

Abstract
Intracellular pathogens are responsible for much of the world-wide morbidity and mortality due to infectious diseases. To colonize their hosts successfully, pathogens must sense their environment and regulate virulence gene expression appropriately. Accordingly, on entry into mammalian cells, the facultative intracellular bacterial pathogen Listeria monocytogenes remodels its transcriptional program by activating the master virulence regulator PrfA. Here we show that bacterial and host-derived glutathione are required to activate PrfA. In this study a genetic selection led to the identification of a bacterial mutant in glutathione synthase that exhibited reduced virulence gene expression and was attenuated 150-fold in mice. Genome sequencing of suppressor mutants that arose spontaneously in vivo revealed a single nucleotide change in prfA that locks the protein in the active conformation (PrfA*) and completely bypassed the requirement for glutathione during infection. Biochemical and genetic studies support a model in which glutathione-dependent PrfA activation is mediated by allosteric binding of glutathione to PrfA. Whereas glutathione and other low-molecular-weight thiols have important roles in redox homeostasis in all forms of life, here we demonstrate that glutathione represents a critical signalling molecule that activates the virulence of an intracellular pathogen.
Listeria monocytogenes is a Gram-positive pathogen of animals and humans that cycles between a saprophytic lifestyle and an intracellular pathogen that escapes from a vacuole and grows in the cytosol of host cells1. The intracellular lifecycle of L. monocytogenes has been well characterized and is entirely dependent on the transcription factor PrfA (refs 2, 3). PrfA directly regulates the transcription of nine virulence factors and is therefore referred to as the master virulence regulator in L. monocytogenes4. In keeping with its central role in pathogenesis, L. monocytogenes strains lacking prfA are completely avirulent1,3. PrfA is a member of the cAMP receptor protein (Crp) family of transcription factors, which are characterized by their allosteric regulation via small-molecule activators. In L. monocytogenes, PrfA is exclusively activated in the cytosol of host cells, leading to the assumption that the activating cofactor for PrfA is specific to this compartment. However, even after decades of study, the biochemical mechanism by which PrfA detects the intracellular environment is not well understood. The goal of this study was to identify how L. monocytogenes recognizes and responds to its intracellular niche of the mammalian cell cytosol.
Genetic selection in macrophages
We devised a genetic selection to isolate bacterial mutants unable to activate virulence genes during intracellular growth. Our strategy took advantage of a L. monocytogenes vaccine strain designed to die in vivo (P.L. et al., manuscript in preparation). Specifically, loxP sites were inserted into the L. monocytogenes chromosome flanking the origin of replication (ori, Fig. 1a). Into this background a codon-optimized cre recombinase gene was inserted under the control of the actA promoter, which is the most exquisitely regulated PrfA-dependent virulence gene in L. monocytogenes and is specifically activated in the host cytosol2,3,5,6. The resulting strain grew like wild type in vitro (Fig. 1b) where actA expression is very low4,5. However, on cytosolic access, Cre-mediated recombination of the loxP sites resulted in excision of the ori, preventing bacterial replication (Fig. 1c). A transposon library was then generated in this ‘suicide’ strain background. Bone-marrow-derived macrophages (BMDMs) were infected with the library of transposon mutants and the surviving bacteria were recovered.
Figure 1. Forward genetic selection to identify factors required for virulence gene activation during infection.
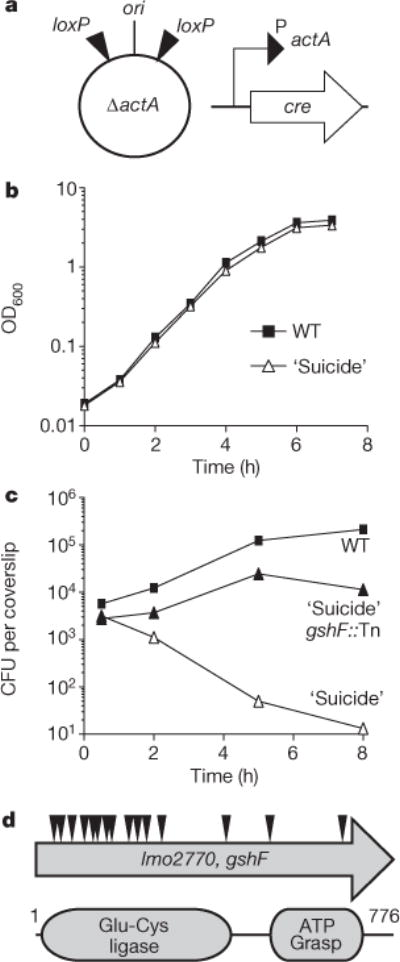
a, Schematic of the ‘suicide’ strain used for genetic selection. See text for description. b, Broth growth curve. Data are representative of three independent experiments. OD600, optical density at 600 nm. c, BMDM growth curve. Data are a combination of three independent experiments. d, Schematic of transposon insertions identified in gshF and the conserved protein domains.
We identified more than 16 independent insertions in a L. monocytogenes gene, previously identified as encoding a bifunctional glutathione synthase (gshF)7, that rescued the death of the ‘suicide’ strain in vivo (Fig. 1c, d). Glutathione is a tripeptide low-molecular-weight (LMW) thiol present in all eukaryotes that contain mitochondria and nearly all Gram-negative bacteria8. L. monocytogenes is one of the few Gram-positive bacteria that synthesize glutathione, whereas many utilize alternative LMW thiols such as bacillithiol and mycothiol9,10. Glutathione was not required for Cre/lox recombination when cre was expressed from a constitutive promoter (data not shown), leading to the hypothesis that glutathione was required specifically for activation of the actA promoter.
Glutathione is required for virulence
To determine the role of gshF in L. monocytogenes, an in-frame deletion strain was generated by allelic exchange (ΔgshF). Consistent with published work7, the gshF-deficient strain was moderately more sensitive to oxidative stress in vitro (Extended Data Fig. 1). However, ΔgshF did not suffer a general loss of fitness, as it exhibited no measurable growth defect in vitro (Fig. 2a) or in BMDMs (Fig. 2b). As expected based on the criteria of the genetic selection, the ΔgshF mutant expressed lower levels of ActA in cells (Fig. 2c), formed very small plaques in tissue culture assays that measure cell-to-cell spread (Fig. 2d), and was greater than 2-logs less virulent in mice (Fig. 2e). Complementation of ΔgshF with its native promoter (ΔgshF + gshF) restored wild-type ActA abundance, wild-type plaque size, and virulence (Fig. 2c–e). Since all mammalian cells have high intracellular levels of glutathione11, we assessed the potential contribution of host glutathione using buthionine sulfoximine (BSO). BSO depletes total cellular glutathione levels >98%12, but had no effect on bacterial growth during infection (Extended Data Fig. 2). Whereas wild-type L. monocytogenes was unaffected, the ΔgshF mutant failed to synthesize detectable ActA in the BSO-treated cells (Fig. 2f). These results demonstrated that the remaining ActA expression in the ΔgshF mutant was due to imported host glutathione and also established that the phenotypes observed for ΔgshF were due to a lack of glutathione and not absence of the GshF protein.
