for Biotechnology Information (NCBI) and European Nucleotide Archive (ENA) assembly database that provides a stable identifier for the collection of sequences and the relationship between these sequences that comprise an assembly [11]. Subsequent minor assembly releases added a number of ‘fix patches’ that could be used to resolve mistakes in the reference sequence, as well as ‘novel patches’ that are new alternative sequence representations [10].
The new assembly model presents significant advances to the genomics community, but, to realize those advances, we must address many technical challenges. The new assembly model is neither haploid nor diploid - instead, it includes additional scaffold sequences, aligned to the chromosome assembly, that provide alternative sequence representations for regions of excess diversity. Widely used alignment programs, variant discovery and analysis tools, as well as most reporting formats, expect reads and features to have a single location in the reference assembly as they were developed using a haploid assembly model. Many alignment and analysis tools penalize reads that align to more than one location under the assumption that the location of these reads cannot be resolved owing to paralogous sequences in the genome. These tools do not distinguish allelic duplication, added by the alternative loci, from paralogous duplication found in the genome, thus confounding repeat and mappability calculations, paired-end placements and downstream interpretation of alignments in regions with alternative loci.
To determine the efforts needed to facilitate use of the full assembly, the GRC organized a workshop in conjunction with the 2014 Genome Informatics meeting in Cambridge, UK (http://www.slideshare.net/GenomeRef). Participants identified challenges presented by the new assembly model and discussed ways forward that we describe here.
Towards the graph of human variation
A graph structure is a natural way to represent a population-based genome assembly, with branches in the graph representing all variation found within the source sequences. Most assembly programs internally use a graph representation to build the assembly, but ultimately produce a flattened structure for use by downstream tools [12-14]. Recently, formal proposals for representing a population-based reference graph have been described [15-17]. The newly formed Global Alliance for Genomics and Health (GA4GH) is leading an effort to formalize data structures for graph-based reference assemblies, but it will likely take years to develop the infrastructure and analysis tools needed to support these new structures and see their widespread adoption across the biological and clinical research communities [18].
The introduction of alternative loci into the assembly model provides a stepping-stone towards a full graph-based representation of a population-based reference genome. The alternative loci provided by the GRC are based on high-quality, finished sequence. Although it is not feasible to represent all known variation using the alternative locus scheme, this model does allow us to better represent regions with extreme levels of diversity. Alternative loci are not meant to represent all variation within a population, but rather provide an immediate solution for adding sequences missing from the chromosome assembly. In practice, alternative locus addition is limited by the availability of high-quality genomic sequence, and the GRC has focused on representing sequence at the most diverse regions, such as the major histocompatibility complex (MHC). The representation of all population variation is better suited to a graph-based representation. The high quality of the sequence at these locations provides robust data to test graph implementations. Additionally, because both NCBI and Ensembl have annotated these sequences, we can also begin to address how to annotate graph structures at these complex loci.
While GRCh37 had only three regions containing nine alternative locus sequences, GRCh38 has 178 regions containing 261 alternative locus sequences, collectively representing 3.6 Mbp of novel sequence and over 150 genes not represented in the primary assembly (Table 1). The increased level of alternative sequence representation intensifies the urgency to develop new analysis methods to support inclusion of these sequences. Inclusion of all sequences in the reference assembly allows us to better analyze these regions with potentially modest updates to currently used tools and reporting structures. Although the addition of the alternative loci to current analysis pipelines might lead to only modest gains in analysis power on a genome-wide scale, some loci will see considerable improvement owing to the addition of significant amounts of sequence that cannot be represented accurately in the chromosome assembly (Figure 1).
Table 1.
Examples of regions with alternative loci, sequences within these regions and genes unique to them
| Region name | Sequence | Chromosome | Unique genes |
|---|---|---|---|
| APOBEC | GL383583.2 | 22 | APOBEC3A_B |
| MHC | GL000250.2 | 6 | HLA-DRB2 |
| GL000251.2 | HLA-DRB3 | ||
| GL000252.2 | HLA-DRB4 | ||
| GL000253.2 | HLA-DRB7 | ||
| GL000254.2 | |||
| GL000255.2 | |||
| GL000256.2 | |||
| KI270758.1 | |||
| CCL5_TBC1D3 | KI270909.1 | 17 | CCL3L1 |
| CCL4L1 | |||
| CCL3P1 | |||
| CYP2D6 | KB663069.1 | 22 | LOC101929829 (CYP2D6 pseudogene) |
| LRC_KIR | KI270938.1, | 19 | LILRA3 |
| GL949747.2 | |||
| GL000209.2, | KIR2DS1 | ||
| GL949747.2 | |||
| KI270882.1, | |||
| KI270887.1, | |||
| KI270890.1, | |||
| KI270916.1, | |||
| KI270921.1 | |||
| TRB | KI270803.1 | 7 | TRBV5-8 |
Figure 1.