Figure 2. Listeria monocytogenes ΔgshF is attenuated in vivo.
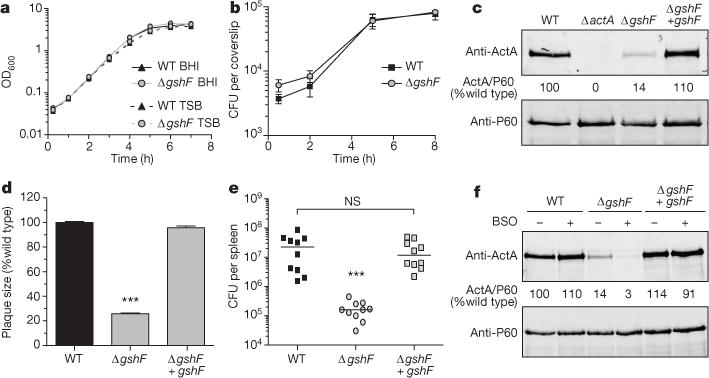
a, Broth growth curve in brain heart infusion (BHI) or tryptic soy broth (TSB). Data are representative of three independent experiments. b, BMDM growth curve. Mean ± standard error of the mean (s.e.m.) for three independent experiments is shown. c, Representative immunoblot of infected BMDMs. Numbers are the mean of four independent experiments and indicate ActA normalized to P60, as a per cent of wild type. d, Plaque size. Mean ± s.e.m. for three independent experiments is shown. e, CD-1 mice were infected intravenously and analysed as described in Methods. Data are a combination of two independent experiments, n = 10 mice per strain. The median of each group is represented as a horizontal line. f, Representative immunoblot of infected BMDMs. Quantification is as described in c. In all panels P values were calculated using Student’s t-test; ***P < 0.001; NS denotes P > 0.05.
Isolation of suppressor mutations in vivo
To elucidate the role of glutathione during infection we sought to isolate suppressor mutations. The selective pressure of the host environment was used to select for compensatory mutations in the ΔgshF background that restored virulence to identify functionally interacting genes and/or pathways. Since previous work identified gshF::Tn mutants as hypo-haemolytic13, we screened for hyper-haemolytic colonies from the liver homogenates of infected animals on blood agar plates. Two hyper-haemolytic colonies were isolated and genome sequencing identified a single nucleotide polymorphism (SNP) common to both strains and absent from the ΔgshF parental strain. The SNP encoded a PrfA G145S mutation, which is the most commonly found spontaneous PrfA* allele14, so called because of its structural similarity to well-characterized Crp* mutants that are constitutively active in the absence of cofactor15. The PrfA G145S allele rescued ActA expression and virulence of ΔgshF, confirming the function of this mutation identified by our in vivo suppressor analysis (Fig. 3a–c). This was not specific to actA, as transcript levels of two other PrfA-dependent genes were also restored by the PrfA G145S mutation (Extended Data Fig. 3). Furthermore, two other previously identified PrfA* alleles16 also rescued the plaque defect of ΔgshF (Fig. 3d), indicating that constitutively activating PrfA completely bypassed the requirement for glutathione during infection. Importantly, these data highlighted that ΔgshF was not attenuated during infection due to a general loss of fitness, but rather, due to a dysregulation of virulence genes.
Figure 3. PrfA* bypasses the requirement for glutathione during infection.
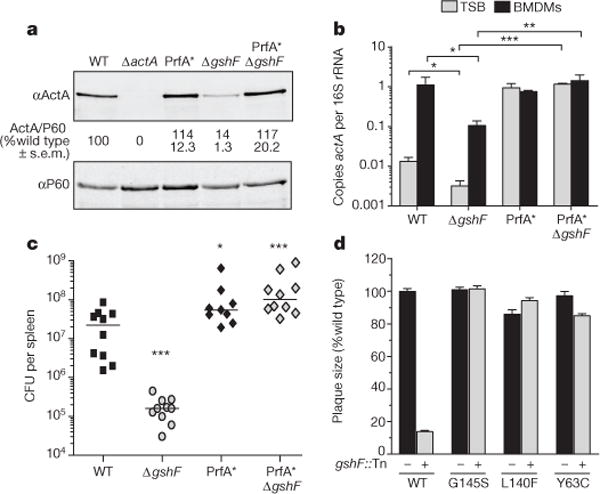
a, Representative immunoblot of infected BMDMs. Quantification is as described in Fig. 2. b, Quantitative reverse transcription polymerase chain reaction (RT–PCR) of actA transcript abundance. Mean ± s.e.m. for three independent experiments is shown. c, Mice were infected as described in Fig. 2. Data area combination of two independent experiments, n = 10 per strain. The median of each group is represented as a horizontal line. d, Plaque size. Mean ± s.e.m. for three independent experiments is shown. In all panels asterisks denote a significant difference compared to wild type, unless otherwise indicated, as determined by Student’s t-test; *P < 0.05, **P < 0.01, ***P<0.001.
PrfA binds glutathione allosterically
In addition to its role in maintaining redox homeostasis, glutathione can be covalently bonded to protein thiols as a post-translational modification, a process referred to as S-glutathionylation17. To determine whether glutathionylation of PrfA is required for its activation, we engineered a PrfA protein in which all four cysteine residues were mutated to alanine (referred to as PrfA(C38A/C144A/C205A/C229A or PrfA C/A)4). Recombinant PrfA(C/A)4 bound DNA with an affinity similar to wild-type PrfA in vitro (Table 1 and Extended Data Fig. 4), establishing that these mutations do not disturb the overall structural integrity of the protein. However, the cysteine residues were found to contribute to DNA binding, as demonstrated by the 25-fold lower affinity of oxidized wild-type PrfA as compared to reduced PrfA (Table 1). Although PrfA(C/A)4 bound DNA in vitro, it was less abundant than wild type when expressed from the native locus on the chromosome of L. monocytogenes (Extended Data Fig. 5). Since PrfA is auto-regulated18, these data suggested that PrfA(C/A)4 was less active in vivo. Indeed, the PrfA(C/A)4 strain synthesized less ActA than the wild-type strain during BMDM infection (Fig. 4a) and was 30-fold less virulent in mice (Fig. 4b). Together, these results suggested that the cysteine residues of PrfA were dispensable for DNA binding in vitro—as the mutant lacking all cysteine residues (PrfA(C/A)4) bound DNA with an affinity similar that of the wild type (Table 1)—but were required for activity in vivo. If glutathionylation of PrfA was important for its activity, then deleting gshF in the PrfA(C/A)4 background would have no effect. Remarkably, combining the PrfA(C/A)4 and gshF::Tn mutations resulted in a strain that was defective for intracellular growth (Extended Data Fig. 6) and completely avirulent in mice (>4-log attenuation, Fig. 4b).
Table 1.
DNA-binding and glutathione-binding affinities of PrfA
| DNA-binding affinity (Kd ± s.e.m.) | ||
|---|---|---|
|
| ||
| Phly (nM) | PactA (nM) | |
| Wild type (oxidized) | 888.5 ± 140.3 | ND |
| Wild type (reduced) | 34.2 ± 4.9 | 96.4 ± 7.3 |
| PrfA(C/A)4 | 32.8 ± 5.5 | 124.9 ± 26.3 |
| PrfA* | 40.8 ± 3.3 | 45.4 ± 3.2 |
| Glutathione-binding affinity (Kd ± s.e.m.) | ||
|---|---|---|
|
| ||
| GSH (mM) | GSSG (mM) | |
| Wild type | 4.37 ± 1.2 | NBD |
| PrfA(C/A)4 | 4.74 ± 1.5 | NBD |
DNA-binding affinity for the hly promoter (Phly) and the actA promoter (PactA), as measured by fluorescence anisotropy, and glutathione-binding affinity, as measured by bio-layer interferometry. The affinity of oxidized PrfA to PactA was not determined (ND). DNA-binding affinities of PrfA(C/A)4 and PrfA* were unaffected by oxidation. For oxidized glutathione (GSSG) no measurable binding was detected (NBD).
Figure 4. Glutathione-dependent PrfA activation is mediated by allosteric binding, not glutathionylation.
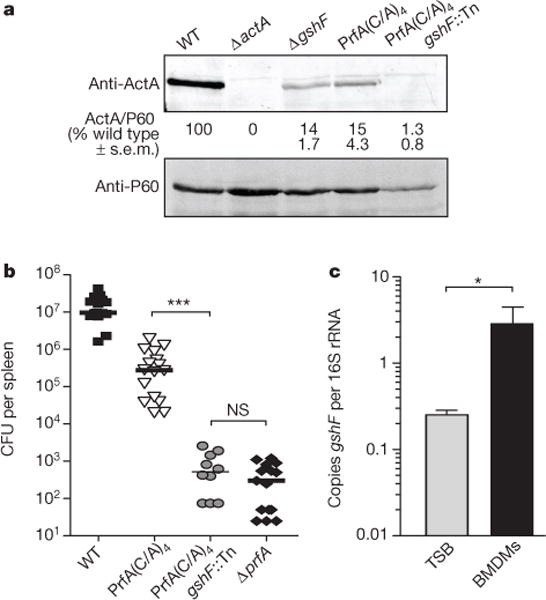
a, Representative immunoblot of infected BMDMs. Quantification is as described in Fig. 2. b, Mice were infected as described in Fig. 2. Data are a combination of at least two independent experiments, n = 10 or 16 per strain. The median of each group is represented as a horizontal line. All strains were significantly different from wild type (P < 0.001). c, Quantitative RT–PCR of gshF transcript abundance. Mean ± s.e.m. for three independent experiments are shown. In all panels asterisks denote a significant difference compared to wild type, unless otherwise indicated, as determined by Student’s t-test; *P < 0.05, ***P < 0.001; NS denotes P < 0.05.
Although we cannot definitively exclude a role for glutathionylation of PrfA in vivo, covalent modification of the protein thiols was not sufficient for PrfA activation, as the PrfA(C/A)4 mutant was only modestly attenuated during infection (Fig. 4b). Therefore, we hypothesized that glutathione may be allosterically binding PrfA, analogous to the interaction of cAMP binding to Crp19. The binding affinity of glutathione for PrfA was measured via bio-layer interferometry. A direct and specific interaction with reduced glutathione (GSH) was detected for both recombinant wild-type and PrfA(C/A)4 with binding affinities of 4.4 ± 1.2 and 4.7 ± 1.5 mM, respectively, whereas nomeasurable interaction was found between either protein and oxidized glutathione (GSSG, Table 1). Although the affinity for reduced glutathione appears to be relatively low, it is well within biologically relevant concentrations of this LMW thiol, as both prokaryotes and eukaryotes have intracellular concentrations of 0.1–10 mM glutathione8,11. This binding affinity would also allow PrfA to be sensitive to varying concentrations of glutathione, rather than beinga simple ON–OFF switch. In support of the hypothesis that glutathione may activate PrfA in vivo, gshF was transcriptionally upregulated tenfold during infection (Fig. 4c). These data demonstrate that reduced glutathione non-covalently binds PrfA and support the model that glutathione is the activating cofactor for PrfA.
Discussion
The results of this study clearly demonstrate that both bacterial and host-synthesized glutathione contribute to expression of L. monocytogenes virulence factors via allosteric binding of the master virulence regulator PrfA. Unlike Crp, PrfA does not require allosteric activation to bind DNA in vitro (Table 1). Indeed, the DNA-binding affinity of PrfA was unchanged in the presence of glutathione (data not shown). In this regard, PrfA is similar to the phylogenetically more closely related Crp family member NtcA from cyanobacteria, which also binds DNA in the absence of its cofactor20,21. Together, our results suggest a model whereby PrfA activation is a two-step process requiring reduced protein thiols for initial DNA binding and allosteric binding of glutathione to PrfA for transcriptional activation (see model in Extended Data Fig. 7). Indeed, eliminating both of these steps, as in the PrfA(C/A)4 gshF::Tn mutant, results in a strain that is as attenuated as a ΔprfA mutant (Fig. 4b).