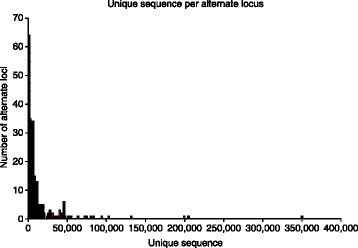
Histogram displaying unique sequence per alternative locus. While the vast majority of alternative loci contribute less than 50 kb of unique sequence, some contribute well over 100 kb of novel sequence.
Omission of the novel sequence contained in the alternative loci can lead to off-target sequence alignments, and thus incorrect variant calls or other errors, when a sample containing the alternative allele is sequenced and aligned to only the primary assembly. Using reads simulated from the unique portion of the alternative loci, we found that approximately 75% of the reads had an off-target alignment when aligned to the primary assembly alone. This finding was consistent using different alignment methods [10]. The 1000 Genomes Project also observed the detrimental effect of missing sequences and developed a ‘decoy’ sequence dataset in an effort to minimize off-target alignments [19,20]. Much of this decoy has now been incorporated into GRCh38, and analysis of reads taken from 1000 Genomes samples that previously mapped only to the decoy shows that approximately 70% of these now align to the full GRCh38, with approximately 1% of these reads aligning only to the alternative loci (Figure 2).
Figure 2.
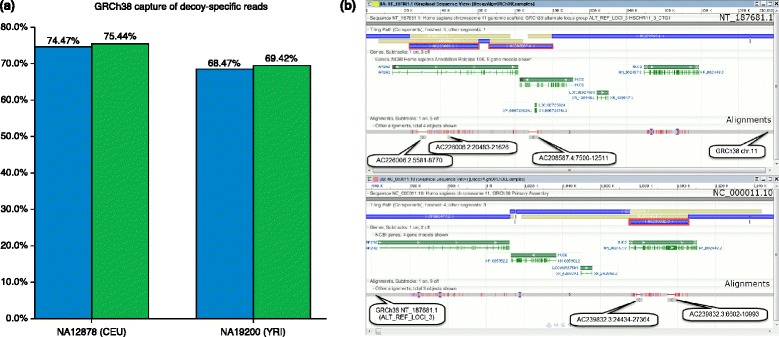
(a) Alignment of decoy-specific reads to GRCh38. To identify decoy-specific reads, the following read sets were used: /panfs/traces01/compress/1KG.p2/NA19200.mapped.ILLUMINA.bwa.YRI.low_coverage.20120522.8levels.csra and /panfs/traces01/compress/1KG.p2/NA12878.mapped.ILLUMINA.bwa.CEU.low_coverage.20120522.8levels.csra. Using SRPRISM [26] (with parameters [p] (force paired/unpaired search): false; [n] (maximum number of allowed errors): 6; [M] (maximum allowed memory usage): 2048.) reads were extracted with MAPQ >20 that uniquely aligned to SN:hs37d5 (the decoy). As a control, these reads were aligned to a target set comprising the GRCh37 primary assembly unit (GCF_000001305.13), chr. MT (GCF_000006015.1) and the decoy. Collectively, this target set is referred to as GRCh37pmd. To assess capture of the decoy sequence in the updated assembly, the reads were aligned to a target set comprising the GRCh38 primary assembly (GCF_000001305.14) and chr. MT (GCF_000006015.1) (referred to as GRCh38pm) or the full GRCh38 assembly (GCF_000001405.26). A captured read was defined as one that aligned only to the decoy (by SRPRISM) when using GRCh37pmd as the target set, and also aligned to GRCh38pm or full. (b) Incorporation of decoy sequences in GRCh38. Upper panel: graphical view of NT_187681.1, a GRCh38 alternative loci scaffold. Blue bars are assembly components; gene annotations are shown in green. Two components (AC226006.2 and AC208587.4, highlighted in red) in this alt scaffold are fosmids from which decoy sequences were derived (AC208587.4:7500–12511, AC226006.2:5581–8770, AC226006.2:20483–21626). The decoy sequences represent variants from the chromosome. The alignment of the decoy fragments and GRCh38 chr. 11 to the alternative scaffold are shown as gray bars (red tick, mismatch; blue tick, deletion; thin red line, insertion). Note that the decoy fragments align to regions that are missing from the reference chromosome. Lower panel: graphical view of NC_000011.10, GRCh38 chr.11. An assembly component (AC239832.2, highlighted in red) added to this region for GRCh38 is a fosmid from which decoy sequence was derived (AC239832.3:6602–10993, AC239832.3:24434–27364). In this case, the decoy adds exonic sequence from MUC2 that is missing from component AC139749.4. The alignments of the decoy fragments and NT_187681.1 to the alternative scaffold are shown as gray bars (red tick, mismatch; blue tick, deletion; thin red line, insertion).
We foresee many computational approaches that allow the inclusion of all assembly sequences in analysis pipelines. To better support exploration in this area, we propose some improvements to standard practices and data structures that will facilitate future development.
Enhancement of standard reporting formats (such as BAM/CRAM, VCF/BCF, GFF3) so that they can accommodate features with multiple locations. Doing so while maintaining the allelic relationship between these features is crucial [21-24].
Adoption of standard sequence identifiers for sequence analysis and reporting. Using shorthand identifiers (for example, ‘chr1’ or ‘1’) to indicate the sequence is imprecise and also ignores the presence of other sequences in the assembly. In many cases, other top-level sequences, such as unlocalized scaffolds, patches and alternative loci, have a chromosome assignment but not chromosome coordinates. These sequences are independent of the chromosome assembly coordinate system and have their own coordinate space. Alternative loci are related to the chromosome coordinates through alignment to the chromosome assembly. Developing a structure that treats all top-level sequences as first-class citizens during analysis is an important step towards adopting use of the full assembly in analysis pipelines.
Curation of multiple sequence alignments of the alternative loci to each other and the primary path. Currently, pairwise alignments of the alternative loci to the chromosome assembly are available to provide the allelic relationship between the alternative locus and the chromosome. However, these pairwise alignments do not allow for the comparison of alternative loci in a given region to each other. These alignments can also be used to develop graph structures. The relationship of the allelic sequences within a region helps define the assembly structure, and the community should work from a single set of alignments. These should be distributed with the GRC assembly releases.
Recently, the GRC has released a track hub [25] that allows for the distribution of GRC data using standard track names and content (http://ngs.sanger.ac.uk/production/grit/track_hub/hub.txt). Additionally, the GRC has created a GitHub page to track development of tools and resources that facilitate use of the full assembly (https://github.com/GenomeRef/SoftwareDevTracking).
Concluding remarks
As we gain understanding of biological systems, we must update the models we use to represent these data. This can be difficult when the model supports common infrastructure and analysis tools used by a large swath of the scientific community. However, this growth is crucial in order to move the scientific community forward. While adoption of this new model will take substantial effort, doing so is an important step for the human genetics and broader genomics communities. We now have an opportunity and imperative to revisit old assumptions and conventions to develop a more robust analysis framework. The use of all sequences included in the reference will allow for improved genomic analyses and understanding of genomic architecture. Additionally, this new assembly model allows us to take a small step towards the realization of a graph-based assembly representation. The evolution of the assembly model allows us to improve our understanding of genomic architecture and provides a framework for boosting our understanding of how this architecture impacts human development and disease.
Acknowledgements
We thank Daniel MacArthur, Jen Harrow and Mike Schatz, the organizers, for Genome Informatics 2014, for facilitating the GRC workshop, and Personalis Inc. for sponsorship. We also thank Laura Clarke for comments on the manuscript.
This research was supported in part by the Intramural Research Program of the NIH, National Library of Medicine (VAS, PAK), the Princess Margaret Cancer Foundation (MMH), the Wellcome Trust (WT095908), the National Human Genome Research Institute (U41HG007234), and the European Molecular Biology Laboratory (BA, PF).
Abbreviations
- ENA
European nucleotide archive
- GA4GH
Global alliance for genomics and health
- GRC
Genome reference consortium
- HGP
Human genome project
- INSDC
International nucleotide sequence database collaboration
- NCBI
National center for biotechnology information
Footnotes
Competing interests
DMC is an employee of Personalis, Inc. VAS, KMS, MCS, ARQ, C-SC, PAK, BA, GTM, MMH, JH, MLZM and PF declare no competing interests.
Authors’ contributions
DMC organized the workshop, moderated the workshop and wrote the manuscript; VAS presented at the GRC workshop, performed the decoy analysis and edited the manuscript; KMS participated in the GRC workshop and edited the manuscript; MCS presented at the GRC workshop and edited the manuscript; ARQ presented at the GRC workshop and edited the manuscript; C-SC presented at the GRC workshop and edited the manuscript; PAK presented at the GRC workshop and edited the manuscript; BA presented at the GRC workshop and edited the manuscript; GTM presented at the workshop and edited the manuscript; MMH summarized the GRC workshop discussion and helped write the manuscript; JH participated in the GRC workshop and edited the manuscript; MLZM provided notes from the GRC workshop and edited the manuscript; PF participated in the GRC workshop and edited the manuscript.