Glutathione is present in the cytosol of all host cells so it is perhaps not surprising that intracellular pathogens import it, as is the case with Francisella tularensis22. What is surprising is that L. monocytogenes imports glutathione from the host and also synthesizes it. The results of this study suggest that L. monocytogenes uses glutathione concentration to regulate its biphasic lifestyle and the switch from saprophyte to pathogen. This may be a reflection of the broad host range of this pathogen and the fact that glutathione is ubiquitous in host cells, making it a reliable signal of the L. monocytogenes cytosolic niche. Perhaps other LMW thiols, such as coenzyme A, mycothiol and bacillithiol have similar activating roles in virulence gene expression in other pathogens.
METHODS
Bacterial strains and cell culture
The L. monocytogenes strains used were all in the 10403S background (Extended Data Table 1). Bacteria were cultured in brain heart infusion (BHI) or tryptic soy broth (TSB), which contains less than 0.5 μM glutathione9. All media were from Becton Dickinson (New Jersey). For broth growth curves, overnight cultures were diluted 1:100 and optical density at 600 nm (OD600) was measured at each time point using a spectrophotometer. gshF (lmo2770) was deleted by allelic exchange using the temperature-sensitive plasmid pKSV7 (refs 23, 24). The ΔgshF complemented strain was generated by inserting a C-terminal 6×His-tagged copy of gshF with its native promoter into the integration vector pPL2 (ref. 25).
Murine L2 fibroblasts were passaged in Dulbecco modified Eagle medium with high glucose (DMEM, Gibco/Invitrogen) supplemented with 1% sodium pyruvate, 1% L-glutamine, and 10% fetal bovine serum (FBS, GemCell) at37°C with 5% CO2. Bone-marrow-derived macrophages (BMDMs) were cultured in DMEM supplemented with 1% sodium pyruvate, 1% L-glutamine, 20% FBS and 10% 3T3-MCSF supernatant at 37°C with 5% CO2.
Transposon library generation and genetic selection
A transposon library was generated in the ‘suicide’ strain using himar1 mariner transposon mutagenesis, as previously described13. BMDMs were then infected with this library of transposon mutants. Cells were collected at various time points after infection, lysed, and surviving bacteria were plated on BHI agar. Individual colonies were then isolated and used to infect BMDMs to confirm the phenotype. To identify the transposon insertion site, colony PCR was performed using primers listed in Extended Data Table 1. A large percentage of the transposon insertion sites were found in the actA promoter, cre, and each loxP site. The fact that we identified multiple transposon insertions in each loxP site, which are less than 40 nucleotides, indicates that the genetic selection approached saturation.
We next screened these mutants by plaque assay and mutants with a plaque size, <90% of wild type were included. Finally, intracellular growth curves were performed to ensure that the mutants had a defect only in actA expression and not in intracellular growth. Greater than 16 independent insertions were identified in lmo2770 (gshF), making it by far the most over-represented hit in the selection.
Intracellular growth curves
BMDMs were harvested as previously described26 and 3 × 106 cells were plated in 60 mm non-TC-treated Petri dishes. Cells were infected with a multiplicity of infection (MOI) of 0.1 and growth curves were performed as described previously27.
Quantitative RT–PCR of bacterial transcripts
For transcript analysis in broth, bacteria were grown overnight in TSB and subcultured 1:100 into 25 ml TSB. Bacteria were harvested at an OD600 = 1.0. For transcript analysis during infection, BMDMs were platedata density of 3 × 107 cellsin150mm TC-treated dishes and infected with an MOI of 10. One hour post-infection the cells were washed and media containing gentamicin (50 μg ml−1) was added. Four hours post-infection the cells were washed and lysed in 0.1% NP-40 containing RNAprotect Bacteria Reagent (Qiagen). Bacteria were harvested by centrifugation.
After harvesting bacteria from either broth or BMDMs they were lysed in phenol: chloroform containing 1% SDS by vortexing with 0.1 mm diameter silica/zirconium beads (BioSpec Products Inc.). Nucleic acids were precipitated from the aqueous fraction overnight at −80°C in ethanol containing 150 mM sodium acetate (pH 5.2). Precipitated nucleic acids were washed with ethanol and treated with TURBO DNase per manufacturer’s specifications (Life Technologies Corporation). RNA was again precipitated overnight and then washed in ethanol. RT–PCR was performed with iScript Reverse Transcriptase (Bio-Rad) and quantitative PCR (qPCR) of resulting cDNA was performed with KAPA SYBR Fast (Kapa Biosystems). Primers used for qPCR are listed in Extended Data Table 1.
Plaque assay
Plaque assays in L2 murine fibroblasts were performed as previously described28. Briefly, bacterial cultures were grown overnight at 30°C, then washed and diluted 1:10 in sterile PBS. Six-well dishes containing 1.2 × 106 L2 cells per well were infected with L. monocytogenes for 1 h, then washed and overlaid with 3 ml of media containing 0.7% agarose and gentamicin (10 μg ml−1) to prevent extracellular growth. After 3 days at 37°C, an overlay containing gentamicin and neutral red dye (Sigma) was added and stained overnight. The plates were then scanned and analysed with ImageJ software29.
Immunoblots of infected BMDMs
BMDMs were plated in 12-well dishes at a density of 106 cells per well and infected with an MOI of 10. One hour postinfection the cells were washed and media containing gentamicin (50 μg ml−1) was added. Where indicated, 2 mM buthionine sulfoximine (Santa Cruz Biotechnology) was added to cells 16 h before infection and included throughout the infection. Four hours post-infection the cells were washed and harvested in LDS buffer containing 5% BME. The samples were then boiled and separated by 10% SDS–PAGE. A rabbit polyclonal antibody against the N terminus of ActA30 and a mouse monoclonal antibody against P60 (Adipogen) were each used at a dilution of 1:5,000. P60 is a constitutively expressed bacterial protein31 used as a loading control.
Virulence experiments
Six-to-eight-week-old female CD-1 mice (The Jackson Laboratory) were infected intravenously with 1 × 105 colony-forming units (CFU). Forty-eight hours post-infection the mice were euthanized and spleens and livers were harvested, homogenized and plated for enumeration of bacterial burdens. Sample size was chosen based on standards within the field32,33. No statistical methods were used to predetermine sample size. Samples were not blinded or randomized. All statistical tests allowed for unequal variance between groups, that is, two-tailed heteroscedastic t-test. All animal work was done in accordance with university regulations. Protocols were reviewed and approved by the Animal Care and Use Committee at the University of California, Berkeley (MAUP# R235-0813B).