Contributor Information
Deanna M Church, Email: deanna.church@personalis.com.
Valerie A Schneider, Email: schneiva@ncbi.nlm.nih.gov.
Karyn Meltz Steinberg, Email: kmeltzst@genome.wustl.edu.
Michael C Schatz, Email: mschatz@cshl.edu.
Aaron R Quinlan, Email: aaronquinlan@gmail.com.
Chen-Shan Chin, Email: jchin@pacificbiosciences.com.
Paul A Kitts, Email: kitts@ncbi.nlm.nih.gov.
Bronwen Aken, Email: ba1@ebi.ac.uk.
Gabor T Marth, Email: gmarth@genetics.utah.edu.
Michael M Hoffman, Email: mhoffman@uhnresearch.ca.
Javier Herrero, Email: javier.herrero@ucl.ac.uk.
M Lisandra Zepeda Mendoza, Email: lisandracady@gmail.com.
Richard Durbin, Email: rd@sanger.ac.uk.
Paul Flicek, Email: flicek@ebi.ac.uk.
References
- 1.Consortium IHGS. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 2.Durbin R. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Consortium T 1000 GP An integrated map of genetic variation. Nature. 2012;491:59–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–5. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins FS. New goals for the U.S. Human Genome Project: 1998–2003. Science. 1998;282:682–9. doi: 10.1126/science.282.5389.682. [DOI] [PubMed] [Google Scholar]
- 7.Xue Y, Sun D, Daly A, Yang F, Zhou X, Zhao M, et al. Adaptive evolution of UGT2B17 copy-number variation. Am J Hum Genet. 2008;83:337–46. doi: 10.1016/j.ajhg.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dennis MY, Nuttle X, Sudmant PH, Antonacci F, Graves TA, Nefedov M, et al. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell. 2012;149:912–22. doi: 10.1016/j.cell.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watson CT, Steinberg KM, Huddleston J, Warren RL, Malig M, Schein J, et al. Complete haplotype sequence of the human immunoglobulin heavy-chain variable, diversity, and joining genes and characterization of allelic and copy-number variation. Am J Hum Genet. 2013;92:530–46. doi: 10.1016/j.ajhg.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Church DM, Schneider VA, Graves T, Auger K, Cunningham F, Bouk N, et al. Modernizing reference genome assemblies. PLoS Biol. 2011;9:e1001091. doi: 10.1371/journal.pbio.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.NCBI Resource Coordinators Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2013;41(Database issue):D8–20. doi: 10.1093/nar/gks1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, et al. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–20. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schatz MC, Delcher AL, Salzberg SL. Assembly of large genomes using second-generation sequencing. Genome Res. 2010;20:1165–73. doi: 10.1101/gr.101360.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paten B, Novak A, Haussler D. Mapping to a reference genome structure. arXiv. 2014:1–26.
- 16.Dilthey A, Cox CJ, Iqbal Z, Cox C, Nelson MR, Mcvean G. Improved genome inference in the MHC using a population reference graph. BioRxiv. 2014. doi: http://dx.doi.org/10.1101/006973. [DOI] [PMC free article] [PubMed]
- 17.Marcus S, Lee H, Schatz MC. SplitMEM: A graphical algorithm for pan-genome analysis with suffix skips. Bioinformatics 2014:1–8. [DOI] [PMC free article] [PubMed]
- 18.Global Alliance for Global Health. http://genomicsandhealth.org/
- 19.1000 Genomes Decoy. ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/
- 20.Genovese G, Handsaker RE, Li H, Altemose N, Lindgren AM, Chambert K, et al. Using population admixture to help complete maps of the human genome. Nat Genet. 2013;45:406–14. doi: 10.1038/ng.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsi-Yang Fritz M, Leinonen R, Cochrane G, Birney E. Efficient storage of high throughput DNA sequencing data using reference-based compression. Genome Res. 2011;21:734–40. doi: 10.1101/gr.114819.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.GFF3. http://www.sequenceontology.org/gff3.shtml
- 25.Raney BJ, Dreszer TR, Barber GP, Clawson H, Fujita PA, Wang T, et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 2014;30:1003–5. doi: 10.1093/bioinformatics/btt637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.SRPRISM. ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/srprism