In vivo suppressor analysis
Female CD-1 mice were infected intravenously with 1 × 107 CFU of ΔgshF for 72 h and the livers were harvested, homogenized and inoculated into broth. New mice were then infected with 1 × 106 CFU of the bacteria from the liver homogenates. Seventy-two hours post-infection the livers were harvested, homogenized and plated on blood agar plates. Two hyper-haemolytic colonies were observed and were chosen for further analysis.
Genome sequencing
L. monocytogenes genomic DNA was extracted (MasterPure Kit, Epicentre) and sequenced by Illumina 50SR (library preparation and sequencing performed by UC Berkeley QB3 Genomic Sequencing Laboratory). Sequencing data were aligned to the 10403S reference genome and SNP/InDel/structural variation was determined (CLC Genomics Workbench, CLC bio) for the ΔgshF parent strain and the two hyper-haemolytic suppressor mutants.
Protein purification and binding analyses
Recombinant PrfA was purified from Escherichia coli BL21(DE3) as previously described34. Glutathione binding to the wild-type or PrfA(C/A)4 protein was measured by bio-layer interferometry on an Octet RED 384 instrument (Pall ForteBio). The buffer used was PBS, pH7.3 containing 2 mM tris(2-carboxyethyl)phosphine (TCEP) when appropriate. Samples or buffer were dispensed into 384-well microtitre plates at a volume of 100 μl per well. Operating temperature was maintained at 26°C with 1,000 rpm rotary agitation. Ni-NTA biosensor tips (Pall FortèBio) were pre-soaked for 10 min with buffer to establish a baseline before protein immobilization. 6×His-taggedproteins diluted in PBS, pH 7.3 were immobilized onto the biosensors for 8 min at a concentration of 35 μg ml−1. The immobilization level attained was 7–8 nm. Binding association of the glutathione with biosensor tips was monitored for 30 s, and subsequent disassociation in buffer was monitored for 30 s. Glutathione was tested at concentrations of 0.5, 1, 1.5, 2, 3, 4, 5 mM. Reduced glutathione (GSH) was diluted in buffer containing TCEP, while oxidized glutathione (GSSG) was diluted in PBS only. To control for background, association and dissociation of GSH/GSSG was measured with biosensor tips loaded with buffer only and biosensor tips loaded with protein were tested for binding in buffer with or without TCEP. The apparent affinities of glutathione and PrfA were calculated from equilibrium measurements and, when appropriate, global fits of the kon and koff values, yielding similar values. Data are mean and s.e.m. of experiments from four independent protein preparations.
Fluorescence polarization
A fluorescence-polarization-based DNA-binding assay was used to determine the affinities of PrfA, PrfA* and the PrfA(C/A)4 mutant for the Phly and PactA promoters. The sequences of the top strands of Phly and PactA used in this study are in Extended Data Table 1. The oligodeoxynucleotides were purchased from IDT (Coralville, Iowa) with a fluorescein label covalently attached to the 5′ end. DNA binding was measured in PBS buffer (11.8 mM Na+/K+ phosphate, 2.7 mM KCl, 137 mM NaCl) at 25°C using 5 nM fluoresceinated target dsDNA, and 1 μg poly(dI-dC) as a nonspecific DNA competitor. In some experiments 1 mM TCEP was included to maintain a reducing environment. PrfA was titrated into the DNA until saturation as denoted by no further change in the millipolarization (mP = units of polarization × 10−3). The fluoresceinated DNA was excited at 490 nm and its parallel and perpendicular emission intensities measured at 530 nm and converted to units of mP using a Beacon 2000 Variable Temperature Fluorescence Polarization System. Data were plotted and analysed with the following equation: mP = {(mPbound − mPfree)[protein]/Kd + [protein]} + mPfree, where mP is the millipolarization measured at a given protein concentration, mPfree is the initial millipolarization of free fluorescein-labelled DNA, mPbound is the maximum millipolarization of specifically bound DNA, and [protein] is the protein concentration. The generated hyperbolic curves are fit by nonlinear least-squares regression analysis, assuming a bimolecular model such that the Kd values represent the protein concentration at half-maximal ligand binding and plotted by using the graphing program, Kaleidograph. The Kd values are expressed in terms of PrfA dimer binding.
Extended Data
Extended Data Figure 1. Listeria monocytogenes ΔgshF is sensitive to hydrogen peroxide.
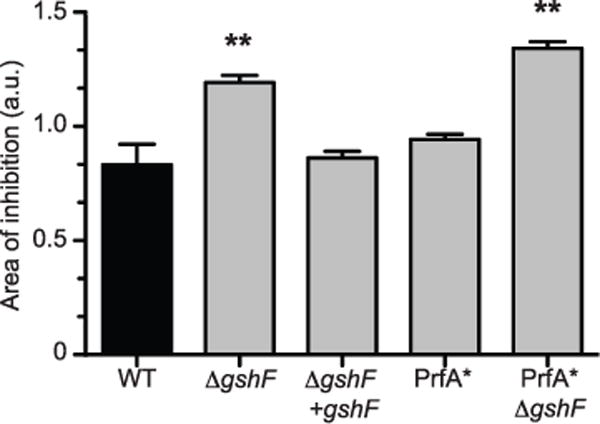
Bacteria were grown overnight in TSB and then inoculated into top agar and spread on tryptic soy agar plates. Sterile disks soaked in 10 μl of 15% H2O2 (Thermo Fisher Scientific) were placed on the agar and incubated overnight. Plates were then scanned and the area of inhibition was measured (in arbitrary units) using ImageJ software (http://rsbweb.nih.gov/ij). The mean ± s.e.m. of four independent experiments is shown. P values were calculated using Student’s t-test; **P < 0.01. a.u., arbitrary units.
Extended Data Figure 2. BSO does not affect L. monocytogenes growth.
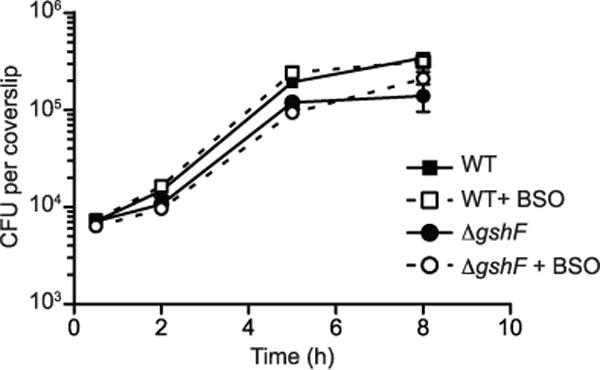
BMDM growth curve in which cells were untreated or treated with 2 mM BSO for 16 h before infection and throughout the infection. The mean ± s.e.m. of three independent experiments is shown.
Extended Data Figure 3. The effect of ΔgshF is not specific to actA regulation.
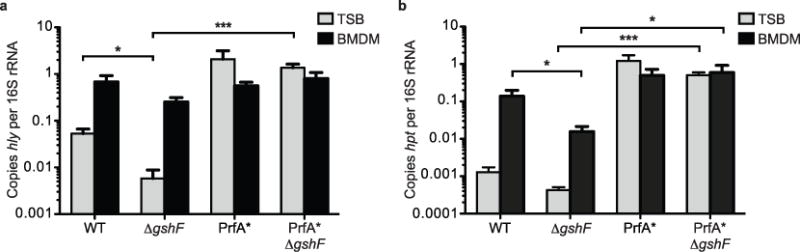
Quantitative RT–PCR of hly (a) or hpt (b) transcript levels. Bacteria were harvested from TSB at mid-log (grey bars) or 4 h post-infection of BMDMs (black bars). Mean ± s.e.m. of three independent experiments is shown. P values were calculated using Student’s t-test.*P < 0.05; ***P < 0.001.
Extended Data Figure 4. Fluorescence polarization binding isotherms.
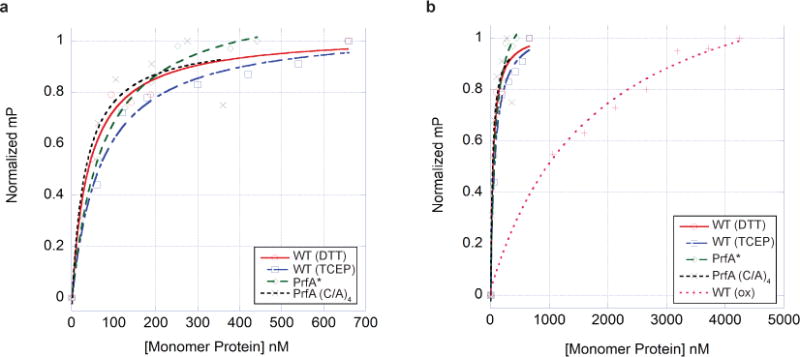
a, Representative binding isotherms of wild-type PrfA plus DTT (circles), wild-type PrfA plus TCEP (squares), PrfA* (diamonds), and PrfA(C/A)4 (crosses), to the PrfA box of Phly. b, Representative binding isotherms of wild-type PrfA plus DTT (circles), wild-type PrfA plus TCEP (squares), PrfA* (diamonds), PrfA(C/A)4 (crosses), and oxidized wild-type PrfA (plus symbols), to the PrfA box of Phly. This plot underscores the very poor binding of oxidized wild-type PrfA to the PrfA box. In both panels the units of millipolarization (mP, y axis) have been normalized to allow the presentation of all binding isotherms on one graph. The protein concentration is shown in terms of protomer on the x axis.
Extended Data Figure 5. PrfA(C/A)4 expression in L. monocytogenes grown in broth.
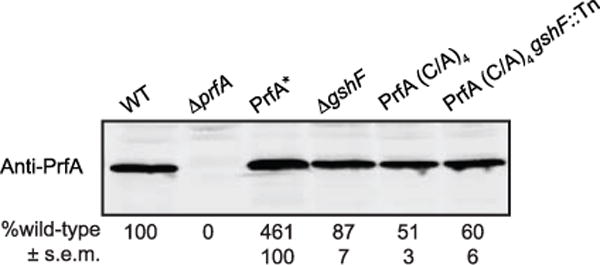
Immunoblot of PrfA in L. monocytogenes lysates harvested at early exponential phase in BHI. Mean ± s.e.m. of four independent experiments is shown.
Extended Data Figure 6. The PrfA(C/A)4 gshF::Tn mutant exhibits a significant intracellular growth defect.
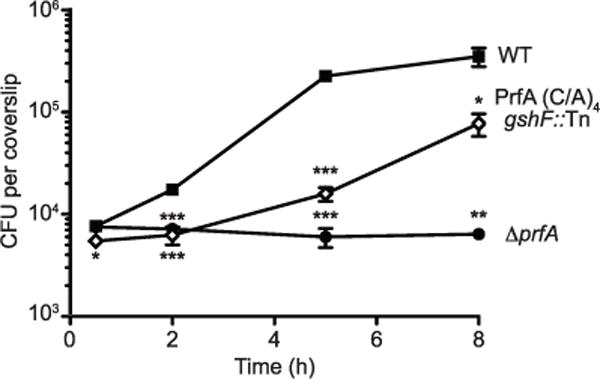
The mean ± s.e.m. of four independent experiments is shown. P values were calculated using Student’s t-test.; *P < 0.05; **P < 0.01; ***P < 0.001.
Extended Data Figure 7. Model of glutathione-dependent PrfA activation.
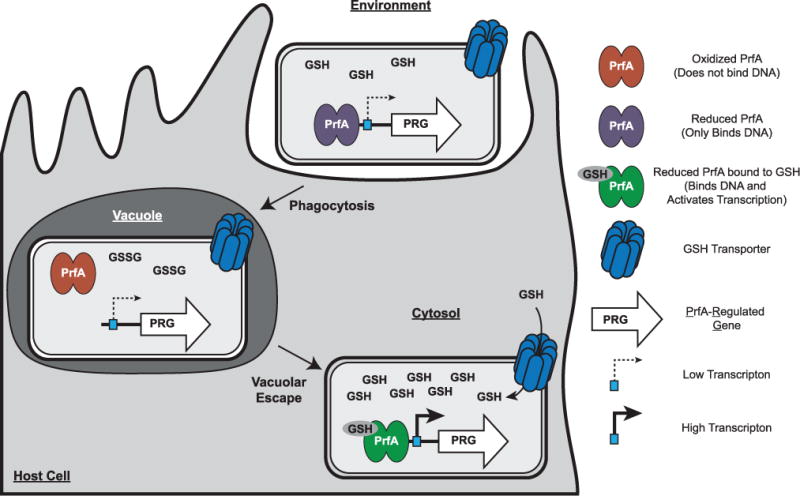
The process of infection or intercellular spread requires that L. monocytogenes inhabit an oxidizing vacuole, which may contain both reactive oxygen and nitrogen species. Upon oxidation, glutathione dimerizes to GSSG, which we have demonstrated does not bind PrfA. In addition, PrfA thiols may be reversibly oxidized, temporarily inactivating the protein by inhibiting DNA binding and leading to a downregulation of PrfA-regulated genes (PRG). L. monocytogenes could then enter the host cytosol, as PrfA activation is dispensable for vacuolar escape in vivo. The host cytosol is a highly reducing environment and upon entry into this compartment, all thiols are expected to be in the reduced form. In the absence of glutathione, it is likely that coenzyme A maintains redox homeostasis in the bacterium, as it is the most abundant LMW thiol in L. monocytogenes. Reduced glutathione could then bind PrfA and activate transcription of PRG. This two-step activation requirement may explain why the mechanism of PrfA activation has been a mystery for over two decades; the redox changes occurring during transit through a vacuole followed by replication in the highly reducing cytosol have yet to be recapitulated in vitro.
Extended Data Table 1.
Strains and primers
| Strain | Escherichia coli | Reference |
|---|---|---|
| NF-E1280 | BL21(DE3) pET100.prfA | 16 |
| NF-E1281 | BL21(DE3) pETIOO.prfA* (G145S) | 16 |
| DP-E6185 | Rosetta pET100.prfA Quad | this work |
|
| ||
| Strain | Listeria monocytogenes | Reference |
|
| ||
| 10403S | wt | 35 |
| DP-L3078 | ΔactA | 36 |
| DP-L4317 | ΔprfA | 37 |
| NF-L1177 | PrfA* (G145S) | 16 |
| BH-3410 | suicide | this work |
| DP-L6187 | suicide gshF::Tn | this work |
| DP-L6188 | ΔgshF | this work |
| DP-L6189 | ΔgshF + gshF | this work |
| DP-L6190 | PrfA*(G145S) ΔgshF | this work |
| DP-L6191 | PrfA* (G145S) gshF::Tn | this work |
| NF-L1166 | PrfA* (L140F) | 16 |
| DP-L6192 | PrfA*(L140F)gsnF::Tn | this work |
| NF-L1214 | PrfA* (Y63C) | 16 |
| DP-L6194 | PrfA* (Y63C) gshF::Tn | this work |
| DP-L6195 | PrfA (C/A)4 | this work |
| DP-L6196 | PrfA (C/A)4 gshF::Tn | this work |
|
| ||
| Primer | Oligonucleotide | Amplicon |
|
| ||
| MLR#123 | CGACATAATATTTGCAGCGAC | actA for qPCR |
| MLR#124 | TGCTTTCAACATTGCTATTAGG | |
| MLR#133 | GACCCTAATCTCCGGAAGC | gshF for qPCR |
| MLR#134 | TACAGAGTCAATCGAGTCCG | |
| MLR#121 | GCGCAACAAACTGAAGCAAAG | hly for qPCR |
| MLR#122 | CATTTGTCACTGCATCTCCG | |
| MLR#125 | CTAACGGTCTATCTTCTAAGG | hpt for qPCR |
| MLR#126 | CAATAATAATTGATATAATAGCGG | |
| MLR#150 | ACCCTTGATTTTAGTTGCCAG | 16S rRNA for qPCR |
| MLR#151 | TGTGTAGCCCAGGTCATAAG | |
| MLR#102 | GCTTCCAAGGAGCTAAAGAGGTCCCTAGCGCC | |
| MLR#103 | CGGGGAATTTGTATCGATAAGGAATAGATTTAAAAATTTCGCTGTTATTTTG | Tn PCR Round#1 |
| MLR#104 | GGCCACGCGTCGACTAGTACNNNNNNNNNNCTTCT | Tn PCR Round#2 |
| MLR#105 | GGCCACGCGTCGACTAGTAC | |
| MLR#106 | ACAATAAGGATAAATTTGAATACTAGTCTCGAGTGGGG | Tn Sequencing |
| KLH#1 | TGAGGCATTAACATTTGTTAACGACGAT | Phly for DNA-binding assays |
| KLH#2 | AACTGATTAACAAATGTTAGAGAAAACT | PactA for DNA-binding assays |
Acknowledgments
We thank N. Freitag for providing strains and P. Hwang (UCSF Biosensor Core Facility) for technical support and advice regarding bio-layer interferometry. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation grants S10RR029668 and S10RR027303 and the UCSF Funding Shared Equipment Award. This work was supported by National Institutes of Health grants 1PO1 AI63302 and 1R01 AI27655 to D.A.P.; M.L.R. is supported by F32AI104247; A.T.W. is supported by the NSF GRFP DGE 1106400; K.L.H. is supported by F32GM008487.
Footnotes
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Author Contributions M.L.R., A.T.W., K.L.H. and S.M.J. performed the experiments; P.L. engineered the ‘suicide’ strain; M.L.R., A.T.W., R.G.B. and D.A.P. designed the study; M.L.R. and D.A.P. wrote the paper. All authors discussed the results and commented on the manuscript.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare competing financial interests: details are available in the online version of the paper. Readers are welcome to comment on the online version of the paper.
References
- 1.Xayarath B, Freitag NE. Optimizing the balance between host and environmental survival skills: lessons learned from Listeria monocytogenes. Future Microbiol. 2012;7:839–852. doi: 10.2217/fmb.12.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freitag NE, Port GC, Miner MD. Listeria monocytogenes—from saprophyte to intracellular pathogen. Nature Rev Microbiol. 2009;7:623–628. doi: 10.1038/nrmicro2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakraborty T, et al. Coordinate regulation of virulence genes in Listeria monocytogenes requires the product of the prfA gene. J Bacteriol. 1992;174:568–574. doi: 10.1128/jb.174.2.568-574.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de las Heras A, Cain RJ, Bielecka MK, Vázquez-Boland JA. Regulation of Listeria virulence: PrfA master and commander. Curr Opin Microbiol. 2011;14:118–127. doi: 10.1016/j.mib.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Moors MA, Levitt B, Youngman P, Portnoy DA. Expression of listeriolysin O and ActA by intracellular and extracellular Listeria monocytogenes. Infect Immun. 1999;67:131–139. doi: 10.1128/iai.67.1.131-139.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shetron-Rama LM, Marquis H, Bouwer HGA, Freitag NE. Intracellular induction of Listeria monocytogenes actA expression. Infect Immun. 2002;70:1087–1096. doi: 10.1128/IAI.70.3.1087-1096.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gopal S, et al. A multidomain fusion protein in Listeria monocytogenes catalyzes the two primary activities for glutathione biosynthesis. J Bacteriol. 2005;187:3839–3847. doi: 10.1128/JB.187.11.3839-3847.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masip L, Veeravalli K, Georgiou G. The many faces of glutathione in bacteria. Antioxid Redox Signal. 2006;8:753–762. doi: 10.1089/ars.2006.8.753. [DOI] [PubMed] [Google Scholar]
- 9.Newton GL, et al. Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J Bacteriol. 1996;178:1990–1995. doi: 10.1128/jb.178.7.1990-1995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newton GL, et al. Bacillithiol is an antioxidant thiol produced in Bacilli. Nature Chem Biol. 2009;5:625–627. doi: 10.1038/nchembio.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 12.Rouzer CA, Scott WA, Griffith OW, Hamill AL, Cohn ZA. Depletion of glutathione selectively inhibits synthesis of leukotriene C by macrophages. Proc Natl Acad Sci USA. 1981;78:2532–2536. doi: 10.1073/pnas.78.4.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zemansky J, et al. Development of a mariner-based transposon and identification of Listeria monocytogenes determinants, including the peptidyl-prolyl isomerase PrsA2, that contribute to its hemolytic phenotype. J Bacteriol. 2009;191:3950–3964. doi: 10.1128/JB.00016-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ripio MT, Domínguez-Bernal G, Lara M, Suárez M, Vázquez-Boland JAA. Gly145Ser substitution in the transcriptional activator PrfA causes constitutive overexpression of virulence factors in Listeria monocytogenes. J Bacteriol. 1997;179:1533–1540. doi: 10.1128/jb.179.5.1533-1540.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eiting M, Hagelüken G, Schubert WD, Heinz DW. The mutation G145S in PrfA, a key virulence regulator of Listeria monocytogenes, increases DNA-binding affinity by stabilizing the HTH motif. Mol Microbiol. 2005;56:433–446. doi: 10.1111/j.1365-2958.2005.04561.x. [DOI] [PubMed] [Google Scholar]
- 16.Miner MD, Port GC, Freitag NE. Functional impact of mutational activation on the Listeria monocytogenes central virulence regulator PrfA. Microbiology. 2008;154:3579–3589. doi: 10.1099/mic.0.2008/021063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Mengaud J, et al. Pleiotropic control of Listeria monocytogenes virulence factors by a gene that is autoregulated. Mol Microbiol. 1991;5:2273–2283. doi: 10.1111/j.1365-2958.1991.tb02158.x. [DOI] [PubMed] [Google Scholar]
- 19.Kolb A, Busby S, Buc H, Garges S, Adhya S. Transcriptional regulation by cAMP and its receptor protein. Annu Rev Biochem. 1993;62:749–797. doi: 10.1146/annurev.bi.62.070193.003533. [DOI] [PubMed] [Google Scholar]
- 20.Valladares A, Flores E, Herrero A. Transcription activation by NtcA and 2-oxoglutarate of three genes involved in heterocyst differentiation in the cyanobacterium Anabaena sp. strain PCC 7120. J Bacteriol. 2008;190:6126–6133. doi: 10.1128/JB.00787-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Körner H, Sofia HJ, Zumft WG. Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol Rev. 2003;27:559–592. doi: 10.1016/S0168-6445(03)00066-4. [DOI] [PubMed] [Google Scholar]
- 22.Alkhuder K, Meibom KL, Dubail I, Dupuis M, Charbit A. Glutathione provides a source of cysteine essential for intracellular multiplication of Francisella tularensis. PLoS Pathog. 2009;5:e1000284. doi: 10.1371/journal.ppat.1000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith K, Youngman P. Use of a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spoIIM gene. Biochimie. 1992;74:705–711. doi: 10.1016/0300-9084(92)90143-3. [DOI] [PubMed] [Google Scholar]
- 24.Camilli A, Tilney LG, Portnoy DA. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol Microbiol. 1993;8:143–157. doi: 10.1111/j.1365-2958.1993.tb01211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lauer P, Chow MYN, Loessner MJ, Portnoy DA, Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol. 2002;184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sauer JD, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. 2011;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Portnoy DA, Jacks PS, Hinrichs DJ. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med. 1988;167:1459–1471. doi: 10.1084/jem.167.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun AN, Camilli A, Portnoy DA. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect Immun. 1990;58:3770–3778. doi: 10.1128/iai.58.11.3770-3778.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lauer P, et al. Constitutive activation of the PrfA regulon enhances the potency of vaccines based on live-attenuated and killed but metabolically active Listeria monocytogenes strains. Infect Immun. 2008;76:3742–3753. doi: 10.1128/IAI.00390-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Köhler S, Bubert A, Vogel M, Goebel W. Expression of the iap gene coding for protein p60 of Listeria monocytogenes is controlled on the posttranscriptional level. J Bacteriol. 1991;173:4668–4674. doi: 10.1128/jb.173.15.4668-4674.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sauer JD, et al. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe. 2010;7:412–419. doi: 10.1016/j.chom.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melton-Witt JA, McKay SL, Portnoy DA. Development of a single-gene, signature-tag-based approach in combination with alanine mutagenesis to identify listeriolysin O residues critical for the in vivo survival of Listeria monocytogenes. Infect Immun. 2012;80:2221–2230. doi: 10.1128/IAI.06196-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Böckmann R, Dickneite C, Middendorf B, Goebel W, Sokolovic Z. Specific binding of the Listeria monocytogenes transcriptional regulator PrfA to target sequences requires additional factor(s) and is influenced by iron. Mol Microbiol. 1996;22:643–653. doi: 10.1046/j.1365-2958.1996.d01-1722.x. [DOI] [PubMed] [Google Scholar]
- 35.Bishop DK, Hinrichs DJ. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J Immunol. 1987;139:2005–2009. [PubMed] [Google Scholar]
- 36.Skoble J, Portnoy DA, Welch MD. Three regions within ActA promote Arp2/3 complex-mediated actin nucleation and Listeria monocytogenes motility. J Cell Biol. 2000;150:527–538. doi: 10.1083/jcb.150.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng LW, Portnoy DA. Drosophila S2 cells: an alternative infection model for Listeria monocytogenes. Cell Microbiol. 2003;5:875–885. doi: 10.1046/j.1462-5822.2003.00327.x. [DOI] [PubMed] [Google Scholar]